

Abstract
Nicotine, the main psychoactive ingredient in tobacco, stimulates dopamine (DA) function, increasing DA neuronal activity and DA release. DA is involved in both motor control and in the rewarding and reinforcing effects of nicotine; however, the complete understanding of its molecular mechanisms is yet to be attained. Substantial evidence indicates that the reinforcing properties of drugs of abuse, including nicotine, can be affected by the nitric oxide (NO) system, which may act by modulating central dopaminergic function. In this study, using single cell recordings in vivo coupled with microiontophoresis and microdialysis in freely moving animals, the role of NO signaling on the hyperactivation elicited by nicotine of the nigrostriatal system was investigated in rats. Nicotine induced a dose‐dependent increase of the firing activity of the substantia nigra pars compacta (SNc) DA neurons and DA and 3,4‐dihydroxyphenylacetic acid (DOPAC) release in the striatum. Pharmacological manipulation of the NO system did not produce any change under basal condition in terms of neuronal discharge and DA release. In contrast, pretreatments with two NO synthase (NOS) inhibitors, N‐ω‐nitro‐l‐arginine methyl ester (l‐NAME) and 7‐nitroindazole (7‐NI) were both capable of blocking the nicotine‐induced increase of SNc DA neuron activity and DA striatal levels. The effects of nicotine in l‐NAME and 7‐NI‐pretreated rats were partially restored when rats were pretreated with the NO donor molsidomine. These results further support the evidence of an important role played by NO on modulation of dopaminergic function and drug addiction, thus revealing new pharmacological possibilities in the treatment of nicotine dependence and other DA dysfunctions.
Keywords: Corpus striatum, Dopamine, Extracellular recording, Microdialysis, Microiontophoresis, Nicotine, Nitric oxide, Substantia nigra pars compacta
Introduction
Nicotine, the major psychoactive agent present in tobacco, acts as a potent addictive drug both in humans and laboratory animals [1, 2, 3, 4]. A large body of evidence indicates that the locomotor activation and the reinforcing effects of nicotine may be related to its stimulatory effects on the mesolimbic and nigrostriatal dopaminergic functions [5, 6, 7]. Moreover, it is now well established that nicotine can increase in vivo dopamine (DA) outflow in the nucleus accumbens and the corpus striatum [8, 9, 10, 11, 12]. The stimulatory effect of nicotine on DA release most probably results from its ability to excite neuronal firing and increase bursting activity of DA neurons in the substantia nigra pars compacta (SNc) and the ventral tegmental area (VTA) [13, 14, 15, 16, 17, 18] and from its stimulatory action on DA terminals in the corpus striatum and the nucleus accumbens [19, 20]. Evidence indicates that the reinforcing properties of nicotine can be affected by several transmitter systems; among them the unorthodox nitric oxide (NO) neuromodulator seems to play a crucial role. In addition, a large body of evidence supports a direct DA/NO interaction under normal and pathological conditions such as Parkinson's disease (PD), schizophrenia, depression, and in drug addiction [21, 22, 23, 24, 25]. Hitherto, the role of NO in DA control is controversial [26]. On the contrary, evidence regarding the interrelationship between NO and drug of addiction such as nicotine is substantial and compelling. NO potentially contributes to nicotine central effects, such as reward, addiction, sensitization, and cognitive enhancement [25, 27], although the exact neuronal mechanism of this modulation is still unclear, especially in the nigrostriatal system.
To this end, in the present study the effect of pharmacological manipulation of the nitrergic system on nicotine‐induced nigrostriatal DA hyperactivity was evaluated by using single‐cell extracellular recordings coupled with microiontophoresis of chloral hydrate‐anesthetized rats and microdialysis in freely moving rats. To test the hypothesis that NO is crucial in nicotine effects, the influence of pretreatment with N‐ω‐nitro‐l‐arginine methyl ester (l‐NAME) and 7‐nitroindazole (7‐NI) on nicotine‐induced excitation of nigrostriatal DA function was evaluated. The reversion of the effect of the NO system inhibition with molsidomine (MOL, a NO donor) cotreatment on nicotine‐induced hyperactivity was also investigated.
Some of this work has been published previously in abstract form (at the IBAGS IX).
Materials and Methods
Male Sprague–Dawley rats, from Charles River Laboratories (Calco, Varese, Italy) were housed at appropriate environmental conditions (21 ± 2°C room temperature, 12‐h light/dark cycle, 40–60% humidity). Water and food was provided ad libitum. Procedures involving animals and their care were conducted in accordance with the institutional guidelines in compliance with national (D.L. no. 116, G.U., suppl. 40, 18 Febbraio 1992) and international laws and policies (EEC Council Directive 86/609, OJ L 358,1, Dec. 12, 1987; NIH Guide for the Care and Use of Laboratory Animals, NIH Publication N. 85–23, 1985 and Guidelines for the Use of Animals in Biomedical Research, Thromb. Haemost. 58, 1078–1084, 1987).
Electrophysiological Recording
Experiments were performed in chloral hydrate‐anesthetized rats (400 mg/kg, i.p.). Nigral DA cells were recorded extracellularly as described previously [28]. Briefly, extracellular recordings were performed using either single‐ or multibarrel micropipettes. The central barrel of microiontophoretic electrode, filled with 2% Pontamine Sky Blue dye in 2 M NaCl, was used for recording (in vitro resistance 4–7 MΩ) while one of the side barrels, filled with 2 M NaCl, was used for continuous automatic current balancing. The remaining barrels contained l‐NAME (50 mM, pH 6.5) and l‐arginine (l‐ARG; 50 mM, pH 5.5–6.0). DA neurons were identified by their location, waveform, firing rate, and pattern [29, 30].
Microdialysis
Microdialysis was performed according to Di Matteo et al. [12]. Experiments were conducted the day after the surgery, in awake, freely moving animals. A microdialysis probe (CMA/10, 3 mm 500 μm outer diameter, Carnegie Medicin, Stockholm, Sweden) was lowered through the guide cannula to reach a depth of 6 and 8 mm below the dura surface [29]. The rat was placed in a CMA/120 system for freely moving animals (Carnegie Medicin, Stockholm, Sweden), and every 20 min samples of perfusate were collected and immediately assayed by high performance liquid chromatography (HPLC) with electrochemical detection. Dialysate samples were analyzed by reversed‐phase HPLC coupled with electrochemical detection (see [12] for extra details).
Drug Administration Protocols
In the electrophysiological experiments, nicotine, apomorphine, l‐NAME, l‐ARG, and MOL were freshly diluted in physiological saline (0.9% NaCl). 7‐NI was dissolved in 200 μL of 10% DMSO, made up to almost required volume with 0.9% saline. Nicotine (25‐400 μg/kg) was administered i.v. (via a lateral tail vein) every 2 min in exponentially increasing doses.
7‐NI (50 mg/kg i.p.) and l‐NAME (50 mg/kg i.p.) were given 10–15 mins before nicotine or saline injections. In some rats, 7‐NI and l‐NAME were coadministrated with MOL before nicotine. In a number of instances, apomorphine (10–30 μg/kg i.v.) was given at the end of the experiment to confirm the dopaminergic identity of the neuron recorded. For the microiontophoresis applications, l‐NAME and l‐ARG were continuously ejected for 5 min with current at +40 nA.
In the microdialysis experiments, all pharmacological treatments were performed following the stabilization of DA levels in the perfusate. 7‐NI or l‐NAME were injected 20 min prior to nicotine administration. MOL was coadministrated with 7‐NI or l‐NAME. The effect of the vehicle of 7‐NI did not differ from saline in both electrophysiological and neurochemical experiments; therefore these data were pooled together and are subsequently referred to as the control vehicle.
Data Analysis
Electrophysiological data acquisition and analysis were accomplished using an integrated software package for electrophysiology (RISI; Symbolic Logic, Dallas, TX, USA). A total of 300–500 consecutive spikes were recorded for each neuron before and at the peak of drug effect. Burst firing, when present, was detected using an algorithm similar to that previously described [30]. The difference (Δ) between the percentage of spikes fired within bursts during the baseline period from the percentage of spikes fired within bursts after drug administration was used as a measure of drug‐induced changes in bursting. Inasmuch as burst firing values did not show a normal distribution, they were analyzed by the nonparametric Mann‐Whitney U test. Drug‐induced changes in neuronal firing rate were analyzed by one‐ or two‐way analysis of variance (ANOVA) with repeated measures followed by post hoc Tukey‐Kramer tests, where appropriate. Differences between basal DA neuronal activity in rats treated with nicotine were analyzed by Student's t‐test.
In dialysis experiments, DA and DOPAC contents in each sample were expressed as the percentage of the average baseline level calculated from three fractions collected before drug administration. Data corresponded to mean ± SEM values of the percentage obtained in each experimental group. Data were analyzed by one‐way or two‐way ANOVA with repeated measures followed by post hoc Tukey‐Kramer test, where appropriate. Differences between basal DA and DOPAC outflow in the different groups were analyzed by Student's t‐test.
All statistical analyses were performed with StatView version 5.0.1 (SAS Institute Inc., Cary, NC, USA).
Histology
Microscopic examination of the sections was carried out to verify that the electrode tip and microdialysis probe position.
Drugs
(–)‐Nicotine hydrogen tartrate salt, 7‐NI, MOL, l‐NAME, l‐ARG were purchased from Sigma (Milano, Italy).
Results
Effects of 7‐NI, MOL, l‐NAME, l‐ARG on the Firing Basal Properties and on Acute Nicotine‐Induced Excitation of DA Neurons in the SNc
In a first series of experiments the effects of two NOS inhibitors, l‐NAME (50 mg/kg, i.p.) and 7‐NI (50 mg/kg, i.p.), were evaluated for at least 30 min or more, at 5‐min intervals (Figure 1A, B)
Figure 1.
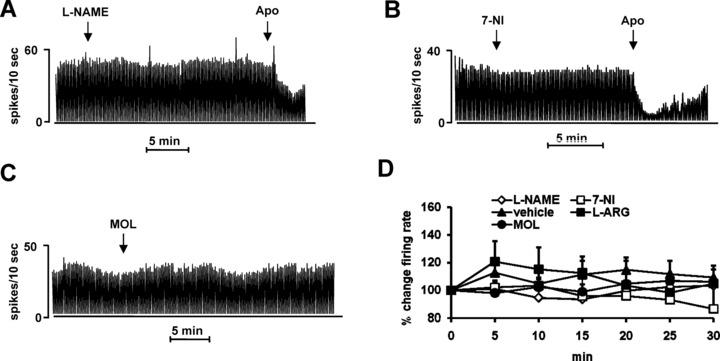
Effect of systemic and local manipulation of NO signaling on the firing rate of dopaminergic SNc neurons. Representative rate histograms showing the effects elicited by i.p. administration (at arrows) of l‐NAME (50 mg/kg) (A), 7‐NI (50 mg/kg) (B), MOL (50 mg/kg) (C). APO, apomorphine administration (10 μg/kg i.v., at arrow). (D) Cumulative dose–response curve showing the mean percentage change (± SEM) in firing rate after vehicle, 7‐NI (50 mg/kg), l‐NAME (50 mg/kg), MOL (50 mg/kg) and l‐ARG (50 mg/kg).
Systemic administration of l‐NAME (Figure 1A, D) did not cause any significant change in the basal firing rate and bursting activity (Table 1) of the SNc DA neurons recorded compared to vehicle effect (n = 10; P > 0.89). The administration of 7‐NI (Figure 1D) elicited a slight decrease of the firing rate that reached the peak at 30 min (–13.5 ± 15.7%, n = 6, P > 0.63). In two cells the pattern mode was strongly affected. A neuron showed a decrease of spikes fired in bursts at 20 min from the drug treatment (Δ=–21.3) and did not recover until the end of the recording. In another neuron, the Δ decrease (≈ –30) was observed 15 min after 7‐NI injection and was followed by a gradual return to the predrug value (at 30 min Δ=−0.4). Overall, the effect of 7‐NI in terms of firing rate (Figure 1D) and pattern (Table 1) was statistically not significant when compared to the vehicle group (n = 10).
Table 1.
Effects of nitric oxide drugs and nicotine on the firing pattern of SNc dopaminergic neurons
| Treatment | Δburst |
|---|---|
| Vehicle | 0.83 ± 1.61 |
| l‐NAME (i.p.) | −0.14 ± 2.9 |
| l‐NAME (iontophoresis) | −0.04 ± 1.6 |
| 7‐NI (i.p.) | −5.70 ± 3.1 |
| l‐ARG (i.p.) | −2.33 ± 6.5 |
| l‐ARG (iontophoresis) | −0.48 ± 1.6 |
| MOL (i.p.) | 1.62 ± 1.5 |
| Nicotine (i.v.) | 20.41 ± 7.51a |
| Nicotine +l‐NAME (i.v.+ i.p.) | 9.9 ± 7.01b |
| Nicotine + 7‐NI (i.v.+ i.p.) | 4.9 ± 2.6b |
| Nicotine +l‐NAME + MOL (i.v.+ i.p.) | 15.2 ± 6.68a |
| Nicotine +7‐NI + MOL (i.v.+ i.p.) | 9.46 ± 11.01b |
a P < 0.05 compared with vehicle group, b P < 0.05 compared to nicotine (Mann‐Whitney U test).
Accordingly, treatment with the NO precursor l‐ARG (50 mg/kg, i.p. n = 6, Figure 1D) and the NO releaser MOL (Figure 1C) (50 mg/kg, i.p.; n = 6) did not produce any significant modification of both the neuronal discharge rate (Figure 1C, D) and pattern (Table 1) when compared to the vehicle group. Similarly, the modification of NO levels within the SNc, by 5 min application of l‐NAME (40 nA, n = 4) or l‐ARG, (40 nA; n = 5) was inactive on the basal electrophysiological activity of DA SNc neurons (Table 1).
Nicotine (25–400 μg/kg, i.v.) significantly caused a dose‐dependent increase of the firing rate (n = 7; P < 0.05; Figure 2A) and the bursting activity (P < 0.05; Table 1) compared to the vehicle group (n = 10). Although all cells were excited by nicotine in terms of firing rate and discharge pattern, the degree of response was not entirely uniform. As shown by the dose–response curve reported in Figure 2D, nicotine reached its maximal effect on the firing rate (+93 ± 19%, above baseline) at the cumulative dose of 775 μg/kg. At the same dose, the spikes fired in bursts increased to a Δ equal to 20.41 ± 7.5 (P < 0.05 compared with vehicle group).
Figure 2.
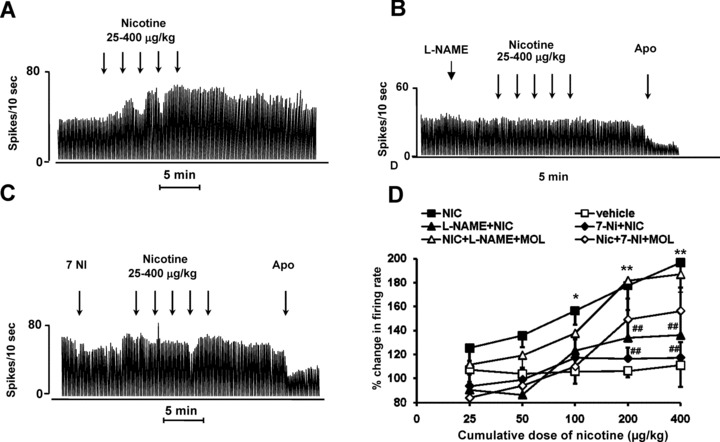
Blockade by 7‐NI and l‐NAME of the excitatory actions of nicotine on the firing rate of SNc neurons. (A) Representative rate histogram showing that i.v. effect of nicotine (25, 50, 100, 200, and 400 μg/kg, at arrows). (B,C) Representative rate histograms showing that pretreatment with 7‐NI and l‐NAME (50 mg/kg, i.p.) prevents the excitatory effect of nicotine. (D) Cumulative dose–response curve showing the mean percentage change (± SEM) in firing rate after nicotine, vehicle, 7‐NI + nicotine, l‐NAME + nicotine, 7‐NI + nicotine + MOL, and l‐NAME + nicotine +MOL. Statistical analysis revealed a significant effect of nicotine (one‐way ANOVA; P < 0.01; n = 7) compared with vehicle group (n = 10). Pretreatment with 7‐NI or l‐NAME (50 mg/kg, i.p.), prevented nicotine‐induced increase in DA firing rate (two‐way ANOVA; nicotine vs. vehicle *P < 0.05; **P < 0.01 and NOS inhibitors vs. nicotine #P < 0.05; ##P < 0.01 by Tukey‐Kramer post hoc test).
In order to evaluate the role of NO on nicotine‐induced excitatory effect of SNc DA neurons, two groups of rats were pretreated with 7‐NI or l‐NAME (50 mg/kg i.p. n = 5 each drug), respectively (Figure 2B, C). Both NOS inhibitors significantly prevented nicotine‐induced increase of DA firing rate and burst firing when compared to the vehicle group (P < 0.05; n = 10). 7‐NI and l‐NAME were injected 10–15 min before administration of cumulative doses of nicotine (25–400 μg/kg, i.v.). In 7 of 10 neurons the effects of the NOS inhibitors and nicotine administration were recorded on the same cells. Three neurons were lost after the NOS inhibitor injection (i.e., two for 7‐NI and one for l‐NAME) and neighbor DA cells were recorded and evaluated for the nicotine effect. In these cases, the interval between the NOS blocker administration and the start of nicotine dose–response was still between 10 and 15 min.
The maximum nicotine‐induced excitation at the cumulative dose of 775 μg/kg after 7‐NI and l‐NAME injection was reduced to 17 ± 13% and 36 ± 20%, respectively (Figure 2D). In terms of burst firing, nicotine was capable of eliciting only a Δ variation of 9.9 ± 7.0 and 4.9 ± 2.6 when the animals were pretreated with 7‐NI and l‐NAME, respectively (Table 1).
The effects of nicotine in l‐NAME and 7‐NI‐pretreated rats could be partially restored when rats were coadministrated with MOL (50 mg/kg i.p; Figure 2D). Thus, in combined l‐NAME + MOL pretreated rats nicotine evoked a significant increase of firing rate group 87.0 ± 15.8% (P < 0.003; n = 7; Figure 2A) that was not different to the nicotine group (P > 0.5; n = 7; Figure 2D). 7‐NI coadministrated with mol did not significantly counteract the excitatory nicotine effect only at the higher dose (P > 0.1). In addition, MOL was able to restore the nicotine effect in the l‐NAME rats in terms of burst firing. The burst firing expressed as Δ at the cumulative dose of 775 μg/kg of nicotine in the rats pretreated with l‐NAME + MOL was 15.2 ± 6.68 and was not different to the effect elicited by nicotine (P > 0.18; n = 7; Table 1). In the pretreated 7‐NI + MOL rats nicotine was capable of increasing the number of spikes fired in burst but the Δ was still significantly different compared to nicotine group (P < 0.05; n = 6; Table 1).
Effects of 7‐NI, MOL, l‐NAME, l‐ARG on Nicotine‐induced DA and DOPAC Release in the Corpus Striatum
Results consistent with the extracellular recordings were obtained with a neurochemical approach. Indeed, 7‐NI or l‐NAME (50 mg/kg, i.p.; n = 5 each) treatment did not modify the DA release in the striatum (Figure 3A); although DOPAC efflux was significantly reduced by the 7‐NI treatment (–38.1 ± 6.8%; P < 0.01) (Figure 4A). Furthermore, neither MOL (50 mg/kg, i.p.; n = 5) nor l‐ARG (50 mg/kg, i.p.; n = 5) modified DA release or DOPAC efflux in the striatum (Figures 3A and 4A).
Figure 3.
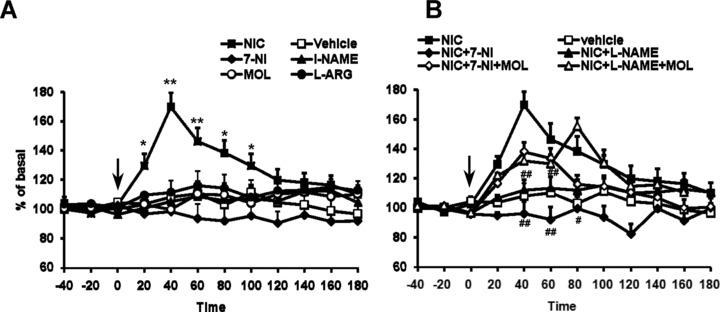
(A) Time course of the effect of acute nicotine (1 mg/kg, i.p.) administration on extracellular levels of DA in the corpus striatum (n = 5). All the drugs and vehicle were injected at the time indicated by vertical arrow. Each data point represents mean ± SEM absolute levels of DA, without considering probe recovery. Statistical analysis shows a significant effect of nicotine (one‐way ANOVA; P < 0.01) as compared with the control group. 7‐NI (50 mg/kg), l‐NAME (50 mg/kg), MOL (50 mg/kg), and l‐ARG (50 mg/kg) did not modify at any time the DA levels. (B) Time course of the blockade by 7‐NI and l‐NAME of the excitatory actions of nicotine (1 mg/kg, i.p.) administration on extracellular levels of DA in the corpus striatum. The dose of 50 mg/kg i.p. 7‐NI and l‐NAME completely prevented nicotine‐induced increase in DA release (two‐way ANOVA; #P < 0.05, ##P < 0.01 by Tukey‐Kramer post hoc test).
Figure 4.
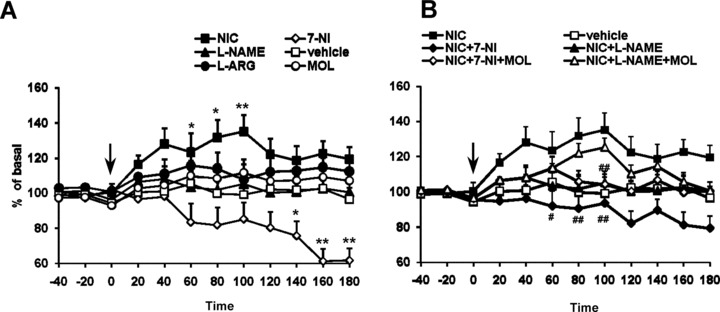
(A) Time course of the effect of acute nicotine (1 mg/kg, i.p.) administration on extracellular levels of DOPAC in the corpus striatum (n = 5). All the drugs and vehicle were injected at the time indicated by vertical arrow. Each data point represents mean ± SEM absolute levels of DA, without considering probe recovery. Statistical analysis shows a significant effect of nicotine and 7‐NI (one‐way ANOVA; P < 0.01) as compared with the control group. l‐NAME (50 mg/kg), MOL (50 mg/kg), and l‐ARG (50 mg/kg) did not modify at any time the DOPAC levels. (B) Time course of the blockade by 7‐NI and l‐NAME of the excitatory actions of nicotine (1 mg/kg, i.p.) administration on extracellular levels of DOPAC in the corpus striatum. The dose of 50 mg/kg i.p. 7‐NI and l‐NAME completely prevented nicotine‐induced increase in DOPAC release (two‐way ANOVA; #P < 0.05, ##P < 0.01 by Tukey‐Kramer post hoc test).
Acute intraperitoneal administration of 1 mg/kg nicotine (n = 5) caused a significant increase in DA and DOPAC outflow in the corpus striatum (Figures 3A and 4A). The effect of nicotine was time‐dependent. DA sharply reached its maximum concentration (+69.7 ± 9.9%, above baseline; P < 0.01) at 40 min after injection, slowly declining to baseline values at 180 min (Figure 1). Striatal DOPAC increase however lasted longer but its concentration was only augmented by 38.5 ± 4.8% (P < 0.01) at 100 min after injection (Figure 4A).
Inhibition of NOS counteracted the nicotine‐induced enhancement of DA and DOPAC release (Figures 3 and 4). l‐NAME and 7‐NI pretreatments completely prevented nicotine‐induced DA and DOPAC release (P < 0.05). Nicotine was still capable of increasing DA in the l‐NAME and 7‐NI‐pretreated rats that received also 50 mg/kg of MOL (P < 0.01). DOPAC levels were instead restored only in the l‐NAME + MOL pretreated group but not in the 7‐NI + MOL group.
Discussion
Our present findings further support the pivotal role played by NO in controlling DA function and give new insights into the modulation of the nigrostriatal DA system under basal and activated conditions. Evidence from studies conducted in our laboratory, in agreement with the wealth of literature available [26, 31], have indeed shown that the NO system plays a prominent role in the control of central nigrostriatal DA function and in the demise of nigral DA cells [21, 22].
The current work extended these findings by examining whether pharmacological inhibition of the NOS induces a modification in the effects of nicotine on the nigrostriatal DA system. Firstly, we failed to reveal any evidence for a tonic NO control of nigrostriatal DA neurotransmission in terms of DA neuronal discharge and striatal DA and DOPAC release, in agreement with some other investigations [17, 32, 33, 34]. Indeed, neither the disruption of NO levels by treatment with l‐NAME, an unselective NOS inhibitor, nor the NO level elevation by NO precursor l‐ARG, and NO donor MOL treatment, were able to produce any changes in the firing rate and burst firing of SNc DA neurons recorded. This lack of effect was obtained both by general and local application of l‐ARG and l‐NAME by microiontophoresis within the SNc, revealing that NO might not play a relevant role under physiological conditions in dopaminergic nigral function outside and within the SNc. Our findings are consistent with the evidence that l‐NAME and l‐ARG treatments did not modify either the firing discharge and pattern of the VTA dopaminergic neurons in vivo[17] or of both VTA and SNc neurons in vitro[17, 32]. Furthermore, our neurochemical data concur with the electrophysiological recordings, likewise showing that variation of endogenous nitrergic tone does not influence basal striatal DA release.
7‐NI electrophysiological and neurochemical effects are more complex and deserve more attention. In our study, we evaluated its effect inasmuch as 7‐NI is a common pharmacological tool used to preferentially inhibit nNOS in vivo[35] with no evident pressor effects [36] in the control of dopaminergic basal function, as well as in the blocking of nicotine‐induced excitation.
Although not significant in the overall statistical analysis, 7‐NI treatment slightly decreased discharge rate and in two DA neurons of six the firing pattern was clearly affected, showing a longlasting decrease in the number of spikes fired in bursts. In addition, 7‐NI slightly reduced DA striatal release while DOPAC concentration was significantly decreased.
These observations are in line with our recent work [22] in which we studied the influence of acute and subchronic NO manipulation, evaluated in population studies of DA cells from multiple‐electrode tracks within the substantia nigra and in HPLC detection of striatal tissue levels of DA and DOPAC. Acute 7‐NI significantly decreased the percentage of action potentials fired in bursts while the number of spontaneously active nigral DA neurons and the mean firing rate of these cells were unaffected. Therefore, it is possible that the tendency to reduction in terms of firing pattern observed in the current study could have been significant if the sample size were larger (in [22] up to 82 cells were recorded).
In the present microdialysis work, 7‐NI pretreatment produced a decrease in striatal DOPAC efflux by 40% when all striata tissue were considered the reduction produced by the same dose reached 80%[22], a variation attributable to methodological differences.
7‐NI‐induced decrease of DOPAC and burst firing are likely to be independent of nNOS inhibition or NO production and rather a consequence of the well known strong monoamine oxidase type B inhibitory activity possessed by 7‐NI [37, 38, 39]. Therefore, the results obtained with 7‐NI should be taken cautiously especially when the dopaminergic system is studied.
Thus, the effects induced by l‐NAME, in our experimental conditions, can be essentially ascribed to the inhibition of nNOS. This is supported by the lack of effect of the other NO‐modulating compounds tested in this current study, administered systemically and locally in terms of firing rate and DA release, in line with the ineffectiveness of these compounds in vivo and in vitro[17, 32, 33, 34].
The major aim of the present study was to demonstrate the possible modulation of nicotine‐induced excitation of nigrostriatal DA function by NOS inhibition. We show that acute intravenous nicotine administration enhances the basal firing rate and bursting activity in presumed DA‐containing neurons in the SNc of chloral hydrate‐anesthetized rats and concurrently increases DA and DOPAC overflow of awake, freely moving rats. These findings are consistent with previous data reported in a number of studies showing that nicotine increases nigrostriatal and mesolimbic DA pathway activity [12, 13, 14, 15, 16, 17, 18]. Nicotine can increase the firing rate and the bursting activity of SNc DA neurons by several mechanisms, including a direct depolarizing effect mediated by the activation of somatodendritic nicotinic receptors [14, 15, 40]. Moreover, nicotine can indirectly increase DA activity by eliciting the release of glutamate (GLU) from nerve terminals synapsing on DA neurons and by depressing the inhibitory gamma‐aminobutyric acid (GABA)‐ergic input to these neurons [41, 42]. In addition to its stimulatory effects on neuronal DA firing rate, nicotine was shown to elicit DA release from DA terminals in the corpus striatum and the nucleus accumbens [11, 19, 20].
As reported under Results, the dose of 50 mg/kg of 7‐NI and l‐NAME was capable of preventing the increase in DA neuronal firing rate and burst firing induced by nicotine administration in the SNc and attenuates nicotine‐induced enhancement of the extracellular levels of DA and DOPAC in the corpus striatum of awake freely moving rats.
Strikingly, despite the inability of NO to modify basal DA function, the inhibition of NOS completely counteracted the stimulation of DA outflow induced by nicotine. Moreover, the critical role played by NO in nigral nicotine/DA interaction is further supported by the evidence of a complete restoration of nicotine effects in rats pretreated with 7‐NI and l‐NAME plus the NO donor MOL.
The mechanism by which inhibition of NO influences the nicotine‐induced activation of DA cells in the SNc observed in vivo is far from simple. It is unlikely that a direct synaptic effect on DA neurons is involved, since it has been shown that l‐NAME does not alter the VTA DA firing rate induced by nicotine in vitro and does not affect nicotine‐induced inward currents in the same area [9]. This assumption concords with the anatomical evidence that only a few DA neurons in the SNc and VTA express NOS machinery [43, 44]. NO might be involved in both nicotine‐induced increase in firing and burst rate of DA neurons modulating GLU and GABA release in an opposite way. Striatal NO may play an important role in this modulation. Indeed, it has been shown that NO striatal levels could be boosted by a direct effect of nicotine on nitrergic, or on large acetylcholine (ACh), striatal interneurons [45], or by the phasic DA activation through activation of D2 receptor [46]. Therefore, NO directly, or via a modulation of GLU, ACh, and GABA striatal levels, might excite a subpopulation of medium spiny neurons projecting to substantia nigra, exciting SNc DA cells by inhibiting substantia nigra pars reticulata neurons, as suggested by West and Grace [31]. It is possible that removal of endogenous NO tone by 7‐NI or l‐NAME treatment might decrease the indirect excitatory pathway through the SNc by balancing the direct inhibitory one, reducing GLU release and leading the SNc neurons to a hypofunctional state. Nicotine in this condition might be unable to exert its excitatory effect.
Therefore, the degree of activity of nigrostriatal DA neurons may constitute a key factor for the expression of the NO/DA interaction, in that enhanced DA synthesis and/or release would be required to permit the occurrence of a NO modulatory control. In line with this hypothesis is the evidence that striatal NO increases only when SNc neurons fire at high frequency and in bursts [46, 47]. NO efflux occurs only when DA transmission is phasically increased thus the information transmitted via the nigrostriatal pathway during DA cell burst firing may be processed and/or amplified by striatal NOS interneurons [46, 47]. Further, it has been shown that the permissive role played by NO in phasic DA activation is not exclusive for nicotine and is applicable to other excitatory stimuli, both in physiological [31] and in pharmacological terms [25]. Thus, NO seems to have a more general role in controlling the DA function, brain reward and motivation circuitries.
Our results are in agreement with substantial evidence on NO/nicotine interactions. It has been shown that 7‐NI blocks nicotine‐induced conditioned place preference [48], NOS inhibitors attenuate symptoms of nicotine abstinence syndrome and withdrawal [49, 50, 51]. NG‐nitro‐L‐arginine (L‐NNA) and l‐NAME prevents behavioral [52], locomotor [53] and DA overflow induced by nicotine‐sensitization [54]. In line with this, NOS inhibitors have been shown to be able to attenuate the development and expression of the abstinence syndrome for such psychostimulant [25].
Furthermore, our results suggest that the NOS represents an important therapeutic target for the development of agents for those neuropsychiatric conditions with an impairment of DA functions, such as depression, schizophrenia, PD, and drug addiction [21, 22, 23, 24, 25]. In particular, we propose that NOS inhibitors might facilitate tobacco smoking cessation by blocking the hedonic spiking dependent nicotine‐induced increase in DA release.
This hypothesis is corroborated by the evidence that bupropion, one of the effective treatments available for the cessation of smoking through the inhibition of DA reuptake [55], seems also to act by inhibiting the l‐ARG‐NO‐cyclic guanosine monophosphate (cGMP) signaling pathway, rather than having a direct effect on the DA system by acting as a DA transporter reuptake inhibitor [56]. The challenge for pharmaceutical research now is to achieve selective inhibition of NOS isoforms, a situation complicated by the possibility that NOS inhibitors can indiscriminately affect beneficial and pathological NO signaling pathways.
Conflict of Interest
All the authors do not have any conflict of interest.
References
- 1. Reavill C, Stolerman IP. Locomotor activity in rats after administration of nicotinic agonists intracerebrally. Br J Pharmacol 1990;99:273–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Corrigall WA, Coen KM. Selective dopamine antagonists reduce nicotine self‐administration. Psychopharmacology 1991;104:171–176. [DOI] [PubMed] [Google Scholar]
- 3. Rose JE, Corrigall WA. Nicotine self‐administration in animals and humans: Similarities and differences. Psychopharmacology 1997;130:28–40. [DOI] [PubMed] [Google Scholar]
- 4. Laviolette SR, Van Der Kooy D. Blockade of mesolimbic dopamine transmission dramatically increases sensitivity to the rewarding effects of nicotine in the ventral tegmental area. Mol Psychiatry 2003;8:50–59. [DOI] [PubMed] [Google Scholar]
- 5. Clarke PBS, Fu DS, Jakubovic A, Fibiger HC. Evidence that Mesolimbic dopaminergic activation underlies the locomotor stimulant action of nicotine in rats. J Pharmacol Exp Ther 1988;246:701–708. [PubMed] [Google Scholar]
- 6. Louis M, Clarke PB. Effect of ventral tegmental 6‐hydroxydopamine lesions on the locomotor stimulant action of nicotine in rats. Neuropharmacology 1998;37:1503–1513. [DOI] [PubMed] [Google Scholar]
- 7. Dani JA, De Biasi M. Cellular mechanisms of nicotine addiction. Pharmacol Biochem Behav 2001;70:439–446. [DOI] [PubMed] [Google Scholar]
- 8. Di Chiara G, Imperato A. Drugs abused by humans preferentially increase synaptic dopamine concentrations in the mesolimbic system of freely moving rats. Proc Natl Acad Sci USA 1988;85:5274–5278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Schilström B, Svensson HM, Svensson TH, Nomikos GG. Nicotine and food induced dopamine release in the nucleus accumbens of the rat: Putative role of α7 nicotinic receptors in the ventral tegmental area. Neuroscience 1998;85:1005–1009. [DOI] [PubMed] [Google Scholar]
- 10. Imperato A, Mulas A, Di Chiara G. Nicotine preferentially stimulates dopamine release in the limbic system of freely moving rats. Eur J Pharmacol 1986;132:337–338. [DOI] [PubMed] [Google Scholar]
- 11. Marshall DL, Redfern PH, Wonnacott S. Presynaptic nicotinic modulation of dopamine release in the three ascending pathways studied by in vivo microdialysis: Comparison of naive and chronic nicotine‐treated rats. J Neurochem 1997;68:1511–1519. [DOI] [PubMed] [Google Scholar]
- 12. Di Matteo V, Pierucci M, Esposito E. Selective stimulation of serotonin2c receptors blocks the enhancement of striatal and accumbal dopamine release induced by nicotine administration. J Neurochem 2004;89:418–429. [DOI] [PubMed] [Google Scholar]
- 13. Clarke PBS, Hommer DW, Pert A, Skirboll LR. Electrophysiological actions of nicotine on substantia nigra single units. Br J Pharmacol 1985;85:827–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Calabresi P, Lacey MG, North RA. Nicotinic excitation of rat ventral tegmental neurones in vitro studied by intracellular recording. Br J Pharmacol 1989;98:135–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sorenson EM, Shiroyama T, Kitai ST. Postsynaptic nicotinic receptors on dopaminergic neurons in the substantia nigra pars compacta of the rat. Neuroscience 1998;87:659–673. [DOI] [PubMed] [Google Scholar]
- 16. Schilström B, Rawal N, Mameli‐Engvall M, Nomikos GG, Svensson TH. Dual effects of nicotine on dopamine neurons mediated by different receptor subtypes. Int J Neuropsychopharmacol 2003;6:1–11. [DOI] [PubMed] [Google Scholar]
- 17. Schilström B, Mameli‐Engvall M, Rawal N, Grillner P, Jardemark K, Svensson TH. Nitric oxide is involved in nicotine‐induced burst firing of rat ventral tegmental area dopamine neurons. Neuroscience 2004;125:957–964. [DOI] [PubMed] [Google Scholar]
- 18. Pierucci M, Di Matteo V, Esposito E. Stimulation of serotonin2C receptors blocks the hyperactivation of midbrain dopamine neurons induced by nicotine administration. J Pharmacol Exp Ther 2004;309:109–118. [DOI] [PubMed] [Google Scholar]
- 19. Mifsud J‐C, Hernandez L, Hoebel BG. Nicotine infused into the nucleus accumbens increases synaptic dopamine as measured by in vivo microdialysis. Brain Res 1989;478:365–367. [DOI] [PubMed] [Google Scholar]
- 20. Ferrari R, Le Nove're N, Picciotto MR, Changeux JP, Zoli M. Acute and long‐term changes in the mesolimbic dopamine pathway after systemic or local single nicotine injections. Eur J Neurosci 2002;15:1810–1818. [DOI] [PubMed] [Google Scholar]
- 21. Di Matteo V, Pierucci M, Benigno A, Crescimanno G, Esposito E, Di Giovanni G. Involvement of nitric oxide in 6‐OHDA‐induced neurodegeneration: A neurochemical study. Ann N Y Acad Sci 2009;1155,309–315. [DOI] [PubMed] [Google Scholar]
- 22. Di Matteo V, Pierucci M, Benigno A, Orbán G, Crescimanno G, Esposito E, Di Giovanni G. Electrophysiological and neurochemical characterization of 7‐Nitroindazole and molsidomine acute and subchronic administration effects in the dopaminergic nigrostriatal system in rats. J Neural Transm Suppl 2009;73,173–182. [DOI] [PubMed] [Google Scholar]
- 23. McLeod TM, López‐Figueroa AL, López‐Figueroa MO. Nitric oxide, stress, and depression. Psychopharmacol Bull 2001;35:24–41. [PubMed] [Google Scholar]
- 24. Yao JK, Leonard S, Reddy RD. Increased nitric oxide radicals in post‐mortem brain from patients with schizophrenia. Schizophr Bull 2004;30:923–934. [DOI] [PubMed] [Google Scholar]
- 25. Tayfun Uzbay I, Oglesby MW. Nitric oxide and substance dependence. Neurosci Biobehav Rev 2001;25:43–52. [DOI] [PubMed] [Google Scholar]
- 26. West AR, Galloway MP, Grace AA. Regulation of striatal dopamine neurotransmission by nitric oxide: Effector pathways and signalling mechanisms. Synapse 2002;44:227–245. [DOI] [PubMed] [Google Scholar]
- 27. Vleeming W, Rambali B, Opperhuizen A. The role of nitric oxide in cigarette smoking and nicotine addiction. Nicotine Tob Res 2002;4:341–348. [DOI] [PubMed] [Google Scholar]
- 28. Di Giovanni G, Shi WX. Effects of scopolamine on dopamine neurons in the substantia nigra: Role of the pedunculopontine tegmental nucleus. Synapse 2009;63:673–680. [DOI] [PubMed] [Google Scholar]
- 29. Paxinos G, Watson C. The Rat brain in stereotaxic coordinates. New York : Academic Press, 1986. [Google Scholar]
- 30. Grace AA, Bunney BS. The control of firing pattern in nigral dopamine neurons: Burst firing. J Neurosci 1984;4:2877–2890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. West AR, Grace AA. Striatal nitric oxide signalling regulates the neuronal activity of midbrain dopamine neurons in vivo. J Neurophysiol 2000;83:1796–1808. [DOI] [PubMed] [Google Scholar]
- 32. Cox BA, Johnson SW. Nitric oxide facilitates N‐methyl‐D‐aspartate‐induced burst firing in dopamine neurons from rat midbrain slices. Neurosci Lett 1998;255:131–134. [DOI] [PubMed] [Google Scholar]
- 33. Nowak P, Brus R, Oświecimska J, Sokoła A, Kostrzewa RM. 7‐Nitroindazole enhances amphetamine‐evoked dopamine release in rat striatum. An in vivo microdialysis and voltammetric study. J Physiol Pharmacol 2002;53:251–263. [PubMed] [Google Scholar]
- 34. Campos F, Alfonso M, Vidal L, Faro LR, Durán R. Mediation of glutamatergic receptors and nitric oxide on striatal dopamine release evoked by anatoxin‐a. An in vivo microdialysis study. Eur J Pharmacol 2006;548:90–98. [DOI] [PubMed] [Google Scholar]
- 35. Southan GJ, Szabo C. Selective pharmacological inhibition of distinct nitric oxide synthase isoforms. Biochem Pharmacol 1996;51:383–394. [DOI] [PubMed] [Google Scholar]
- 36. Meyer RC, Spangler EL, Patel N, London ED, Ingram DK. Impaired learning in rats in a 14‐unit T‐maze by 7‐nitroindazole, a neuronal nitric oxide synthase inhibitor, is attenuated by the nitric oxide donor, molsidomine. Eur J Pharmacol 1998;341:17–22. [DOI] [PubMed] [Google Scholar]
- 37. Castagnoli K, Palmer S, Anderson A, Bueters T, Castagnoli N Jr. The neuronal nitric oxide synthase inhibitor 7‐nitroindazole also inhibits the monoamine oxidase‐B‐catalyzed oxidation of 1‐methyl‐4‐phenyl‐1,2,3,6‐tetrahydropyridine. Chem Res Toxicol 1997;10:364–368. [DOI] [PubMed] [Google Scholar]
- 38. Boireau A, Dubedat P, Bordier F, Imperato A, Moussaoui S. The protective effect of riluzole in the MPTP model of Parkinson's disease in mice is not due to a decrease in MPP(+) accumulation. Neuropharmacology 2000;39:1016–1020. [DOI] [PubMed] [Google Scholar]
- 39. Thomas B, Saravanan KS, Mohanakumar KP. In vitro and in vivo evidences that antioxidant action contributes to the neuroprotective effects of the neuronal nitric oxide synthase and monoamine oxidase‐B inhibitor, 7‐nitroindazole. Neurochem Int 2008;52:990–1001. [DOI] [PubMed] [Google Scholar]
- 40. Klink R, De Kerkhove d’Exaerde A, Zoli M, Changeux J‐P. Molecular and physiological diversity of nicotinic acetylcholine receptors in the midbrain dopaminergic nuclei. J Neurosci 2001;21:1452–1463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Mansvelder HD, McGehee DS. Cellular and synaptic mechanisms of nicotine addiction. J Neurobiol 2002;53:606–617. [DOI] [PubMed] [Google Scholar]
- 42. Mansvelder HD, Keath JR, McGehee DS. Synaptic mechanisms underlie nicotine‐induced excitability of brain reward areas. Neuron 2002;33:905–919. [DOI] [PubMed] [Google Scholar]
- 43. Klejbor I, Domaradzka‐Pytel B, Ludkiewicz B, Wójcik S, Moryś J. The relationships between neurons containing dopamine and nitric oxide synthase in the ventral tegmental area. Folia Histochem Cytobio 2004;42:83–87. [PubMed] [Google Scholar]
- 44. Del Bel E, Bermúdez‐Echeverry M, Salum C, Raisman‐Vozari R. Nitric oxide system and basal ganglia physiopathology In: Di Giovanni G, editor. The basal ganglia pathophysiology: Recent advances. Kerala : Transworld Research Network, 2007;129–158. [Google Scholar]
- 45. Exley R, Cragg SJ. Presynaptic nicotinic receptors: A dynamic and diverse cholinergic filter of striatal dopamine neurotransmission. Br J Pharmacol 2008;153(Suppl 1):S283–S297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Sammut S, Bray KE, West AR. Dopamine D2 receptor‐dependent modulation of striatal NO synthase activity. Psychopharmacology 2007;191:793–803. [DOI] [PubMed] [Google Scholar]
- 47. Sammut S, Dec A, Mitchell D, Linardakis J, Ortiguela M, West AR. Phasic dopaminergic transmission increases NO efflux in the rat dorsal striatum via a neuronal NOS and a dopamine D(1/5) receptor‐dependent mechanism. Neuropsychopharmacology 2006;31:493–505. [DOI] [PubMed] [Google Scholar]
- 48. Martin JL, Itzhak Y. 7‐Nitroindazole blocks nicotine‐induced conditioned place preference but not LiCl‐induced conditioned place aversion. Neuroreport 2000;11:947–949. [DOI] [PubMed] [Google Scholar]
- 49. Malin DH, Lake JR, Shenoi M, Upchurch TP, Johnson SC, Schweinle WE, Cadle CD. The nitric oxide synthesis inhibitor nitro‐L‐arginine (L‐NNA) attenuates nicotine abstinence syndrome in the rat. Psychopharmacology (Berl) 1998;140:371–377. [DOI] [PubMed] [Google Scholar]
- 50. Adams ML, Cicero T. Nitric oxide mediates mecamylamine‐ and naloxone‐precipitated nicotine withdrawal. Eur J Pharmacol 1998;345:R1–R2. [DOI] [PubMed] [Google Scholar]
- 51. Jain R, Mukherjee K, Mohan D. Effects of nitric oxide synthase inhibitors in attenuating nicotine withdrawal in rats. Pharmacol Biochem Behav 2008;88:473–480. [DOI] [PubMed] [Google Scholar]
- 52. Shim I, Kim HT, Kim YH, et al Role of nitric oxide synthase inhibitors and NMDA receptor antagonist in nicotine‐induced behavioral sensitization in the rat. Eur J Pharmocol 2002;443:119–124. [DOI] [PubMed] [Google Scholar]
- 53. Ulusu U, Uzbay IT, Kayir H, Alici T, Karakas S. Evidence for the role of nitric oxide in nicotine‐induced locomotor sensitization in mice. Psychopharmacology (Berl) 2005;178:500–504. [DOI] [PubMed] [Google Scholar]
- 54. Hong SK, Jung IS, Bang SA, Kim SE. Effect of nitric oxide synthase inhibitor and NMDA receptor antagonist on the development of nicotine sensitization of nucleus accumbens dopamine release: An in vivo microdialysis study. Neurosci Lett 2006;409:220–223. [DOI] [PubMed] [Google Scholar]
- 55. Wilkes S. The use of bupropion SR in cigarette smoking cessation. Int J Chron Obstruct Pulmon Dis 2008;3:45–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Dhir A, Kulkarni SK. Involvement of nitric oxide (NO) signaling pathway in the antidepressant action of bupropion, a dopamine reuptake inhibitor. Eur J Pharmacol 2007;568:177–185. [DOI] [PubMed] [Google Scholar]