

Abstract
Amyotrophic lateral sclerosis (ALS) is a devastating and fatal neurodegenerative disease of adults which preferentially attacks the neuromotor system. Riluzole has been used as the only approved treatment for amyotrophic lateral sclerosis since 1995, but its mechanism(s) of action in slowing the progression of this disease remain obscure. Searching PubMed for “riluzole” found 705 articles published between January 1996 and June 2009. A systematic review of this literature found that riluzole had a wide range of effects on factors influencing neural activity in general, and the neuromotor system in particular. These effects occurred over a large dose range (<1 μM to >1 mM). Reported neural effects of riluzole included (in approximate ascending order of dose range): inhibition of persistent Na+ current = inhibition of repetitive firing < potentiation of calcium‐dependent K+ current < inhibition of neurotransmitter release < inhibition of fast Na+ current < inhibition of voltage‐gated Ca2+ current = promotion of neuronal survival or growth factors < inhibition of voltage‐gated K+ current = modulation of two‐pore K+ current = modulation of ligand‐gated neurotransmitter receptors = potentiation of glutamate transporters. Only the first four of these effects commonly occurred at clinically relevant concentrations of riluzole (plasma levels of 1–2 μM with three‐ to four‐fold higher concentrations in brain tissue). Treatment of human ALS patients or transgenic rodent models of ALS with riluzole most commonly produced a modest but significant extension of lifespan. Riluzole treatment was well tolerated in humans and animals. In animals, despite in vitro evidence that riluzole may inhibit rhythmic motor behaviors, in vivo administration of riluzole produced relatively minor effects on normal respiration parameters, but inhibited hypoxia‐induced gasping. This effect may have implications for the management of hypoventilation and sleep‐disordered breathing during end‐stage ALS in humans.
Keywords: Hypoxia, Motor neuron disease, Neuronal excitability, Neurotransmitter release, Persistent sodium current, Respiration
Introduction
Riluzole (2‐amino‐6‐trifluoromethoxy benzothiazole, RP 54274, Rilutek™) was originally developed in the 1950s [1] as a centrally acting muscle relaxant and was later investigated as an anticonvulsant and neuroprotective agent [reviewed in 2]. In 1995, the FDA narrowly approved the use of riluzole for the treatment of amyotrophic lateral sclerosis (ALS, motor neuron disease). More than a decade later, riluzole still remains the only approved treatment for this fatal neurodegenerative condition [3]. Searching PubMed for the term “riluzole” returned 705 articles published from January 1996 to June 2009, in contrast to the 60 articles published from 1985 to the end of 1995. Clearly, our investigation of the actions and clinical usage of riluzole has increased dramatically in a decade, and a review of key research findings in animal and human settings would be a timely addition to this large body of literature.
The purpose of this review is therefore to survey and summarize progress in the last 13 years in understanding the mechanisms of action of riluzole on neural activity, particularly in the context of the neuromotor system, and the clinical efficiency of riluzole treatment in animal models of ALS and in human ALS patients. Reviews of pathophysiological changes in human ALS or in vitro and in vivo animal models of ALS were excluded, except where they specifically addressed actions of riluzole. In the interest of brevity, numerous references related to the off‐label use of riluzole in treatment of other neurodegenerative diseases, such as Huntington's or Parkinson's diseases [e.g., 4, 5], or of mood and anxiety disorders in humans or in animal models of these disorders [e.g., 6, 7] were excluded from this review; interested readers are referred to these recent reviews of this literature. Readers may also wish to consult previous reviews on the pharmacology and actions of riluzole which summarize earlier work up to 1996 [2, 8, 9, 10, 11]; literature prior to 1996 will only be referred to when it provides relevant background to subsequent investigations or where no new data has emerged.
Effects of Riluzole on Neuronal Firing and Membrane Properties
Since an initial brief note that riluzole decreased firing in cultured rat cerebellar granule cells [2], there have been a wealth of observations in the last decade reporting that riluzole significantly decreased repetitive firing of action potentials in many types of neuron (Table 1), including rat striatal neurons [12], mouse and rat cortical neurons [13, 14, 15], rat hippocampal pyramidal neurons [16], cultured rat spinal motoneurons [17], neonatal rat spinal cord interneurons [18], acutely isolated rat brainstem dorsal column nuclei neurons [19], neonatal rat mesencephalic V brainstem neurons [20, 21], spinal motoneurons in adult rat and neonatal mouse and rat [17, 22, 23, 24], adult rat facial motoneurons [25], and hypoglossal motoneurons from neonatal mouse [26], neonatal rat [27], and juvenile rat (M.C. Bellingham, unpublished observations).
Table 1.
Riluzole concentration for effects on neuronal firing
| Current/channel | Effect (%) | Concentration (μM) | Tissue | Reference |
|---|---|---|---|---|
| Neuronal excitability | ||||
| Firing frequency | Inhibition | 6a | Cultured rat hippocampal neurons | [16] |
| Firing frequency | Inhibition | 0.3a | Rat striatal neurons | [12] |
| Firing frequency | Inhibition | 1.1a | Cultured embryonic rat spinal motor neurons | [17] |
| Firing frequency | Inhibition | 0.52a | Rat neocortical neurons | [14] |
| Firing frequency | Inhibition | 2–5 | Rat facial motoneurons | [25] |
| Firing frequency | Inhibition (87%) | 5 | Rat carotid body chemoreceptor neurons | [52] |
| Firing frequency | Inhibition (25%) | 0.75 | Cultured embryonic G93A SOD1 mouse cortical neurons | [15] |
| Firing frequency | Inhibition (50%) | 1 | GH3 neuroendocrine cells | [35] |
| Evoked axonal fiber volley | Inhibition | 7.5a | Rat CA3 hippocampal neurons | [89] |
aIC50.
Effects of riluzole on current‐induced firing were not usually associated with changes in resting (i.e., subthreshold) membrane properties, such as resting membrane potential or input resistance, as noted for rat striatal neurons [12], cultured embryonic rat spinal motoneurons [28], neonatal rat lumbar spinal motoneurons [24], and neonatal mouse hypoglossal motoneurons [26].
Riluzole reduced repetitive neuronal firing in response to sustained current injection, without blocking single action potentials generated by transient current injection, at concentrations ranging from ≤1 μM in rat striatal neurons [12], mouse spinal neurons [17], and rat cortical neurons [14, 15], 3 μM in rat cortical neurons [13] and ≤5 μM in rat brainstem neurons [20] or rat and mouse spinal interneurons [18, 29]. Riluzole also reduced spontaneous or tonic firing in some neurons, an effect sometimes (but not always) accompanied by membrane hyperpolarization [19, 21, 25, 30, 31, 32, 33, 34, 35]. In some cases, riluzole also decreased action potential amplitude, increased action potential duration [35, 36] and increased the threshold for action potential initiation [17, 36].
In putative spinal motoneurons, cultured from embryonic rat spinal cord, riluzole (0.5–10 μM; Table 1) caused decreased repetitive firing during sustained current injection, but had little effect on firing in response to transient inputs [17]. At low concentrations (0.1–1 μM), riluzole decreased the slope of the F–I relationship and increased the threshold for firing, while at higher concentrations (2–10 μM), repetitive firing was abolished (Fig. 1). These effects on firing were likely due to inhibition of the persistent Na+ current in these motoneurons, as riluzole produced a relatively small decrease in action potential amplitude and no change in afterhyperpolarization amplitude. Similar effects on repetitive versus transient firing have been reported in neonatal mouse [22] and adult rat spinal motoneurons with 10 μM riluzole application [23].
Figure 1.
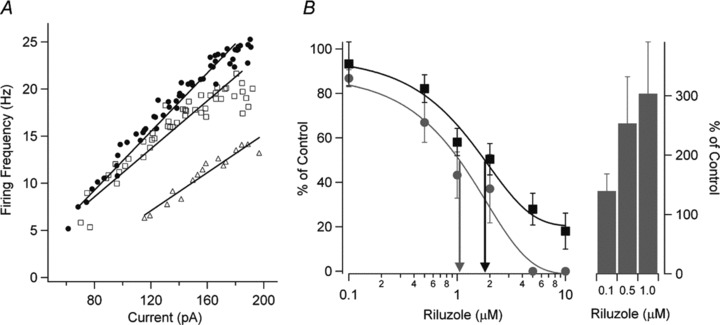
Riluzole inhibits neuronal excitability and the persistent Na+ current (INaP) in cultured spinal neurons in a dose‐dependent way. (A) The relationship between firing frequency and current injection (F–I) and linear regressions are shown for control (•), 0.1 μM riluzole (□), and 1.0 μM riluzole (▵). The F–I gain is reduced and the current threshold for the onset of firing is increased with increasing riluzole concentrations. (B) The dose–response curve for all cells is shown for the effect of riluzole on the F–I gain (gray circle) for 0.1 μM riluzole (n = 11), 0.5 μM riluzole (n = 10), 1 μM riluzole (n = 8), 2 μM riluzole (n = 5), 5 μM riluzole (n = 5), and 10 μM riluzole (n = 5). The effect of riluzole on INaP (▪) is also shown for 0.1 μM riluzole (n = 6), 0.5 μM riluzole (n = 5), 1 μM riluzole (n = 7), 2 μM riluzole (n = 5), 5 μM riluzole (n = 5), and 10 μM riluzole (n = 5). The EC50 for riluzole inhibition of the F–I gain (gray arrow) was 1.1 μM and the EC50 for inhibition of INaP (black arrow) was 1.8 μM. Riluzole also dose‐dependently increased the current threshold for firing (right side, bars; mean ± SEM shown for [B]). Threshold amplitudes could not be measured above 2 μM riluzole because spiking behavior became very irregular. Reprinted from [17], copyright (2006), with permission from John Wiley & Sons.
As outlined later and in 1, 2, 3, 4, the similarity in dose–response relationships between the action of riluzole on repetitive firing and the persistent Na+ current, and the higher riluzole concentrations usually required to inhibit voltage‐dependent K+ or Ca2+ currents suggests that riluzole primarily decreases repetitive firing by suppressing the sub‐threshold depolarizing influence of the persistent Na+ current [37, 38]; however, the possibility that riluzole also acts to increase Ca2+‐dependent K+ currents at low concentrations (see later and Table 2), with a consequent increase in inter‐spike interval, should be born in mind and tested where possible.
Table 2.
Effects of riluzole on voltage‐gated Na+ currents and channels
| Current/channel | Effect ([μM] Max%) | Concentration (μM) | Tissue | Reference |
|---|---|---|---|---|
| Fast (inactivating) sodium currents | ||||
| Fast Na+ current | Inhibition | 51a | Cultured embryonic rat cortical neurons | [41] |
| Fast Na+ current | Inhibition (51%) | 1 | Dissociated rat cortical pyramidal neurons | [43] |
| Inhibition (max 80%) | 10 | |||
| Fast Na+ current | Inhibition | 11a | Dissociated neonatal rat cerebellar Purkinje neurons | [45] |
| Fast Na+ current | Inhibition | 90b | Frog myelinated nerve fiber | [39] |
| Fast Na+ current | Inhibition | 90b (TTX sensitive) | Cultured rat dorsal root ganglion neurons | [44] |
| 143b (TTX resistant) | ||||
| Fast Na+ current | Inhibition (200 μM >80%) | 51a | Neonatal rat mesencephalic V brainstem neurons | [20] |
| Fast Na+ current | Inhibition | 50a | Rat neocortical neurons | [14] |
| Fast Na+ current | Inhibition (35%) | 10 | GH3 neuroendocrine cells | [35] |
| Fast Na+ current | Inhibition | 33.7a | Mouse sympathetic superior cervical ganglion neurons | [48] |
| Sodium channels | ||||
| Rat NaV 1.2 Na+ current | Inhibition | 30a | Rat brain clone expressed in oocytes | [40] |
| Nav 1.2 Na+ current | Inhibition | 15a | N1E‐115 neuroblastoma cells | [47] |
| Human NaV 1.4 Na+ current | Inhibition (2%) | 1 | HEK‐293 cells | [49] |
| Inhibition (26%) | 1 mM | |||
| Human NaV 1.4 Na+ current | Inhibition | 9a | HEK‐293 cells | [47] |
| Guinea pig NaV 1.4 and 1.5 Na+ current | Inhibition (20%) | 100 | Isolated cardiac Purkinje neurons | [2] |
| Rat NaV 1.5 Na+ current | Inhibition | 8a | COS‐7 cells | [47] |
| Human NaV 1.5 Na+ current | Inhibition | 2.3a | Cultured skeletal muscle cells | [50] |
| Inactivated Na+ channels | Inhibition | 0.29b | Frog myelinated nerve fiber | [39] |
| Inactivated Na+ channels | Inhibition | 2.0b (TTX sensitive) | Cultured rat dorsal root ganglion neurons | [44] |
| 3.0b (TTX resistant) | ||||
| Inactivated Na+ channels | Inhibition | 0.3 | Isolated rat brainstem neurons | [42] |
| Inactivated NaV 1.2 Na+ channels | Inhibition | 0.2a | Rat brain clone expressed in oocytes | [40] |
| Veratridine induced activation of Na+ channels | Inhibition | 1.3a | Cultured embryonic rat motor neurons | [80] |
| Veratridine induced activation of Na+ channels | Inhibition | 10 | Cultured bovine adrenal chromaffin cells | [81] |
| Persistent (non‐inactivating) sodium currents | ||||
| Persistent Na+ current | Inhibition | 0.55a | Isolated rat cortical pyramidal neurons | [51] |
| Persistent Na+ current | Inhibition (53%) | 2 | Neonatal rat mesencephalic V brainstem neurons | [20] |
| Inhibition (81%) | 5 | |||
| Persistent Na+ current | Inhibition (max 80%) | 1.8a | Cultured embryonic rat spinal motor neurons | [17] |
| Persistent Na+ current | Inhibition (25 μM, 100%) | 1–2a | Cultured rat suprachiasmatic nucleus neurons | [31] |
| Persistent Na+ current | Inhibition | 2.8a | Neonatal mouse lumbar spinal cord motor neurons | [54] |
| Persistent Na+ current | Inhibition (100%) | 10 | Rat lumbar spinal cord interneurons and motor neurons | [24] |
| Mouse lumbar spinal cord interneurons | [29] | |||
| Persistent Na+ current | Inhibition | 2.4a | Isolated rat brainstem neurons | [42] |
| Persistent Na+ current | Inhibition (78%) | 10 | Rat carotid body chemoreceptor neurons | [52] |
| Persistent Na+ current | Inhibition (86%) | 10 | Mouse nucleus accumbens medium spiny neurons | [55] |
| Persistent Na+ current | Inhibition (69.5%) | 5 | Rat spinal cord neurons | [18] |
| Persistent Na+ current | Inhibition | 3a | Rat brainstem neurons | [36, 53] |
| Persistent Na+ current | Inhibition | 5a | Rat brainstem neurons | [56] |
| Persistent Na+ current | Inhibition | 2a | Rat neocortical neurons | [14] |
| Persistent Na+ current | Inhibition | 2–5 | Rat facial motoneurons | [25] |
| Persistent Na+ current | Inhibition | 2.7a | Mouse sympathetic superior cervical ganglion neurons | [48] |
| Persistent Na+ current | Inhibition (46%) | 1 | Cultured embryonic G93A SOD1 mouse cortical neurons | [15] |
aIC50.
bApparent dissociation constant.
Table 3.
Effects of riluzole on voltage‐gated and other K+ currents and channels
| Current/channel | Effect (%) | Concentration (μM) | Tissue | Reference |
|---|---|---|---|---|
| Potassium currents | ||||
| Fast 1 component | Inhibition | 21a | Frog myelinated nerve fiber | [58] |
| Fast 2 component | Inhibition | 24a | Frog myelinated nerve fiber | [58] |
| Slow component | Inhibition | 413a | Frog myelinated nerve fiber | [58] |
| Delayed rectifier K+ current | Inhibition | 88a | Cultured embryonic rat cortical neurons | [41] |
| Voltage‐gated potassium channels | ||||
| Mouse Kv1.1 | Inhibition | 92a | NIH/NT3 cells | [47] |
| Mouse Kv1.3 | Inhibition | 50a | NIH/NT3 cells | [47] |
| Rat Kv1.5 | Inhibition | 40a | CHO cells | [64] |
| Kv1.5 | Inhibition (30%) | 100 | Rat vascular smooth muscle | [62] |
| Human Kv1.5 | Inhibition | 95a | MEL cells | [47] |
| Rat Kv3.1 | Inhibition | 124a | CHO cells | [64] |
| Human Kv3.1 | Inhibition | 95a | HEK‐293 cells | [47] |
| Rat Kv3.2 | Inhibition | 100a | COS‐7 cells | [47] |
| Kv1.4 | Inhibition | 70 | Bovine adrenal zona fasiculata cells | [61] |
| Rat Kv4.2 | Inhibition | 130a | LTK cells | [47] |
| Rat Kv4.3 | Inhibition | 116.5a | CHO cells | [63] |
| HERG Kv11.1 | Inhibition | 50a | HEK‐293 cells | [47] |
| Two pore potassium channels | ||||
| TRAAK | Activation (3.9 fold increase) | 100 | COS cells | [72] |
| TREK‐1 | Activation (∼2 fold increase) | 100 | COS cells | [71] |
| TREK‐1 | Activation | 110b | Not stated | [47] |
| TREK‐1 | Activation (3.3 fold increase) | 100 | Bovine adrenal zona fasciulata cells | [74] |
| THIK‐1 | Inhibition (36%) | 500 | Mouse cerebellar Purkinje neurons | [75] |
| Calcium‐dependent potassium currents and channels | ||||
| After hyperpolarization current | No effect | 10 | Cultured embryonic rat spinal motor neurons | [17] |
| After hyperpolarization current | Potentiation (70%) | 3 | Rat hippocampal pyramidal neurons | [16] |
| After hyperpolarizaton | No effect (amplitude) | 1 | GH3 neuroendocrine cells | [35] |
| Increase (four‐fold in recovery time) | ||||
| BK KCa | Activation | 5b | GH3 neuroendocrine cells | [66] |
| BK KCa | Activation (30%) | 10 | GH3 neuroendocrine cells | [35] |
| BK KCa | Activation (2 fold increase) | 5 | Human retinal pigment epithelial cells | [67] |
| BK KCa | Activation (2 fold increase) | 10 | Human skeletal muscle cells | [50] |
| Human BK KCa1.1 | Activation (2 fold increase) | 100 | HEK‐293 cells | [47] |
| Human SK1 KCa2.1 | Activation (250 nM Ca2+) | 21b | HEK‐293 cells | [47] |
| Rat SK2 KCa2.2 | Activation (250 nM Ca2+) | 12.8b | HEK‐293 cells | [47] |
| Human SK3 KCa2.3 | Activation (250 nM Ca2+) | 12.5b | COS‐7 cells | [47] |
| Rat SK3 | Activation (2.3 fold increase, 100 nM Ca2+) | 3 | HEK‐293 cells | [68] |
| Human SK4 KCa3.1 | Activation (250 nM Ca2+) | 1.9b | HEK‐293 cells | [47] |
| Rat SK2 K(Ca) | Activation (Ca2+ free) | 43b | HEK‐239 cells | [16] |
| (100 nM Ca2+) | 0.11b | |||
aIC50.
bEC50.
Table 4.
Effects of riluzole on voltage‐gated Ca2+ currents and channels
| Current/channel | Effect (%) | Concentration (μM) | Tissue | Reference |
|---|---|---|---|---|
| Voltage‐gated calcium currents and channels | ||||
| HVA Ca2+ current | No effect | 10 | Neonatal rat hypoglossal motor neurons | [76] |
| HVA Ca2+ current | No effect | 10 | GH3 neuroendocrine cells | [35] |
| Human N‐type HVA Ca2+ current | No effect | 200 | HEK‐293 cells | [45] |
| HVA Ca2+ current | No effect | 100, 300 | Cultured embryonic rat cortical neurons | [41] |
| HVA Ca2+ current | Inhibition (20%) | 10 | Dissociated rat cortical neurons | [43] |
| LVA Ca2+ current | Inhibition (12%) | 10–20 | Dissociated rat cortical neurons | [43] |
| L‐type HVA Ca2+ current | Inhibition | 22 | Cultured embryonic rat motor neurons | [80] |
| Transient HVA Ca2+ current | Inhibition | 43 | Dissociated neonatal rat DRG neurons | [82] |
| Sustained HVA Ca2+ current | Inhibition | 40 | Dissociated neonatal rat DRG neurons | [82] |
| L‐type HVA CaV1.2 | Inhibition | 30a | HEK‐293 cells | [47] |
aIC50.
Effects of Riluzole on Specific Ion Channels
Voltage‐Gated Sodium Currents and Channels
Rapidly Inactivating Voltage‐Dependent Na+ Current
Prior to 1996, two studies reported that riluzole decreased peak Na+ current and shifted the voltage‐dependence of Na+ channel inactivation to lower voltages in frog myelinated nerve fibers [39] or rat brain type IIA (the NaV 1.2 isoform) Na+ channels expressed in Xenopus oocytes [40]. This action occurred at concentrations of 0.1–1 mM (Table 2) and was not associated with open channel block or changes in the voltage‐dependence of Na+ channel activation [39, 40]. Since then, numerous studies have largely replicated these findings in neurons [14, 18, 20, 35, 41, 42, 43, 44, 45, 46, 47], with the main difference being that lower concentrations of riluzole were sometimes able to reduce fast inactivating Na+ currents (Table 2). Riluzole (10 μM) decreased the amplitude of a tetrodotoxin (TTX)‐sensitive fast Na+ current by 20 to 30% in immortalized GH3 or GT1 neuroendocrine cells, but did not alter the voltage dependence of current activation or inactivation [35]. In cultured embryonic rat cortical neurons, riluzole decreased peak fast Na+ currents and shifted the voltage dependence of inactivation to more negative levels [41]. Stefani et al. [43] found that riluzole inhibited the fast inactivating Na+ current in dissociated mature rat cortical neurons, with a maximal inhibition of peak Na+ current of >80% at concentrations of 10 or 30 μM. Similarly, the peak fast Na+ current in dissociated cerebellar Purkinje neurons from neonatal rat was strongly inhibited by riluzole [45]. In dissociated mouse sympathetic superior cervical ganglion neurons, riluzole inhibited peak fast Na+ current with an IC50 of 33.7 μM, more than 10‐fold higher than the IC50 of 2.7 μM for inhibition of persistent Na+ current in the same neurons [48].
A study by Song et al. [44] revealed differences in the actions of riluzole on TTX‐sensitive (TTX‐S) and TTX‐resistant (TTX‐R) Na+ currents native to cultured rat dorsal root ganglion cells. At 3 μM, riluzole blocked 50% of the TTX‐S current without effect on its activation or inactivation time constant, while 30 μM riluzole was required to block 50% of the TTX‐R current and the effect of riluzole was associated with a faster inactivation time constant (Fig. 2), an effect not observed in previous studies [39, 40] but also present in dissociated rat brainstem neurons [42]. Riluzole caused a negative shift in the voltage‐dependence of Na+ channel inactivation which was larger for TTX‐S current than for TTX‐R current, but there was a relatively small difference in the affinity of riluzole for inactivated TTX‐S and TTX‐R Na+ channels (Table 2). In contrast to previous studies [39, 40], Song et al. [44] observed that riluzole also caused a positive shift in the voltage dependence of Na+ channel activation, with this shift being more pronounced for TTX‐R Na+ channels. Song et al. [44] concluded that the preferential inhibition of TTX‐S Na+ channels by riluzole was due to the greater proportion of TTX‐S Na+ channels in the inactive state at negative potentials.
Figure 2.
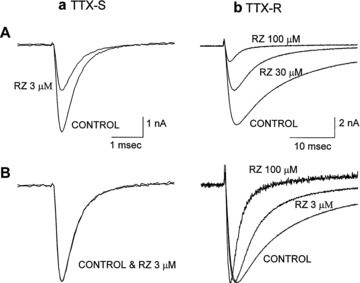
Effects of riluzole (RZ) on tetrodotoxin‐sensitive (TTX‐S) and tetrodotoxin‐resistant (TTX‐R) sodium channel currents of rat dorsal root ganglion neurons. Currents were evoked by depolarizing steps to 0 mV from a holding potential of −80 mV. TTX‐S currents activated and inactivated rapidly and were selectively blocked by 200 nM TTX, whereas TTX‐R currents activated and inactivated more slowly and were unaffected by 200 nM TTX. (a) TTX‐sensitive sodium channel. (b) TTX‐resistant sodium channel. (A) Riluzole blocks TTX‐sensitive sodium channel currents more potently than TTX‐resistant sodium channel currents when the membrane was held at −80 mV. (B) Peak current amplitude in the presence of riluzole is normalized to the control current. Riluzole did not alter the activation and inactivation kinetics of TTX‐S currents, while the time course of inactivation of TTX‐R currents was accelerated. Reprinted from [44], copyright (1997), with permission from the American Society for Experimental Pharmacology and Therapeutics.
These effects of riluzole may be largely confined to Na+ channel isoforms found in neurons. Doble [2] noted that riluzole had only small inhibitory effects on Na+ channel isoforms (NaV 1.4 and 1.5) in isolated guinea pig cardiac Purkinje fibers, decreasing peak action potential amplitude by ∼20% at 100 μM. Riluzole had no effect at 0.01 mM on human skeletal muscle Na+ channels (NaV 1.4) expressed in HEK‐293 cells and only caused a relatively small decrease in peak current (22%) at higher concentration (up to 1 mM) [49]. However, currents from human NaV 1.4 channels expressed in HEK‐293 cells, rat NaV 1.5 channels expressed in COS‐7 cells, and native human NaV 1.5 channels in cultured human skeletal muscle have been subsequently reported to be inhibited by low concentrations of riluzole [47, 50] (see Table 2).
These latter studies suggest that asthenia (a general feeling of weakness), one of the more common side‐effects of riluzole reported in ALS patients [11], may be due in part to decreased muscular tissue excitability, as riluzole has minor effects of riluzole on muscle nicotinic acetylcholine receptors [49, see later and Table 5]. However, as similar doses of riluzole will also reduce central motoneuronal activity, a dose study of riluzole effects on nerve–muscle coupling would be useful.
Table 5.
Effects of riluzole on neurotransmission and ligand‐gated neurotransmitter receptors
| Current/channel | Effect (%) | Concentration (μM) | Tissue | Reference |
|---|---|---|---|---|
| Excitatory neurotransmission and transmitter release | ||||
| Evoked population spike and field EPSP | Inhibition | 5a | Rat CA1 hippocampal neurons | [89] |
| Evoked cortical field potential | Inhibition | 29.5a | Rat cortex | [13] |
| Evoked excitatory transmission | Inhibition | 6a | Rat striatal neurons | [12] |
| Evoked excitatory transmission | Inhibition (45%) | 10 | Rat lumbar spinal cord interneurons and motor neurons | [24] |
| Evoked glutamate EPSC amplitude | Inhibition (8%) | 0.5 | Cultured neonatal rat hippocampal neurons | [46, 93] |
| (33%) | 10 | |||
| (85%) | 20 | |||
| Excitatory miniature EPSCs | No effect | 10 | Rat lumbar spinal cord interneurons and motor neurons | [24] |
| Excitatory miniature EPSCs | No effect | 10 | Neonatal rat hypoglossal motor neurons | [27] |
| Release of [3H]glutamate | Inhibition (77%) | 19.5a | Human, rat and mouse neocortex slices | [88] |
| Release of [3H]dopamine | Inhibition (72%) | 6.8a | Human, rat and mouse neocortex slices | [88] |
| Release of [3H]dopamine | Inhibition | 1–10a | Rat striatum synaptosomes | [197] |
| Release of [3H]acetylcholine | Inhibition (92%) | 3.3a | Human, rat and mouse neocortex slices | [88] |
| Release of [3H]serotonin | Inhibition (53%) | 39.8a | Human, rat and mouse neocortex slices | [88] |
| Evoked glutamate EPSC amplitude | Inhibition (11%) | 1 | Adult mouse hippocampal CA1 neurons | [92] |
| Excitatory amino acid receptors | ||||
| Glutamate‐elicited depolarization | Inhibition (20%) | 30 | Rat striatal neurons | [12] |
| (40%) | 100 | |||
| Glutamate‐elicited depolarization | No effect | 30 | Rat cortical neurons | [13] |
| Radioligand binding at glutamate receptors | Inhibition (30%) | 100 μM | Rat brain | [85] |
| Radioligand binding at glutamate receptors | No effect | 100 μM | Rat brain | [47] |
| Kainate‐elicited depolarization | Inhibition | 101a | Cultured rat cortical neurons | [97] |
| Kainate‐elicited depolarization | No effect | 20 | Cultured neonatal rat hippocampal neurons | [93] |
| Rat Kainate glutamate receptor | Inhibition | 167a | Xenopus oocytes | [95] |
| Radioligand binding at kainate glutamate receptors | No effect | 100 μM | Rat brain | [47] |
| Rat NMDA glutamate receptor | Inhibition | 18a | Xenopus oocytes | [95] |
| Radioligand binding at NMDA glutamate receptors | No effect | 100 μM | Rat brain | [47] |
| NMDA‐elicited depolarization | No effect | 20 | Cultured neonatal rat hippocampal neurons | [93] |
| Evoked NMDA glutamate receptor EPSC | Inhibition (37%) | 20 | Cultured neonatal rat hippocampal neurons | [93] |
| Inhibitory amino acid receptors | ||||
| Radioligand binding at GABAA receptors | No effect | 100 μM | Rat brain | [85] |
| Rat GABAA receptors | No effect | 10 μM to 1 mM | HEK‐293 cells | [103] |
| Rat GABAA receptors | Inhibition (54%, 1 mM GABA) | 30 | HEK‐293 cells | [105] |
| Potentiation (2.3‐fold, 3 μM GABA) | 30 | |||
| GABAA receptor | Potentiation | 53b | Cultured rat hippocampal neurons and oocytes | [104] |
| Evoked GABA IPSC amplitude | Inhibition (24%) | 20 | Cultured neonatal rat hippocampal neurons | [93] |
| Radioligand binding at GABAB receptors | No effect | 100 μM | Rat brain | [85] |
| Evoked glycinergic IPSC | Inhibition (87%) | 10 | Neonatal rat hypoglossal motor neurons | [76] |
| Miniature glycinergic IPSC | No effect | 10 | Neonatal rat hypoglossal motor neurons | [76] |
| Rat glycine receptors | No effect | 30 μM to 1 mM | HEK‐293 cells | [103] |
| Radioligand binding at glycine receptors | No effect | 100 μM | Rat brain | [47, 85] |
| Other ligand‐gated neurotransmitter receptors | ||||
| 5‐HT3 receptor | Inhibition | 3.6a | NCB‐20 neuroblastoma cells | [106] |
| Human skeletal muscle nicotinic acetylcholine receptors | Inhibition (32%) | 1 mM | HEK‐293 cells | [49] |
| Nicotinic acetylcholine receptors | No effect | 10 | Neonatal rat hypoglossal motor neurons | [27] |
| Nicotinic acetylcholine receptors | No effect | 1–100 | Cultured bovine adrenal chromaffin cells | [81] |
| Glutamate transporters | ||||
| [H3]‐glutamate uptake | Potentiation (16%) | 100 | Rat cortical synaptosomes | [101] |
| [H3]‐glutamate uptake | Potentiation (27%) | 100 | GLAST glutamate transporter expressed in HEK‐293 cells | [101] |
| [H3]‐glutamate uptake | Potentiation (37%) | 100 | GLT1 glutamate transporter expressed in HEK‐293 cells | [101] |
| [H3]‐glutamate uptake | Potentiation (39%) | 100 | EAAC1 glutamate transporter expressed in HEK‐293 cells | [101] |
| [H3]‐glutamate uptake | Potentiation (67%) | 0.1 | Rat spinal cord synaptosomes | [99] |
| (47%) | 1 | |||
| No effect | >10 | |||
| [H3]‐glutamate uptake | Potentiation (25–30%) | 10–300 | Rat spinal cord synaptosomes | [100] |
aIC50.
bEC50.
Persistent Na+ Current
Early studies reported that riluzole at low concentrations (200–300 nM) bound selectively to voltage‐gated Na+ channels in an inactive state. This action occurred at 150–300 times lower concentration than for binding to resting Na+ or K+ channels [39, 40]. Since the observation by Urbani and Bellozi [14] that riluzole selectively inhibited the persistent Na+ current and repetitive firing at much lower concentration than that which inhibited the fast inactivating Na+ current (Fig. 3), many studies have shown that the persistent Na+ current in neurons was usually more sensitive to riluzole than the fast inactivating Na+ current [20, 25, 43, 48, 51, 52]. In rat carotid body neurons, 10 μM riluzole suppressed the persistent Na+ current evoked by slow voltage ramps but did not inhibit fast Na+ current evoked by voltage steps [52]. Many other studies have shown that the persistent Na+ current in different neurons was strongly inhibited by relatively low (<1–10 μM; see Table 2) concentrations of riluzole [15, 18, 20, 21, 24, 25, 29, 31, 36, 48,53, 54, 55, 56].
Figure 3.
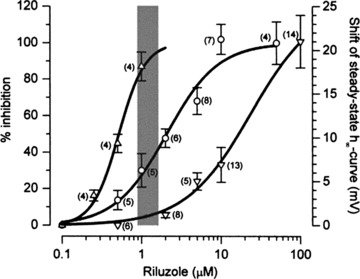
Overlaid view of the dose–response curves of riluzole on firing rate, INaP, and fast sodium current shows that decreases in firing are most likely due to inhibition of INaP. From left to right: concentration–response curves showing the effects of riluzole on firing rate (▵, left vertical axis), on INaP amplitude (○, left vertical axis), and on the shift of steady‐state inactivation curve for fast sodium current (∇, right vertical axis). The effect on firing rate was computed as the percentage ratio of the total number of events in 30 successive stimulations in tests over controls. For all concentrations INaP was recorded at the test potential of −15 mV (holding potential of −75 mV). Data relative to INaP were pooled after they had been normalized within each cell with respect to controls. For the shifts of the steady‐state inactivation curve (actually negative), absolute values are given. The gray band represents the putative plasma concentrations of riluzole [177] at the suggested therapeutic dose (2 × 50 mg/day). For all curves, data points are given as mean ± SEM, n within parentheses. Reprinted from [14], copyright (2000), with permission from John Wiley & Sons.
However, it should be noted that higher concentrations of riluzole (≥10 μM) did not completely suppress the persistent Na+ current in some neurons [57] and that low doses of riluzole did sometimes also inhibit fast Na+ currents [42, 47, 48]. It is therefore important to independently verify dose‐dependent effects of riluzole on persistent and fast Na+ currents rather than assume that riluzole selectively blocks the persistent Na+ current at low concentrations.
Potassium Currents and Channels
There is only one report of effects of riluzole on K+ currents prior to 1996. Benoit and Escande [58] found that kinetically distinct components of total K+ current in frog myelinated axon were differentially inhibited by riluzole, with two fast activating components exhibiting half‐inhibition at approximately 20‐fold lower concentrations (∼20 μM) than for the slow activating component (∼400 μM) (Table 3). Since 1996, riluzole has been reported to modulate many types of K+ currents. The effects of riluzole are highly dependent on the type of K+ current, the dose of riluzole used, and the tissue or cell type; the actions of riluzole on functionally identified K+ currents, or specific K+ channel isoforms, are summarized later and in Table 3.
A‐type K+ Currents
A‐type K+ currents are rapidly activated by depolarization and show varying rates of inactivation [59]. Riluzole inhibited different voltage‐gated K+ channel types producing A‐type K+ currents at concentrations usually ≥100 μM (see later and Table 3), although Zona et al. [41] reported that riluzole (up to 500 μM) had no effect on a rapidly inactivating A‐type K+ current present in cultured embryonic rat cortical neurons. Effects of riluzole on specific K+ channel isoforms producing A‐type K+ currents are briefly described here.
-
1
Kv1.1 and Kv1.3 produce voltage‐dependent rapidly activating and slowly inactivating A‐type currents [59, 60]. Current produced by mouse Kv1.1 channels stably expressed in NIH/NT3 cells was inhibited by riluzole with an IC50 of 92 μM, whereas mouse Kv1.3 channel currents were inhibited with an IC50 of 50 μM [47] (Table 3).
-
2
Kv1.4 produces a voltage‐dependent rapidly activating and slowly inactivating A‐type K+ current often present in axons and synaptic terminals [59]. Riluzole reversibly inhibited this current in dissociated bovine adrenal zona fasiculata cells (IC50 70 μM) and irreversibly slowed its rate of inactivation (Table 3) [61]. Riluzole did not alter the voltage‐dependence of activation or inactivation of this current, while slowing of the inactivation rate was blocked by intracellular application of reducing agents, suggesting that this effect was due to interactions of riluzole with an intracellular portion of the channel protein susceptible to oxidation [61].
-
3
Kv1.5 produces a voltage‐dependent rapidly activating and slowly inactivating A‐type K+ current in vascular smooth muscle, where riluzole (100 μM) inhibited peak Kv1.5 current by ∼30% (Table 3) [62].
-
4
Kv4.2 and Kv4.3 produce classical voltage‐dependent, rapidly activating and inactivating A‐type K+ currents [59]. Riluzole inhibited rat Kv4.3 channels stably expressed in Chinese Hamster Ovary cells with an IC50 of 115.6 μM, and shifted the steady‐state inactivation curve for this current in the hyperpolarizing direction (Table 3) [63]. The inhibitory action of riluzole was use‐dependent, suggesting that riluzole acted by binding to the closed inactivated state of these channels, and that depolarization was needed to unbind riluzole and allow Kv4.3 channels to enter the open state [63]. Riluzole also inhibited rat Kv4.2 expressed in LTK cells with an IC50 of 130 μM [47].
Delayed Rectifier K+ Currents
Delayed rectifier K+ currents are slowly activated by depolarization and show varying levels of slow inactivation or no inactivation [59]. Many K+ channel genes can contribute to production of delayed rectifier K+ current in native cells, making the molecular identity of this native current often difficult to ascertain. Zona et al. [41] reported that riluzole (3–100 μM) selectively inhibited a slowly activating delayed rectifier K+ current evoked by depolarizing steps in cultured embryonic rat cortical neurons (Table 3). Effects of riluzole on specific K+ channel isoforms producing delayed rectifier K+ currents are briefly described here.
-
1
Kv1.5 produces a voltage‐dependent rapidly activating and slowly inactivating delayed rectifier K+ current [59]. Riluzole reversibly inhibited Kv1.5 current stably expressed in CHO cells (Table 3); this action was not use‐dependent, and did not require G proteins as it was not blocked by preincubation with pertussis toxin [64]. Riluzole did not alter the voltage dependence of activation for this current, but did shift the voltage dependence of inactivation to more negative voltages and accelerated the kinetics of inactivation [64]. MEL cells transfected with human Kv1.5 channels also express a rapidly activating delayed rectifier K+ current [60], which was inhibited by riluzole with an IC50 of 95 μM [47] (Table 3).
-
2
Kv3.1 and Kv3.2 produces rapidly activating and slowly inactivating delayed rectifier currents [59]. Riluzole reversibly inhibited the current (Table 3) produced by Kv3.1 channels stably expressed in CHO cells, and shifted the inactivation curve to more negative values [64]. Currents produced by human Kv3.1 channels stably expressed in HEK‐293 cells were inhibited with an IC50 of 95 μM, whereas currents produced by rat Kv3.2 channels stably expressed in COS‐7 cells were inhibited with an IC50 of 100 μM [47].
-
3
hERG (Kv11.1) produces a rapidly activating or constitutively open delayed rectifier K+ current in cardiac muscle and some neurons [59]. Current from hERG channels expressed in HEK‐293 cells was inhibited by riluzole with an IC50 of 50 μM [47].
Calcium‐Dependent Potassium Channels
Ca2+‐dependent K+ channels (K(Ca)) are activated by increases in intracellular Ca2+ and contribute to the amplitude and time course of the after‐hyperpolarization following action potentials [65]. Riluzole either has no effect [17] or enhances the action potential after‐hyperpolarization [16, 35]. Effects of riluzole on native or expressed K(Ca) isoforms are summarized later and in Table 3.
Large Conductance (BK) K(Ca) Channels
Several studies have reported that riluzole stimulated large conductance (BK) K(Ca) channels at concentrations between 1 and 100 μM. Riluzole increased the amplitude of the BK K(Ca) current in rat neuroendocrine GH3 and PC12 cells, via an intracellular action which increased the open channel probability without changing the single channel conductance [66]. A similar direct increase in BK K(Ca) channel activity has also been reported for human retinal pigment epithelial cells [67], and for native BK K(Ca) channels recorded from cultured human skeletal muscle cells [50]. Iberiotoxin, a specific blocker of BK K(Ca) current [65], blocked the effect of riluzole (1–10 μM) in slowing the rate of recovery of the action potential afterhyperpolarization in immortalized neuroendocrine GH3 cells [35], whereas riluzole (10 μM) increased an iberiotoxin‐sensitive steady state K+ current in GH3 and GT1 cells [35]. Current produced by human KCa1.1 channels expressed in HEK‐293 cells was doubled in amplitude with 100 μM riluzole [47].
Small and Intermediate Conductance (SK) K(Ca) Channels
Riluzole also enhances small conductance (SK) K(Ca) current in recombinant expression systems and cultured hippocampal neurons. Human SK1 channels expressed in HEK‐293 cells are activated by riluzole with an EC50 of 21 μM [47]. Riluzole caused a shift in the calcium dependence of recombinant rat SK2 channel activity to lower intracellular Ca2+ concentrations [16], and activated SK2 current with an EC50 of 12.8 μM [47]. Riluzole application (30 and 100 μM) to cultured rat hippocampal neurons increased the IAHP following long depolarizing voltage steps and decreased tonic firing frequency [16]. Rat SK3 channels expressed in HEK293 cells were also activated by riluzole at concentrations higher than 3 μM (Table 3); this action of riluzole required the presence of intracellular Ca2+[68]. In COS‐7 cells transfected with human KCa2.3 (SK3), riluzole had no effect on current slope conductance at 1 μM, but increased slope conductance 11 fold at 10 μM and 30 fold at 100 μM; EC50 was 12.5 μM [47].
Riluzole is also highly potent in stimulating intermediate conductance K (Ca) channels in recombinant systems. Human KCa3.1 (SK4) channels expressed in COS‐7 cells showed a six‐fold increase in current slope conductance at 1 μM riluzole, and 30‐fold increases at both 10 and 100 μM riluzole; EC50 was 1.9 μM [47]. Riluzole inhibited efflux of Rb+ and proliferation of human prostate cancer cells by blocking an intermediate conductance K(Ca) [69].
Two Pore K+ Channels
Riluzole can activate members of the two pore K+ (2PK+) channel family of voltage‐independent K+ channels that contribute significantly to the resting “leak” conductance of many neurons [70] (Table 3). Riluzole caused a sustained dose‐dependent activation of whole cell TRAAK 2PK+ current, or of individual TRAAK channels transiently expressed in COS cells, without inactivation during prolonged riluzole application [71, 72]. In contrast, activation of whole cell TREK‐1 or TREK‐2 2PK+ currents by riluzole was only transient, and was rapidly followed by an inhibition of these currents in COS cells [71, 73] and bovine adrenocortical cells [74]. This inhibition required a soluble intracellular second messenger, as it was not present in TREK‐1 currents recorded from excised membrane patches [71]. The difference in the action of riluzole on these 2PK+ channels was due to the generation of intracellular cAMP and PKA‐mediated phosphorylation of a intracellular site present on the TREK‐1 but not the TRAAK channel protein [71]. Current produced by TREK‐1 stably expressed in an unidentified cell line was activated by riluzole with an EC50 of 110 μM [47]. In contrast to these reports of activation of 2PK+ currents by riluzole, a TEA‐insensitive, voltage‐independent outward K+ current (thought by the authors to be most like the THIK‐1 member of the 2PK+ family) in mouse cerebellar Purkinje neurons was inhibited by high concentrations (500 μM) of riluzole [75].
Voltage‐Gated Calcium Currents and Channels
Umemiya and Berger [76] first found that riluzole (10 μM) did not alter voltage‐gated Ca2+ currents in neonatal rat hypoglossal motoneurons. Since this report, several groups have found that riluzole can act to directly decrease voltage‐gated Ca2+ currents or to inhibit neuronal processes dependent on these currents. The pertinent results are summarized later and in Table 4.
Neuronal Processes Dependent on Voltage‐Gated Ca2+ Currents
The inhibitory effect of riluzole (1 μM) on glutamate release from rat cerebral cortex synaptosomes was blocked by ω‐agatoxin‐IV [77], a specific blocker of P/Q‐type high voltage activated (HVA) Ca2+ channels [78], suggesting that riluzole may block this Ca2+ channel type. In IMR32 neuroblastoma cells, reduction of intracellular Ca2+ concentration by riluzole (100 μM to 1 mM) was mediated partly by blockade of L‐type HVA Ca2+ channels and partly by blockade of release from intracellular Ca2+ stores [79]. In cultured embryonic rat motor neurons, intracellular Ca2+ transients resulting from Ca2+ entry through HVA L‐type Ca2+ channels were blocked by riluzole [80]. In cultured bovine adrenal chromaffin cells, catecholamine secretion mediated by depolarization‐induced opening of HVA Ca2+ channels was not blocked by riluzole (1–100 μM), but veratridine‐induced catecholamine secretion was, indicating that riluzole acted to inhibit Na+ channels and not voltage‐gated Ca2+ channels [81].
Direct Effects on Voltage‐Gated Ca2+ Currents
Riluzole did not block HVA Ca2+ currents in immortalized neuroendocrine GH3 cells at 10 μM [35], at 20 μM in cultured hippocampal neurons [46] or at higher concentrations (100–300 μM) in cultured embryonic rat cortical neurons [41]. In contrast, Stefani et al. [43] reported that riluzole (10–20 μM) inhibited both HVA and low voltage‐activated (LVA) Ca2+ currents in dissociated neonatal or mature rat cortical neurons (Table 4). Maximal inhibition of HVA Ca2+ peak current was 20%, while maximal inhibition of LVA Ca2+ current was 12%, at a riluzole concentration of 10 or 30 μM, respectively [43]. The N‐type HVA Ca2+ current expressed in HEK‐293 cells showed 17% inhibition at 100 μM and 60% inhibition at 300 μM [45]. L‐type HVA Ca2+ current produced by Cav1.2 channels expressed in HEK‐293 cells was inhibited by riluzole with an IC50 of 30 μM [47].
Huang et al. [82] did a detailed study of the effects of riluzole on different components of the HVA Ca2+ current in dissociated dorsal root ganglion neurons from neonatal rat. Riluzole inhibited both the transient and sustained components of the HVA Ca2+ current with similar potency (Table 4). Riluzole had no effect on the voltage dependence of activation, and had differing effects on the two kinetic components of inactivation of the HVA Ca2+ current. The inhibitory effects of riluzole on the HVA current were due to selective inhibition of N‐type and P/Q‐type, but not L‐type HVA Ca2+ currents, as the inhibitory effect of riluzole was significantly less in the presence of ω‐conotoxin‐GVIA or ω‐agatoxin‐IV but not nimodipine, which are specific blockers of these respective Ca2+ channel types [78, 83].
Effects on Neurotransmission
Excitatory Amino Acid Neurotransmission
Studies prior to 1996 suggested that riluzole inhibited the release of excitatory amino acids from brain tissue, as it decreased glutamate receptor‐dependent convulsions [84], decreased glutamate receptor‐dependent second messenger formation [85], reduced spontaneous glutamate release [86] and depressed glutamate receptor agonist‐induced motoneuron firing [2, 87]. As noted by Doble [2], none of these early studies addressed the question of whether or not riluzole directly blocked neuronal responses to excitatory amino acids. Significant doubt remains over this issue to the current time; results since 1996 are summarized later and in Table 5.
In a recent study, riluzole inhibited electrically evoked release of glutamate, acetylcholine, dopamine, and, to a lesser extent, serotonin (but not noradrenaline) from human, rat, and mouse cortex slices [88]. In rat hippocampal slices, riluzole depressed the extracellular population spike and field EPSP, consistent with a presynaptic depression of glutamate release [89]. Centonze et al. [12] showed that riluzole (3–100 μM) reduced the amplitude of non‐NMDA glutamate receptor‐mediated EPSCs in rat striatal neurons (Fig. 4; Table 5). These authors reported that riluzole did not change paired pulse facilitation of the evoked EPSCs but reduced the response to direct postsynaptic activation of glutamate receptors (Fig. 4), and concluded that the effect on EPSC amplitude was due to direct inhibition of non‐NMDA glutamate receptor responses by riluzole. In contrast, riluzole (2–10 μM) greatly reduced both non‐NMDA and NMDA evoked EPSC amplitude to the same extent in cultured hippocampal neurons and increased paired pulse depression, but did not alter responses of directly activated glutamate receptors [46]; these responses are consistent with a presynaptic reduction of glutamate release [90, 91]. Similarly, evoked glutamatergic EPSC amplitude and charge in CA1 neurons of adult mouse hippocampus were reduced by 1 and 10 μM riluzole, with similar reduction of the non‐NMDA and NMDA receptor mediated components [92], again consistent with presynaptic reduction of glutamate release. In vivo administration of riluzole significantly reduced glutamate levels in rat spinal cord dorsal horn (see later and Fig. 6). This is consistent with a 45% reduction in the area of monosynaptic EPSPs evoked by dorsal root stimulation and recorded in neonatal rat lumbar spinal motoneurons in vitro by application of 10 μM riluzole; this reduction was due to a presynaptic reduction in excitability or in transmitter release, as miniature glutamatergic EPSCs in the same neurons were not reduced in either amplitude or frequency by the same dose of riluzole [24]. In contrast, miniature glutamatergic EPSC frequency and amplitude recorded in neonatal rat hypoglossal motoneurons were reduced by 10 μM when miniature EPSC frequency was high, but not when frequency was low [27]; this effect of riluzole was occluded by prior treatment with a protein kinase C inhibitor or with an NMDA glutamate receptor antagonist.
Figure 4.
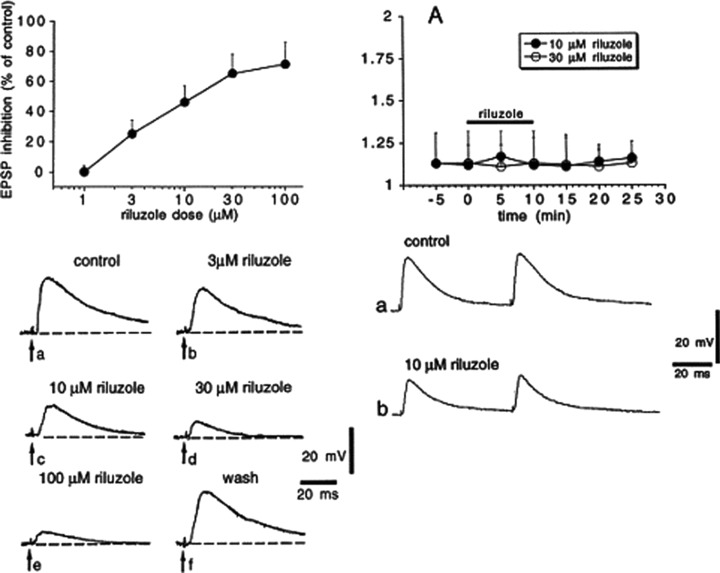
Riluzole inhibits non‐NMDA glutamate receptor excitatory postsynaptic potentials (EPSPs) in rat striatal spiny neurons but does not alter paired pulse facilitation. (Left panel) Riluzole inhibits EPSPs evoked by cortical stimulation. The graph in the upper part of the figure shows the dose–response curve obtained at various concentrations of riluzole on the amplitude of corticostriatal EPSPs. Each data point was obtained from at least four single experiments; the IC50 for EPSP inhibition was 6 μM. The lower part of the left panel shows averages (four single sweeps) of EPSPs recorded from a striatal spiny neuron under control condition, during the application of four different concentrations of riluzole and after 30 min washout. Each concentration was applied for 10 min. The resting membrane potential of the cell was −87 mV and was constant throughout the experiment. (Right panel) Riluzole does not alter paired pulse facilitation. (A) The graph shows the amplitude ratio of the second EPSP response to the first EPSP response (EPSP2:EPSP1) before, during, and after the application of two different concentrations of riluzole (black bar). Traces in the lower part of the figure show synaptic responses to paired stimulation under control condition (a) and after 10 min application of 10 μM riluzole (b). Reprinted from [12], copyright (1998), with permission from Elsevier.
Figure 6.
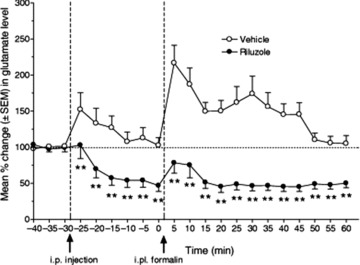
Pretreatment with riluzole (12 mg/kg i.p.) produced significant antinociceptive effects following hind paw injection of formalin. The percentage change in glutamate content of in vivo microdialysate from rat thoracic spinal cord was measured by reverse phase HPLC with fluorescence detection. Riluzole (•) significantly reduced the formalin‐induced increase in spinal glutamate both in the first phase (from 5 to 15 min following i.p. injection of riluzole) and in the second phase (from 20 to 60 min) after formalin injection (**p < 0.01), compared to sham‐injected animals (○). Riluzole also significantly reduced basal spinal glutamate levels throughout the testing period. Reprinted from [129], copyright (2007), with permission of John Wiley & Sons.
Apart from the results of Centonze et al. [12], published data so far largely supports presynaptic reduction of excitatory amino acid release by riluzole. The dose range from these studies is consistent with a reduction of presynaptic excitability by inhibition of presynaptic voltage‐gated Na+ channels (either persistent or fast inactivating) [46, 93, 94], although inhibition of presynaptic voltage‐gated Ca2+ channels cannot be ruled out as a contributing factor.
Direct Effects of Riluzole on Neurotransmitter Receptors
Glutamate Receptors
Work prior to 1996 found that riluzole inhibited NMDA (IC50 of 18 μM) or kainic acid (IC50 of 167 μM) evoked—currents evoked in Xenopus oocytes expressing rat NMDA or kainate glutamate receptors [95]. However, radioligand‐binding studies failed to demonstrate any interaction of riluzole (100 μm) with the NMDA‐, glycine‐, or phencyclidine‐binding sites of the NMDA glutamate receptor, of non‐NMDA glutamate receptors labeled with radioactive glutamate, AMPA or kainic acid, or of metabotropic glutamate receptors coupled to inositol phosphate metabolism [85, 95, 96]. Doble [2] concluded that “These findings indicate that the interaction of riluzole with excitatory amino acid‐mediated transmission may indeed be indirect.” Data published since then continues to suggest that direct effects of riluzole on glutamate receptors are limited, and usually require high concentrations.
Depolarization induced in rat cortical neurons in brain slices by brief bath applications of glutamate (300 μM to 1 mM) was not blocked by the presence of riluzole (30 μM) [13]. Non‐NMDA and NMDA glutamate receptors directly activated by application of kainic acid or NMDA to cultured hippocampal neurons were not altered by 20 μM riluzole [46]. Kainate glutamate receptor currents in cultured rat cortical neurons were reduced by riluzole (5–1000 μM; Table 1) by a noncompetitive mechanism, which decreased the open probability of kainate glutamate receptor channels without changing channel conductance [97]. Non‐NMDA glutamate receptors activated in rat striatal neurons in brain slices by brief bath applications of glutamate (300 μM to 1 mM) were reduced by up to 40% by riluzole (30–100 μM) [12]. Radioligand binding to the NMDA site of NMDA glutamate receptors or to kainate glutamate receptors in rat brain was not inhibited by riluzole (100 μM) [47].
Chronic application of riluzole may also alter glutamate receptor expression. Treatment of cultured mouse hippocampal neurons with 20 or 100 μM riluzole significantly increased surface expression of the GluR1 and GluR2 AMPA glutamate receptors [98]. This increase was correlated with enhanced membrane depolarization in response to AMPA application, increased phosphorylation of the S845 protein kinase A site of the GluR1 AMPA glutamate receptor and increased total GluR1 and GluR2 AMPA glutamate receptor protein levels in hippocampal neuron cultures, while chronic in vivo treatment of mice with riluzole (10 mg/kg i.p.) increased phosphorylation of the S845 protein kinase A site of the GluR1 AMPA receptor, without changes in total GluR1 or GluR2 AMPA glutamate receptor protein levels [98].
Effects of Riluzole on Glutamate Transporters
Glutamate uptake by rat spinal cord synaptosomes was increased by low concentrations (0.1–1 μM) of riluzole through a pertussis toxin‐sensitive mechanism [99], whereas higher concentrations of riluzole (10–300 μM) were needed to increase glutamate uptake in rat spinal cord synaptosomes in wild type and G93A SOD1 transgenic rats in a later study [100]. In both of these studies, maximal increase in uptake was 25–30% (Table 5). The three major glutamate tranporters, GLAST, EAAC1 and GLT1, stably expressed in HEK‐293 cells, all showed a dose‐dependent increase in activity up to a maximum of 30% with riluzole (0.01–100 μM) [101] (Table 5); this study also briefly reported that 100 μM riluzole increased glutamate uptake in rat cortical synaptosomes by a maximum of 16%. A microdialysis study in adult rat hippocampus found that riluzole (1 mM in dialysate) completely inhibited increases in extracellular glutamate levels evoked by iodoacetate, an inhibitor of glycolysis, but did not alter basal glutamate levels when perfused alone [102].
Effects of Riluzole on Glycine and GABA Receptors
GABA Receptors
Early work found that riluzole did not have a high affinity for GABAA, GABAB or glycine receptors, as 100 μM riluzole did not significantly displace radiolabeled ligands for these receptors [85]. Evidence since 1996 suggests that low concentrations of riluzole may enhance GABAA currents, while higher concentrations will inhibit these currents (Table 5).
GABAergic IPSCs recorded from autaptic inhibitory neurons in neonatal rat hippocampus cultures were reduced in amplitude by 24% by 20 μM riluzole; the IPSC decay time constant was also significantly increased [93]. Steady state currents evoked by application of 1 mM GABA to HEK‐293 cells expressing heteromeric GABAA receptors (α1β2γ2) were substantially inhibited by high concentrations of riluzole (0.1–1 mM) [103]. This inhibitory effect was accompanied by enhancement of GABAA current desensitization, without change in peak current amplitude, suggesting a mechanism other than open channel block. In contrast, He et al. [104] reported that GABAA currents evoked by application of lower concentrations of GABA (2 μM) to hippocampal neuron cultures or to Xenopus oocytes expressing heteromeric GABAA receptors (α1β2γ2) were potentiated by lower concentrations of riluzole (20–300 μM; Table 5; Fig. 5). A desensitization of the GABAA current was first observed at 300 μM riluzole (Fig. 5), compatible with the inhibitory effects observed by Mohammadi et al. [103]. Riluzole was capable of directly gating GABAA receptors in both hippocampal neurons and oocytes at concentrations ≥50 μM (Fig. 5), doubled the affinity of GABA for the GABAA receptor at 100 μM, and prolonged the time course of miniature GABAergic IPSCs at 50 μM [104]. Rat GABAA receptors (α1β2γ2s) expressed in cotransfected HEK‐293 cells did not show changes in single channel conductance with coapplication of 1 mM GABA and 100 μM riluzole, but did show shorter open channel durations and accelerated desensitization [105]. Macroscopic GABA currents for the same cotransfection were not significantly reduced in peak amplitude by coapplication of 1 mM GABA and riluzole between 30 and 1000 μM riluzole, but currents elicited by coapplication of 3 μM GABA and between 3 and 30 μM riluzole were potentiated [105]. These authors concluded that riluzole may influence GABAA channels by an open channel block mechanism.
Figure 5.
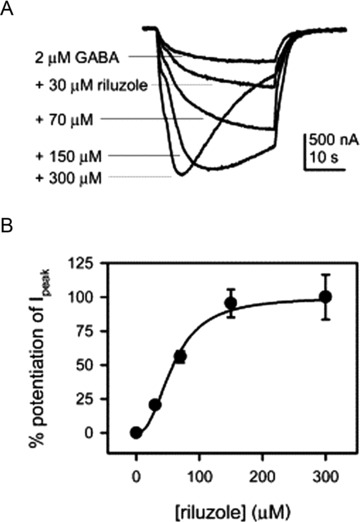
Riluzole concentration‐dependently potentiated GABA responses from heterologously expressed receptors in Xenopus oocytes. (A) Sample recordings showing the potentiation of the response to 2 μM GABA induced by different concentrations of riluzole. With high concentrations of riluzole, GABA responses usually displayed apparent desensitization. (B) Concentration dependence of the effect of riluzole on the responses to 2 μM GABA. Normalized potentiation relative to the peak potentiation by 300 μM riluzole is plotted against riluzole concentration. Smooth curves are fit to the Hill equation. The Hill coefficient is 2.4, and riluzole EC50 (the concentration of riluzole that induces half‐maximal potentiation) is 58.7 μM (n = 5). Before normalization, the amplitudes of potentiation caused by different concentrations of riluzole were: 75.7 ± 7.1% from 30 μM riluzole, 208.0 ± 16.1% from 70 μM riluzole, 353.6 ± 38.1% from 150 μM riluzole, and 370.8 ± 61.5% from 300 μM riluzole. Reprinted from [104], copyright (2002), with permission from Elsevier.
Glycine Receptors
An earlier study reported that in rat hypoglossal motoneurons, the amplitude of glycinergic IPSCs evoked by electrical stimulation was reduced by 83% by 10 μM riluzole but the amplitude of minature glycinergic IPSCs was not altered by riluzole, indicating that riluzole did not directly modulate postsynaptic glycine receptors [76] (Table 5). Radioligand‐binding studies (Table 5) have found no effects of riluzole (100 μM) on glycine receptors from rat brain [47, 85]. High concentrations of riluzole (0.5–1 mM) inhibited steady state currents evoked by application of 1 mM glycine to HEK‐293 cells expressing heteromeric glycine receptors (α1β), albeit to a lesser extent than the inhibition of GABAA receptors in the same study [103].
Nicotinic Acetylcholine Receptors
Riluzole did not inhibit nicotine‐induced Na+ influx through nicotinic acetylcholine receptors in cultured bovine adrenal chromaffin cells [81] and caused a relatively small (22.2%) inhibition of human nicotinic acetylcholine receptor currents in HEK‐293 cells at a concentration of 1 mM [49]. Nicotine‐elicited currents in neonatal mouse hypoglossal motoneurons were not inhibited by 10 μM riluzole [27].
5‐HT3 Ligand‐Gated Ion Channels
The 5‐HT3 receptor is a ligand‐gated ion channel which responds to both serotonin and dopamine. Rapid application of either agonist to NCB‐20 neuroblastoma cells elicited rapid inward currents which were reduced in amplitude and rise slope by riluzole. The IC50 for reducing the rise slope of serotonin‐elicited currents was 3.6 μM, and 4.8 μM for dopamine‐elicited currents [106], suggesting that riluzole directly slowed 5HT‐3 channel activation, as blockade of G protein modulation did not alter responses to riluzole. As serotonin acting on 5HT‐3 receptors is thought to promote gut motility [107], functional antagonism of 5HT‐3 receptor responses by riluzole should inhibit gut motility. It is thus unlikely that direct effects of riluzole on 5HT‐3 receptors would cause diarrhea, one side‐effect of riluzole treatment in humans [108].
Effects of Riluzole Treatment in Transgenic Animal Models of ALS
The identification of inherited gene mutations in the Cu–Zn superoxide dismutase 1 (SOD1) enzyme in familial ALS [109] rapidly led to the development of several transgenic animal models which express or over‐express different human SOD1 mutations and display similar symptoms to those seen in human ALS [110, 111, 112, 113]. All mouse or rat strains expressing human SOD1 mutations display symptoms of peripheral motor axonal degeneration and central motor neuron loss, albeit with different latencies to symptom onset and rates of symptom progression to end‐stage between strains [113]. The most commonly used transgenic strain for research or therapeutic testing is the B6SJL‐Tg(SOD1*G93A)1Gur/J strain, which expresses up to 25 copies of the G93A SOD1 mutation [114]. Most studies reporting effects of riluzole treatment have utilized this transgenic strain. It should be noted that as the copy number of the mutant G93A SOD1 gene increases, the age of symptom onset decreases and rate of symptom progression increases, thus shortening lifespan [113]. The B6SJL‐Tg(SOD1*G93A)1Gur/J strain displays symptom onset at 3 months and symptom progression for 1–2 months, with an average lifespan of 134 ± 10 days (range 104–179 days) [115]. Within this strain, transgene copy number variation and gender have significant effects on lifespan, with lower copy number extending life, and females living an additional 4 days [115]. It is important to control for gender and copy number in therapeutic studies, and it is also important to control for animal deaths not due to ALS symptoms, and to match litter‐mates carefully [115].
In studies using small treatment groups (∼10–15 animals) controlled for some (gender, copy number) but not all of the above variables, riluzole alone modestly (∼10%) prolonged the lifespan of mutant SOD1 transgenic animals (Table 6) [116, 117, 118, 119, 120, 121]. Riluzole treatment alone delayed the onset of motor symptoms by 0–30 days, prolonged mean lifespan by 14–21 days, and delayed declines in motor activity compared to wild‐type controls [116, 120]. Differences in effects between these studies most likely reflected the dosage or route of administration of riluzole, the age at which treatment began, or animal gender [113, 118]. In contrast, a recent critical reevaluation, using rigorous matching of larger groups (∼35 animals per group) by gender and litter, with controls for copy number, reported no significant effects of riluzole treatment on survival, and raised significant doubt about the validity of published riluzole effects on animal survival [115]. Allowing for the fact that large clinical trials with hundred to thousands of human patients are required to detect an approximately 2 month increase in lifespan (∼0.33% at 50 years of age), equivalent to less than a day in the mouse model, this result suggests that future animal treatment trials will require larger groups, careful control of variables, and preferably multiple measures of outcome.
Table 6.
Studies of riluzole effects in transgenic mice
| G93A strain | Scott et al. (2008) High copy number | Guerney et al. (1996) High copy number | Guerney et al. (1998) High copy number | Snow et al. (2003) Low copy number | Waibel et al. (2004) High copy numbera | Del Signore et al. (2009) High copy number (?) |
|---|---|---|---|---|---|---|
| Dose | 44 mg/kg in water | 100 μg/mL in water | 44 mg/kg in food | 100 μg/mL in water | 30 mg/kg in water | 16 mg/kg in water |
| Started at (days) | 50 | 50 | 42 | 40 | 60 | 30 |
| Group size | 35 (riluzole) | 9 (riluzole) | 11 | 14 (riluzole) | 15 | 10 |
| 34 (control) | 8 (control) | 17 (control) | ||||
| Groups controlled for | ||||||
| Copy number | Yes | Yes | Yes | No | No | No |
| Gender | Yesb | Yes | Yesc | No | No | Yes (M) |
| Litter mates | Yes | No | No | No | Noa | d |
| Exclusion criteria | Yes | Yese | Yes | Nof | No | No |
| Onset (days) | ||||||
| Control | ND | 95 ± 12 | ND | 161 ± 8f | ND | ND |
| G93A | ND | 98 ± 11 | ND | 185 ± 5f | ND | ND |
| Significance | N.S. | <0.05 | ||||
| Survival (days) | ||||||
| Control | 132 | 134 ± 8 | 127 ± 6 | ND | 210 ± 7.5 | 126 ± 3 |
| G93A | 135 | 148 ± 14 | 139 ± 3 | ND | 234 ± 13 | 135 ± 4 |
| Significance | NS | 0.039e | 0.047c | N.S. | <0.05 | |
aAll animals used were second generation crosses, with much longer survival times (200 days), cf. first generation (∼130 days) which were high copy number G93A mutants.
bAll treatment groups had equal numbers of males and females.
cNumbers of males and females in control (five males, six females) and riluzole treatment (four males and seven females) were different; Student's t‐test used for comparison.
dAll animals used were male and born within 4 days of each other; litter mate balancing not specified.
eReported deaths due to non‐ALS symptoms; Student's t‐test used for comparison.
fAll animals sacrificed at 199 days; none had reached end‐stage disease at this age. Age of onset measured by author from Figure 1 of reference.
Several studies have also investigated the effect of combining riluzole treatment with other treatments or supplements. Combined treatment with riluzole plus rasagaline resulted in significant extension of lifespan compared to riluzole alone [120]. Combination of riluzole with creatinine or vitamin E supplements did not delay symptom onset or extend lifespan compared to riluzole alone [117], but did improve a panel of motor symptoms [118]. Combination treatment with riluzole, minocycline, and nimodipine significantly delayed onset of muscle weakness, hind–limb paralysis, and animal lifespan by 4–6 weeks compared to untreated controls [119]. Combination treatment with sodium phenylbutyrate, a histone deacetylase inhibitor, increased mean survival time in G93A SOD1 mutant mice by 21.5%, compared to the separate administration of riluzole alone (7.5%) or sodium phenylbutyrate alone (12.8%); all forms of treatment improved grip strength and reduced body weight loss [121]. Again, these studies used small group sizes, and did not control for all significant variables; repetition of treatment with creatine, minocycline, or sodium phenylbutyrate alone to larger and carefully controlled groups did not replicate previous findings of significant effects [115].
Effects of Riluzole on Neuronal Survival and Neural Growth Factors
Riluzole treatment in vivo or in vitro appears to promote neuronal survival. Chronic treatment of organotypic cortical slices from neonatal rat with malonate, an inhibitor of mitochondrial energy production [122] or glutamate uptake inhibitors [123], caused significant loss of large pyramidal neurons in layers V and II/III, as occurs in human cortex at early (layer V) and more advanced stages (layers II/III) of ALS [124]. This neuronal death was inhibited by cotreatment with riluzole (10 μM) [122]. In adult rats, spinal cord ventral root avulsion normally leads to massive motoneuron death, even after ventral root reimplantation [125]. Following ventral root reimplantation, riluzole treatment (4 mg/kg i.p., once daily for 7 days followed by once every 2 days for 7 days), either immediately after avulsion [126] or up to 10 days postavulsion [125] rescued the majority of motor neurons from death and facilitated axon regrowth into the avulsed ventral root. Similarly, riluzole (5 and 10 mg/kg i.p. daily for 14 days) also enhanced motor neuron survival following peripheral nerve section in neonatal mice [127]. In a mouse line displaying motor neuron death due to progressive motor neuronopathy, riluzole (8 mg/kg/day by gavage) also delayed the onset of symptoms (hind–limb weakness), increased lifespan and improved tests of motor performance (grip strength and electromyography) [128]. In vivo treatment of rats with riluzole (6–12 mg/kg i.p.) following chronic constriction of the sciatic nerve significantly reduced the development of mechanical and cold hypersensitivity, and reduced both baseline and formalin‐induced glutamate (Fig. 6) and aspartate release in the spinal dorsal horn [129]; this effect of riluzole on excitatory transmitter release was particularly significant, as it established the in vivo efficacy of riluzole as an inhibitor of glutamate release, as shown in vitro for synaptosomes [77] and brain slices [88]. Finally, riluzole administered by intra‐cochlear infusion or intraperitoneal injection protected guinea pig cochlear hair cells against noise trauma‐induced cell death with an ED50 of 17 μM [130].
Riluzole also enhances production of various neural growth factors, an action which may explain at least some of its survival‐promoting effects. Caumont et al. [131] found that riluzole (1 μM) strongly up‐regulated GDNF mRNA and protein levels in cultured rat glioma cells via a mitogen‐activated protein kinase extracellular signal‐related kinase‐regulated transcription pathway. Upregulation of BDNF production in dentate granule neurons, hilus, and stratum radiatum of the CA3 regions of the mouse hippocampus was stimulated by a single intraperitoneal injection of riluzole (19 mg/kg) in adult mice, and repeated injections produced a sustained increase in BDNF production with enhanced production of new dentate gyrus cells [132]; this increase in BDNF production also required activation of the p38 mitogen‐activated protein kinase via N‐type Ca2+ channels and adenosine A1 receptors [133]. In cultured embryonic rat cortical neurons, glutamate decreased axonal transport of neurofilaments and induced phosphorylation of both ERK and p38 mitogen‐activated protein kinase; these effects of glutamate were attenuated by riluzole (25–100 μM) [134]. In cultured mouse astrocytes, riluzole (100 μM) strongly up‐regulated NGF, GDNF, and BDNF mRNA levels [135]. Riluzole increased heat shock induction of the hsp70 protein in cultured HeLa cells in a dose‐dependent manner with maximal effect at 1–3 μM; this increase was due to posttranslational mechanisms, as hsp70 mRNA expression was not altered [136].
Other In Vivo Effects of Riluzole in Animals
In wild‐type mice dosed orally by inclusion of 200 μg/mL in drinking water (approximately 40 mg/kg), high performance liquid chromatography measured plasma levels of 0.5 μg/mL and brain tissue levels of 2.3 μg/g, while G93A SOD1 mutant littermate mice had lower brain tissue levels of 0.2 μg/g [137].
There have been relatively few recent in vivo studies of the acute effects of riluzole on normal physiological parameters. In adult rats, riluzole decreased motor coordination and behavior in a dose‐dependent manner (1–4 mg/kg i.p.) and had anticataleptic properties, counteracting effects of dopamine receptor antagonists [138]. Coadministration of NMDA or non‐NMDA receptor ligands produced effects which suggested that riluzole did not act as an NMDA receptor antagonist in these neurobehavioral tasks [138].
Respiratory Regulation and Rhythm Generation
The central pattern generator of respiratory muscle movements has been hypothesized to be critically dependent on pacemaker properties of ventrolateral medullary neurons in an area called the pre‐Bötzinger complex [139]. Two significantly reduced preparations—rhythmically active rodent medullary slices in vitro[139] and in situ arterially perfused rodent brainstem–spinal cord preparations [140] generate inspiratory motor activity and allow good pharmacological access to brain tissue, but are physically and functionally reduced compared to the in vivo situation. This section later reviews the effects of riluzole on rhythmic activity in these reduced preparations and in vivo.
In Vivo Effects of Riluzole
Riluzole (2 mg/kg) decreased minute ventilation, metabolic rate, and inspiratory motor activity, and blunted respiratory responses to hypoxia but not hypercapnia in rats [52]. Intracisternal administration of riluzole (0.4–6 nmol) in neonatal mice under normoxia increased inspiratory frequency at all doses, but decreased inspiratory half duration only at the highest dose [141]. In contrast, riluzole (3 or 6 mg/kg, i.v.) decreased respiratory frequency and increased the amplitude of inspiratory motor bursts in unanesthetized adult rats [142], while anaesthetized adult rats given the same dose showed no change in respiratory frequency but hypoxia‐induced gasping was abolished [56]. Under severe hypoxia (3% O2), riluzole decreased the frequency of respiratory gasps at all doses, and reduced the ability of mice to autoresuscitate from apnea in a dose‐dependent way, following restoration to normoxic conditions [141].
In Vitro Effects of Riluzole
Several studies have reported that riluzole (5–20 μM) reduced persistent Na+ current in rhythmically active pre‐Bötzinger complex neurons [34, 36, 56]. Riluzole also blocked rhythmic neuronal activity in some [143, 25 or 50 μM] but not other cases [34, at 10–20 μM; 53, at up to 200 μM; 144, at 10 μM]. In the rhythmically active neonatal mouse brainstem slice, riluzole (30 μM) also substantially reduced intracellular increases in Na+ and Ca2+ concentration evoked by a brief period of hypoxia, as well as decreasing the membrane depolarization and diminishing rhythmic synaptic drive to neurons firing during the inspiratory phase [145].
In an in situ arterially perfused juvenile rat preparation with intact pontomedullary structures (precollicular decerebration), phrenic nerve discharge increased in frequency and decreased in amplitude when riluzole (1–10 μM) was added to the perfusate, and respiratory gasps were eliminated [142]; a similar change was seen when riluzole was administered after blockade of flufenamic acid [146], a drug which has been shown to eliminate pacemaker activity in pre‐Bötzinger complex neurons in vitro which are insensitive to riluzole [34]. In the same preparation, riluzole (1–5 μM) eliminated intrinsic neuronal bursting activity in pre‐Bötzinger complex neurons when excitatory and inhibitory synaptic transmission was blocked; when synaptic transmission was intact, riluzole (1–5 μM) blocked respiratory gasps elicited by hypoxia, but had no effect on normoxic rhythmic motor output at concentrations up to 20 μM [56]. In contrast, rhythmic phrenic nerve discharge in an in situ arterially perfused juvenile mouse preparation without pontine respiratory structures was completely blocked by addition of riluzole (10 or 20 μM) to the perfusate [147].
Current evidence suggests that, while rhythmic bursting activity in most pre‐Bötzinger complex neurons may be driven by a riluzole‐sensitive persistent Na+ current, this pacemaker activity does not appear to be essential for generation of the respiratory rhythm under normoxic conditions, although it may assume importance under hypoxic conditions which promote respiratory gasping.
Locomotion Rhythm Generation
In brainstem–spinal cord preparations from neonatal mouse or rat, riluzole at low concentrations (5–10 μM), which had been previously shown to suppress persistent Na+ currents in motoneurons and interneurons, reduced or abolished rhythmic bursts of motor activity or burst activity in individual motoneurons and interneurons [24, 29, 148].
Clinical Effects of Riluzole in Human ALS
Clinical Trials of Riluzole Effects in Treating ALS
Since the original clinical trials which led to the licensing of riluzole for use in treating ALS [149, 150], several studies have consistently confirmed that riluzole has small but significant beneficial effects in prolonging life span and moderating some declines in motor function of ALS patients [151, 152, 153, 154].
A meta‐analysis of three randomized double blind clinical studies of riluzole treatment in ALS [149, 150, 166], using death or tracheostomy as an endpoint, concluded that riluzole at 100 mg/day had a modest effect, prolonging survival to endpoint of ALS patients by about 2–3 months [155]. It should be emphasized that effects of riluzole on survival curves in the original clinical trials analyzed were small. Meta‐analysis also concluded that, while individual clinical trials did not show any effect of riluzole on motor function, the meta‐analysis revealed that riluzole had small but significant effects on bulbar and limb function, but not on muscle strength [155]. Quality of life scores and ALSFRS‐R scores were not assessed in these controlled studies; as the ALSFRS‐R is significantly correlated with survival, a controlled trial using this measure would provide useful data [155].
Other open label, nonrandomized studies have suggested that treatment with riluzole at earlier disease stages may have greater benefits [153, 156, 157]. Patients with bulbar onset ALS may also benefit more from riluzole treatment [153, 158], and lifespan increase may be greater in ALS patients with symptom onset at older ages [158]. Individual studies suggest that riluzole therapy produced prolongation of the time spent in the initial or milder stages of ALS but had little effect on time spent in advanced or severe disease stages [157].
Combination treatment of human ALS patients with riluzole plus other agents has not led to significantly better clinical outcomes, compared to treatment with riluzole alone. Studies using low [159] or high dose vitamin E [160] found no benefit of coadministered vitamin E compared to riluzole alone.
Side Effects of Riluzole Treatment in ALS Patients
A meta‐analysis of controlled double blind clinical trials concluded that drug safety was not a major concern in treatment, with nausea, asthenia, and elevated serum alanine transaminase being the only side effects which were significantly increased in patients receiving riluzole, compared to placebo [155]. Asthenia is of particular interest, as clinically relevant levels of riluzole could act to decrease motor neuron excitability and neurotransmitter release. The fact that riluzole usually blocks repetitive firing at therapeutic concentrations, but requires higher than therapeutic concentrations to block single action potential generation, suggests that riluzole could contribute to asthenia by lowering individual motor neuron firing rate, thus delaying fusion of muscle twitches in a single motor unit to tetany. Quantitative studies on effects of riluzole on neuromuscular junction transmission in animals or humans are needed to address this question.
Subsequent study has shown that riluzole administration in ALS patients was well tolerated for periods of up to 7 years [161], and riluzole can be administered to all patients except those with elevated serum transaminase levels or liver disease. Adverse effects most commonly observed were somnolence, nervousness, anorexia, asthenia, circumoral paraesthesia, headache, itching, diarrhea, depression, nausea, and reversible increases in transaminase levels [108, 162, 163, 164, 165]. Incidence of these adverse drug effects were not related to riluzole dosage in the range from 50 to 200 mg/day [161, 163]. Only the incidence of diarrhea was positively correlated with serum concentration of riluzole, while muscle fasiculations and cramps showed decreased incidence as plasma concentrations of riluzole increased [108]. Administration of riluzole to elderly patients or advanced stage patients did not increase the incidence of adverse effects [163, 166]. An increased risk of neutropenia during riluzole therapy has also been reported [162, 167]. Other case reports of possible adverse reactions to riluzole therapy in ALS include hypersensitivity pneumonitis [168], pancreatitis [169], methemoglobinemia [170], reversible granulocytopenia [171], and hypertension [172].
Pharmacokinetics of Rilzuole in Humans
Human plasma concentrations of riluzole at clinically effective concentrations are in the range between 0.5 and 2 μM [108, 173]. Relatively high interindividual variations in plasma concentrations have been reported [108, 174]. It is thought that this may be related to individual variations in the rate of metabolism of cytochrome P450 isoenzyme 1A2 responsible for riluzole metabolism [175], although a recent study found no correlation between riluzole plasma concentration and polymorphisms in cytochrome P450 isoenzymes in ALS patients treated with riluzole [176]. It should be noted that animal pharmacokinetic studies indicate that levels of riluzole in brain tissue may be up to four‐fold higher than plasma levels [137], so that plasma levels may not be a good guide to effective therapeutic levels.
There is no gender difference in plasma pharmacokinetics, but mean terminal elimination half life was longer in healthy elderly adults [163, 177]. In a study of plasma concentrations of riluzole in ALS patients, riluzole clearance was independent of dosage (from 25 to 100 mg/kg twice daily), treatment duration, age, and renal function, but was significantly lower in women compared to men, and in smokers compared to nonsmokers [178], consistent with a report that mean plasma concentrations were higher in ALS patients who were women or smokers [108]. Most recently, survival in a group of ALS patients was found not to be correlated with individual plasma levels of riluzole [179].
Neurophysiological Effects of Riluzole Treatment in Humans
Treatment with riluzole has been reported to have no effects on clinical EMG tests commonly used to measure the progression of ALS, including fasciculations, polyphasic motor unit potentials, increased motor fiber jitter and density, and increased amplitude and area of motor units [180]. In normal human subjects, riluzole at clinical doses had no effect on the motor threshold or intracortical inhibition, but decreased intracortical facilitation, as assessed by paired transcranial magnetic stimulation [173, 181], and did not alter F‐wave amplitudes or latencies assessed by peripheral nerve stimulation [181]. ALS patients often showed deficient paired pulse inhibition, presumably due to cortical hyper‐excitability [182, 183]. One study showed that riluzole treatment partially restored deficient paired pulse inhibition but had no effect on paired pulse facilitation [184], while others report that riluzole treatment was without effect on motor evoked potentials or paired pulse inhibition [182, 185].
Summary
Riluzole has wide‐ranging neural effects on a number of factors which can influence neuronal activity and survival. These effects occur over a large dose range (<1 to >1000 μM) and only some of these factors are influenced at doses (<5 μM) likely to be relevant to the clinical effects of riluzole in ALS patients. The use of riluzole in ALS treatment has now been well‐established as consistently extending lifespan (albeit by a modest time span) and appears relatively free of side effects, with similar effects observed in animal models of ALS.
At low concentrations of riluzole (<1–10 μM), the most prominent and consistent effects are depression of repetitive firing frequency and suppression of the persistent Na+ current. This is likely that these two effects are causally linked. Several studies have reported nerve or neuronal hyper‐excitability in human ALS patients [186, 187] and neuronal hyper‐excitability is also associated with increased persistent Na+ current in motoneurons and cortical neurons in the G93A SOD1 mutant mouse model of ALS [15, 26, 188]. Evaluation of other drugs which selectively suppress the persistent Na+ current as ALS treatments may be a useful avenue for future investigation.
Enhancement of calcium‐dependent K+ currents by riluzole (2–20 μM) may also contribute to slowing of firing frequency, by increasing interspike interval. In contrast, inhibition of various voltage‐dependent A‐type or delayed rectifier K+ currents, which can modulate neural excitability, requires higher concentrations of riluzole (20–100 μM), and this effect is much less likely to influence neuronal firing frequency.
Riluzole consistently and strongly suppresses the persistent Na+ current in a wide variety of neurons at concentrations <10 μM. However, some caution should be exercised when using riluzole as a pharmacological tool to selectively suppress this current. Where possible, the effects of riluzole (at concentrations used to suppress persistent Na+ current) on fast Na+ and K(Ca) currents should be tested and lack of effect verified, particularly in neurons where no data on riluzole effects on these currrents exist.
Other low dose effects of riluzole include reduction of transmitter release (1–20 μM) and inhibition of voltage‐gated Ca2+ channels (10–40 μM). Current evidence is most consistent with an action of riluzole which causes a presynaptic reduction of transmitter release, rather than direct suppression of postsynaptic neurotransmitter receptor responses. Inhibition of presynaptic Ca2+ channels is a parsimonious explanation for the effects of riluzole on evoked neurotransmission. While the effect of riluzole on Ca2+ channels is limited in degree and also tends to require higher doses of riluzole, the highly nonlinear relationship between presynaptic Ca2+ influx and transmitter release means that small reductions in Ca2+ influx can lead to larger reductions in transmitter release. An alternative (or complementary) mechanism for reducing neurotransmitter release is inhibition of presynaptic terminal excitability by riluzole, due to fast and/or persistent Na+ current suppression [93, 94, 189]. A careful study of the dose–response relationship for inhibition of Na+ and Ca2+ currents, and reduction of transmitter release in the same neuron type would be a useful addition to our knowledge of the actions of riluzole on synaptic transmission.
The direct effect of riluzole on ligand‐gated neurotransmitter receptors appears to be relatively limited. One report found that glutamate receptor responses are depressed by riluzole (≥30 μM) but several other studies have found no effects on glutamate receptors at ≤100 μM. GABAA receptors show potentiation at low concentrations (up to 30 μM) of riluzole and depression at higher concentrations (≥100 μM). Riluzole (<100 μM) had no effect on glycine receptors, while the kinetics of 5HT3 receptors were altered by riluzole (∼3 μM). It is likely that depression of synaptic responses by riluzole is principally mediated by reduction of transmitter release, rather than depression of neurotransmitter responses. Evidence of effects of riluzole on G protein‐coupled neurotransmitters is very limited, despite some evidence that riluzole could modulate G protein‐coupled signaling pathways.
Riluzole enhances neuronal survival in vivo and in vitro at concentrations between 1 and ∼100 μM. This action is often associated with enhanced production of various neurotrophic factors, such as GDNF or BDNF, in neurons and glia via activation of the MAPK signaling pathway. How riluzole acts to activate MAPK signaling and regulate downstream neurotrophic factors remains unclear. As neuronal survival and neurotrophic factor production is often dependent on activity‐dependent synaptic interactions between presynaptic neurons and their postsynaptic targets of other neurons or muscle [190, 191, 192, 193], investigation of the interplay between riluzole's effects on neuronal firing and regulation of synaptic transmitter release, and the production of these neurotrophic factors should be a fruitful avenue of investigation.
In whole animals, riluzole produces some sedation and inhibition of movement and slight changes in respiratory rate and amplitude. Rhythmic motor activity, such as breathing and locomotion, has been hypothesized to require neuronal pacemaker activity dependent on persistent Na+ current, based on in vitro experiments using reduced preparations. Riluzole has been widely used as a selective blocker of the persistent Na+ current in these preparations, but interpretation of results remains uncertain without careful controls for other low dose effects of riluzole.
In animals, riluzole consistently inhibited respiratory gasping induced by hypoxia and may prevent autoresuscitation of respiratory rhythm following a hypoxic challenge. End‐stage disease in human ALS patients often involves chronic daytime hypoventilation due to respiratory muscle insufficiency and intermittent nocturnal hypoxia associated with sleep‐disordered breathing [194, 195, 196]. It would be worthwhile to investigate whether voluntary ventilation and/or sleep‐disordered breathing could be improved in end‐stage ALS patients by withdrawal of riluzole treatment.
In human ALS patients and rodent models of ALS, riluzole treatment produces modest extensions of lifespan without significant side effects in the majority of cases. As plasma concentrations of riluzole following clinical doses is 1–2 μM, with brain tissue concentrations probably three‐ to four‐fold higher, the effects of riluzole most likely to be relevant in slowing the progression of ALS are those present at <10 μM: reduction of neuronal firing, reduction of the persistent Na+ current, potentiation of K(Ca) currents and presynaptic reduction of neurotransmitter release.
References
- 1. Domino EF, Unna KR, Kerwin J. Pharmacological properties of benzazoles. I. Relationship between structure and paralyzing action. J Pharmacol Exp Ther 1952;105:486–497. [PubMed] [Google Scholar]
- 2. Doble A. The pharmacology and mechanism of action of riluzole. Neurology 1996;47:233S–241S. [DOI] [PubMed] [Google Scholar]
- 3. Bruijn LI, Miller TM, Cleveland DW. Unraveling the mechanisms involved in motor neuron degeneration in ALS. Annu Rev Neurosci 2004;27:723–749. [DOI] [PubMed] [Google Scholar]
- 4. Bensimon G, Ludolph A, Agid Y, et al Riluzole treatment, survival and diagnostic criteria in Parkinson plus disorders: The NNIPPS study. Brain 2009;132:156–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Landwehrmeyer GB, Dubois B, de Yebenes JG, et al Riluzole in Huntington's disease: A 3‐year, randomized controlled study. Ann Neurol 2007;62:262–272. [DOI] [PubMed] [Google Scholar]
- 6. Pittenger C, Coric V, Banasr M, et al Riluzole in the treatment of mood and anxiety disorders. CNS Drugs 2008;22:761–786. [DOI] [PubMed] [Google Scholar]
- 7. Zarate CA, Manji HK. Riluzole in psychiatry: A systematic review of the literature. Expert Opin Drug Metab Toxicol 2008;4:1223–1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Mantz J. Rilzuole. CNS Drug Rev 1996;2:40–51. [Google Scholar]
- 9. Wokke J. Riluzole. Lancet 1996;348:795–799. [DOI] [PubMed] [Google Scholar]
- 10. Stutzmann JM, Wahl F, Pratt J, et al Neuroprotective profile of riluzole in in vivo models of acute neurodegenerative diseases. CNS Drug Rev 1997;3:83–101. [Google Scholar]
- 11. Bryson HM, Fulton B, Benfield P. Riluzole. A review of its pharmacodynamic and pharmacokinetic properties and therapeutic potential in amyotrophic lateral sclerosis. Drugs 1996;52:549–563. [DOI] [PubMed] [Google Scholar]
- 12. Centonze D, Calabresi P, Pisani A, et al Electrophysiology of the neuroprotective agent riluzole on striatal spiny neurons. Neuropharmacol 1998;37:1063–1070. [DOI] [PubMed] [Google Scholar]
- 13. Siniscalchi A, Bonci A, Mercuri NB, et al Effects of riluzole on rat cortical neurones: An in vitro electrophysiological study. Br J Pharmacol 1997;120:225–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Urbani A, Belluzzi O. Riluzole inhibits the persistent sodium current in mammalian CNS neurons. Eur J Neurosci 2000;12:3567–3574. [DOI] [PubMed] [Google Scholar]
- 15. Pieri M, Carunchio I, Curcio L, et al Increased persistent sodium current determines cortical hyperexcitability in a genetic model of amyotrophic lateral sclerosis. Exp Neurol 2009;215:368–379. [DOI] [PubMed] [Google Scholar]
- 16. Cao YJ, Dreixler JC, Couey JJ, et al Modulation of recombinant and native neuronal SK channels by the neuroprotective drug riluzole. Eur J Pharmacol 2002;449:47–54. [DOI] [PubMed] [Google Scholar]
- 17. Kuo JJ, Lee RH, Zhang L, et al Essential role of the persistent sodium current in spike initiation during slowly rising inputs in mouse spinal neurones. J Physiol (Lond) 2006;574:819–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Theiss RD, Kuo JJ, Heckman CJ. Persistent inward currents in rat ventral horn neurones. J Physiol (Lond) 2007;580:507–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Reboreda A, Sanchez E, Romero M, et al Intrinsic spontaneous activity and subthreshold oscillations in neurones of the rat dorsal column nuclei in culture. J Physiol (Lond) 2003;551:191–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wu N, Enomoto A, Tanaka S, et al Persistent sodium currents in Mesencephalic V Neurons participate in burst generation and control of membrane excitability. J Neurophysiol 2004;93:2710–2722. [DOI] [PubMed] [Google Scholar]
- 21. Hsiao CF, Gougar K, Asai J, et al Intrinsic membrane properties and morphological characteristics of interneurons in the rat supratrigeminal region. J Neurosci Res 2007;85:3673–3686. [DOI] [PubMed] [Google Scholar]
- 22. Miles GB, Dai Y, Brownstone RM. Mechanisms underlying the early phase of spike frequency adaptation in mouse spinal motoneurones. J Physiol (Lond) 2005;566:519–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Harvey PJ, Li Y, Li X, et al Persistent sodium currents and repetitive firing in motoneurons of the sacrocaudal spinal cord of adult rats. J Neurophysiol 2006;96:1141–1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Tazerart S, Viemari JC, Darbon P, et al Contribution of persistent sodium current to locomotor pattern generation in neonatal rats. J Neurophysiol 2007;98:613–628. [DOI] [PubMed] [Google Scholar]
- 25. Cramer NP, Li Y, Keller A. The whisking rhythm generator: A novel mammalian network for the generation of movement. J Neurophysiol 2007;97:2148–2158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. van Zundert B, Peuscher MH, Hynynen M, et al Neonatal neuronal circuitry shows hyperexcitable disturbance in a mouse model of the adult‐onset neurodegenerative disease amyotrophic lateral sclerosis. J Neurosci 2008;28:10864–10874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lamanauskas N, Nistri A. Riluzole blocks persistent Na+ and Ca2+ currents and modulates release of glutamate via presynaptic NMDA receptors on neonatal rat hypoglossal motoneurons in vitro. Eur J Neurosci 2008;27:2501–2514. [DOI] [PubMed] [Google Scholar]
- 28. Kuo JJ, Siddique T, Fu R, et al Increased persistent Na+ current and its effect on excitability in motoneurones cultured from mutant SOD1 mice. J Physiol (Lond) 2005;563:843–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ziskind‐Conhaim L, Wu L, Wiesner EP. Persistent sodium current contributes to induced voltage oscillations in locomotor‐related hb9 interneurons in the mouse spinal cord. J Neurophysiol 2008;100:2254–2264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Dorval AD Jr., White JA. Channel noise is essential for perithreshold oscillations in entorhinal stellate neurons. J Neurosci 2005;25:10025–10028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kononenko NI, Shao LR, Dudek FE. Riluzole‐sensitive slowly inactivating sodium current in rat suprachiasmatic nucleus neurons. J Neurophysiol 2004;91:710–718. [DOI] [PubMed] [Google Scholar]
- 32. Del Negro CA, Koshiya N, Butera RJ Jr., et al Persistent sodium current, membrane properties and bursting behavior of pre‐Bötzinger complex inspiratory neurons in vitro. J Neurophysiol 2002;88:2242–2250. [DOI] [PubMed] [Google Scholar]
- 33. Darbon P, Yvon C, Legrand JC, et al INaP underlies intrinsic spiking and rhythm generation in networks of cultured rat spinal cord neurons. Eur J Neurosci 2004;20:976–988. [DOI] [PubMed] [Google Scholar]
- 34. Pena F, Parkis MA, Tryba AK, et al Differential contribution of pacemaker properties to the generation of respiratory rhythms during normoxia and hypoxia. Neuron 2004;43:105–117. [DOI] [PubMed] [Google Scholar]
- 35. Beltran‐Parrazal L, Charles A. Riluzole inhibits spontaneous Ca2+ signaling in neuroendocrine cells by activation of K+ channels and inhibition of Na+ channels. Br J Pharmacol 2003;140:881–888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Del Negro CA, Morgado‐Valle C, Feldman JL. Respiratory rhythm: An emergent network property? Neuron 2002;34:821–830. [DOI] [PubMed] [Google Scholar]
- 37. Crill WE. Persistent sodium current in mammalian central neurons. Annu Rev Physiol 1996;58:349–362. [DOI] [PubMed] [Google Scholar]
- 38. Goldin AL. Resurgence of sodium channel research. Annu Rev Physiol 2001;63:871–894. [DOI] [PubMed] [Google Scholar]
- 39. Benoit E, Escande D. Riluzole specifically blocks inactivated Na channels in myelinated nerve fibre. Pflügers Arch 1991;419:603–609. [DOI] [PubMed] [Google Scholar]
- 40. Hebert T, Drapeau P, Pradier L, et al Block of the rat brain IIA sodium channel alpha subunit by the neuroprotective drug riluzole. Mol Pharmacol 1994;45:1055–1060. [PubMed] [Google Scholar]
- 41. Zona C, Siniscalchi A, Mercuri NB, et al Riluzole interacts with voltage‐activated sodium and potassium currents in cultured rat cortical neurons. Neurosci 1998;85:931–938. [DOI] [PubMed] [Google Scholar]
- 42. Ptak K, Zummo GG, Alheid GF, et al Sodium currents in medullary neurons isolated from the pre‐Bötzinger complex region. J Neurosci 2005;25:5159–5170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Stefani A, Spadoni F, Bernardi G. Differential inhibition by riluzole, lamotrigine, and phenytoin of sodium and calcium currents in cortical neurons: Implications for neuroprotective strategies. Exp Neurol 1997;147:115–122. [DOI] [PubMed] [Google Scholar]
- 44. Song JH, Huang CS, Nagata K, et al Differential action of riluzole on tetrodotoxin‐sensitive and tetrodotoxin‐resistant sodium channels. J Pharmacol Exp Ther 1997;282:707–714. [PubMed] [Google Scholar]
- 45. O’Neill MJ, Bath CP, Dell CP, et al Effects of Ca2+ and Na+ channel inhibitors in vitro and in global cerebral ischaemia in vivo. Eur J Pharmacol 1997;332:121–131. [DOI] [PubMed] [Google Scholar]
- 46. He Y, Zorumski CF, Mennerick S. Contribution of presynaptic Na+ channel inactivation to paired‐pulse synaptic depression in cultured hippocampal neurons. J Neurophysiol 2002;87:925–936. [DOI] [PubMed] [Google Scholar]
- 47. Sankaranarayanan A, Raman G, Busch C, et al Naphtho1,2‐d]thiazol‐2‐ylamine (SKA‐31), a new activator of KCa2 and KCa3.1 potassium channels, potentiates the endothelium‐derived hyperpolarizing factor response and lowers blood pressure. Mol Pharmacol 2009;75:281–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Lamas J, Romero M, Reboreda A, et al A riluzole‐ and valproate‐sensitive persistent sodium current contributes to the resting membrane potential and increases the excitability of sympathetic neurones. Pflügers Arch 2009;458:589–599. [DOI] [PubMed] [Google Scholar]
- 49. Mohammadi B, Lang N, Dengler R, et al Interaction of high concentrations of riluzole with recombinant skeletal muscle sodium channels and adult‐type nicotinic receptor channels. Muscle Nerve 2002;26:539–545. [DOI] [PubMed] [Google Scholar]
- 50. Wang YJ, Lin MW, Lin AA, et al Riluzole‐induced block of voltage‐gated Na+ current and activation of BKCa channels in cultured differentiated human skeletal muscle cells. Life Sci 2008;82:11–20. [DOI] [PubMed] [Google Scholar]
- 51. Spadoni F, Hainsworth AH, Mercuri NB, et al Lamotrigine derivatives and riluzole inhibit INa,P in cortical neurons. NeuroRep 2002;13:1167–1170. [DOI] [PubMed] [Google Scholar]
- 52. Faustino EV, Donnelly DF. An important functional role of persistent Na+ current in carotid body hypoxia transduction. J Appl Physiol 2006;101:1076–1084. [DOI] [PubMed] [Google Scholar]
- 53. Del Negro CA, Morgado‐Valle C, Hayes JA, et al Sodium and calcium current‐mediated pacemaker neurons and respiratory rhythm generation. J Neurosci 2005;25:446–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Zhong G, Masino MA, Harris‐Warrick RM. Persistent sodium currents participate in fictive locomotion generation in neonatal mouse spinal cord. J Neurosci 2007;27:4507–4518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. D’Ascenzo M, Podda MV, Fellin T, et al Activation of mGluR5 induces spike afterdepolarization and enhanced excitability in medium spiny neurons of the nucleus accumbens by modulating persistent Na+ currents. J Physiol (Lond) 2009;587:3233–3250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Paton JF, Abdala AP, Koizumi H, et al Respiratory rhythm generation during gasping depends on persistent sodium current. Nat Neurosci 2006;9:311–313. [DOI] [PubMed] [Google Scholar]
- 57. Niespodziany I, Klitgaard H, Margineanu DG. Is the persistent sodium current a specific target of anti‐absence drugs? NeuroRep 2004;15:1049–1052. [DOI] [PubMed] [Google Scholar]
- 58. Benoit E, Escande D. Fast K channels are more sensitive to riluzole than slow K channels in myelinated nerve fibre. Pflügers Arch 1993;422:536–538. [DOI] [PubMed] [Google Scholar]
- 59. Coetzee WA, Amarillo Y, Chiu J, et al Molecular diversity of K+ channels. Ann N Y Acad Sci 1999;868:233–285. [DOI] [PubMed] [Google Scholar]
- 60. Grissmer S, Nguyen AN, Aiyar J, et al Pharmacological characterization of five cloned voltage‐gated K+ channels, types Kv1.1, 1.2, 1.3, 1.5 and 3.1, stably expressed in mammalian cell lines. Mol Pharmacol 1994;45:1227–1234. [PubMed] [Google Scholar]
- 61. Xu L, Enyeart JA, Enyeart JJ. Neuroprotective agent riluzole dramatically slows inactivation of Kv1.4 potassium channels by a voltage‐dependent oxidative mechanism. J Pharmacol Exp Ther 2001;299:227–237. [PubMed] [Google Scholar]
- 62. McGahon MK, Dawicki JM, Arora A, et al Kv1.5 is a major component underlying the A‐type potassium current in retinal arteriolar smooth muscle. Am J Physiol Heart Circ Physiol 2007;292:H1001–H1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Ahn HS, Kim SE, Jang HJ, et al Interaction of riluzole with the closed inactivated state of Kv4.3 channels. J Pharmacol Exp Ther 2006;319:323–331. [DOI] [PubMed] [Google Scholar]
- 64. Ahn HS, Choi JS, Choi BH, et al Inhibition of the cloned delayed rectifier K+ channels, Kv1.5 and Kv3.1, by riluzole. Neurosci 2005;133:1007–1019. [DOI] [PubMed] [Google Scholar]
- 65. Sah P, Faber ESL. Channels underlying neuronal calcium‐activated potassium currents. Prog Neurobiol 2002;66:345–353. [DOI] [PubMed] [Google Scholar]
- 66. Wu SN, Li HF. Characterization of riluzole‐induced stimulation of large‐conductance calcium‐activated potassium channels in rat pituitary GH3 cells. J Invest Med 1999;47:484–495. [PubMed] [Google Scholar]
- 67. Sheu SJ, Wu SN, Hu DN. Stretch‐stimulated activity of large conductance calcium‐activated potassium channels in human retinal pigment epithelial cells. J Ocul Pharmacol Ther 2005;21:429–435. [DOI] [PubMed] [Google Scholar]
- 68. Grunnet M, Jespersen T, Angelo K, et al Pharmacological modulation of SK3 channels. Neuropharmacol 2001;40:879–887. [DOI] [PubMed] [Google Scholar]
- 69. Parihar AS, Coghlan MJ, Gopalakrishnan M, et al Effects of intermediate‐conductance Ca2+‐activated K+ channel modulators on human prostate cancer cell proliferation. Eur J Pharmacol 2003;471:157–164. [DOI] [PubMed] [Google Scholar]
- 70. Lesage F. Pharmacology of neuronal background potassium channels. Neuropharmacol 2003;44:1–7. [DOI] [PubMed] [Google Scholar]
- 71. Duprat F, Lesage F, Patel AJ, et al The neuroprotective agent riluzole activates the two P domain K+ channels TREK‐1 and TRAAK. Mol Pharmacol 2000;57:906–912. [PubMed] [Google Scholar]
- 72. Fink M, Lesage F, Duprat F, et al A neuronal two P domain K+ channel stimulated by arachidonic acid and polyunsaturated fatty acids. EMBO J 1998;17:3297–3308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Lesage F, Terrenoire C, Romey G, et al Human TREK2, a 2P domain mechano‐sensitive K+ channel with multiple regulations by polyunsaturated fatty acids, lysophospholipids, and Gs, Gi, and Gq protein‐coupled receptors. J Biol Chem 2000;275:28398–28405. [DOI] [PubMed] [Google Scholar]
- 74. Enyeart JJ, Xu L, Danthi S, et al An ACTH‐ and ATP‐regulated background K+ channel in adrenocortical cells is TREK‐1. J Biol Chem 2002;277:49186–49199. [DOI] [PubMed] [Google Scholar]
- 75. Bushell T, Clarke C, Mathie A, et al Pharmacological characterization of a non‐inactivating outward current observed in mouse cerebellar Purkinje neurones. Br J Pharmacol 2002;135:705–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Umemiya M, Berger AJ. Riluzole inhibits glycinergic postsynaptic currents in rat hypoglossal motoneurons. Br J Pharmacol 1995;116:3227–3230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Wang SJ, Wang KY, Wang WC. Mechanisms underlying the riluzole inhibition of glutamate release from rat cerebral cortex nerve terminals (synaptosomes). Neurosci 2004;125:191–201. [DOI] [PubMed] [Google Scholar]
- 78. Turner TJ, Adams ME, Dunlap K. Calcium channels coupled to glutamate release identified by omega‐Aga‐IVA. Science 1992;258:310–313. [DOI] [PubMed] [Google Scholar]
- 79. Wang JL, Lee KC, Tang KY, et al Effect of the neuroprotective agent riluzole on intracellular Ca2+ levels in IMR32 neuroblastoma cells. Arch Toxicol 2001;75:214–220. [DOI] [PubMed] [Google Scholar]
- 80. Hubert JP, Burgevin MC, Terro F, et al Effects of depolarizing stimuli on calcium homeostasis in cultured rat motoneurones. Br J Pharmacol 1998;125:1421–1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Yokoo H, Shiraishi S, Kobayashi H, et al Selective inhibition by riluzole of voltage‐dependent sodium channels and catecholamine secretion in adrenal chromaffin cells. Naunyn Schmiedebergs Arch Pharmacol 1998;357:526–531. [DOI] [PubMed] [Google Scholar]
- 82. Huang CS, Song JH, Nagata K, et al Effects of the neuroprotective agent riluzole on the high voltage‐activated calcium channels of rat dorsal root ganglion neurons. J Pharmacol Exp Ther 1997;282:1280–1290. [PubMed] [Google Scholar]
- 83. Takahashi T, Momiyama A. Different types of calcium channels mediate central synaptic transmission. Nature 1993;366:156–158. [DOI] [PubMed] [Google Scholar]
- 84. Mizoule J, Meldrum B, Mazadier M, et al 2‐Amino‐6‐trifluoromethoxy benzothiazole, a possible antagonist of excitatory amino acid neurotransmission–I: Anticonvulsant properties. Neuropharmacol 1985;24:767–773. [DOI] [PubMed] [Google Scholar]
- 85. Benavides J, Camelin JC, Mitrani N, et al 2‐Amino‐6‐trifluoromethoxy benzothiazole, a possible antagonist of excitatory amino acid neurotransmission–II: Biochemical properties. Neuropharmacol 1985;24:1085–1092. [DOI] [PubMed] [Google Scholar]
- 86. Chéramy A, Barbeito L, Godeheu G, et al Riluzole inhibits the release of glutamate in the caudate nucleus of the cat in vivo. Neurosci Lett 1992;147:209–212. [DOI] [PubMed] [Google Scholar]
- 87. Girdlestone D, Dupuy A, Coston A, et al Riluzole antagonizes excitatory amino acid evoked firing in rat facial motoneurons in vivo. Br J Pharmacol 1989;97:583P. [Google Scholar]
- 88. Jehle T, Bauer J, Blauth E, et al Effects of riluzole on electrically evoked neurotransmitter release. Br J Pharmacol 2000;130:1227–1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. MacIver MB, Amagasu SM, Mikulec AA, et al Riluzole anesthesia: Use‐dependent block of presynaptic glutamate fibers. Anesthesiology 1996;85:626–634. [DOI] [PubMed] [Google Scholar]
- 90. Bellingham MC, Walmsley B. A novel presynaptic inhibitory mechanism underlies paired pulse depression at a fast central synapse. Neuron 1999;23:159–170. [DOI] [PubMed] [Google Scholar]
- 91. Ireland MF, Noakes PG, Bellingham MC. P2×7‐like receptor subunits enhance excitatory synaptic transmission at central synapses by presynaptic mechanisms. Neurosci 2004;128:269–280. [DOI] [PubMed] [Google Scholar]
- 92. Rammes G, Zieglgansberger W, Parsons CG. The fraction of activated N‐methyl‐D‐aspartate receptors during synaptic transmission remains constant in the presence of the glutamate release inhibitor riluzole. J Neural Transm 2008;115:1119–1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Prakriya M, Mennerick S. Selective depression of low‐release probability excitatory synapses by sodium channel blockers. Neuron 2000;26:671–682. [DOI] [PubMed] [Google Scholar]
- 94. Huang H, Trussell LO. Control of presynaptic function by a persistent Na+ current. Neuron 2008;60:975–979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Debono M‐W, Le Guern J, Canton T, et al Inhibition by riluzole of electrophysiological responses mediated by rat kainate and NMDA receptors expressed in Xenopus oocytes. Eur J Pharmacol 1993;235:283–289. [DOI] [PubMed] [Google Scholar]
- 96. Doble A, Perrier ML. Pharmacology of excitatory amino acid receptors coupled to inositol phosphate metabolism in neonatal rat striatum. Neurochemistry International 1989;15:1–8. [DOI] [PubMed] [Google Scholar]
- 97. Zona C, Cavalcanti S, De Sarro G, et al Kainate‐induced currents in rat cortical neurons in culture are modulated by riluzole. Synapse 2002;43:244–251. [DOI] [PubMed] [Google Scholar]
- 98. Du J, Suzuki K, Wei Y, et al The anticonvulsants lamotrigine, riluzole, and valproate differentially regulate AMPA receptor membrane localization: Relationship to clinical effects in mood disorders. Neuropsychopharmacol 2007;32:793–802. [DOI] [PubMed] [Google Scholar]
- 99. Azbill RD, Mu X, Springer JE. Riluzole increases high‐affinity glutamate uptake in rat spinal cord synaptosomes. Brain Res 2000;871:175–180. [DOI] [PubMed] [Google Scholar]
- 100. Dunlop J, Beal MH, She Y, et al Impaired spinal cord glutamate transport capacity and reduced sensitivity to riluzole in a transgenic superoxide dismutase mutant rat model of amyotrophic lateral sclerosis. J Neurosci 2003;23:1688–1696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Fumagalli E, Funicello M, Rauen T, et al Riluzole enhances the activity of glutamate transporters GLAST, GLT1 and EAAC1. Eur J Pharmacol 2008;578:171–176. [DOI] [PubMed] [Google Scholar]
- 102. Camacho A, Montiel T, Massieu L. The anion channel blocker, 4,4’‐dinitrostilbene‐2,2’‐disulfonic acid prevents neuronal death and excitatory amino acid release during glycolysis inhibition in the hippocampus in vivo. Neuroscience 2006;142:1005–1017. [DOI] [PubMed] [Google Scholar]
- 103. Mohammadi B, Krampfl K, Moschref H, et al Interaction of the neuroprotective drug riluzole with GABA(A) and glycine receptor channels. Eur J Pharmacol 2001;415:135–140. [DOI] [PubMed] [Google Scholar]
- 104. He Y, Benz A, Fu T, et al Neuroprotective agent riluzole potentiates postsynaptic GABA A receptor function. Neuropharmacol 2002;42:199–209. [DOI] [PubMed] [Google Scholar]
- 105. Jahn K, Schlesinger F, Jin LJ, et al Molecular mechanisms of interaction between the neuroprotective substance riluzole and GABA(A)‐receptors. Naunyn Schmiedebergs Arch Pharmacol 2008;378:53–63. [DOI] [PubMed] [Google Scholar]
- 106. Kim KJ, Cho HS, Choi SJ, et al Direct effects of riluzole on 5‐hydroxytryptamine (5‐HT)3 receptor‐activated ion currents in NCB‐20 neuroblastoma cells. J Pharmacol Sci 2008;107:57–65. [DOI] [PubMed] [Google Scholar]
- 107. Spiller R. Role of motility in chronic diarrhoea. Neurogastroenterol Motil 2006;18:1045–1055. [DOI] [PubMed] [Google Scholar]
- 108. Groeneveld GJ, Van Kan HJ, Kalmijn S, et al Riluzole serum concentrations in patients with ALS: Associations with side effects and symptoms. Neurology 2003;61:1141–1143. [DOI] [PubMed] [Google Scholar]
- 109. Rosen DR, Siddique T, Patterson D, et al Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 1993;362:59–62. [DOI] [PubMed] [Google Scholar]
- 110. Dal Canto MC, Gurney ME. Neuropathological changes in two lines of mice carrying a transgene for mutant human Cu,Zn SOD, and in mice overexpressing wild type human SOD: A model of familial amyotrophic lateral sclerosis (FALS). Brain Res 1995;676:25–40. [DOI] [PubMed] [Google Scholar]
- 111. Gurney ME, Pu H, Chiu AY, et al Motor neuron degeneration in mice that express a human Cu, Zn superoxide dismutase mutation. Science 1994;264:1772–1775. [DOI] [PubMed] [Google Scholar]
- 112. Howland DS, Liu J, She Y, et al Focal loss of the glutamate transporter EAAT2 in a transgenic rat model of SOD1 mutant‐mediated amyotrophic lateral sclerosis (ALS). Proc Natl Acad Sci USA 2002;99:1604–1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Turner BJ, Talbot K. Transgenics, toxicity and therapeutics in rodent models of mutant SOD1‐mediated familial ALS. Prog Neurobiol 2008;85:94–134. [DOI] [PubMed] [Google Scholar]
- 114. Chiu AY, Zhai P, Dal Canto MC, et al Age‐dependent penetrance of disease in a transgenic mouse model of familial amyotrophic lateral sclerosis. Mol Cell Neurosci 1995;6:349–362. [DOI] [PubMed] [Google Scholar]
- 115. Scott S, Kranz JE, Cole J, et al Design, power, and interpretation of studies in the standard murine model of ALS. Amyotroph Lateral Scler Other Motor Neuron Disord 2008;9:4–15. [DOI] [PubMed] [Google Scholar]
- 116. Gurney ME, Fleck TJ, Himes CS, et al Riluzole preserves motor function in a transgenic model of familial amyotrophic lateral sclerosis. Neurology 1998;50:62–66. [DOI] [PubMed] [Google Scholar]
- 117. Gurney ME, Cutting FB, Zhai P, et al Benefit of vitamin E, riluzole, and gabapentin in a transgenic model of familial amyotrophic lateral sclerosis. Ann Neurol 1996;39:147–157. [DOI] [PubMed] [Google Scholar]
- 118. Snow RJ, Turnbull J, da Silva S, et al Creatine supplementation and riluzole treatment provide similar beneficial effects in copper, zinc superoxide dismutase (G93A) transgenic mice. Neurosci 2003;119:661–667. [DOI] [PubMed] [Google Scholar]
- 119. Kriz J, Gowing G, Julien JP. Efficient three‐drug cocktail for disease induced by mutant superoxide dismutase. Ann Neurol 2003;53:429–436. [DOI] [PubMed] [Google Scholar]
- 120. Waibel S, Reuter A, Malessa S, et al Rasagiline alone and in combination with riluzole prolongs survival in an ALS mouse model. J Neurol 2004;251:1080–1084. [DOI] [PubMed] [Google Scholar]
- 121. Del Signore SJ, Amante DJ, Kim J, et al Combined riluzole and sodium phenylbutyrate therapy in transgenic amyotrophic lateral sclerosis mice. Amyotroph Lateral Scler Other Motor Neuron Disord 2009;10:85–94. [DOI] [PubMed] [Google Scholar]
- 122. Van Westerlaak MG, Joosten EA, Gribnau AA, et al Differential cortico‐motoneuron vulnerability after chronic mitochondrial inhibition in vitro and the role of glutamate receptors. Brain Res 2001;922:243–249. [DOI] [PubMed] [Google Scholar]
- 123. Young KC, McGehee DS, Brorson JR. Glutamate receptor expression and chronic glutamate toxicity in rat motor cortex. Neurobiol Dis 2007;26:78–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Nihei K, McKee AC, Kowall NW. Patterns of neuronal degeneration in the motor cortex of amyotrophic lateral sclerosis patients. Acta Neuropathol (Berl) 1993;86:55–64. [DOI] [PubMed] [Google Scholar]
- 125. Nogradi A, Szabo A, Pinter S, et al Delayed riluzole treatment is able to rescue injured rat spinal motoneurons. Neuroscience 2007;144:431–438. [DOI] [PubMed] [Google Scholar]
- 126. Nogradi A, Vrbova G. The effect of riluzole treatment in rats on the survival of injured adult and grafted embryonic motoneurons. Eur J Neurosci 2001;13:113–118. [DOI] [PubMed] [Google Scholar]
- 127. Iwasaki Y, Ikeda K. Prevention by insulin‐like growth factor‐I and riluzole in motor neuron death after neonatal axotomy. J Neurol Sci 1999;169:148–155. [DOI] [PubMed] [Google Scholar]
- 128. Kennel P, Revah F, Bohme GA, et al Riluzole prolongs survival and delays muscle strength deterioration in mice with progressive motor neuronopathy (pmn). J Neurol Sci 2000;180:55–61. [DOI] [PubMed] [Google Scholar]
- 129. Coderre TJ, Kumar N, Lefebvre CD, et al A comparison of the glutamate release inhibition and anti‐allodynic effects of gabapentin, lamotrigine, and riluzole in a model of neuropathic pain. J Neurochem 2007;100:1289–1299. [DOI] [PubMed] [Google Scholar]
- 130. Wang J, Dib M, Lenoir M, et al Riluzole rescues cochlear sensory cells from acoustic trauma in the guinea‐pig. Neurosci 2002;111:635–648. [DOI] [PubMed] [Google Scholar]
- 131. Caumont AS, Octave JN, Hermans E. Specific regulation of rat glial cell line‐derived neurotrophic factor gene expression by riluzole in C6 glioma cells. J Neurochem 2006;97:128–139. [DOI] [PubMed] [Google Scholar]
- 132. Katoh‐Semba R, Asano T, Ueda H, et al Riluzole enhances expression of brain‐derived neurotrophic factor with consequent proliferation of granule precursor cells in the rat hippocampus. FASEB J 2002;16:1328–1330. [DOI] [PubMed] [Google Scholar]
- 133. Katoh‐Semba R, Kaneko R, Kitajima S, et al Activation of p38 mitogen‐activated protein kinase is required for in vivo brain‐derived neurotrophic factor production in the rat hippocampus. Neurosci 2009;163:352–361. doi: 10.1016/j.neuroscience.2009.06.011. [DOI] [PubMed] [Google Scholar]
- 134. Stevenson A, Yates DM, Manser C, et al Riluzole protects against glutamate‐induced slowing of neurofilament axonal transport. Neurosci Lett 2009;454:161–164. [DOI] [PubMed] [Google Scholar]
- 135. Mizuta I, Ohta M, Ohta K, et al Riluzole stimulates nerve growth factor, brain‐derived neurotrophic factor and glial cell line‐derived neurotrophic factor synthesis in cultured mouse astrocytes. Neurosci Lett 2001;310:117–120. [DOI] [PubMed] [Google Scholar]
- 136. Yang J, Bridges K, Chen KY, et al Riluzole increases the amount of latent HSF1 for an amplified heat shock response and cytoprotection. PLoS ONE 2008;3:e2864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Colovic M, Zennaro E, Caccia S. Liquid chromatographic assay for riluzole in mouse plasma and central nervous system tissues. J Chromatogr B Analyt Technol Biomed Life Sci 2004;803:305–309. [DOI] [PubMed] [Google Scholar]
- 138. Kretschmer BD, Kratzer U, Schmidt WJ. Riluzole, a glutamate release inhibitor, and motor behavior. Naunyn Schmiedebergs Arch Pharmacol 1998;358:181–190. [DOI] [PubMed] [Google Scholar]
- 139. Smith JC, Ellenberger HH, Ballanyi K, et al Pre‐Bötzinger Complex: A brainstem region that may generate respiratory rhythm in mammals. Science 1991;254:726–729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Paton JFR. The ventral medullary respiratory network of the mature mouse studied in a working heart‐brainstem preparation. J Physiol (Lond) 1996;493:819–831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Pena F, Aguileta MA. Effects of riluzole and flufenamic acid on eupnea and gasping of neonatal mice in vivo. Neurosci Lett 2007;415:288–293. [DOI] [PubMed] [Google Scholar]
- 142. St John WM, Waki H, Dutschmann M, et al Maintenance of eupnea of in situ and in vivo rats following riluzole: A blocker of persistent sodium channels. Respir Physiol Neurobiol 2007;155:97–100. [DOI] [PubMed] [Google Scholar]
- 143. Rybak IA, Shevtsova NA, St John WM, et al Endogenous rhythm generation in the pre‐Botzinger complex and ionic currents: Modelling and in vitro studies. Eur J Neurosci 2003;18:239–257. [DOI] [PubMed] [Google Scholar]
- 144. Pace RW, Mackay DD, Feldman JL, et al Role of persistent sodium current in mouse preBotzinger Complex neurons and respiratory rhythm generation. J Physiol (Lond) 2007;580:485–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Mironov SL, Langohr K. Mechanisms of Na+ and Ca2+ influx into respiratory neurons during hypoxia. Neuropharmacology 2005;48:1056–1065. [DOI] [PubMed] [Google Scholar]
- 146. St John WM. Eupnea of in situ rats persists following blockers of in vitro pacemaker burster activities. Respir Physiol Neurobiol 2008;160:353–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Ramirez JM, Viemari JC. Determinants of inspiratory activity. Respir Physiol Neurobiol 2005;147:145–157. [DOI] [PubMed] [Google Scholar]
- 148. Tazerart S, Vinay L, Brocard F. The persistent sodium current generates pacemaker activities in the central pattern generator for locomotion and regulates the locomotor rhythm. J Neurosci 2008;28:8577–8589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Bensimon G, Lacomblez L, Meininger V. A controlled trial of riluzole in amyotrophic lateral sclerosis. ALS/Riluzole Study Group. N Engl J Med 1994;330:585–591. [DOI] [PubMed] [Google Scholar]
- 150. Lacomblez L, Bensimon G, Leigh PN, et al Dose‐ranging study of riluzole in amyotrophic lateral sclerosis. Amyotrophic Lateral Sclerosis/Riluzole Study Group II. Lancet 1996;347:1425–1431. [DOI] [PubMed] [Google Scholar]
- 151. Mitchell JD, O’brien MR, Joshi M. Audit of outcomes in motor neuron disease (MND) patients treated with riluzole. Amyotroph Lateral Scler Other Motor Neuron Disord 2006;7:67–71. [DOI] [PubMed] [Google Scholar]
- 152. Miller RG, Mitchell JD, Lyon M, et al Riluzole for amyotrophic lateral sclerosis (ALS)/motor neuron disease (MND). Amyotroph Lateral Scler Other Motor Neuron Disord 2003;4:191–206. [PubMed] [Google Scholar]
- 153. Traynor BJ, Alexander M, Corr B, et al An outcome study of riluzole in amyotrophic lateral sclerosis–a population‐based study in Ireland, 1996–2000. J Neurol 2003;250:473–479. [DOI] [PubMed] [Google Scholar]
- 154. Chio A, Mora G, Leone M, et al Early symptom progression rate is related to ALS outcome: A prospective population‐based study. Neurology 2002;59:99–103. [DOI] [PubMed] [Google Scholar]
- 155. Miller RG, Mitchell JD, Lyon M, et al Riluzole for amyotrophic lateral sclerosis (ALS)/motor neuron disease (MND). Cochrane Database Syst Rev 2007;Issue 1, Article No. CD001447, DOI: 10.1002/14651858.CD001447.pub2. [DOI] [PubMed] [Google Scholar]
- 156. Zoing MC, Burke D, Pamphlett R, et al Riluzole therapy for motor neurone disease: An early Australian experience (1996–2002). J Clin Neurosci 2006;13:78–83. [DOI] [PubMed] [Google Scholar]
- 157. Riviere M, Meininger V, Zeisser P, et al An analysis of extended survival in patients with amyotrophic lateral sclerosis treated with riluzole. Arch Neurol 1998;55:526–528. [DOI] [PubMed] [Google Scholar]
- 158. Zoccolella S, Beghi E, Palagano G, et al Riluzole and amyotrophic lateral sclerosis survival: A population‐based study in southern Italy. Eur J Neurol 2007;14:262–268. [DOI] [PubMed] [Google Scholar]
- 159. Desnuelle C, Dib M, Garrel C, et al A double‐blind, placebo‐controlled randomized clinical trial of a‐tocopherol (vitamin E) in the treatment of amyotrophic lateral sclerosis. Amyotroph Lateral Scler Other Motor Neuron Disord 2001;2:9–18. [DOI] [PubMed] [Google Scholar]
- 160. Graf M, Ecker D, Horowski R, et al High dose vitamin E therapy in amyotrophic lateral sclerosis as add‐on therapy to riluzole: Results of a placebo‐controlled double‐blind study. J Neural Trans 2005;112:649–660. [DOI] [PubMed] [Google Scholar]
- 161. Lacomblez L, Bensimon G, Leigh PN, et al Long‐term safety of riluzole in amyotrophic lateral sclerosis. Amyotroph Lateral Scler Other Motor Neuron Disord 2002;3:23–29. [DOI] [PubMed] [Google Scholar]
- 162. Bensimon G, Doble A. The tolerability of riluzole in the treatment of patients with amyotrophic lateral sclerosis. Expert Opin Drug Saf 2004;3:525–534. [DOI] [PubMed] [Google Scholar]
- 163. Le Liboux A, Cachia JP, Kirkesseli S, et al A comparison of the pharmacokinetics and tolerability of riluzole after repeat dose administration in healthy elderly and young volunteers. J Clin Pharmacol 1999;39:480–486. [PubMed] [Google Scholar]
- 164. Pongratz D, Neundorfer B, Fischer W. German open label trial of riluzole 50 mg b.i.d. in treatment of amyotrophic lateral sclerosis (ALS). J Neurol Sci 2000;180:82–85. [DOI] [PubMed] [Google Scholar]
- 165. Debove C, Zeisser P, Salzman PM, et al The Rilutek (riluzole) Global Early Access Programme: An open‐label safety evaluation in the treatment of amyotrophic lateral sclerosis. Amyotroph Lateral Scler Other Motor Neuron Disord 2001;2:153–158. [DOI] [PubMed] [Google Scholar]
- 166. Bensimon G, Lacomblez L, Delumeau JC, et al A study of riluzole in the treatment of advanced stage or elderly patients with amyotrophic lateral sclerosis. J Neurol 2002;249:609–615. [DOI] [PubMed] [Google Scholar]
- 167. Weber G, Bitterman H. Riluzole‐induced neutropenia. Neurology 2004;62:1648. [DOI] [PubMed] [Google Scholar]
- 168. Cassiman D, Thomeer M, Verbeken E, et al Hypersensitivity pneumonitis possibly caused by riluzole therapy in ALS. Neurology 2003;61:1150–1151. [DOI] [PubMed] [Google Scholar]
- 169. Rodrigo L, Moreno M, Calleja S, et al Riluzole‐induced acute pancreatitis. Am J Gastroenterol 2001;96:2268–2269. [DOI] [PubMed] [Google Scholar]
- 170. Viallon A, Page Y, Bertrand JC. Methemoglobinemia due to riluzole. N Engl J Med 2000;343:665–666. [DOI] [PubMed] [Google Scholar]
- 171. North WA, Khan AM, Yamase HT, et al Reversible granulocytopenia in association with riluzole therapy. Ann Pharmacother 2000;34:322–324. [DOI] [PubMed] [Google Scholar]
- 172. Scelsa SN, Khan I. Blood pressure elevations in riluzole‐treated patients with amyotrophic lateral sclerosis. Eur Neurol 2000;43:224–227. [DOI] [PubMed] [Google Scholar]
- 173. Schwenkreis P, Liepert J, Witscher K, et al Riluzole suppresses motor cortex facilitation in correlation to its plasma level. A study using transcranial magnetic stimulation. Exp Brain Res 2000;135:293–299. [DOI] [PubMed] [Google Scholar]
- 174. Groeneveld GJ, Van Kan HJ, Torano JS, et al Inter‐ and intraindividual variability of riluzole serum concentrations in patients with ALS. J Neurol Sci 2001;191:121–125. [DOI] [PubMed] [Google Scholar]
- 175. Sanderink GJ, Bournique B, Stevens J, et al Involvement of human CYP1A isoenzymes in the metabolism and drug interactions of riluzole in vitro. J Pharmacol Exp Ther 1997;282:1465–1472. [PubMed] [Google Scholar]
- 176. Ajroud‐Driss S, Saeed M, Khan H, et al Riluzole metabolism and CYP1A1/2 polymorphisms in patients with ALS. Amyotroph Lateral Scler 2007;8:1–5. [DOI] [PubMed] [Google Scholar]
- 177. Le Liboux A, Lefebvre P, Le Roux Y, et al Single‐ and multiple‐dose pharmacokinetics of riluzole in white subjects. J Clin Pharmacol 1997;37:820–827. [DOI] [PubMed] [Google Scholar]
- 178. Bruno R, Vivier N, Montay G, et al Population pharmacokinetics of riluzole in patients with amyotrophic lateral sclerosis. Clin Pharmacol Ther 1997;62:518–526. [DOI] [PubMed] [Google Scholar]
- 179. Groeneveld GJ, Van Kan HJ, Lie AH, et al An association study of riluzole serum concentration and survival and disease progression in patients with ALS. Clin Pharmacol Ther 2007;83:718–722. [DOI] [PubMed] [Google Scholar]
- 180. Desai J, Sharief M, Swash M. Riluzole has no acute effect on motor unit parameters in ALS. J Neurol Sci 1998;160:S69–S72. [DOI] [PubMed] [Google Scholar]
- 181. Liepert J, Schwenkreis P, Tegenthoff M, et al The glutamate antagonist riluzole suppresses intracortical facilitation. J Neural Trans 1997;104:1207–1214. [DOI] [PubMed] [Google Scholar]
- 182. Caramia MD, Palmieri MG, Desiato MT, et al Pharmacologic reversal of cortical hyperexcitability in patients with ALS. Neurology 2000;54:58–64. [DOI] [PubMed] [Google Scholar]
- 183. Desiato MT, Palmieri MG, Giacomini P, et al The effect of riluzole in amyotrophic lateral sclerosis: A study with cortical stimulation. J Neurol Sci 1999;169:98–107. [DOI] [PubMed] [Google Scholar]
- 184. Stefan K, Kunesch E, Benecke R, et al Effects of riluzole on cortical excitability in patients with amyotrophic lateral sclerosis. Ann Neurol 2001;49:536–539. [PubMed] [Google Scholar]
- 185. Sommer M, Tergau F, Wischer S, et al Riluzole does not have an acute effect on motor thresholds and the intracortical excitability in amyotrophic lateral sclerosis. J Neurol 1999;246(Suppl 3):III22–III26. [DOI] [PubMed] [Google Scholar]
- 186. Mogyoros I, Kiernan MC, Burke D, et al Strength‐duration properties of sensory and motor axons in amyotrophic lateral sclerosis. Brain 1998;121(Pt 5):851–859. [DOI] [PubMed] [Google Scholar]
- 187. Vucic S, Nicholson GA, Kiernan MC. Cortical hyperexcitability may precede the onset of familial amyotrophic lateral sclerosis. Brain 2008;131:1540–1550. [DOI] [PubMed] [Google Scholar]
- 188. Kuo JJ, Schonewille M, Siddique T, et al Hyperexcitability of cultured spinal motoneurons from presymptomatic ALS mice. J Neurophysiol 2004;91:571–575. [DOI] [PubMed] [Google Scholar]
- 189. Tamura N, Kuwabara S, Misawa S, et al Increased nodal persistent Na+ currents in human neuropathy and motor neuron disease estimated by latent addition. Clin Neurophysiol 2006;117:2451–2458. [DOI] [PubMed] [Google Scholar]
- 190. Banks GB, Kanjhan R, Wiese S, et al Glycinergic and GABAergic synaptic activity differentially regulate motoneuron survival and skeletal muscle innervation. J Neurosci 2005;25:1249–1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191. Oppenheim RW, Houenou LJ, Parsadanian AS, et al Glial cell line‐derived neurotrophic factor and developing mammalian motoneurons: Regulation of programmed cell death among motoneuron subtypes. J Neurosci 2000;20:5001–5011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192. Oppenheim RW. Cell death during development of the nervous system. Annu Rev Neurosci 1991;14:453–501. [DOI] [PubMed] [Google Scholar]
- 193. Bohn MC. Motoneurons crave glial cell line‐derived neurotrophic factor. Exp Neurol 2004;190:263–275. [DOI] [PubMed] [Google Scholar]
- 194. Bourke SC, Tomlinson M, Williams TL, et al Effects of non‐invasive ventilation on survival and quality of life in patients with amyotrophic lateral sclerosis: A randomised controlled trial. Lancet Neurol 2006;5:140–147. [DOI] [PubMed] [Google Scholar]
- 195. Carratu P, Spicuzza L, Cassano A, et al Early treatment with noninvasive positive pressure ventilation prolongs survival in Amyotrophic Lateral Sclerosis patients with nocturnal respiratory insufficiency. Orphanet J Rare Dis 2009;4:10, DOI: 10.1186/1750-1172-4-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196. Ferguson KA, Strong MJ, Ahmad D, et al Sleep‐disordered breathing in amyotrophic lateral sclerosis. Chest 1996;110:664–669. [DOI] [PubMed] [Google Scholar]
- 197. Keita H, Lepouse C, Henzel D, et al Riluzole blocks dopamine release evoked by N‐methyl‐D‐aspartate, kainate, and veratridine in the rat striatum. Anesthesiology 1997;87:1164–1171. [DOI] [PubMed] [Google Scholar]