

SUMMARY
Recent studies support the notion that statins, widely prescribed cholesterol‐lowering agents, may target key elements in the immunological cascade leading to inflammation and tissue damage in the pathogenesis of multiple sclerosis (MS). Compelling experimental and observational clinical studies highlighted the possibility that statins may also exert immunomodulatory synergy with approved MS drugs, resulting in several randomized clinical trials testing statins in combination with interferon‐beta (IFN‐β). Some data, however, suggest that this particular combination may not be clinically beneficial, and might actually have a negative effect on the disease course in some patients with MS. In this regard, a small North American trial indicated that atorvastatin administered in combination with IFN‐β may increase disease activity in relapsing‐remitting MS. Although other trials did not confirm this finding, the enthusiasm for studies with statins dwindled. This review aims to provide a comprehensive overview of the completed clinical trials and reports of the interim analyses evaluating the combination of IFN‐β and statins in MS. Moreover, we try to address the evident question whether usage of this combination routinely requires caution, since the number of IFN‐β‐treated MS patients receiving statins for lowering of cholesterol is expected to grow.
Keywords: Immunomodulatory therapy, Interferon‐beta, Multiple sclerosis, Statin
Introduction
Multiple sclerosis (MS) is considered a chronic autoimmune disease with complex genetic background in which autoreactive T cells infiltrate the central nervous system (CNS) and initiate inflammatory and destructive processes leading to permanent neurological disability [1]. In light of the initial clinical manifestation in early adulthood, uncertainty of prognosis, and limited impact of disease‐modifying drugs (DMD) on disability progression, this, often devastating, disease poses a significant burden on patients, families, and caregivers. Over the years, several DMDs have been approved for the treatment of MS, including interferon‐beta‐(IFN‐β)‐1a (Avonex®, Rebif®), IFN‐β‐1b (Betaseron/Betaferon®), glatiramer‐acetate (GA; Copaxone®), mitoxantrone (Novantrone®), and natalizumab (Tysabri®). Although the arsenal of treatment options is constantly growing, insufficient response in a subgroup of patients, considerable side effects, and tedious regular and parenteral application have been challenging for some patients [2]. Thus, one strategy to increase efficacy is the combination of partially effective agents [3].
Statins are orally administered inhibitors of the 3‐hydroxy‐3‐methyl‐glutaryl (HMG)‐CoA reductase, an enzyme that catalyzes the rate‐limiting step of cholesterol biosynthesis (Figure 1). These substances are well established in the treatment of cardiovascular disease and have attracted significant interest in autoimmune disorders due to an expanding knowledge of additional immunomodulatory, antiinflammatory, and neuroprotective effects. Indeed, both in vivo and in vitro experiments demonstrated pleiotropic effects on the immune system that might be beneficial in the treatment of MS [4, 5, 6]. In this regard, two small open‐label trials involving a total of 35 patients with relapsing‐remitting MS (RRMS) confirmed that a statin monotherapy is safe and indicated potential efficacy on short‐term clinical and magnetic resonance imaging (MRI) measures. The first study (2003), in which 7 RRMS patients were treated with 40 mg lovastatin for 12 months noticed a decrease of the mean annual relapse rate and no adverse events [7]. In the second study (2004) the 80 mg simvastatin treatment for 6 months was associated with a lowering of mean number (−44%) and volume of contrast‐enhancing lesions (CELs) by (−41%) compared to pretreatment scans [8]. A very recent double‐blind, placebo‐controlled trial evaluated atorvastatin in prevention of progression from clinically isolated syndrome (CIS) to MS [9]. While the primary endpoint with development of >3 new T2 lesions or one clinical exacerbation by 12 months was not met, patients in the atorvastatin group were more likely to remain T2 lesion‐free compared with placebo (odds ratio 3.93; P= 0.012). Based on these findings, considerable enthusiasm developed to also investigate the effect of statins in combination with IFN‐β. Unexpectedly, combination trials to date generated preliminary data indicating that the concomitant administration of statins and IFN‐β may not provide a superior efficacy over IFN monotherapy. In this regard, a placebo‐controlled randomized study in which 28 RRMS patients were treated with 40 or 80 mg atorvastatin for 6 months in combination with high‐dose IFN‐β‐1a even suggested a potential increase of clinical and MRI activity [10]. Evidence for a divergent action of the two substances on immune mechanism had already been shown in vitro[5, 11]. The investigators presented further data, which suggested that statins interfere with the phosphorylation of the transcription factor STAT1, which also medicates the transcription of interferon beta response genes [12, 13]. Based on these observations, a lively discussion emerged whether further combination studies of IFN‐β and statins should be halted. Remarkably, interim and final reports of additional combination studies were presented since then and could not confirm potential adverse effects on clinical and MRI measures (Table 1).
Figure 1.
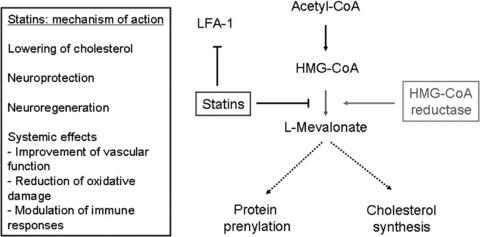
Statins exhibit different mechanisms of action by interfering with cholesterol synthesis and protein prenylation. Statins inhibit the conversion of 3‐hydroxy‐3‐methylglutaryl coenzyme A (HMG‐CoA) to L‐mevalonate through competitive inhibition of the rate‐limiting enzyme HMG‐CoA reductase. This inhibition results in a decrease in the downstream biosynthesis of cholesterol and other intermediate metabolites. The latter are involved in the isoprenylation of proteins which serve as essential adjuncts in the posttranslational modification of numerous key proteins including Ras, Rac, Rab, cdc42, RhoB, and Rho. Less cholesterol also impairs the lipid raft formation and thus has impact on expression of molecules on the cell surface and cell proliferation.
Table 1.
Lineup of clinical studies evaluating effects interferon‐β (IFN‐β) in combination with statins in patients with clinically isolated syndrome (CIS) and relapsing‐remitting multiple sclerosis (RRMS)
| First author, year of publication | Study type | Patients | Allocation | Interferon‐β (IFN‐β) | Statin and dosage per day | Primary endpoint | Secondary endpoints | |
|---|---|---|---|---|---|---|---|---|
| Original articles | ||||||||
| 1 | Paul F et al. [14], 2008 | Phase II | RRMS | IFN‐β+ statin (n = 16), statin (n = 25) | IFN‐β‐1a 22 μg s.c. thrice weekly or IFN‐β‐1b s.c. every other day | Atorvastatin 80 mg | CEL at months 6–9: decrease/trend for combitherapy in number and volume of CEL | Changes in EDSS and MSFC: not stated |
| 2 | Birnbaum G et al. [10], 2008 | Safety study | RRMS | IFN‐β (n = 9), IFN‐β+ statin (n = 17) | IFN‐β‐1a 44 μg s.c. thrice weekly | Atorvastatin 40 mg (n = 7) and 80 mg (n = 10) | EDSS change, CEL or new lesion: greater clinical and MRI disease activity for patients on combitherapy | |
| 3 | Rudick RA et al. [15], 2009 | Post‐hoc analysis of other trial (SENTINEL) | RRMS | IFN‐β (n = 542), IFN‐β+ statin (n = 40) | IFN‐β‐1a 30 μg i.m. once weekly | Most frequently atorvastatin (65%) and simvastatin (32.5%) | Annualized relapse rate, disability progression, number CEL, number of new/enlarging T2‐lesions after 2 years: no differences | |
| 4 | Lanzillo R et al. [16] 2010 | Open‐label randomized study | RRMS | IFN‐β (n = 24), IFN‐β+ statin (n = 21) | IFN‐β‐1a 44 μg s.c. thrice weekly | Atorvastatin 20 mg | Number of CEL after 24 months: reduction comparable between the groups. Combitherapy; significantly reduced when compared to baseline | Relapse rate: significantly lower for combitherapy. EDSS and laboratory data: no difference |
| Communications | ||||||||
| 1 | Sörensen PS et al. [17] 2007 | Safety study, interim analysis | RRMS | Total (n = 8), IFN‐β, IFN‐β+ statin | IFN‐β‐1a 30 μg i.m. once weekly | Simvastatin 80 mg | First time to documented relapse after a mean of 6.9 months: no differences | Relapses, new/enlarging T2‐lesions: n.c. |
| 2 | Markovic‐Plese et al. [18] 2007 | Safety study | CIS | IFN‐β (n = 9), IFN‐β+ statin (n = 10) | IFN‐β‐1a 30 μg i.m. once weekly | Simvastatin 80 mg | Clinical and MRI activity: no differences | |
| 3 | Oztekin NS et al. 2009 [19] | Preliminary data at 18 months (of 24) | RRMS | IFN‐β (n = 11), IFN‐β+ statin (n = 7) | IFN‐β‐1a 44 μg s.c. thrice weekly | Atorvastatin 20 mg | MRI activity: comparable between the groups | Relapses, EDSS, safety laboratory data: n.c. |
RRMS, relapsing‐remitting MS; CEL, contrast‐(Gadolinium) enhancing lesions; EDSS, expanded disability status scale; MSFC, multiple sclerosis functional composite score; s.c., subcutaneous application; i.m., intramuscular application; n.c., not communicated.
This review provides a comprehensive overview on the current knowledge of statin‐IFN‐β combination therapy in patients with MS. Specifically, clinical trials and potential obstacles of this combination therapy will be discussed.
Statins Are Well‐Tolerated Oral Agents with Immunomodulatory and Neuro‐Protective Properties
The most common side effects of statins are gastrointestinal symptoms and muscle ache. Hepatotoxicity, indicated by increases in serum amino transaminase levels, occurs in less than 1% of patients even at high dosages, but the risk of liver toxicity and rhabdomyolysis increases under combination therapies [20]. Other side effects include myopathy, rash, peripheral neuropathy, insomnia, and cognitive problems. Systemic HMG‐CoA inhibition was shown to affect brain cholesterol production but not brain cholesterol content [21]. Moreover, due to the long half‐life of brain cholesterol, only extended usage of statins was able to reduce cholesterol levels in the cerebrospinal fluid (CSF) [22]. It should also be noted that cholesterol is an indispensable component of myelin membranes and cholesterol availability in oligodendrocytes is a rate‐limiting factor for brain maturation [23].
The source of effector mechanisms on the immune system can be generally divided into HMG‐CoA reductase dependent and independent pathways [24, 25, 26]. To this end statins were shown to exert neuroprotection by activating neuroprotective‐signaling pathways and as a consequence of different systemic effects [27].
HMG‐CoA Reductase‐Dependent Effects
The majority of statin‐mediated effects on the immune system appear to be related to the competitive inhibition of HMG‐CoA reductase (Figure 1). The subsequent decrease in the production of its substrate l‐mevalonate and its metabolites interferes with gene regulation and posttranslational modification of proteins that are associated with proliferation and differentiation of various cells and tissues. In this regard, the synthesis of isoprenoid metabolites is downregulated, which serve as lipid attachements for a number of intracellular signaling molecules including the GTP‐binding proteins Ras, Rac, and Rho [28]. Apart from influencing GTP‐binding proteins, isoprenyelation of these molecules also interferes with transcription factors such as nuclear factor (NF); statins have shown to limit TNF‐related NF‐kB accumulation and the increase of inhibitor IkB [29]. The effects of HMG‐CoA reductase inhibition on the immune system were recently summarized in a review by Greenwood and colleagues as follows: there is a decrease of (1) leukocyte motility, (2) antigen uptake, processing, and presentation, (3) leukocyte activation, proliferation, and function, (4) phagocytosis, (5) leukocyte transvascular migration, and (6) endothelial‐cell immune function [30] under statin therapy. The effects evident from various experimental studies potentially beneficial in MS include the inhibition of expression and secretion of proinflammatory cytokines, inhibition of major histocompatibility complex (MHC) class II expression on antigen‐presenting cells (APCs), and costimulatory molecules, and the suppression of Th1 differentiation. Indeed, simvastatin intake had an inhibitory effect on the differentiation and maturation of dendritic cells from patients with optic neuritis (ON), and selectively reduced T‐cell proliferation [31]. Likewise, simvastatin treatment in RRMS was associated with inhibition of peripheral‐blood mononuclear cell (PBMC) proliferation, antigen presentation by blocking expression of MHC class II DR molecules in CD14+ monocytes, activation and differentiation of T cells, and attenuation of gene expression of early proinflammatory cytokines via inhibition of T‐bet, a master controller of the Th1 cytokine pathway [32]. The effect of statins on Th1 differentiation has been consistent throughout many published experimental autoimmune encephalomyelitis (EAE) studies [4, 33, 34, 35, 36]. There is also a reduced activation of the transcription factor STAT (signal transducer and activator of transcription)‐4, which is required for IL12‐dependent Th1‐differentiation [4, 25]. The induction of an antiinflammatory Th2 phenotype, which is associated with induction and secretion of antiinflammatory Th2 cytokines (IL4, IL5, and IL10) is less consistent and was related to the enhanced activation of STAT6, which is involved in IL4‐dependent Th2 differentiation [4]. Most interestingly, atorvastatin was shown to enhance the Th2‐promoting effects of glatiramer‐acetate in EAE, indicating that a combination of statins with an established immunomodulator may be an exciting concept for future clinical trials [36]. Further studies in MS and healthy controls revealed that simvastatin inhibits Th17 cell differentiation, a recently identified CD4+ T‐cell subset supposed to play a critical role in autoimmunity [37]. Immunomodulatory effects of statins on T‐cell activation and differentiation were indeed related to inhibition of prenylation of regulatory proteins [28]. Simvastatin however interferes with remyelination by directly impacting oligodendrocyte progenitor cell function and affecting mature oligodendrocyte numbers at immunomodulatory concentration by interference with Ras and Rho signaling [38, 39]. Another in vitro study revealed that the inhibition of the mevalonate pathway by atorvastatin was associated with reduced length of neurites and ultimatively cell death of primary cortical neurons [40]. In contrast, Paintilla reported that lovastatin promoted myelin repair by inhibition of Rho and augments survival and differentiation of oligodendrocyte progenitors [41, 42]. To this end, the combination of IFN‐β and atorvastatin lowered serum levels of high‐sensitivity C‐reactive protein (CRP) in RRMS, pointing at the potential additional antiinflammatory effect of statins [43].
HMG‐CoA Reductase‐Independent Effects
Among the HMG‐CoA reductase‐independent effects of statins is the binding and inhibition of β2‐integrin leukocyte function antigen 1 (LFA‐1), which is also known as αL‐β2 or CD11a/CD18. LFA‐1 is constitutively expressed on the surface of leukocytes and binds to intercellular adhesion molecule (ICAM‐1 or CD54) with subsequent leukocyte recirculation and infiltration of inflamed tissue [44]. Likewise, in EAE, an animal model of MS, treatment with lovastatin lead to reduced immune activation, leukocyte infiltration in the brain and subsequent paralysis [33, 45]. In vitro, simvastatin was shown to inhibit the expression of ICAM‐1 on PBMCs, whereas VLA‐4 and LFA‐1 were unaltered [5]. Eventually, ex vivo treatment with statins impede the migration of monocytes and lymphocytes taken from MS patients across a blood‐brain barrier model due to reduced secretion of chemokines CCL2 and CXCL10 by endothelial cells [46].
Neuroprotective Action of Statins
Several systemic effects of statins have been described which are likely to contribute to neuroprotection. These effects include (1) reduction of oxidative damage, (2) improvement of vascular function by regulation of nitric oxide production, inhibition of coagulation, and effects on angiogenesis, and (3) modulation of the peripheral inflammatory response [27, 47]. Further observations suggest that statins provide neuroprotection by attenuation of inflammation‐induced glutamate/calcium excitotoxicity, an important component of axonal injury in MS [48, 49]. In addition, treatment of rodents with statins following brain injury increased neurogenesis and synaptogenesis, most likely via the release of neurotrophic factors such as brain‐derived neurotrophic factor (BDNF) [50, 51, 52]. Neuroprotective pathways directly involved in statin‐mediated neuroprotection are protein kinase B (PKB/Akt) and the Ras‐(extracellular‐signal‐regulated cascade) ERK signaling cascade [35, 53, 54]. However, several in vitro studies indicate that particularly lipophilic statins exert neurotoxic action and induce cell death in neurons and glial cells. Yet, the concentrations required for these effects are not expected in the CNS and were achieved under cholesterol‐ or LDL‐depleted medium, which do not mirror physiological conditions [27].
Clinical Trials Evaluating the Combination of IFN‐β and Statins in MS
Previously, various placebo‐controlled, randomized clinical trials in CIS and RRMS had shown the positive impact of IFN‐β on modifying the disease course, with short‐term trials altering relapse rate, disability progression, and MRI measures, and long‐term treatment delaying secondary progression [2]. Thus, the interest in the combination of statins with IFN‐β was reflected by the aim to improve the efficacy of IFN‐β on the one hand and the difficulties to perform treatment trials versus IFN‐β in MS on the other. Indeed, placebo‐controlled trials in MS have been becoming increasingly difficult to perform since the establishment of immunomodulatory treatment, both for ethical and practical reasons [55]. The precise IFN‐β mechanisms of action, however, remain unclear. Several biological effects have been described such as attenuation of proliferation of leukocytes and APCs, the modulation of cytokine and chemokine production toward an antiinflammatory phenotype, and the potential to inhibit T‐cell migration across the blood‐brain barrier [56].
Numerous MS trials that tested different IFN‐βs and statins were presented at European and North American conferences between 2005 and 2009 and reflected the lively interest in evaluating this drug combination in MS. F. Paul and colleagues had published the encouraging results of their phase II trial evaluating 80 mg atorvastatin (40 mg twice daily) with or without additional subcutaneous (s.c.) IFN‐β in 41 RRMS patients (n = 16 with comedication). A peculiarity of this study was the inclusion criteria of at least one CEL and the baseline‐to‐treatment concept, which involved a baseline period of 3 months prior to start of HMG‐CoA reductase inhibition, followed by a 9‐month treatment duration. A nonsignificant reduction in CEL number and volume in the group receiving the combination was observed in a multivariate analysis, providing further evidence for a potential immunomodulatory synergy. The authors reported that the combined treatment of IFN‐β with high‐dose atorvastatin was safe and well tolerated in the majority of the patients. A temporary mild elevation of liver enzymes with no consistent timeframe of occurrence after initiation of statin treatment was reported in 16 out of 41 patients. In 5 patients treatment with statins had to be discontinued temporarily, and was resumed after liver enzymes returned to normal. The most frequent side effects, however, were respiratory tract infections including rhinitis, sinusitis, and bronchitis.
The report by Birnbaum and colleagues in 2008 on IFN‐β−statin combination therapy, however, lead to a critical rethinking of this therapeutic approach. This double‐blind, placebo‐controlled trial evaluated 26 RRMS patients who had been clinically stable on IFN‐β‐1a; the treatment groups consisted of placebo (n = 9) or 40 (n = 7) or 80 mg (n = 10) atorvastatin daily (Table 1). Perhaps unexpectedly, atorvastatin‐treated subjects were at greater risk for experiencing either clinical and MRI disease activity relative to controls (P= 0.019). Of the 17 patients treated with atorvastatin, 10 developed either new lesions on MRI or had clinical relapses, contrasting 1 in 9 placebo‐treated patients. Noteworthy, some relapses occurred after years of stable disease and a cox‐proportional hazard model analysis rebutted that group differences in baseline demographics influenced the risk of disease activity. Certainly, the study participants were relatively old (group mean age 38.4, 40.1, and 45.1 years) with a mean disease duration of around 7 years and relatively short time on IFN‐β (mean 1.8, 2.0, and 2.2 years, respectively). In this study, no significant changes of liver enzymes and creatine kinase (CK) was found between the three treatment groups, whereas total cholesterol levels were reduced in subjects receiving atorvastatin.
The third study was published in 2009 and represented a post‐hoc analysis of the SENTINEL trial, a prospective study which determined the effects of natalizumab plus intramuscular (i.m.) IFN‐β 1a in RRMS. The IFN‐β‐1a arm included 40 patients who received statins to treat hyperlipidemia; clinical and MRI outcomes of 542 patients who were not treated with statins served as reference. No significant differences were observed between the groups with regard to adjusted annualized relapse rate, disability progression, number of CEL, or number of new or enlarging T2‐hyperintense lesions after 2 years. The authors concluded that statin therapy did not affect clinical effects of i.m. IFN‐β‐1a in RRMS patients. The incidence of muscle‐related pain was higher in patients of the statin group. Other commonly reported adverse events of the statin group were fatigue, headache, back or extremity pain, arthralgia, depression, and asthenia.
In the most recent study, the ACTIVE trial by Lanzillo and colleagues, patients with RRMS who continued to have CEL or relapses while on therapy with IFN‐β‐1a for 12 months were randomized to a combination therapy with 20 mg atorvastatin (n = 21) or remained on IFN‐β‐1a (n = 24) [16]. The analysis of the primary endpoint, the number of CEL at 24 months, revealed that both groups had a decrease in the number of CEL. The difference between baseline and 24‐month follow‐up was significant for the combination therapy (P= 0.007) but not in the monotherapy group. However, a statistical analysis between the groups did not show differences. Secondary outcome measures were number of relapses, expanded disability status scale (EDSS) variation, and laboratory safety data. Patients treated with the combination therapy had a significantly lower relapse rate (P < 0.005), while comparison of the EDSS after 24 months did not show differences between the groups. In either groups laboratory parameters such as CK and liver enzymes remained unchanged, and no muscle pain or cramps were reported. The authors concluded that “low‐dose atorvastatin may be beneficial as add‐on therapy in poor responders to IFN‐β‐1a alone.”
Information on further combination trials is available but is restricted to interim analyses in abstract form (n = 2) and a letter to the editor (n = 1). Among these three mostly safety trials (Table 1), no major concerns of the IFN‐β and statin combination were noted in general and with regard to clinical or MRI outcomes. Taken together, seven trial reports evaluating a combination therapy of IFN‐β and statins are available for analysis, even though three need to be regarded as too preliminary being interim study reports and only being published as conference proceedings. Yet, both the study by Paul et al. and Lanzillo et al. suggested a trend for an additive effect on MRI measures, whereas the Rudick et al. trial did not find differences with regard to their outcome parameters. Lanzillo even reported a significantly lower relapse rate with the combination therapy compared to the two prerandomization years. Most importantly, among the trials no further study indicated a potential detrimental effect of this combination. Indeed, in an accompanying editorial, Goldman and Cohen raise the possibility that the results of Birnbaum et al. may be an artifact [57]. This argumentation is based on the small sample size, which could magnify potential group imbalances, differences in compliance with assigned treatment, unblinding, differences in event ascertainment, or outliers in on‐study disease activity. Further issues that make comparisons between the trials difficult include the usage of different IFN‐β and statins, as well as different statin dosages.
Potential Pharmacological Interference of a Combination Therapy
An immunoregulatory effect by IFN‐β in the context of recently described Th17‐cell‐mediated autoimmune response has been attributed to the STAT1‐induced decrease in the frequency of IL17‐producing CD4+ cells [58, 59]. Accordingly, in vitro studies suggested that the increase of clinical and MRI disease activity in the Birnbaum study may have been related to abrogation of IFN‐β signaling by statins (Figure 2). This potential loss of therapeutic efficacy was shown in cell culture experiments to be induced by blocking tyrosine phosphorylation of the STAT1 transcription factor (P‐Tyr STAT1) by statins, which is essential for type I IFN‐(α/β) signaling [13, 61].
Figure 2.
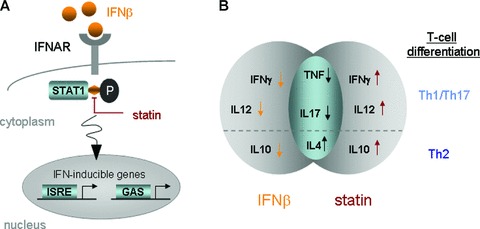
Two potential grounds for attenuation of IFN‐β bioactivity by statins. (A) Blocking of IFN‐β induced phosphorylation of STAT1 and (B) differential impact on cytokine secretion and subsequent T‐cell differentiation. (A) Simplified scheme of the IFN‐α/β signaling pathway: binding of IFN‐α/β with the receptor complex IFNAR leads to activation of the receptor associated Tyk2 and Janus kinase (Jak1) [60]. This is followed by the tyrosine phosphorylation (P) of STAT1 and STAT2, which can be blocked by statins in vitro[12, 13]. Activation of the STATs leads to formation of two transcriptional‐activator complexes which subsequently activate ISRE and GAS, respectively, in the nucleus. (B) IFN‐β and statins exert differential impact on modulation of cytokine responses and subsequently T‐cell differentiation. A diverse action of IFN‐β and statins was shown in vitro for Th1 cytokines IFN‐γ and IL12 and Th2 cytokine IL10 [5]. IFN, interferon; IFNAR, IFN‐α/β receptor; STAT1, member of the Signal transducer and activators of transcription family of transcription factors; ISRE, IFN‐stimulated response element; GAS, IFN‐γ activated site; IL, interleukin; TNF, tumor‐necrosis factor.
A study by Zhang et al. evaluating the effects of simvastatin on monocytes derived from MS patients reported the inhibition of IL6 and IL23 and induction of IFN‐γ, IL4, and IL27 resulting from increased SOCS3 protein expression and inhibition of STAT1 and STAT3 phosphorylation [62]. Of note, simvastatin inhibited the expansion of Th17 cells in vitro but enhanced the differentiation of Foxp3(+) CD4(+) T cells [63]. Additional experiments revealed that in Jurkat cells stimulated with different IFN‐β preparations, atorvastatin starts to inhibit P‐Tyr STAT1 activation and subsequent IFN‐α/β responses after 3 h for a duration of 24 h [12]. The inhibitory effect was more pronounced in monocytes (25–100%) than in T cells (15–40%). Subsequently, amelioration of IFN‐β effects by statins was also determined in vivo in a subgroup of RRMS patients receiving the combination therapy [12]. Moreover, whether the increase of infectious complications in the German trial are related to an attenuation of antiviral immune responses by statins via the STAT1 signaling pathway remains speculative. To this end, studies evaluating interference of IFN‐β signaling by statins are currently only published in abstract form.
In contrast, four independent studies evaluated markers of IFN‐β activity including IFN‐induced genes MxA and TRAIL and could not confirm a loss of IFN‐β signaling in patients cotreated with statins in vivo (Table 2) [14, 15]. Paul et al. even determined supraadditive effects on inhibition of MBP‐specific T‐cell proliferation in vitro but could not confirm these data in vivo[14]. Unfortunately, the Birnbaum study did not include an evaluation of IFN‐β bioactivity. The other four studies did not show a substantial inhibition of IFN‐β signaling by statins in vivo.
Table 2.
In vivo evaluation of IFN‐β bioactivity and potential alterations in combination with statins
| Reference | Measures | Clinical Trial | Specimen | Method | Findings: IFN‐β versus IFNβ+ statin |
|---|---|---|---|---|---|
| Marker of IFN‐β activity | |||||
| [15] | IFN‐stimulated gene | [15] | PBMC | cDNA macroarray | No differences |
| [14] | TRAIL | [14] | PBMC | rtPCR | No alteration by atorvastatin |
| [17] | MxA, TRAIL | [17] | PBMC | Affymetrix gene chip | No differences |
| [64] | IFN‐β induced genes | [18] | PBMC | Affymetrix gene chip | No differences |
| Modulation of immune system | |||||
| [14] | TNF, IFN‐γ, IL4, IL10 | [14] | Supernatant of ConA‐stimulated PBMC | Multiplex bead array | Atorvastatin: increase of IL10 |
| [11] | IL1β, IL2, IL6, IL12p70, TNF, IFN‐γ, IL4, IL5, IL10 | [65] | Serum | Multiplex bead array | combination: increase of IL12p70 |
| [66] | MMP9, TIMP1 | [65] | Serum | ELISA | No differences |
| [67] | soluble Fas (CD95), soluble FasL (CD95L) | [65] | Serum | Multiplex bead array | No differences |
| [14] | T‐cell proliferation | [14] | PBMC | 3H thymidine incorporation assay | Atorvastatin: no anti‐proliferative effect |
| Leukocyte migration | |||||
| [68] | Transendothelial migration | [68] | T cells | In vitro BBB model | Combination therapy: migrational capacity decreases |
| [46] | Transendothelial migration | treatment ex vivo | Monocytes/lymphocytes | In vitro BBB model | Statin treatment ex vivo: restricts migration |
| Antiinflammatory effects | |||||
| [43] | High sensitivity CRP | [65] | Serum | ELISA | Combination therapy: reduces hs‐CRP |
BBB, blood‐brain barrier; ConA, concanavalin A; CRP, C‐reactive protein; TRAIL, TNF‐related apoptosis‐inducing ligand; TNF, tumor necrosis factor; hs, high sensitivity; IFN, interferon; IL, interleukin; MMP, matrix‐metalloproteinase; PBMC, peripheral‐blood mononuclear cells; TIMP, tissue inhibitor of MMP.
The immunomodulatory action of statins is reflected by modification of the expression of several molecules crucially implicated in the pathogenesis of MS. Both, similarities and differences of statins and IFN‐β with regard to their immunomodulatory actions and potency were observed in vitro[5]. These differences particularly refer to an increase of proinflammatory cytokines such as IFN‐γ and IL12 and decrease of the antiinflammatory IL10 (Figure 2). While IFN‐β‐1b reduces and simvastatin increases the expression IFN‐γ and IL12 in vitro[5], patients on a combination therapy of IFN‐β‐1b and atorvastatin had significantly increased IL12p70 levels [11]. Likewise, in vitro IL10 expression is raised by IFN‐β‐1b and decreased by simvastatin [5], and a trend for an increase of IL10 serum levels was found in vivo by the combination treatment [11]. The role of Th1/Th2/Th17 immunity in EAE has become more apparent during recent years. However, the situation in MS is more complex and the exact role of immunomodulatory treatments such as IFN‐β and statins are yet to be determined. The evaluation of soluble CD95 and CD95L confirmed previously described effects by IFN‐β and no further alteration by additional treatment with atorvastatin [67]. To this end, it was reported that simvastatin may increase the proteolytic activity MMP9, a protease essential for degradation of the extracellular matrix and subsequent migration of leukocyte to the brain [69, 70]. Indeed, treatment with statins increased influx of leukocytes to the inflamed peritoneum [71]. In vivo we could confirm that MMP9 activity is attenuated by IFN‐β but the net effect is not altered after joint treatment with atorvastatin [66]. Treatment with IFN‐β was shown to enhance gene expression of certain chemokines in peripheral blood including CCL1, CCL2, CCL7, CXCL10, CXCL11, and this peripheral upregulation was suggested to reduce chemoattraction of leukocytes to the CNS [72]. Statins, however, were reported to restrict leukocyte migration by attenuation of chemokine secretion (CCL2, CXCL10) by endothelial cells [46].
Hence, many of the in vitro findings pointing at a potential interference of IFN‐β and statins or a differential action are only partially confirmed in vivo. Additional mechanisms may be involved in supraadditive or antagonizing effects and further evaluations are required. Particularly whether certain statins are more likely to affect IFN‐β bioactivity due to different pharmacodynamic characteristics and immunomodulatory potency and interfere with physiological and regenerative pathways within the CNS due to lowering of cholesterol are important questions that need to be addressed.
Treatment of Hyperlipidemia in Patients with MS
At this time, no clear statement can be made on the value of statins as potential DMDs in MS. However another important issue is certain to emerge in clinical practice. Treatment of hyperlipidemia is an essential component of primary and secondary prevention of cardiovascular events and can be achieved through HMG‐CoA reductase inhibition. Hypercholesterolemia is among the most frequent comorbidities in MS (37%) [73] and a substantial proportion of these MS patients will require pharmacological treatment for lowering cholesterol with statins. In many patients, lowering of cholesterol will likely be in concert with IFN‐β. Based on all available data there is no rationale to stop IFN‐β in these patients but a higher rate of adverse events including elevation of liver enzymes, CK, and muscle pain can be expected and a close clinical follow‐up including laboratory examinations is indicated. Yet, whether a certain statin is better tolerated when used together with IFN‐β and whether lower statin dosages should be preferred in this case still need to be elucidated.
Conclusions
The approval of immunomodulatory drugs in the early 1990s was a major therapeutic advance and while it is accepted that IFN‐β modifies the inflammatory disease phase of MS, little is known about their exact mechanisms of action. Statins, the well‐established therapeutic agents in cardiovascular medicine have been considered a potentially interesting add‐on agent for many years. Compelling experimental and preliminary clinical background provided the rationale for several small Phase II trials evaluating different combinations of IFN‐β preparations and statins in CIS and RRMS. The combined treatments were generally well tolerated; the side effects with most adverse events related to hepatic and muscle problems were in the expected range and need to be kept in mind for both further clinical trials and patients on IFN‐β with the need of treating hyperlipidemia by HMG‐CoA reductase inhibition. A single trial, however, raised concerns toward this combination by reporting a possible abrogation of IFN‐β effects by statins. These findings illustrate the problematic issue of translating in vitro and animal studies into clinical practice and more importantly how to draw conclusions from small trials evaluating of short‐term effects. While additional trials, admittedly mostly interim and safety studies, did not confirm these findings and particularly did not detect a loss of IFN‐β bioactivity in vivo, further clinical and experimental interest in this direction have almost certainly been significantly diminished. Partly, the approval of natalizumab and the introduction of other oral DMDs including cladibrine (Leustatin®) and FTY720 (fingolimod) as well as highly specific and effective biologics such as alemtuzumab (Campath®) or rituximab (MabThera®) may have been involved in this development. The use of statins as DMDs outside of controlled MS trials or beyond the treatment of hyperlipidemia in MS patients, regardless if mono‐ or combination therapy cannot be advised until further study evidence is available. A large, prospective, randomized, double‐blind, placebo‐controlled trial will be required to make a definite statement with regard to the value of this potential treatment strategy of IFN‐β and statins and may subsequently rehabilitate this drug combination. However, such a trial is currently not scheduled and it can be hoped that a critical analysis of the shortly finished trials will shed light on the potential impact of combining IFN‐β and statins on the course of RRMS.
Disclosures
JS and PV: none.
MSW has received research funding from Teva Pharmaceutical Industries Ltd.
HPM received honoraria and research funding from Bayer‐Schering, Merck‐Serono/Biogen‐Idec, and Sanofi‐Aventis.
BH Editorial/Advisory board and speaker's fees from Bayer Schering, Biogen Idec, Merck Serono, Novartis, Teva. Travel grants from Bayer, Biogen Idec, Merck Serono. Research Grants from Bayer, BiogenIdec, MerckSerono, Novartis.
OS serves on scientific advisory boards for Novartis and Teva Pharmaceutical Industries Ltd., serves on editorial boards for Archives of Neurology and Therapeutic Advances in Neurological Disorders, has received honoraria from Teva Pharmaceutical Industries Ltd., Genzyme Corporation, and Bayer Schering Pharma, and has received research support from the US Department of Veterans Affairs (Merit Review Grant).
Conflict of Interest
The authors declare no conflict of interests.
Acknowledgments
Author Contributions: All authors were involved in the following steps: concept/design, data analysis/interpretation, drafting article, critical revision of article, and approval of article.
Funding: JS was supported by a KKF fellowship provided by the Technische Universität München.
References
- 1. McFarland HF, Martin R. Multiple sclerosis: A complicated picture of autoimmunity. Nat Immunol 2007;8:913–919. [DOI] [PubMed] [Google Scholar]
- 2. Kieseier BC, Wiendl H, Leussink VI, Stüve O. Immunomodulatory treatment strategies in multiple sclerosis. J Neurol 2008;255:15–21. [DOI] [PubMed] [Google Scholar]
- 3. Soos JM, Stüve O, Youssef S, Bravo M, Johnson HM, Weiner HL,, Zamvil SS. Cutting edge: Oral type I IFN‐tau promotes a Th2 bias and enhances suppression of autoimmune encephalomyelitis by oral glatiramer acetate. J Immunol 2002;169:2231–2235. [DOI] [PubMed] [Google Scholar]
- 4. Youssef S, Stuve O, Patarroyo JC, et al The HMG‐CoA reductase inhibitor, atorvastatin, promotes a Th2 bias and reverses paralysis in central nervous system autoimmune disease. Nature 2002;420:78–84. [DOI] [PubMed] [Google Scholar]
- 5. Neuhaus O, Strasser‐Fuchs S, Fazekas F, Kieseier BC, Niederwieser G, Hartung HP, Archelos JJ. Statins as immunomodulators: Comparison with interferon‐beta 1b in MS. Neurology 2002;59:990–997. [DOI] [PubMed] [Google Scholar]
- 6. Aktas O, Waiczies S, Smorodchenko A, et al Treatment of relapsing paralysis in experimental encephalomyelitis by targeting Th1 cells through atorvastatin. J Exp Med 2003;197:725–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Sena A, Pedrosa R, Graca Morais M. Therapeutic potential of lovastatin in multiple sclerosis. J Neurol 2003;250:754–755. [DOI] [PubMed] [Google Scholar]
- 8. Vollmer T, Key L, Durkalski V, et al Oral simvastatin treatment in relapsing‐remitting multiple sclerosis. Lancet 2004;363:1607–1608. [DOI] [PubMed] [Google Scholar]
- 9. Waubant E, Pelletier D, Mass M, et al Atorvastatin therapy in patients with clinically isolated syndrome and high‐risk for conversion to multiple sclerosis: The STAyCIS study. Mult Scler 2009;15:S272. [Google Scholar]
- 10. Birnbaum G, Cree B, Altafullah I, Zinser M, Reder AT. Combining beta interferon and atorvastatin may increase disease activity in multiple sclerosis. Neurology 2008;71:1390–1395. [DOI] [PubMed] [Google Scholar]
- 11. Sellner J, Greeve I, Findling O, et al Effect of interferon‐beta and atorvastatin on Th1/Th2 cytokines in multiple sclerosis. Neurochem Int 2008;53:17–21. [DOI] [PubMed] [Google Scholar]
- 12. Feng X, Han D, Kilaru B, TB N, Reder AT. Inhibitory effect of high‐dose atorvastatin on interferon‐beta signalling in multiple sclerosis. Mult Scler 2009;15:S78. [Google Scholar]
- 13. Dhawan N, Reder AT. Statins block interferon signaling in human immune cells: Potential loss of the therapeutic effect of IFN beta in multiple sclerosis. Neurology 2007;68:A364. [Google Scholar]
- 14. Paul F, Waiczies S, Wuerfel J, et al Oral high‐dose atorvastatin treatment in relapsing‐remitting multiple sclerosis. PLoS One 2008;3:e1928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Rudick RA, Pace A, Rani MR, et al Effect of statins on clinical and molecular responses to intramuscular interferon beta‐1a. Neurology 2009;72:1989–1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lanzillo R, Orefice G, Quarantelli M, et al Atorvastatin combined to interferon to verify the efficacy (ACTIVE) in relapsing‐remitting active multiple sclerosis patients: A longitudinal controlled trial of combination therapy. Mult Scler 2010;16:450–454. [DOI] [PubMed] [Google Scholar]
- 17. Sörensen PS, Frederikson IL, Lycke J, Sellebjerg E. Does simvastatin antagonise the effect of interferon beta? Interim safety analysis of the ongoing SIMCOMBIN study. Mult Scler 2007;13:S25. [Google Scholar]
- 18. Markovic‐Plese S, Speer D, Jin J, Chen J, Smrtka J, Ingram L, Jewells V. Statin and intramuscular interferon beta‐1a combination therapy is safe and well tolerated in patients with clinically isolated syndrome suggestive of multiple sclerosis: A pilot study. Mult Scler 2007;13:S270. [Google Scholar]
- 19. Oztekin NS, Oztekin FM, Munis OB. Atorvastatin combined with interferon beta 1a in relapsing‐remitting multiple sclerosis: Preliminary results of a 24 month randomized open‐label clinical trial. Mult Scler 2008;14:S171. [Google Scholar]
- 20. Clark LT. Treating dyslipidemia with statins: The risk‐benefit profile. Am Heart J 2003;145:387–396. [DOI] [PubMed] [Google Scholar]
- 21. Lutjohann D, Stroick M, Bertsch T, et al High doses of simvastatin, pravastatin, and cholesterol reduce brain cholesterol synthesis in guinea pigs. Steroids 2004;69:431–438. [DOI] [PubMed] [Google Scholar]
- 22. Evans BA, Evans JE, Baker SP, et al Long‐term statin therapy and CSF cholesterol levels: Implications for Alzheimer's disease. Dement Geriatr Cogn Disord 2009;27:519–524. [DOI] [PubMed] [Google Scholar]
- 23. Saher G, Brugger B, Lappe‐Siefke C, et al High cholesterol level is essential for myelin membrane growth. Nat Neurosci 2005;8:468–475. [DOI] [PubMed] [Google Scholar]
- 24. Weber MS, Youssef S, Dunn SE, et al Statins in the treatment of central nervous system autoimmune disease. J Neuroimmunol 2006;178:140–148. [DOI] [PubMed] [Google Scholar]
- 25. Nath N, Giri S, Prasad R, Singh AK, Singh I. Potential targets of 3‐hydroxy‐3‐methylglutaryl coenzyme A reductase inhibitor for multiple sclerosis therapy. J Immunol 2004;172:1273–1286. [DOI] [PubMed] [Google Scholar]
- 26. Neuhaus O, Hartung HP. Evaluation of atorvastatin and simvastatin for treatment of multiple sclerosis. Expert Rev Neurother 2007;7:547–556. [DOI] [PubMed] [Google Scholar]
- 27. Van Der Most PJ, Dolga AM, Nijholt IM, Luiten PG, Eisel UL. Statins: Mechanisms of neuroprotection. Prog Neurobiol 2009;88:64–75. [DOI] [PubMed] [Google Scholar]
- 28. Dunn SE, Youssef S, Goldstein MJ, Prod’homme T, Weber MS, Zamvil SS, Steinman L. Isoprenoids determine Th1/Th2 fate in pathogenic T cells, providing a mechanism of modulation of autoimmunity by atorvastatin. J Exp Med 2006;203:401–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ahn KS, Sethi G, Aggarwal BB. Reversal of chemoresistance and enhancement of apoptosis by statins through down‐regulation of the NF‐kappaB pathway. Biochem Pharmacol 2008;75:907–913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Greenwood J, Steinman L, Zamvil SS. Statin therapy and autoimmune disease: From protein prenylation to immunomodulation. Nat Rev Immunol 2006;6:358–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Tsakiri A, Tsiantoulas D, Frederiksen J, Svane IM. Increased immunopotency of monocyte derived dendritic cells from patients with optic neuritis is inhibited in vitro by simvastatin. Exp Neurol 2010;221:320–328. [DOI] [PubMed] [Google Scholar]
- 32. Peng X, Jin J, Giri S, et al Immunomodulatory effects of 3‐hydroxy‐3‐methylglutaryl coenzyme‐A reductase inhibitors, potential therapy for relapsing remitting multiple sclerosis. J Neuroimmunol 2006;178:130–139. [DOI] [PubMed] [Google Scholar]
- 33. Paintlia AS, Paintlia MK, Singh AK, Stanislaus R, Gilg AG, Barbosa E, Singh I. Regulation of gene expression associated with acute experimental autoimmune encephalomyelitis by Lovastatin. J Neurosci Res 2004;77:63–81. [DOI] [PubMed] [Google Scholar]
- 34. Stanislaus R, Gilg AG, Singh AK, Singh I. Immunomodulation of experimental autoimmune encephalomyelitis in the Lewis rats by Lovastatin. Neurosci Lett 2002;333:167–170. [DOI] [PubMed] [Google Scholar]
- 35. Waiczies S, Prozorovski T, Infante‐Duarte C, Hahner A, Aktas O, Ullrich O, Zipp F. Atorvastatin induces T cell anergy via phosphorylation of ERK1. J Immunol 2005;174:5630–5635. [DOI] [PubMed] [Google Scholar]
- 36. Stuve O, Youssef S, Weber MS, et al Immunomodulatory synergy by combination of atorvastatin and glatiramer acetate in treatment of CNS autoimmunity. J Clin Inves 2006;116:1037–1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zhang X, Markovic‐Plese S. Statins’ immunomodulatory potential against Th17 cell‐mediated autoimmune response. Immunol Res 2008;41:165–174. [DOI] [PubMed] [Google Scholar]
- 38. Miron VE, Zehntner SP, Kuhlmann T, et al Statin therapy inhibits remyelination in the central nervous system. Am J Pathol 2009;174:1880–1890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Klopfleisch S, Merkler D, Schmitz M, et al Negative impact of statins on oligodendrocytes and myelin formation in vitro and in vivo. J Neurosci 2008;28:13609–13614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Schulz JG, Bosel J, Stoeckel M, Megow D, Dirnagl U, Endres M. HMG‐CoA reductase inhibition causes neurite loss by interfering with geranylgeranylpyrophosphate synthesis. J Neurochem 2004;89:24–32. [DOI] [PubMed] [Google Scholar]
- 41. Paintlia AS, Paintlia MK, Khan M, Vollmer T, Singh AK, Singh I. HMG‐CoA reductase inhibitor augments survival and differentiation of oligodendrocyte progenitors in animal model of multiple sclerosis. Faseb J 2005;19:1407–1421. [DOI] [PubMed] [Google Scholar]
- 42. Paintlia AS, Paintlia MK, Singh AK, Singh I. Inhibition of Rho family functions by lovastatin promotes myelin repair in ameliorating experimental autoimmune encephalomyelitis. Mol Pharmacol 2008;73:1381–1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Sellner J, Greeve I, Mattle HP. Atorvastatin decreases high‐sensitivity C‐reactive protein in multiple sclerosis. Mult Scler 2008;14:981–984. [DOI] [PubMed] [Google Scholar]
- 44. Weitz‐Schmidt G. Lymphocyte function‐associated antigen‐1 blockade by statins: Molecular basis and biological relevance. Endothelium 2003;10:43–47. [DOI] [PubMed] [Google Scholar]
- 45. Stanislaus R, Singh AK, Singh I. Lovastatin treatment decreases mononuclear cell infiltration into the CNS of Lewis rats with experimental allergic encephalomyelitis. J Neurosci Res 2001;66:155–162. [DOI] [PubMed] [Google Scholar]
- 46. Ifergan I, Wosik K, Cayrol R, et al Statins reduce human blood‐brain barrier permeability and restrict leukocyte migration: Relevance to multiple sclerosis. Ann Neurol 2006;60:45–55. [DOI] [PubMed] [Google Scholar]
- 47. Stepien K, Tomaszewski M, Czuczwar SJ. Neuroprotective properties of statins. Pharmacol Rep 2005;57:561–569. [PubMed] [Google Scholar]
- 48. Zacco A, Togo J, Spence K, Ellis A, Lloyd D, Furlong S, Piser T. 3‐hydroxy‐3‐methylglutaryl coenzyme A reductase inhibitors protect cortical neurons from excitotoxicity. J Neurosci 2003;23:11104–11111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Dolga AM, Granic I, Nijholt IM, Nyakas C, Van Der Zee EA, Luiten PG, Eisel UL. Pretreatment with lovastatin prevents N‐methyl‐D‐aspartate‐induced neurodegeneration in the magnocellular nucleus basalis and behavioral dysfunction. J Alzheimers Dis 2009;17:327–336. [DOI] [PubMed] [Google Scholar]
- 50. Chen J, Zhang ZG, Li Y, et al Statins induce angiogenesis, neurogenesis, and synaptogenesis after stroke. Ann Neurol 2003;53:743–751. [DOI] [PubMed] [Google Scholar]
- 51. Lu D, Goussev A, Chen J, Pannu P, Li Y, Mahmood A, Chopp M. Atorvastatin reduces neurological deficit and increases synaptogenesis, angiogenesis, and neuronal survival in rats subjected to traumatic brain injury. J Neurotrauma 2004;21:21–32. [DOI] [PubMed] [Google Scholar]
- 52. Lu D, Qu C, Goussev A, et al Statins increase neurogenesis in the dentate gyrus, reduce delayed neuronal death in the hippocampal CA3 region, and improve spatial learning in rat after traumatic brain injury. J Neurotrauma 2007;24:1132–1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Dolga AM, Nijholt IM, Ostroveanu A, Ten Bosch Q, Luiten PG, Eisel UL. Lovastatin induces neuroprotection through tumor necrosis factor receptor 2 signaling pathways. J Alzheimers Dis 2008;13:111–122. [DOI] [PubMed] [Google Scholar]
- 54. Cerezo‐Guisado MI, Garcia‐Marin LJ, Lorenzo MJ, Bragado MJ. Lovastatin inhibits the growth and survival pathway of phosphoinositide 3‐kinase/protein kinase B in immortalized rat brain neuroblasts. J Neurochem 2005;94:1277–1287. [DOI] [PubMed] [Google Scholar]
- 55. Polman CH, Reingold SC, Barkhof F, et al Ethics of placebo‐controlled clinical trials in multiple sclerosis: A reassessment. Neurology 2008;70:1134–1140. [DOI] [PubMed] [Google Scholar]
- 56. Stuve O, Bennett JL, Hemmer B, et al Pharmacological treatment of early multiple sclerosis. Drugs 2008;68:73–83. [DOI] [PubMed] [Google Scholar]
- 57. Goldman MD, Cohen JA. Statins to treat multiple sclerosis: Friend or foe? Neurology 2008;71:1386–1387. [DOI] [PubMed] [Google Scholar]
- 58. Durelli L, Conti L, Clerico M, et al T‐helper 17 cells expand in multiple sclerosis and are inhibited by interferon‐beta. Ann Neurol 2009;65:499–509. [DOI] [PubMed] [Google Scholar]
- 59. Ramgolam VS, Sha Y, Jin J, Zhang X, Markovic‐Plese S. IFN‐beta inhibits human Th17 cell differentiation. J Immunol 2009;183:5418–5427. [DOI] [PubMed] [Google Scholar]
- 60. Taniguchi T, Takaoka A. A weak signal for strong responses: Interferon‐alpha/beta revisited. Nat Rev Mol Cell Biol 2001;2:378–386. [DOI] [PubMed] [Google Scholar]
- 61. Singh P, Kohr D, Kaps M, Blaes F. Influence of statins on MHC class I expression. Ann N Y Acad Sci 2009;1173:746–751. [DOI] [PubMed] [Google Scholar]
- 62. Zhang X, Jin J, Peng X, Ramgolam VS, Markovic‐Plese S. Simvastatin inhibits IL‐17 secretion by targeting multiple IL‐17‐regulatory cytokines and by inhibiting the expression of IL‐17 transcription factor RORC in CD4+ lymphocytes. J Immunol 2008;180:6988–6996. [DOI] [PubMed] [Google Scholar]
- 63. Kagami S, Owada T, Kanari H, et al Protein geranylgeranylation regulates the balance between Th17 cells and Foxp3+ regulatory T cells. Int Immunol 2009;21:679–689. [DOI] [PubMed] [Google Scholar]
- 64. Markovic‐Plese S, Jewells V, Speer D. Combining beta interferon and atorvastatin may increase disease activity in multiple sclerosis. Neurology 2009;72:1965; author reply 1965–1966. [DOI] [PubMed] [Google Scholar]
- 65. Kamm CP, Mattle HP. SWiss Atorvastatin and interferon Beta‐1b trial in multiple sclerosis (SWABIMS): Rationale, design and methodology. Trials 2009;10:115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Sellner J, Greeve I, Leib SL, Mattle HP. Atorvastatin does not alter interferon Beta‐induced changes of serum matrix metalloproteinase 9 and tissue inhibitor of metalloproteinase 1 in patients with multiple sclerosis. Arch Neurol 2008;65:672–674. [DOI] [PubMed] [Google Scholar]
- 67. Sellner J, Greeve I, Findling O, Grandgirard D, Leib SL, Mattle HP. Atorvastatin does not alter serum levels of sCD95 and sCD95L in multiple sclerosis. Clin Exp Immunol 2008;152:280–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Greeve I, Sellner J, Minten C, et al Effects of immunomodulatory treatment in multiple sclerosis on the transendothelial migration of T‐cells across the blood‐brain barrier. Mult Scler 2009;15:S237. [Google Scholar]
- 69. Kieseier BC, Archelos JJ, Hartung HP. Different effects of simvastatin and interferon beta on the proteolytic activity of matrix metalloproteinases. Arch Neurol 2004;61:929–932. [DOI] [PubMed] [Google Scholar]
- 70. Sellner J, Simon F, Meyding‐Lamade U, Leib SL. Herpes‐simplex virus encephalitis is characterized by an early MMP‐9 increase and collagen type IV degradation. Brain Res 2006;1125:155–162. [DOI] [PubMed] [Google Scholar]
- 71. Kiener PA, Davis PM, Murray JL, Youssef S, Rankin BM, Kowala M. Stimulation of inflammatory responses in vitro and in vivo by lipophilic HMG‐CoA reductase inhibitors. Int Immunopharmacol 2001;1:105–118. [DOI] [PubMed] [Google Scholar]
- 72. Cepok S, Schreiber H, Hoffmann S, et al Enhancement of chemokine expression by interferon beta therapy in patients with multiple sclerosis. Arch Neurol 2009;66:1216–1223. [DOI] [PubMed] [Google Scholar]
- 73. Marrie R, Horwitz R, Cutter G, Tyry T, Campagnolo D, Vollmer T. Comorbidity, socioeconomic status and multiple sclerosis. Mult Scler 2008;14:1091–1098. [DOI] [PubMed] [Google Scholar]