

SUMMARY
The second generation atypical antipsychotic, asenapine (Saphris), was approved by the US FDA (August 2009) for the acute treatment of manic or mixed episodes with or without psychotic features associated with bipolar I disorder in adults as well as the acute treatment of schizophrenia. Asenapine exhibits a high affinity for and antagonism at several serotonergic (5‐HT2A‐C, 5HT5A, 5HT6, 5HT7), dopaminergic (D2, D3), alpha‐adrenergic (α1 and α2), and histaminergic (H1, H2) receptor subtypes. Asenapine is the first atypical antipsychotic formulated as a fast‐dissolving, rapidly absorbed sublingual tablet. Asenapine was evaluated in adults with bipolar I disorder, manic or mixed episodes with or without psychotic features. Two identically designed 3‐week registration trials confirmed the efficacy of asenapine relative to placebo in studies that included olanzapine as an active control. The placebo‐subtracted rate of EPS (excluding akathisia) is 5% whereas the placebo‐subtracted rate of akathisia was 2%. The placebo‐subtracted rate of clinically significant weight gain (≥7%) with asenapine was approximately 5% during the 3‐week acute mania trials. A 9‐ extension trial indicated that 19% of asenapine patients will experience clinically significant weight gain. Clinically significant metabolic abnormalities were not observed during the acute and/or extension trials. Asenapine can be associated with somnolence (asenapine 24%, placebo 6%) and does not appear to be associated with clinically significant changes in vital signs, laboratory parameters, or electrocardiographic changes. Bipolar depression and recurrence prevention studies are required to fully characterize this novel agent's position in the treatment of bipolar disorder.
Keywords: Acute mania, Asenapine, Bipolar disorder, Maintenance
Introduction
During the past decade, numerically more agents have received US FDA approval for bipolar disorder than the previous five decades combined. Atypical antipsychotics have been the class of agents most studied across various phases of bipolar disorder. The acute treatment of mania has been the primary focus with bipolar depression and recurrence prevention studies a more recent emphasis of clinical drug development. Notwithstanding the disparate options available to practitioners, most individuals with bipolar disorder fail to functionally recover from an acute manic episode whereas others are intolerant and/or have treatment discontinued because of safety concerns [1, 2, 3]. Taken together, a modest proportion of individuals with bipolar disorder are able to tolerate and achieve symptomatic, syndromal, and functional recovery with available agents, underscoring the need for alternative treatment avenues [4].
Asenapine was approved by the US FDA for the acute treatment of manic or mixed episodes, with or without psychotic features, associated with bipolar I disorder (and the acute treatment of schizophrenia). Orally administered sublingual asenapine is formulated as a fast‐dissolving, rapidly absorbed tablet. This review summarizes study results from two identically designed acute studies enrolling individuals with bipolar mania or mixed episodes with or without psychotic features as well as a 9‐week extension.
Asenapine: Pharmacology
Pharmodynamics
Asenapine exhibits high affinity for serotonin 5‐HT1A, 5‐HT1B, 5‐HT2A, 5‐HT2B, 5‐HT2C, 5‐HT5, 5‐HT6, and 5‐HT7 receptors, dopamine D2, D3, D4, and D1 receptors, α1 and α2‐adrenergic receptors, and histamine H1 receptors, and moderate affinity for H2 receptors. Asenapine has no appreciable affinity for muscarinic cholinergic receptors [5].
Pharmacokinetics
Asenapine is rapidly absorbed; the absolute bioavailability of sublingual asenapine 5 mg is 35%. Increasing the dose from 5 to 10 mg BID results in nonlinear increases in maximum concentration [5, 6]. The absolute bioavailability of asenapine when swallowed is low (i.e., less than 2% with oral tablet formation). The intake of water within 5 min after asenapine administration decreases asenapine exposure by up to 20%; an observation that has provided the rationale for recommending no eating or drinking for a minimum of 10 min after administration. Oral hypoesthesia (i.e., numbing of the tongue) was spontaneously reported in 4% of subjects enrolled in the acute mania trials. The rate of discontinuation due to oral hypoesthesia was 0.27% across the short‐term acute mania (and schizophrenia) trials. Dysgeusia (i.e., distortion of the sense of taste) was spontaneously reported in 3% of subjects in the acute mania trials with a discontinuation rate of 0%[5, 6].
Asenapine is rapidly distributed and has a large volume of distribution (approximately 20/25 L/kg) and is highly bound to circulating proteins (e.g., albumin and alpha‐1‐acid glycoprotein). Steady‐state concentrations of asenapine are reached within 3 days of BID dosing. Asenapine is a substrate for CYP1A2, UGT1A4 and to a lesser extent CYP3A4 and CYP2D6. Asenapine is a weak inhibitor of CYP2D6 and does not induce CYP1A2 or CYP3A4. Smoking is not known to affect the absorption kinetics of asenapine. It is not known, however, if chronic smoking alters the pharmacokinetics of asenapine (i.e., via induction of hepatic enzymes). Consumption of food prior to administration decreases asenapine exposure by 20%; consumption of food up‐to 4 h after administration decreases exposure by 10%. The coadministration of asenapine with the CYP1A2 inhibitor, fluvoxamine 25 mg BID for 8 days, increased the maximum concentration of asenapine (Cmax) 13%, and the area under the concentration curve (AUC) 29%. No known clinically significant changes in the bioavailability of asenapine were noted when it was coprescribed with paroxetine, imipramine, cimetidine, carbamazepine, and valproate [5, 6, 7]. Nevertheless, as asenapine relies principally on CYP1A2, and to a lesser extent, CYP2D6 and UGT1A4 for clearance, as a therapeutic principle, caution is warranted when co‐prescribing agents that are either substrates inducers and/or inhibitors of these enzymes.
Asenapine: Efficacy in Acute Mania
The efficacy of asenapine in acute mania was evaluated in two identically designed trials in adults (≥18 years) presenting with acute manic or mixed episode associated with bipolar I disorder (methodological details are presented in the published manuscripts for each of these studies [8, 9, 10]). Both studies were similar insofar as they were multicentre, international 3‐week, randomized, double‐blind, placebo‐controlled, parallel‐group studies, with olanzapine used to assess assay sensitivity. Eligible patients presented with a current Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition (DSM‐IV) primary diagnosis of bipolar I disorder experiencing a manic or mixed episode, with a Young Mania Rating Scale (YMRS) total score ≥20 at screening and baseline. The current manic or mixed bipolar I episode had to begin ≤3 months before screening, and patients needed a documented history of ≥1 previous moderate to severe mood episode with or without psychotic features. Rapid cycling bipolar disorder during the past year was an exclusion criterion. Asenapine was initiated in both studies at 10 mg BID on day 1 with an opportunity to decrease the dose to 5 mg BID from day 2 onward. Olanzapine was initiated at 15 mg QD on day 1 with an opportunity to titrate to 5–20 mg from day 2 onward.
Results from study one (N = 488 subjects were randomized) are presented in Figure 1 (Ares 7501004; NCT0015974) [10]. Mean daily doses were 18.4 mg asenapine and 15.9 mg olanzapine. Least squares mean changes in YMRS total score on day 21 were significantly greater with asenapine than placebo (−11.5 vs. −7.8; P < 0.007), with advantage seen at the first postrandomization observation day (i.e., day 2) (−3.2 vs. −1.7; P= 0.022). YMRS response and remission rates with olanzapine (54.7%; 46.3%, respectively) but not asenapine (42.6%; 35.5%, respectively), were significantly greater than placebo (34.0%; 30.9%, respectively). The mean change from baseline using last observation carried forward (LOCF) analysis at day 21 on the Montgomery Asberg Depression Rating Scale (MADRS) was −3.0 with asenapine (P= NS vs. placebo) and −4.1 (<0.03 vs. placebo) with olanzapine.
Figure 1.
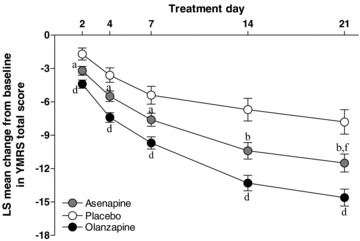
Mean change from baseline in YMRS total score (Study 1 Ares7501004).
Study discontinuations due to treatment‐related adverse events were recorded in 9.2% of asenapine‐treated patients, 4.1% of placebo patients, and 3.4% of olanzapine‐treated patients. Incidence of EPS‐related adverse events was 10.3%, 3.1%, and 6.8% with asenapine, placebo, and olanzapine, respectively; the incidence of clinically significant weight gain (i.e., ≥ 7%) was 7.2%; 1.2%; 19.0%, respectively. Mean weight change (baseline to endpoint) was 0.9, 0.1, and 2.6 kg with asenapine, placebo, and olanzapine, respectively.
Results from study two (N = 489 subjects were randomized) are presented in Figure 2 (Ares 7501005, NCT 00159796) [8]. The mean daily doses were asenapine 18.2 mg and olanzapine 15.8 mg. Significantly greater least squares mean ± standard error changes in YMRS scores were observed on day 2 with asenapine (−3.0 ± 0.4) and olanzapine (−3.4 ± 0.4) versus placebo (−1.5 ± 0.5, bother P < 0.01) and were maintained until day 21 (−10.8 ± 0.8 with asenapine, −12.6 ± 0.8 with olanzapine; both P≤ 0.0001 vs. placebo, −5.5 ± 1.1) with LOCF. Asenapine‐treated subjects exhibited significantly greater response (42.3%) and remission (40.2%) compared with placebo (25.2% and 22.3%, respectively; P < 0.01). Olanzapine‐treated subjects also exhibited significantly greater response (50.0%, P < 0.0001) versus placebo and remission (39.4%, P= 0.0041).
Figure 2.
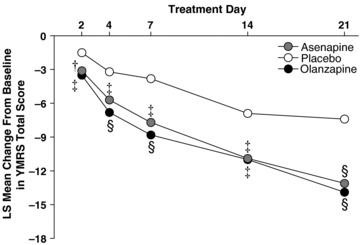
Mean change from baseline in YMRS total score (Study 2 Ares7501005).
Post hoc LOCF analysis in patients with a diagnosis of a manic episode indicated that the mean change from baseline to day 21 in the total YMRS score was significantly greater with asenapine and olanzapine than with placebo (−11.0, −12.9, and −4.8, respectively). In comparison, LOCF analysis indicated that the change from baseline to treatment day 21 in mixed patients, as measured by the YMRS, was not significant for asenapine (−11.3) versus placebo (−7.4) but was significantly greater with olanzapine (−12.7; P= 0.006).
Study discontinuations due to treatment‐related adverse events were recorded in 6.2% of asenapine‐treated patients, 3.8% of placebo patients, and 4.2% of olanzapine‐treated patients. Incidence of EPS‐related adverse events was 7.2%, 2.9%, and 7.9% with asenapine, placebo, and olanzapine, respectively. The incidence of clinically significant weight gain was 6.0%; 0.0%, and 12.9%, respectively. Mean weight change (baseline to endpoint) was 1.6, 0.3 and 1.9 kg [Correction added after online publication: 15th October 2010 The values 0.9, 0.1 and 2.6 were amended to 1.6, 0.3 and 1.9] with asenapine, placebo, and olanzapine, respectively. In both studies changes from baseline in metabolic parameters, laboratory values, and vital signs were not considered clinically significant.
Asenapine Extension Phase Efficacy
A multicentre, 9‐week noninferiority double‐blind extension of both (Ares 7501004, Ares 7501005) 3‐week double‐blind trials in patients with acute manic or mixed episodes as part of bipolar I disorder was completed (Ares 7501006, NCT00143182) [9]. This 9‐week extension did not include a placebo arm and instead was a direct comparison of asenapine versus olanzapine. The 9‐week extension study began at the end of the 3‐week acute trials without breaking the blind occurring at week 12. There was no rerandomization at the beginning of the extension study and no identification of patients who had received active treatment versus placebo during the 3‐week trial. In the extension study, patients who had received acute treatment with asenapine or olanzapine continued the same regimen and were evaluated at the end of week 12 for efficacy and safety. Patients who received placebo during the acute treatment were blindly switched to asenapine 10 mg BID on day 1 with flexible dosing thereafter and included in the safety analysis only.
A total of 504 patients were enrolled in the extension study [9]. At day 84, the mean change from baseline in YMRS total score was −24.4 (8.7) for asenapine and −23.9 (7.9) for olanzapine (Figure 3). Noninferiority statistical analysis indicated no significant difference between asenapine and olanzapine. The rates of discontinuation due to adverse events were 19.1%, 13.3%, and 9.6% in the placebo/asenapine, asenapine, and olanzapine groups, respectively. The incidence of EPS of any type was 10% with placebo/asenapine, 15% with asenapine, and 13% with olanzapine treatment. The percentage of patients with clinically significant weight gain was greater with olanzapine (31%) than asenapine (19%). Similar to the acute trials, there were no clinically significant changes in laboratory measures, metabolic parameters or vital signs.
Figure 3.
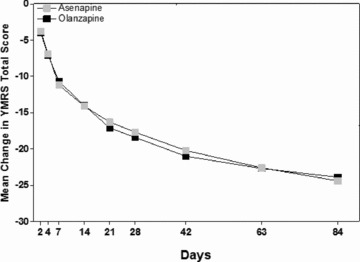
Mean change from baseline in YMRS total score. Results from the 9‐week extension phase.
Patients completing the extension phase (i.e., 12‐week study) were eligible for a 40‐week extension double‐blind study. Results of this study have not been published and as such will not be reviewed in detail herein.
Conclusion
Asenapine is established as an effective agent in the acute treatment of mania associated with bipolar I disorder. The overall effect of asenapine in reducing manic severity as measured by the YMRS is not dissimilar from any other FDA‐approved antimanic agent [4, 11]. Moreover, the trial design herein, and with other antimanic agents, does not provide adequate insight regarding relative onset of action. It is currently unknown which, if any, antimanic agents have a faster onset of action. Asenapine's tmax of 30–90 min provides the basis for hypothesizing that it may have a unique rapidity in its onset of antimanic action. This however remains a testable hypothesis rather than a proven fact as the studies herein have their first observation point on day 2 of the treatment trial.
Changes in weight observed in this clinical trial program were greater in the asenapine‐treated group when compared to the placebo‐treated subjects. As expected, changes in body composition, as well as metabolic parameters were significant in the olanzapine‐treated group relative to placebo. Olanzapine was included for assay sensitivity and as such, this is not the ideal trial design to establish comparative efficacy and tolerability profiles. Nevertheless it is notable that olanzapine's efficacy, presented as either a total YMRS reduction or as response and remission rates, appear to be greater than with asenapine.
Asenapine did not exert any clinically significant effects on metabolic or laboratory parameters (e.g., prolactin), vital signs or the electrocardiographic measures. Asenapine is associated with EPS when compared to placebo; there is insufficient data to determine whether or not there is a hazard for tardive dyskinesia over and above placebo. The induction and/or intensification of depressive symptoms in manic patients was not observed and instead an improvement in total depression scores was noted coincident with improved manic symptoms.
Clinicians and patients have several options available for the acute and extension treatment of bipolar mania. Asenapine exhibits a placebo‐subtracted improvement on manic symptoms (i.e., total YMRS score) comparable to other agents FDA approved for acute mania [11]. The sublingual formulation is an advantage in terms of acceptance for some patients. In addition, patients who have difficulty swallowing (and/or do not like to swallow tablets), may prefer a sublingual alternative. Disadvantages of asenapine are the need to wait up to 10 min before consuming food or drink after administration. It is reassuring that the rates of hypoesthesia and dysgeusia are relatively low. Initial impressions of this agent amongst clinicians is that the taste alteration might be more common than was exprienced in the clinical trials. The recent approval of the black cherry flavour formulation may partially assuage this concern. As such, all patients should be informed of this possible adverse event. Moreover, asenapine is recommended to be prescribed in multiple daily doses, without question, patients and providers prefer once a day dosing. It is reasonable to surmise that asenapine will be prescribed, or defaulted, to once a day dosing. The effectiveness and acceptability of this “off‐label” approach has not been studied. Results in bipolar depression and maintenance studies will “round out” the therapeutic portrait of this agent.
Conflict of Interest
The author is on the advisory board for Astra Zeneca, Bristol‐Myers Squibb, France Foundation, GlaxoSmithKline, Janssen‐Ortho, Solvay/Wyeth, Eli Lilly, Organon, Lundbeck, Biovail, Pfizer, Shire, Schering‐Plough and Merck. He is also on the speaker's bureau for Janssen‐Ortho, AstraZeneca, Eli Lilly, Lundbeck, Biovail and Merck and has received research grants from Eli Lilly, Janssen‐Ortho, Shire, Astra‐Zeneca and Pfizer.
References
- 1. McIntyre RS, Konarski JZ. Tolerability profiles of atypical antipsychotics in the treatment of bipolar disorder. J Clin Psychiatry 2005;66(Suppl 3):28–36. [PubMed] [Google Scholar]
- 2. Tohen M, Zarate CA, Jr ., Hennen J, et al The McLean‐Harvard First‐Episode Mania Study: Prediction of recovery and first recurrence. Am J Psychiatry 2003;160:2099–2107. [DOI] [PubMed] [Google Scholar]
- 3. Perlis RH, Ostacher MJ, Patel JK, et al Predictors of recurrence in bipolar disorder: Primary outcomes from the systematic treatment enhancement program for bipolar disorder (STEP‐BD). Am J Psychiatry 2006;163:217–224. [DOI] [PubMed] [Google Scholar]
- 4. Yatham LN, Kennedy SH, Schaffer A, et al Canadian Network for Mood and Anxiety Treatments (CANMAT) and International Society for Bipolar Disorders (ISBD) collaborative update of CANMAT guidelines for the management of patients with bipolar disorder: Update 2009. Bipolar Disord 2009;11:225–255. [DOI] [PubMed] [Google Scholar]
- 5. Shahid M, Walker GB, Zorn SH, Wong EH. Asenapine: A novel psychopharmacologic agent with a unique human receptor signature. J Psychopharmacol 2009;23:65–73. [DOI] [PubMed] [Google Scholar]
- 6. Tarazi FI, Shahid M. Asenapine maleate: A new drug for the treatment of schizophrenia and bipolar mania. Drugs Today (Barc) 2009;45:865–876. [DOI] [PubMed] [Google Scholar]
- 7. McIntyre RS. Pharmacology and efficacy of asenapine for manic and mixed states in adults with bipolar disorder. Expert Rev Neurother 2010;10:645–649. [DOI] [PubMed] [Google Scholar]
- 8. McIntyre RS, Cohen M, Zhao J, Alphs L, Macek TA, Panagides J. A 3‐week, randomized, placebo‐controlled trial of asenapine in the treatment of acute mania in bipolar mania and mixed states. Bipolar Disord 2009;11:673–686. [DOI] [PubMed] [Google Scholar]
- 9. McIntyre RS, Cohen M, Zhao J, Alphs L, Macek TA, Panagides J. Asenapine versus olanzapine in acute mania: A double‐blind extension study. Bipolar Disord 2009;11:815–826. [DOI] [PubMed] [Google Scholar]
- 10. McIntyre RS, Cohen M, Zhao J, Alphs L, Macek TA, Panagides J. Asenapine in the treatment of acute mania in bipolar I disorder: A randomized, double‐blind, placebo‐controlled trial. J Affect Disord 2010;122:27–38. [DOI] [PubMed] [Google Scholar]
- 11. Perlis RH, Welge JA, Vornik LA, Hirschfeld RM, Keck PE, Jr . Atypical antipsychotics in the treatment of mania: A meta‐analysis of randomized, placebo‐controlled trials. J Clin Psychiatry 2006;67:509–516. [DOI] [PubMed] [Google Scholar]