

Abstract
The purpose of this study was to determine whether lomerizine, a Ca2+ channel blocker, protects against neuronal degeneration within the dorsal lateral geniculate nucleus (dLGN) and superior colliculus (SC) after the induction of retinal damage by intravitreal injection of N‐methyl‐d‐aspartate (NMDA) in mice. NMDA (20 mM/2 μL) was injected into the vitreous body of the left eye in mice (DAY 0). Lomerizine at 30 mg/kg, p.o. was administered daily from immediately after the injection of NMDA (DAY 0) to 90 days after (DAY 90). To investigate the neuroprotective effects of lomerizine, the retina, dLGN, and SC were examined using histochemistry and immunohistochemistry. Lomerizine reduced the retinal damage induced by NMDA and partially prevented the transsynaptic neuronal degeneration within dLGN and SC on the contralateral side. Moreover, lomerizine reduced the intravitreal NMDA induced decrease in the light‐induced expression of c‐Fos in the contralateral dLGN (used in this study to evaluate residual vision). These results indicate that lomerizine affords some protection against transsynaptic neuronal degeneration within the visual center of the mouse brain.
Keywords: Glaucoma, Lateral geniculate nucleus, Lomerizine, N‐methyl‐d‐aspartate, Superior colliculus
Abbreviations:
- dLGN
dorsal lateral geniculate nucleus
- GCL
ganglion cell layer
- NMDA
N‐methyl‐d‐aspartate
- NTG
normal tension glaucoma
- ONH
optic nerve head
- RGC
retinal ganglion cell
- SC
superior colliculus
- VDCCs
voltage‐dependent Ca2+ channels
Introduction
Lomerizine, 1‐[bis(4‐fluorophenyl)methyl]‐4‐(2,3,4‐trimethoxybenzyl)‐piperazine dihydrochloride, is a Ca2+ channel blocker, which was developed as a potential agent for the selective improvement of the ocular or cerebrovascular circulation with minimal adverse cardiovascular effects [1, 2]. Lomerizine selectively relaxes smooth muscle cells by inhibiting Ca2+ influx, and by doing so, it can reduce tone and increase blood flow in cerebral vessels [3]. Currently, lomerizine is used clinically for the oral prophylaxis of migraine in Japan [1]. Migraine, an inherited or acquired clinical syndrome consisting of moderate‐to‐severe pulsatile headache, has traditionally been considered to be associated with changes in the caliber of blood vessels in the head.
The development of ocular disorders such as glaucoma is attributed to an insufficient blood flow [4, 5, 6, 7]. In glaucoma, it is believed that insufficient blood flow leads to retinal ganglion cell (RGC) death [6, 8, 9]. Certainly, there have been many reports of Ca2+ channel blockers being beneficial therapeutic agents for glaucoma (following both clinical and experimental studies) [5, 10, 11, 12, 13]. Several investigators have reported beneficial effects of Ca2+ channel blockers against RGC death [14, 15, 16], while systemic administration of Ca2+ channel blockers reportedly retards the progression of visual field loss, at least temporarily, in the subset of glaucoma patients with normal tension glaucoma (NTG) [10, 17, 18]. Although the mechanisms underlying these effects are not fully understood, lomerizine has been reported to (1) increase blood flow in ocular neuronal tissue [the retina and optic nerve head (ONH)] in conscious rabbits, without changing systemic blood pressure [19], and (2) have beneficial effects against the RGC death induced by ischemia/reperfusion (high intraocular pressure) injury in the rat retina [20]. These findings indicate that the protective effects of lomerizine against retinal ischemic injury may be partly due to improvements in the ocular circulation.
Recent evidence indicates that glaucomatous damage extends from the retina to the visual center of the brain [21, 22]. Such data suggest that the visual field loss induced by glaucoma may result from not only RGC loss but also neuronal degeneration within the visual center of the brain. Visual information entering the human eye is processed in the retina and then transmitted via the optic nerve to the lateral geniculate nucleus (LGN), the major relay center between the eye and the visual cortex. It is generally believed that in rodent, most axonal projections from the retina pass to the superior colliculus (SC). Protection of neurons located within the visual center of the brain, as well as protection of RGC, could be effective for the prevention of blindness in glaucoma. However, little is known about the kinds of drugs that might offer protection against neuronal degeneration within the visual center after retinal damage. In this study, we investigated whether lomerizine might protect against the neuronal degeneration within LGN and SC, which occurs as a results of RGC damage in mice.
Materials and Methods
All experiments were performed in accordance with the ARVO Statement for the Use of Animals in Ophthalmic and Vision Research, and they were approved and monitored by the Institutional Animal Care and Use Committee of Gifu Pharmaceutical University.
Animal Subjects
Male adult C57BL/6J mice weighing 20–28 g (Clea Japan Inc., Fujimiya, Japan) were kept under lighting conditions of 12 h light and 12 h dark. They were anesthetized with 3.0% isoflurane (Merck, Osaka, Japan) and maintained with 1.5% isoflurane in 70% N2O and 30% O2 via an animal general anesthesia apparatus (Soft Lander; Sin‐ei Industry Co. Ltd., Saitama, Japan). Retinal damage was induced (on DAY 0) by the intravitreal injection (2 μL/eye) of NMDA (Sigma‐Aldrich, St. Louis, MO, USA) dissolved at 20 mM in 0.01 M phosphate‐buffered saline (PBS) at pH 7.4. This was injected into the vitreous body of the left eye, under the above anesthesia. One drop of levofloxacin ophthalmic solution (Santen Pharmaceuticals Co. Ltd., Osaka, Japan) was applied topically to the treated eye immediately after the intravitreal injection. Mice that received intravitreal NMDA were also treated either with lomerizine (30 mg/kg, p.o.; Schering‐Plough, Osaka, Japan) or with the vehicle for lomerizine (distilled water containing 5% arabic gum, p.o.) by oral gavage for the periods detailed below. Mice receiving an intravitreal injection of saline instead of NMDA formed the sham‐treated group. Mice were euthanized on DAY 90. At 90 days after NMDA injection, three groups of mice were evaluated and compared: (1) sham‐treated group, n = 11, (2) vehicle‐treated group (mice receiving an intravitreal injection of NMDA and also [from DAY 0 to DAY 90; p.o.] the vehicle for lomerizine), n = 11, and (3) lomerizine‐treated group (mice receiving an intravitreal injection of NMDA and also a daily administration of lomerizine at 30 mg/kg, p.o., from DAY 0 to DAY 90), n = 11. The body weight of each mouse was monitored twice a week, from DAY 0 to DAY 90. For the c‐Fos immunohistochemistry experiment, mice (n = 6 in each group) were kept under dark conditions for 36 h, then exposed to light stimuli (800–1200 lux) for 1 h before euthanasia.
Tissue Processing
At the end of their survival period, mice were anesthetized with sodium pentobarbital (80 mg/kg, i.p.; Nembutal; Dainippon, Osaka, Japan), then perfused with 2% (w/v) paraformaldehyde solution in 0.01 M PBS. The brains were removed after 15‐min perfusion at 4°C, immersed in the same fixative solution for 24 h, soaked in 25% (w/v) sucrose for 1 day, and then frozen in embedding compound (Tissue‐Tek; Sakura Finetechnical Co. Ltd., Tokyo, Japan). Serial coronal sections through the levels of LGN (bregma −1.70 to −2.80 mm) and SC (bregma −3.30 to −3.96 mm) were cut at 20‐μm thickness, then stained with cresyl violet. Each eye was enucleated at the time of brain removal, and 2% paraformaldehyde solution was injected into the vitreous body. The eye was then kept immersed for at least 24 h in the same fixative solution at 4°C. Six paraffin‐embedded sections (thickness, 5 μm) cut through the optic disc of each eye were prepared in a standard manner and then stained with hematoxylin and eosin.
Histological Analysis of Mouse Retina
Retinal damage was evaluated as previously described [23], three of the six sections stained with hematoxylin and eosin from each eye being used for the morphometric analysis. Light‐microscope photographs were taken using a digital camera (Coolpix 4500; Nikon, Tokyo, Japan), and the cell counts in the ganglion cell layer (GCL) at a distance between 375 and 625 μm from the optic disc were measured on the images in a masked fashion by a single observer (Y. I.). Data from three sections (selected randomly from the six sections) were averaged for each eye, and the values obtained were used to evaluate the GCL cell count.
Neuron Numbers in dLGN and SC
To assess the protective effects of lomerizine against neuronal loss in dorsal LGN (dLGN) and SC, sections stained with cresyl violet were used for neuronal cell counting: six sections for dLGN (L1: bregma −1.70 mm, L2: −1.92 mm, L3: −2.14 mm, L4: −2.36 mm, L5: −2.58 mm, and L6: −2.80 mm) and four for SC (S1: bregma −3.30 mm, S2: −3.52 mm, S3: −3.74 mm, and S4: −3.96 mm) in each mouse (Figure 1). The area of each field used for this cell counting was 0.036 mm2 of dLGN (Figure 1B & D) and 0.036 mm2 of the superficial layer of SC (Figure 1C & E). Cell counts were carried out under a microscope at 400× magnification in a masked fashion by a single observer (Y. I.). Care was taken to count only neurons with clearly visible nuclei and cytoplasm.
Figure 1.
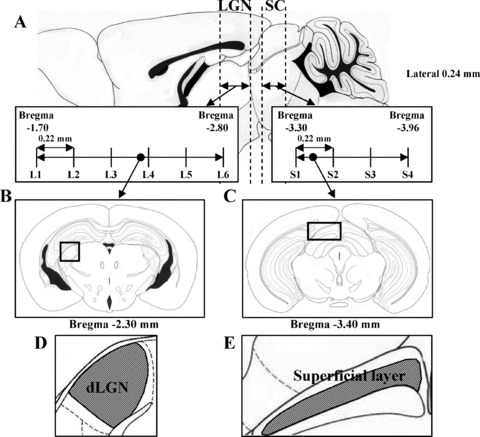
Illustrations showing (A) serial coronal sections through the levels of LGN (bregma −1.70 to −2.80 mm) and SC (bregma −3.30 to −3.96 mm) in mice. Coronal sections through LGN level (bregma −2.30 mm) (B) and SC level (bregma −3.40 mm) (C) in mice [boxed areas are shown diagrammatically in (D) and (E), respectively].
Volume of dLGN
Measurements of the surface area of dLGN were made in four equally spaced sections (interval, 220 μm) from section L1 to section L6 (i.e., the sections covering the bulk of dLGN). These area measurements were carried out under a microscope at 100× magnification in a masked fashion by a single observer (Y. I.), recorded as images using a digital camera (Coolpix 4500; Nikon, Tokyo, Japan), and then quantitated using Image J. Cavalieri's estimator of volume was used to calculate the volume (VT) of dLGN (bregma −1.70 to −2.80 mm) using the following formula: VT=∑ai× sf, where ai is the cross‐sectional area of the dLGN in the i‐th profile and sf is the mean distance between sections (section thickness multiplied by the inverse of the periodicity of sections in the series) [24].
Immunohistochemistry
For immunohistochemistry, coronal sections containing LGN and SC were obtained from sham‐, vehicle, and lomerizine‐treated mice, and placed on slides (MASCOAT; Matsunami, Osaka, Japan). In the immunostaining procedures for c‐Fos, coronal sections containing LGN and SC were washed with 0.01 M PBS, then treated with 0.3% hydrogen peroxidase in 0.01 M PBS. Next, they were preincubated with 10% normal goat serum (Vector, CA, USA) in 0.01 M PBS for 30 min and then incubated for 1 day at 4°C with specific rabbit anti‐c‐Fos polyclonal antibody (1: 20,000 dilution; PC38, Calbiochem, CA, USA) in the following solution: 10% normal goat serum in 0.01 M PBS containing 0.3% (v/v) Triton X‐100. They were washed with PBS and then incubated with biotinylated anti‐rabbit IgG, before being incubated with the avidin‐biotin‐peroxidase complex for 30 min at room temperature. Finally, for visualization, DAB was used as a peroxidase substrate.
To visualize colocalization of c‐Fos with NeuN, double immunofluorescence was performed on dLGN and SC sections from the three groups (sham‐, vehicle‐, and lomerizine‐treated mice). Coronal sections containing dLGN and SC were washed with 0.01 M PBS. Next, they were preincubated with 10% normal goat serum in 0.01 M PBS for 30 min, then incubated overnight at 4°C with rabbit anti‐c‐Fos polyclonal antibody (1: 20,000 dilution) in the following solution: 10% normal goat serum in 0.01 M PBS with 0.3% (v/v) Triton X‐100. Sections were blocked with M.O.M. blocking reagent (M.O.M. immunodetection kit; Vector), then incubated with mouse anti‐NeuN monoclonal antibody (1: 250 dilution; MAB377, Chemicon, CA, USA) for one day at 4°C. They were washed with 0.01 M PBS and then incubated for 3 h at room temperature with a mixture of Alexa Fluor 488 F(ab')2 fragment of goat anti‐rabbit IgG (H + L) (1: 1000 dilution; Molecular Probes, OR, USA) and Alexa Fluor 546 F(ab')2 fragment of goat anti‐mouse IgG (H + L) (1: 1000 dilution; Molecular Probes).
NeuN‐Immunopositive Neurons and c‐Fos‐Immunopositive Cells in dLGN and SC
For the quantitative analysis of NeuN‐immunopositive neurons and c‐Fos‐immunopositive cells in dLGN and SC, sections immunostained with NeuN and c‐Fos were used (two sections each for the dLGN and SC of each mouse). The area of each field used for this cell counting was 0.036 mm2 (in both dLGN and the superficial layers of SC). Cell counts were carried out under a microscope at 400× magnification in a masked fashion by a single observer (Y. I.). Data from two sections were averaged for each region, and the values obtained were used to evaluate the NeuN‐immunopositive neurons and c‐Fos‐immunopositive cells count.
Statistical Analysis
Data are presented as means ± SEM statistical comparisons were made using a by a one‐way analysis of variance (ANOVA), followed by Dunnett's t‐test with SPSS (Ver. 16.0J, SPSS Japan, Tokyo, Japan). A value of P < 0.05 was considered to indicate statistical significance.
Results
Animals
There were no significant differences among the three groups (sham‐, vehicle‐, and lomerizine‐treated mice) in body‐weight changes from DAY 0 to DAY 90 (data not shown).
Protective Effect of Lomerizine against Retinal Damage
Intravitreal injection of NMDA at 40 nmol/eye decreased the GCL cell count (33.4 ± 2.3 cells/mm, n = 8) in the retina versus that in the sham‐treated retina (127.6 ± 1.2 cells/mm, n = 8; P < 0.01) at 90 days after the injection. The lomerizine‐treated group displayed a significant suppression of this decrease (45.8 ± 2.1 cells/mm, n = 8; P < 0.01) (Figure 2A–C, Table 1).
Figure 2.
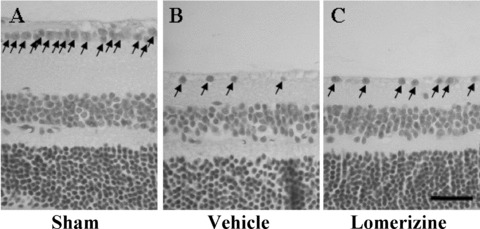
Protective effects of lomerizine against retinal damage (as assessed at 90 days after NMDA intravitreous injection). Representative photographs of retinas obtained from sham‐ (A), vehicle‐ (B), and lomerizine‐treated (C) groups. Arrows indicate live cells within GCL. Scale bar = 30 μm.
Table 1.
Protective effects of lomerizine against retinal damage at 90 days after NMDA intravitreous injection
| Groups | Cell number in GCL | % of sham |
|---|---|---|
| Sham | 127.6 ± 1.2 | 100.0 |
| Vehicle | 33.4 ± 2.3 ## | 26.2 |
| Lomerizine | 45.8 ± 2.1 ** | 35.9 |
Cells number in GCL was measured. Each eye was enucleated, together with the optic nerve on DAY 90 (NMDA injection = DAY 0). Three of the six sections stained with hematoxylin and eosin (thickness, 5 μm) from each eye were used for the morphometric analysis. Mice received a daily administration of lomerizine at 30 mg/kg, p.o., from DAY 0 to DAY 90. Each value represents the mean ± SEM for 8 eyes.
**P < 0.01 versus vehicle, ## P < 0.01 versus sham (Dunnett's t‐test).
Protective Effect of Lomerizine against dLGN Damage
In the contralateral dLGN, the neuron counts obtained for the vehicle‐treated group were 40.2 ± 2.5 (n = 11), 38.6 ± 1.8 (n = 11), 34.0 ± 2.2 (n = 11), 33.4 ± 2.7 (n = 11), 41.1± 4.1 (n = 11), and 36.2 ± 2.3 (n = 11) in sections L1, L2, L3, L4, L5, and L6, respectively. Each of these counts was smaller than the corresponding one for in the sham‐treated group (54.6 ± 2.5, 46.6 ± 2.6, 54.4 ± 2.1, 56.8 ± 3.9, 62.2 ± 4.4, and 56.0 ± 2.8, respectively, each for n = 9, P < 0.01 in each case). The lomerizine‐treated group (48.5 ± 2.5, 48.9 ± 2.5, 47.9 ± 2.7, 47.3 ± 3.5, 55.6 ± 4.6, and 48.0 ± 3.3, respectively, each for n = 9) exhibited significantly greater contralateral dLGN neuron numbers than the vehicle‐treated group (Figures 3 & 4A).
Figure 3.
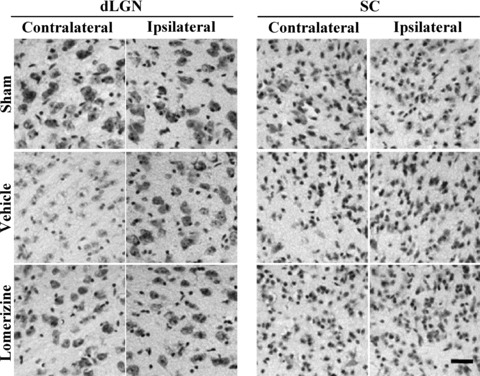
Protective effects of lomerizine against secondary neuronal degeneration in two brain regions (LGN and SC) at 90 days after NMDA intravitreous injection. Representative photographs of parts of cresyl violet‐stained coronal sections containing LGN of SC (contralateral or ipsilateral side). Photographs are shown for sham‐treated, vehicle‐treated, and lomerizine‐treated groups. Scale bar = 30 μm.
Figure 4.
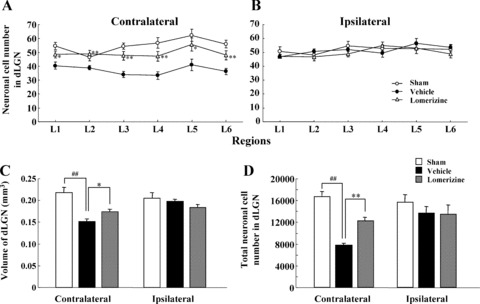
Protective effects of lomerizine against neuronal loss and volume shrinkage in dLGN at 90 days after NMDA intravitreous injection. Neuronal cell number within dLGN was measured on the contralateral (A) and ipsilateral (B) sides. Coronal sections through level of dLGN (L1: bregma −1.70 mm, L2: −1.92 mm, L3: −2.14 mm, L4: −2.36 mm, L5: −2.58 mm, and L6: −2.80 mm). In each field, the tissue area of dLGN examined was 0.036 mm2. On both the contralateral and ipsilateral sides, dLGN volume (C) and total neuronal cell number (D) were measured. Each value represents the mean ± SEM for 9–11 brains. *P < 0.05, **P < 0.01 versus vehicle; ## P < 0.01 versus sham (Dunnett's t‐test).
The volume of the contralateral dLGN was smaller in the vehicle‐treated group (0.152 ± 0.197 mm3, n = 11) than in the sham‐treated group (0.218 ± 0.205 mm3, n = 9; P < 0.01), while the value for the lomerizine‐treated group (0.174 ± 0.183 mm3, n = 9) was significantly greater (P < 0.05) than that for the vehicle‐treated group (Figure 4C).
Likewise, the total neuronal cell number in the contralateral dLGN was smaller in the vehicle‐treated group (7790.4 ± 406.2, n = 11) than in the sham‐treated group (16705.8 ± 956.5, n = 9; P < 0.01), while the value for the lomerizine‐treated group (12189.8 ± 697.3, n = 9) was significantly greater (P < 0.01) than that for the vehicle‐treated group (Figure 4D).
In contrast, in the ipsilateral dLGN there were no significant differences in numbers of neurons or volume among the three groups (sham‐, vehicle‐, and lomerizine‐treated mice; Figures 3 & 4B–D).
Protective Effect of Lomerizine against SC Damage
In the contralateral SC, the neuron counts obtained for in the vehicle‐treated group were 63.2 ± 6.3 (n = 9), 48.3 ± 3.4 (n = 9), 56.1 ± 5.1 (n = 9), and 73.3 ± 7.3 (n = 9) in sections S1, S2, S3, and S4, respectively. Each of these counts was smaller than the corresponding one for the sham‐treated group (69.1 ± 4.5, 73.7 ± 3.3, 79.0 ± 5.8, and 85.9 ± 8.1, respectively, each for n = 9, P < 0.01 in each case). The lomerizine‐treated group (78.1 ± 3.2 for S1, 76.0 ± 1.7 for S2, 74.9 ± 3.2 for S3, and 74.0 ± 1.5 for S4, each for n = 8) exhibited significantly greater contralateral SC neuron numbers than the vehicle‐treated group (Figures 3 & 5A).
Figure 5.
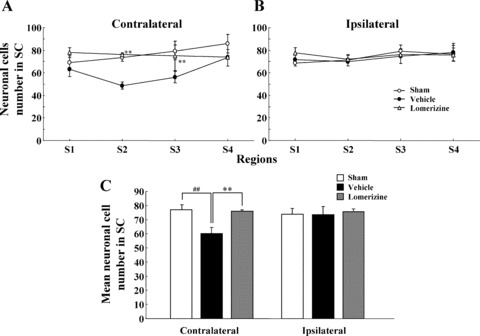
Protective effects of lomerizine against neuronal loss in SC at 90 days after NMDA intravitreous injection. Neuronal cell number within SC was measured on the contralateral (A) and ipsilateral (B) sides. Coronal sections through level of SC (S1: bregma −3.30 mm, S2: −3.52 mm, S3: −3.74 mm, and S4: −3.96 mm). In each field, the tissue area of SC examined was 0.036 mm2. On both the contralateral and ipsilateral sides, mean neuronal cell number in SC was measured (C). Each value represents the mean ± SEM for 8–10 brains. **P <0.01 versus vehicle; ## P < 0.01 versus sham (Dunnett's t‐test).
The mean neuronal cell number in the contralateral SC was smaller in the vehicle‐treated group (60.3 ± 4.1, n = 9) than in the sham‐treated group (76.9 ± 3.7, n = 9; P < 0.01), while the value was significantly greater (P < 0.01) for the lomerizine‐treated group (75.8 ± 1.0, n = 8) than for the vehicle‐treated group (Figure 5C).
In contrast, in the ipsilateral SC there were no significant differences in numbers of neurons among the three groups (sham‐, vehicle‐, and lomerizine‐treated mice; 3, 5).
c‐Fos‐Immunopositive Cells in dLGN and SC
At 90 days after the intravitreal injections, the c‐Fos‐immunopositive cell number in the contralateral dLGN was smaller in the vehicle‐treated group (38.5 ± 1.7, n = 6) than in the sham‐treated group (45.6 ± 2.1, n = 6; P < 0.05), while the value for the lomerizine‐treated group (46.3 ± 1.6, n = 6) was significantly greater (P < 0.01) than that for the vehicle‐treated group (Figure 6A–D). In contrast, in the ipsilateral dLGN there were no significant differences in numbers of c‐Fos‐immunopositive cells among the three groups (sham‐, vehicle‐, and lomerizine‐treated mice) (Figure 6D).
Figure 6.
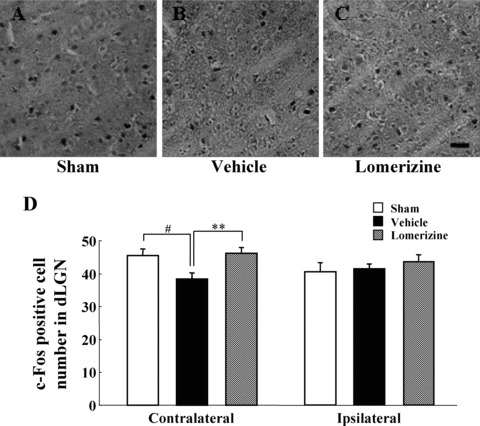
Effects of lomerizine on intravitreal NMDA‐induced attenuation of light‐induced c‐Fos expression in dLGN. c‐Fos immunostaining of d LGN sections obtained from sham‐, vehicle‐, and lomerizine‐treated mice. Representative microphotographs are shown for contralateral dLGN of sham‐ (A), vehicle‐ (B), and lomerizine‐treated mice (C). Scale bar = 30 μm. (D) Average number of c‐Fos immunopositive cell counts is shown for dLGN (per 0.036 mm2 tissue area). Each value represents the mean ± SEM for 6 brains. **P < 0.01 versus vehicle, # P < 0.05 versus sham (Dunnett's t‐test).
Concerning the SC on the contralateral and ipsilateral sides, there were no significant differences in numbers of c‐Fos‐immunopositive cells among the three groups (sham‐, vehicle‐, and lomerizine‐treated mice; data not shown).
Double Immunofluorescence
To identify c‐Fos‐positive cells, double immunofluorescence was performed for c‐Fos and NeuN using LGN sections. NeuN‐positive neuronal cells were found to express c‐Fos in sections from all three groups (sham‐, vehicle‐, and lomerizine‐treated mice (Table 2); Fig. 7 shows the representative results for the sham‐treated group).
Table 2.
Cell counts for c‐Fos/NeuN double‐labeling in dLGN at 90 days after NMDA intravitreous injection
| Groups | Contralateral | Ipsilateral |
|---|---|---|
| Sham | 40.4 ± 2.7 | 36.0 ± 3.0 |
| Vehicle | 26.3 ± 3.2 ## | 37.8 ± 2.0 |
| Lomerizine | 36.0 ± 1.5 * | 35.6 ± 1.4 |
c‐Fos/NeuN double‐immunostaining of d LGN sections obtained from sham‐, vehicle‐, and lomerizine‐treated mice. Average number of c‐Fos/NeuN immunopositive cell counts is shown for dLGN (per 0.036 mm2 tissue area). Each value represents the mean ± SEM for 5–6 brains.
*P < 0.05 versus vehicle, ## P < 0.01 versus sham (Dunnettt's t‐test).
Figure 7.
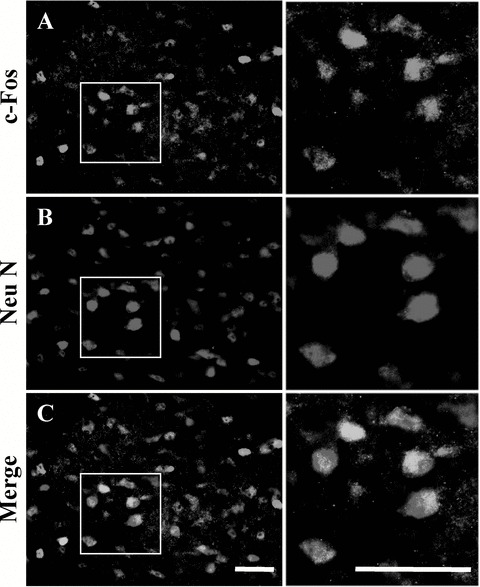
Representative photographs showing c‐Fos (A) and NeuN (B) immunostaining, and c‐Fos/NeuN (C) double‐immunostaining of dLGN from sham‐treated mice. Some c‐Fos‐expressing cells (A, green) were colocalized with NeuN‐labeled neurons (B, red), as indicated by yellow color in C (merge of A and B). Boxed areas in (A), (B), and (C) are shown at higher magnification in the right‐hand panels. Scale bars = 30 μm.
Discussion
The purpose of this study was to determine whether lomerizine exerts protective effects against the neuronal degeneration that occurs within dLGN and SC following retinal damage in mice. We found that systemic administration of lomerizine (for 90 days, p.o.) attenuated both the retinal and brain (dLGN and SC) damage seen at 90 days after an intravitreal injection of NMDA.
Within the visual center of the brain, neuronal loss was evident in the contralateral dLGN (number of neurons and volume) and the contralateral SC (number of neurons) on DAY 90 after intravitreal NMDA injection (3, 4, 5). Using the present mouse model, we showed previously that systemic administration of memantine, an NMDA‐receptor antagonist, protected against secondary neuronal degeneration within the visual center of the brain after retinal damage [25]. Furthermore, Yücel et al. have noted that monkeys with experimental glaucoma that had been treated systemically with memantine showed significantly less neuronal shrinkage in LGN than the vehicle‐treated glaucoma group [26]. Presumably, these neuronal cell‐death mechanisms partly involve the NMDA‐receptor subtype expressed by the relay neurons within the LGN [27, 28]. Indeed, NMDA‐receptor activation and subsequent excessive Ca2+ influx through voltage‐dependent Ca2+ channels (VDCCs) induces neuronal death [29, 30]. In previous in vitro studies, lomerizine (0.1–10 μM) reduced glutamate‐induced neurotoxicity in rat hippocampal primary cell and retinal cell culuture by blocking excess Ca2± entry through VDCCs [20, 31, 32, 33]. In vivo studies, brain concentration (Cmax) of lomerizine (2 mg/kg, p.o.) in rats was about 0.43 μM [34]. Therefore, we can estimate that the brain concentration of lomerizine (30 mg/kg, p.o.) in mice is about 6.5 μM. The concentration of lomerizine exhibited protective effects on RGC death after optic nerve injury and cerebral ischemic neuronal damage in the rat [13, 35]. Taken together, these data suggest that lomerizine may block the influx of Ca2+ via VDCCs located both in RGC and within the brain and, by this means, may protect RGC and the neurons of the visual center.
The light‐stimulated expression of transcriptional regulatory protein (c‐Fos) was used in this study to evaluate the residual vision of NMDA‐treated mice [36, 37], with conventional immunohistochemistry being employed to detect c‐Fos‐immunopositive cells within the visual center of the brain after retinal damage. It has been reported that a basal expression of c‐Fos is seldom observed, and that c‐Fos‐labeled nuclei are expressed in the LGN and SC of C57BL/6J mice only after light‐stimulation [37]. NeuN‐positive neuronal cells expressing c‐Fos are thought to retain their sensitivity to light. In the present study, the lomerizine‐treated group exhibited a greater c‐Fos‐immunopositive cell number in the contralateral dLGN than the vehicle‐treated group (Figure 6). In addition, there were no significant differences among the three groups (sham‐, vehicle‐, and lomerizine‐treated mice) in the c‐Fos/NeuN double‐labeled cell number when this was expressed as a percentage of the total c‐Fos‐labeled cell number in the contralateral dLGN (data not shown). In a previous report we demonstrated that glial fibrillary acid protein (GFAP) and brain‐derived neurotrophic factor (BDNF) expressions were also increased before neuronal loss within LGN in our mouse model. Furthermore, the GFAP‐positive astroglial cells partly exhibited BDNF [38]. In previous reports, reactive astrocytes expressing BDNF have promoted neuronal rescue and generate functional recovery [39]. Taken together, these factors will be involved in the relation to the retinal to central cell activity mechanism after NMDA injection. Since the cell activity mechanism was not determined in this study, further studies will be needed to clarify the precise mechanism. However, our findings indicate that lomerizine may help to maintain visual function in dLGN after retinal damage, and as a consequence it may help to prevent visual disturbances.
Anterograde and retrograde axonal transport processes are essential for the delivery of both extracellular components (such as growth factors) and intracellular components (such as recycled synaptic vesicles) between the nerve terminals and the cell bodies of neurons. Disturbances of such transport can deprive the somata of various essential substances, eventually leading to the death of the neuron. It has been demonstrated that impairments of axonal transport can be induced by numerous pathological conditions, including transient ischemia, acute intraocular pressure elevation, and glaucoma [40, 41, 42]. In glaucoma, neuronal loss within LGN may result from a reduction in the visual stimuli transmitted from RGCs, as well as from the depletion of neurotrophins that is associated with a dysfunction of anterograde axonal transport from RGCs [22]. These effects may lead to “Wallerian degeneration,” a process in which the part of the axon separated from the neuron's cell nucleus will degenerate [43]. A certain minimal level of Ca2+ is necessary to maintain both anterograde and retrograde transport [44, 45], and an impairment of Ca2+ homoeostasis is thought to play a pivotal role in triggering neuronal vulnerability in animal models [46]. Indeed, an elevation of the Ca2+ concentration inhibits both the anterograde and the retrograde transport of proteins [47]. Taken together, we suggest that lomerizine may rescue RGCs and neurons within the visual center by maintaining or improving axonal transport [13].
Lomerizine, which is used clinically for the oral prophylaxis of migraine in Japan, selectively reduces vascular tone by inhibiting Ca2+ influx and thereby increases cerebral blood flow [1, 3]. It has been reported that a decreasing cerebral blood flow is closely involved with glaucoma [7, 48]. In fact, epidemiologic studies suggest that migraine is significantly more common in patients with NTG [49] and both migraine and NTG involve reductions in blood flow (cerebral and ocular, respectively). These findings indicate that migraine and NTG may share, at least in part, a common pathogenesis, and that some Ca2+ channel blockers may be useful for the treatments of both diseases. In addition, it has been reported that cerebrovascular insufficiency has been observed in certain primary open‐angle glaucoma (POAG) and NTG patients' diminished central visual function [50, 51, 52]. These reports suggest that cerebrovascular insufficiency involved in the visual field defect in glaucoma patients. Therefore, lomerizine, a selective cerebral vasodilator, may have a therapeutic benefit for preventing or reducing visual field defect in glaucoma patients.
In clinical studies, a loss of more than 50% of RGC has been reported to induce visual field loss. However, the initial loss of RGC does not lead to visual field loss in humans [53]. These data suggest that the visual field loss induced by glaucoma may result not only from RGC loss but also from neuronal degeneration within LGN [21]. Therefore, we also performed daily administration of lomerizine from 7 to 90 days after NMDA injection (posttreatmet), because retinal damage in our model occurred dramatically until 7 days after NMDA injection, and then the degree was gradually increased by 180 days after NMDA injection [38]. Lomerizine protected against transsynaptic neuronal degeneration in some regions (L3, L4, and L6) of LGN but not total, and the potency of posttreatment was weaker than that of lomerizine treatment (data not shown).
In conclusion, our results demonstrate that long‐term oral repeated administration of lomerizine partially protects neurons within dLGN and SC against the secondary degeneration induced by retinal damage in mice. The protective mechanism may involve a suppression of NMDA receptor‐mediated neurotoxicity and an increase in blood flow in the brain (via a blockage of Ca2+ channels in neuronal cells and endothelial cells). In addition to therapy aimed at rescuing RGCs, a neuroprotective strategy aimed at rescuing neurons within the visual center of the brain, may be of therapeutic benefit in preventing or reducing visual field loss after retinal damage.
Conflict of Interest
None.
Acknowledgments
This study was supported in part by Grants‐in‐Aid for exploratory research from the Ministry of Education, Culture, Sports, Science, and Technology, Japan (Nos.18209053 and 18210101), and by a Grant‐in‐Aid from the Japan Society for the Promotion of Science (No. 20·10786).
References
- 1. Hara H, Morita T, Sukamoto T, Cutrer FM. Lomerizine (KB‐2796), a new antimigraine drug. CNS Drug Rev 1995;1:204–226. [Google Scholar]
- 2. Hara H, Toriu N, Shimazawa M. Clinical potential of lomerizine, a Ca2+ channel blocker as an anti‐glaucoma drug: Effects on ocular circulation and retinal neuronal damage. Cardiovasc Drug Rev 2004;22:199–214. [DOI] [PubMed] [Google Scholar]
- 3. Kanazawa T, Nakasu Y, Masuda M, Handa J. Acute effect of 1‐[bis(4‐fluorophenyl)methyl]‐4‐(2,3,4‐trimethoxybenzyl)‐piperazine dihydrochloride, KB‐2796, on the cerebral blood flow in unanesthetized cats. Arch Jpn Chir 1986;55:682–688. [PubMed] [Google Scholar]
- 4. Fechtner R, Weinreb RN. Mechanism of optic nerve damage in primary open angle glaucoma. Surv Ophthalmol 1994;39:23–42. [DOI] [PubMed] [Google Scholar]
- 5. Flammer J. The vascular concept of glaucoma. Surv Ophthalmol 1994;38:S3–S6. [DOI] [PubMed] [Google Scholar]
- 6. Martinez A, Sanchez M. Ocular blood flow and glaucoma. Br J Ophthalmol 2008;1301:1301–1302. [PubMed] [Google Scholar]
- 7. Yücel YH, Gupta N. Paying attention to the cerebrovascular system in glaucoma. Can J Ophthalmol 2008;43:342–346. [DOI] [PubMed] [Google Scholar]
- 8. Flammer J, Orgül S. Optic nerve blood‐flow abnormalities in glaucoma. Prog Retin Eye Res 1998;17:267–289. [DOI] [PubMed] [Google Scholar]
- 9. Flammer J, Orgül S, Costa VP, et al The impact of ocular blood flow in glaucoma. Prog Retin Eye Res 2002;21:359–393. [DOI] [PubMed] [Google Scholar]
- 10. Kitazawa Y, Shirai H, Go FJ. The effect of Ca2+ antagonist on visual field in low‐tension glaucoma. Graefes Arch Clin Exp Ophthalmol 1989;227:408–412. [DOI] [PubMed] [Google Scholar]
- 11. Netland PA, Chaturvedi N, Dreyer EB. Calcium channel blockers in the management of low‐tension and open‐angle glaucoma. Am J Ophthalmol 1993;115:608–613. [DOI] [PubMed] [Google Scholar]
- 12. Sawada A, Kitazawa Y, Yamamoto T, Okabe I, Ichien K. Prevention of visual field defect progression with brovincamine in eyes with normal‐tension glaucoma. Ophthalmology 1996;103:283–288. [DOI] [PubMed] [Google Scholar]
- 13. Karim Z, Sawada A, Kawakami H, Yamamoto T, Taniguchi T. A new calcium channel antagonist, lomerizine, alleviates secondary retinal ganglion cell death after optic nerve injury in the rat. Curr Eye Res 2006;31:273–283. [DOI] [PubMed] [Google Scholar]
- 14. Takahashi K, Lam TT, Edward DP, Buchi ER, Tso MO. Protective effects of flunarizine on ischemic injury in the rat retina. Arch Ophthalmol 1992;110:862–870. [DOI] [PubMed] [Google Scholar]
- 15. Jensen RJ. Effects of Ca2+ channel blockers on directional selectivity of rabbit retinal ganglion cells. J Neurophysiol 1995;74:12–23. [DOI] [PubMed] [Google Scholar]
- 16. Adachi K, Kashii S, Masai H, et al Mechanism of the pathogenesis of glutamate neurotoxicity in retinal ischemia. Graefes Arch Clin Exp Ophthalmol 1998;236:766–774. [DOI] [PubMed] [Google Scholar]
- 17. Gaspar AZ, Flammer J, Hendrickson P. Influence of nifedipine on the visual fields of patients with optic‐nerve‐head diseases. Eur J Ophthalmol 1994;4:24–28. [DOI] [PubMed] [Google Scholar]
- 18. Koseki N, Araie M, Yamagami J, Shirato S, Yamamoto S. Effects of oral brovincamine on visual field damage in patients with normal‐tension glaucoma with low‐normal intraocular pressure. J Glaucoma 1999;8:117–123. [PubMed] [Google Scholar]
- 19. Shimazawa M, Sugiyama T, Azuma I, et al Effect of lomerizine, a new Ca2+ channel blocker, on the microcirculation in the optic nerve head in conscious rabbits: A study using a laser speckle technique. Exp Eye Res 1999;69:185–193. [DOI] [PubMed] [Google Scholar]
- 20. Toriu N, Akaike A, Yasuyoshi H, Zhang S, Kashii S, Honda Y, Shimazawa M, Hara H. Lomerizine, a Ca2+ channel blocker, reduces glutamate‐induced neurotoxicity and ischemia/reperfusion damage in rat retina. Exp Eye Res 2000;70:475–484. [DOI] [PubMed] [Google Scholar]
- 21. Gupta N, Ang LC, Noel‐de TL, Bidaisee L, Yücel YH. Human glaucoma and neural degeneration in intracranial optic nerve, lateral geniculate nucleus, and visual cortex. Br J Ophthalmol 2006;90:674–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Vrabec JP, Levin LA. The neurobiology of cell death in glaucoma. Eye 2007;21:S11–S14. [DOI] [PubMed] [Google Scholar]
- 23. Yoneda S, Tanihara H, Kido N, Honda Y, Goto W, Hara H, Miyawaki N. Interleukin‐1beta mediates ischemic injury in the rat retina. Exp Eye Res 2001;73:661–667. [DOI] [PubMed] [Google Scholar]
- 24. Mooney SM, Miller MW. Postnatal generation of neurons in the ventrobasal nucleus of the rat thalamus. J Neurosci 2007;27:5023–5032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ito Y, Nakamura S, Tanaka H, Shimazawa M, Araie M, Hara H. Memantine protects against secondary neuronal degeneration in lateral geniculate nucleus and superior colliculus after retinal damage in mice. CNS Neurosci Ther 2008;14:192–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Yücel YH, Gupta N, Zhang Q, Mizisin AP, Kalichman MW, Weinreb RN. Memantine protects neurons from shrinkage in the lateral geniculate nucleus in experimental glaucoma. Arch Ophthalmol 2006;124:217–225. [DOI] [PubMed] [Google Scholar]
- 27. Jones EG, Tighilet B, Tran BV, Huntsman MM. Nucleus‐ and cell‐specific expression of NMDA and non‐NMDA receptor subunits in monkey thalamus. J Comp Neurol 1998;397:371–393. [DOI] [PubMed] [Google Scholar]
- 28. Jaubert‐Miazza L, Green E, Lo FS, Bui K, Mills J, Guido W. Structural and functional composition of the developing retinogeniculate pathway in the mouse. Vis Neurosci 2005;22:661–676. [DOI] [PubMed] [Google Scholar]
- 29. Lipton SA, Rosenberg PA. Excitatory amino acids as a final common pathway for neurologic disorders. N Engl J Med 1994;330:613–622. [DOI] [PubMed] [Google Scholar]
- 30. Sucher NJ, Lipton SA, Dreyer EB. Molecular basis of glutamate toxicity in retinal ganglion cells. Vision Res 1997;37:3483–3493. [DOI] [PubMed] [Google Scholar]
- 31. Hara H, Yokota K, Shimazawa M, Sukamoto T. Effect of KB‐2796, a new diphenylpiperazine Ca2+ antagonist, on glutamate‐induced neurotoxicity in rat hippocampal primary cell cultures. Jpn J Pharmacol 1993;61: 361–365. [DOI] [PubMed] [Google Scholar]
- 32. Hara H, Ozaki A, Yoshidomi M, Sukamoto T. Protective effect of KB‐2796, a new calcium antagonist, in cerebral hypoxia and ischemia. Arch Int Pharmacodyn Ther 1990;304:206–218. [PubMed] [Google Scholar]
- 33. Yamada H, Chen YN, Aihara M, Araie M. Neuroprotective effect of calcium channel blocker against retinal ganglion cell damage under hypoxia. Brain Res 2006;1071: 75–80. [DOI] [PubMed] [Google Scholar]
- 34. Kawashima T, Awata N, Satomi O, et al Absorption, distribution and excretion of 1‐[Bis (4‐fluorophenyl) methyl]‐4 (2, 3, 4‐trimethoxybenzyl) piperazine dihydrochloride (KB‐2796) after single oral administration in rats. Drug Metab Pharmacokinet 1990;5:723–737. [Google Scholar]
- 35. Yamashita A, Ozaki A, Ikegami A, Hayashi A, Hara H, Sukamoto T, Ito K. Effects of a new diphenylpiperazine calcium antagonist, KB‐2796, on cerebral ischemic neuronal damage in rats. Gen Pharmacol 1993;24:1473–1480. [DOI] [PubMed] [Google Scholar]
- 36. Oelschläger HH, Nakamura M, Herzog M, Burda H. Visual system labeled by c‐Fos immunohistochemistry after light exposure in the ‘blind’ subterranean zambian mole‐rat (Cryptomys anselli). Brain Behav Evol 2000;55:209–220. [DOI] [PubMed] [Google Scholar]
- 37. Lima AG, Britto LR, Hossokawa NM, Hamassaki‐Britto DE. Static, but not optokinetic visual stimuli induce Fos expression in the retina and brain of retinal degeneration mice. Neurosci Lett 2003;342:9–12. [DOI] [PubMed] [Google Scholar]
- 38. Ito Y, Shimazawa M, Inokuchi Y, Fukumitsu H, Furukawa S, Araie M, Hara H. Degenerative alterations in the visual pathway after NMDA‐induced retinal damage in mice. Brain Res 2008;1212:89–101. [DOI] [PubMed] [Google Scholar]
- 39. Young YM, Kim YJ, Lee U. Expression of mRNAs for BDNF and NT‐3 in Reactive Astrocytes. J Korean Neurosurg 2003;33:572–575. [Google Scholar]
- 40. Minckler DS, Bunt AH, Johanson GW. Orthograde and retrograde axoplasmic transport during acute ocular hypertension in the monkey. Invest Ophthalmol Vis Sci 1977;16:426–441. [PubMed] [Google Scholar]
- 41. Quigley HA, Addicks EM, Green WR, Maumenee AE. Optic nerve damage in human glaucoma, II: The site of injury and susceptibility to damage. Arch Ophthalmol 1981;99:635–649. [DOI] [PubMed] [Google Scholar]
- 42. Zhang L, Ino‐ue M, Dong K, Yamamoto M. Retrograde axonal transport impairment of large‐ and medium‐sized retinal ganglion cells in diabetic rat. Curr Eye Res 2000;20:131–136. [PubMed] [Google Scholar]
- 43. Finn JT, Weil M, Archer F, Siman R, Srinivasan A, Raff MC. Evidence that Wallerian degeneration and localized axon degeneration induced by local neurotrophin deprivation do not involve caspases. J Neurosci 2000;20:1333–1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Chan SY, Ochs S, Worth RM. The requirement for calcium ions and the effect of other ions on axoplasmic transport in mammalian nerve. J Physiol 1980;301:477–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Johansson JO. Influence of low IOP and low calcium on retrograde axoplasmic transport in rat optic nerve in vitro . Exp Eye Res 1985;41:739–744. [DOI] [PubMed] [Google Scholar]
- 46. Taft WC, Clifton GL, Blair RE, DeLorenzo RJ. Phenytoin protects against ischemia‐produced neuronal cell death. Brain Res 1989;483:143–148. [DOI] [PubMed] [Google Scholar]
- 47. Lees GJ. Inhibition of the retrograde axonal transport of dopamine‐beta‐hydroxylase antibodies by the calcium ionophore A23187. Brain Res 1985;345:62–67. [DOI] [PubMed] [Google Scholar]
- 48. Gupta VK. Systemic hypertension, headache, and ocular hemodynamics: A new hypothesis. Med Gen Med 2006;8:63. [PMC free article] [PubMed] [Google Scholar]
- 49. Cursiefen C, Wisse M, Cursiefen S, Jünemann A, Martus P, Korth M. Migraine and tension headache in high‐pressure and normal‐pressure glaucoma. Am J Ophthalmol 2000;129:102–104. [DOI] [PubMed] [Google Scholar]
- 50. Harris A, Zarfati D, Zalish M, et al Reduced cerebrovascular blood flow velocities and vasoreactivity in open‐angle glaucoma. Am J Ophthalmol 2003;135:144–147. [DOI] [PubMed] [Google Scholar]
- 51. Harris A, Siesky B, Zarfati D, Haine CL, Catoira Y, Sines DT, McCranor L, Garzozi HJ. Relationship of cerebral blood flow and central visual function in primary open‐angle glaucoma. J Glaucoma 2007;16:159–163. [DOI] [PubMed] [Google Scholar]
- 52. Sugiyama T, Utsunomiya K, Ota H, Ogura Y, Narabayashi I, Ikeda T. Comparative study of cerebral blood flow in patients with normal‐tension glaucoma and control subjects. Am J Ophthalmol 2006;141:394–396. [DOI] [PubMed] [Google Scholar]
- 53. Quigley HA, Sanchez RM, Dunkelberger GR, L'Hernault NL, Baginski TA. Chronic glaucoma selectively damages large optic nerve fibers. Invest Ophthalmol Vis Sci 1987;28:913–920. [PubMed] [Google Scholar]