

SUMMARY
Introduction: Sevoflurane is well known to exert a neuroprotective effect through anesthetic preconditioning. However, its effects on postconditioning, a neuroprotective phenomenon following an insult, have not been well studied. Aims: In this study, we examined the ability of sevoflurane to induce postconditioning in rat hippocampal slices, in vitro. Results: 2%, 4%, and 6% sevoflurane reduced neurophysiologic and morphologic neuronal injury following oxygen–glucose deprivation (OGD) and reperfusion. The quantity of damaged neurons was significantly reduced on immunofluorescence staining; excitatory amino acids (Asp, Glu) increased and inhibitory amino acids (GABA) decreased significantly. The effect was concentration‐dependent. Conclusion: Postconditioning with sevoflurane reduces neuronal damage after OGD—reperfusion injury in the CA1 area of rat hippocampus, in vitro.
Keywords: Hippocampal slice, Hypoxia, Neuronal damage, Postconditioning, Sevoflurane
Introduction
Stroke is one of the leading causes of morbidity and mortality in the United States. Interventions to reduce brain injury following stroke and brain trauma play an important role in reducing not only the morbidity and mortality, but also the health care costs. Ischemic preconditioning refers to a phenomenon that involves a reduction in total ischemic injury in various organ systems, including brain, heart, liver, kidneys, lungs, and intestines. Ischemic preconditioning involves application of brief, repeated periods of sublethal ischemia to induce a robust protection against the deleterious effects of subsequent, prolonged periods of lethal ischemia and reperfusion [1, 2]. In addition to brief episodes of ischemia, many other stimuli, including volatile anesthetics, such as sevoflurane [3] and Isoflurane [4], also can induce preconditioning effects in the brain, leading to protection in ischemic and hypoxic neurologic injury. Preconditioning is clinically feasible only when the occurrence of brain ischemia is predictable, e.g., carotid endarterectomy, intracranial aneurysm surgery, and other procedures requiring temporary occlusion of the blood vessels supplying the brain. In postconditioning, the brief episodes of ischemia‐reperfusion are employed during reperfusion after a prolonged ischemic insult. Postconditioning may be more clinically important as the onset of brain ischemia in patients with stroke and brain trauma often occurs before admission to the hospital. Postconditioning effects of Isoflurane on ischemic neurologic injury were demonstrated by a recent study in which isoflurane administrated after oxygen–glucose deprivation (OGD) or brain ischemia provides neuroprotection [5]. Sevoflurane postconditioning has been shown to offer protection against myocardial reperfusion injury [6] and to reduce expression of TNF‐alpha, CINC‐1, MIP‐2, and MCP‐1, leading to diminished neutrophil chemotaxis [7]. However, the postconditioning effects of Sevoflurane on neurologic injury have not been studied. In this study, we examined the postconditioning ability of sevoflurane in a rat neuronal OGD injury model.
Materials and Methods
Animals, Slice Preparation, and Maintenance
All experimental procedures and protocols used in this investigation were reviewed and approved by the Animal Care and Use Committee of SuZhou University.
Male Sprague–Dawley rats (200–250 g) were housed in community cages under a 12‐h light/dark cycle at room temperature (22°C) and fed ad lib. The procedures for preparation of hippocampal slices for electrophysiologic recording and analysis were followed as described in detail elsewhere [8]. In brief, following decapitation under ethyl ether anesthesia, hippocampi were dissected out, rapidly placed in ice‐cold artificial cerebrospinal fluid (aCSF) bubbled with 5% CO2 and 95% O2 and sliced transversely at 400 μm with a McIlwain tissue slicer. The slices were placed in a linear‐flow, interface incubation/recording chamber. The chamber was supplied with a humidified gas mixture (95%O/5%CO) and artificial cerebrospinal fluid (aCSF) via a peristaltic pump (1 mL/min). The temperature of the incubation chamber was held at 34 ± 0.3°C
Experimental Protocols and Hypoxia Experiments
The study group included the slices in one compartment of the dual incubation/recording chamber that were exposed to 13 minutes of OGD (95% N2/5% CO2) after 90 minutes of incubation, followed by 60 minutes of reoxygenation (95% O2/5% CO2). The slices in the study group were exposed to 0% (OGD‐reperfusion/hypoxia group), 2%, 4%, and 6% concentrations of sevoflurane during the 60‐min reoxygenation period. The concentration of sevoflurane was measured at the gas outlet of the incubation/recording chamber by an infrared anesthetic analyzer that was calibrated with known standards before and during experimentation. The control group included the slices in the second compartment of the dual incubation/recording chamber that were not exposed to OGD and reoxygenation.
Neuronal Morphology Under Light Microscopy
After 60 min of reoxygenation, the slices were fixed using 10% formaldehyde, dehydrated in increasing grades of ethanol, embedded in parafin and sectioned to a thickness of about 5 μm. The sections were then stained with hematoxylin–eosin (H&E), cleared in xylene and sealed with neutral gum. Neuronal morphology in the CA1 area was observed under light microscopy.
Neuronal Morphology Under Electron Microscopy
After perfusion, the slices were fixed in precooled 4% glutaraldehyde solution for 24 h. They were then washed with a phosphate buffer (pH = 7.4), 1% osmium acid postfixation, washed in a phosphate buffer again and dehydrated in increasing grades of ethanol overnight with uranyl acetate solution. Specimens were embedded in epon 812, sectioned and stained with uranium and aluminum for the observation of neuronal ultrastructure.
Neuronal Damage and Propidium Iodide Staining
8 slices were randomly selected in each group and stained with propidium iodide (PI). Mean fluorescence intensity (MFI) was used to demonstrate the degree of neuronal damage, consistent with the method reported by Cronberg et al. [9], by adding PI (2 μg/mL) in aCSF to fluorescence staining. Photoshop CS3 image processing software was used to analyze fluorescence intensity in a standardized CA1 area.
Electrophysiologic Neuronal Function
Extracellular potentials were investigated using glass microelectrode recording. The Schaffer lateral branch in the CA3 region was simulated (orthodromic stimulation, stimulation intensity 0.6 mA, stimulating frequency 0.1 Hz, duration 100 μs and stimulating interval 10 seconds) and extracellular potential changes were recorded in the CA1 region. Orthodromic population spike (OPS) traces were measured using glass microelectrodes (tip diameter 1–2 μm). OPS disappearance time, OPS recovery degree (amplitude percentage of reoxygenated/ischemic tissue) and OPS recovery rate (percentage of recovered slices/total slices) were recorded before OGD, at the end of OGD and 60 min after reoxygenation. Statistical analysis of data on OPS recovery rate was performed using Fisher's exact test (SPSS 12.0).
Measurement of Amino Acid Neurotransmitters in aCSF
Perfusate was collected before OGD and following 60 minutes of reoxygenation. The perfusion rate was 0.3 mL/min in each group and direction from bottom to top. Samples were collected and refrigerated at −20°C. After centrifugation, the supernatant was collected for high performance liquid chromatography (HPLC). Concentrations of five amino acids: glutamic acid (Glu), aspartic acid (Asp), γ‐aminobutyric Acid (GABA), glutaminate (Gln), and glutamic acid (Gly) were measured. Peak area score was calculated by using Prostar software and a standard curve was created relative to standard amino acid concentration.
Statistical Analysis
Statistical analysis of data was performed with one‐way analysis of variance (anova) followed by post hoc Duncan test. Changes within and between the groups were considered statistically significant when the P‐value was less than 0.05. All data are expressed as mean ± SD.
Results
Twenty‐four rats were used to obtain 24 successful experiments. Amplitudes of evoked CA1 population spikes were quantified (10–15 slices/hemisphere/rat).
Morphologic Changes Under Light Microscopy
In the control group, the neurons were seen to have a lamellar arrangement. Neurons were arranged closely and parallel‐aligned dendrites formed a clear layer. The Pyramidal cells were irregular polygons or ovals, neuronal plasma was rich and stained homogeneously, nuclei were on the core and the nucleoli were visible (Figure 1A). In the OGD group without treatment, neurons had unclear layers, were swollen and the membrane edges were blurred. Their nuclei were densely stained and shrunken (Figure 1B). In groups exposed to 4% sevoflurane and 6% sevoflurane, neuronal swelling was less evident, many cells of normal morphology were observed and nucleoli were visible. Fewer cells were smaller, irregularly shaped and intercellular distance was not seen. (Figure 1D and E).
Figure 1.
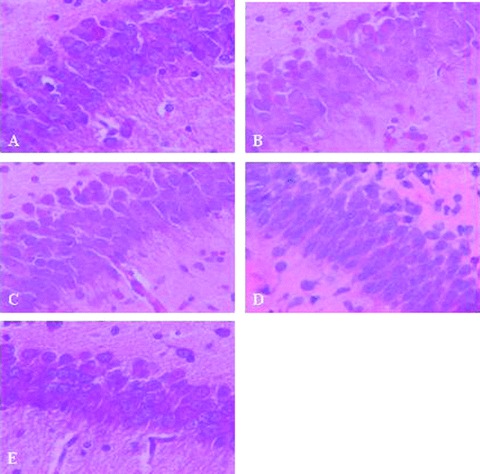
HE staining of hippocampal CA1 pyramidal neurons in group normal (A). hypoxia (B), 2% Sevo (C), 4% Sevo (D), 6% Sevo (E).(40 × 10).
Morphologic Changes Under Electron Microscopy
In the control group, the neurons and glial cells had nearly normal ultramorphology in the CA1 area. Cell membranes were intact, nuclear membranes were clearly seen, and euchromatin was granular and homogeneously distributed within the nucleus. The cell nuclei were oval in their overall shape, gathered along the edge. Mitochondria, rough endoplasmic reticulum, and Golgi complexes were observed in cytoplasm; mitochondrial cristae were regular and well defined. Synaptic structure was clearly visible; the presynaptic and postsynaptic membranes were slightly thickened and synaptic space was relatively narrow (Figure 1A). In the OGD group, neurons and glial cells demonstrated edema and vacuole formation in the CA1 area. Cell membranes were ruptured and nuclear membranes were incomplete, broken and wrinkled with a concave‐convex boundary. Nuclear chromatin was gathered into a block. The organelles were densely gathered inside the cell; mitochondria were swollen with broken ridges or missing entirely (Figure 2B).
Figure 2.
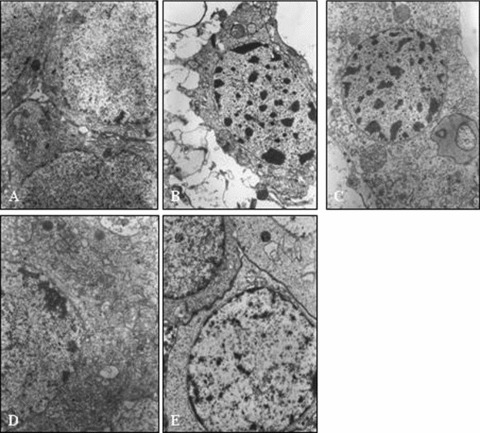
Electron micro graph of the hippocaittpal CA1 pyramidal neurons in normal (A), hypora (B),2% Sevo (C), 4% Sevo (D), 6% Sevo (E).(×10,000).
In the group exposed to 2% sevoflurane, there were no significant differences in the basic forms of neurons and glial cells, compared with the OGD group (Figure 2C).
In groups exposed to 4% sevoflurane and 6% sevoflurane, neurons and glial cells were slightly edematous in the CA1 area, but cell membranes and nuclear membranes were still clearly seen and complete. The main changes in neuronal cytoplasm were some mild mitochondrial swelling, fewer broken ridges, and a mild expansion of rough endoplasmic reticulum (Figure 2D and E).
Mean Fluorescence Intensity of PI Stained Images
In the control group, no fluorescence appeared in the CA1 region; but MFI in the OGD group was very strong (62.3 ± 12.0). All groups exposed to sevoflurane demonstrated significantly weaker MFI than the OGD group. Specimens exposed to 6% sevoflurane (17.5 ± 5.0) had significantly weaker MFI than the groups exposed to 4% sevoflurane (33.0 ± 9.6) and 2% sevoflurane (39 ± 9.9), but between the 4% sevoflurane and 2% sevoflurane groups there was no difference (P < 0.05) (Figure 3).
Figure 3.
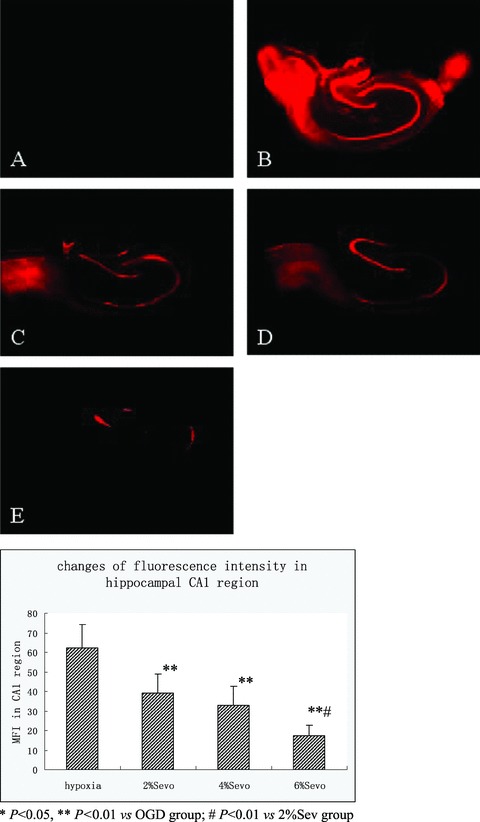
Examples of propidium iodide fluorescence images of hippocampal slices. Images were acquired ĞOmin after the simulated ischemia. Bright areas indicate propidiurn iodide fluorescence (dead neurons). Bar = 500 fim. Group normal (A), hypoxia (B), 2% Sevo (C), 4% Sevo (D), 6% Sevo (E).
Changes in Extra Cellular Evoked Potentials (Figure 4)
Figure 4.
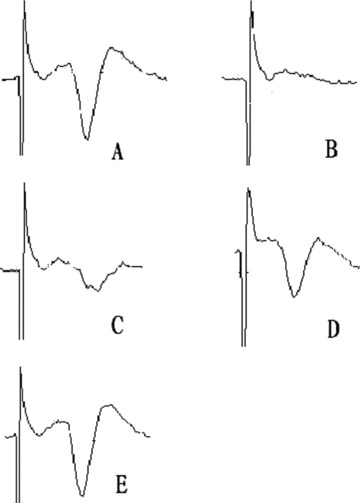
Examples öf extracellular evoked potential images, before OGD (A), at the end of OGD (B), 60 mitt after reoxygen and exposed to 2%Seı™ (C), 4%Seı™ (D), 6%Se™ (E).
Exposure to 2% sevoflurane did not significantly improve the degree of recovery of the OPS, but delayed the disappearance of OPS after OGD (3.9 ± 0.9 min, P < 0.01). 4% Sevoflurane not only delayed the disappearance of OPS after OGD (4.8 ± 1.5 min, P < 0.01), but also significantly improved the degree of OPS recovery (58.9%± 17.1%, P < 0.01). Similarly, 6% sevoflurane significantly delayed disappearance of OPS (5.0 ± 1.1 min) and improved recovery degree (67.9 ± 21.8%), (P < 0.01). Both the 4% sevoflurane and 6% sevoflurane groups had an OPS recovery rate of 62.5%, which was significant compared to the OGD group (P < 0.05) (Table 1).
Table 1.
Changes in extra cellular evoked potentials(mean ± SD, n = 8)
| Group | OPS disappearing time (min) | OPS recovery degree (%) | OPS recovery rate (%) |
|---|---|---|---|
| OGD | 2.9 ± 0.7 | 9.5%± 19.2 | 0%(0/8) |
| 2%Sev | 3.9 ± 0.9** | 31.2%± 29.8 | 12.5%(1/8) |
| 4%Sev | 4.8 ± 1.5** | 58.9%± 17.1** | 62.5%(5/8)* |
| 6%Sev | 5.0 ± 1.1** | 67.9%± 21.8**,# | 62.5%(5/8)* |
*P < 0.05, **P < 0.01 vs. OGD group; # P < 0.01 vs. 2% Sev group.
Amino acid Neurotransmitters in Perfusion Fluid (aCSF)
Compared to the control group, five amino acid concentrations were higher in the OGD group. Asp, Glu and GABA increased as much as 51.9%, 58.8%, and 29.3%, respectively. Exposure to 4% sevoflurane and 6% sevoflurane reduced Asp, Glu and Gly release and further increased the release of GABA, but there was no significant impact on the release of Gln. 2% Sevoflurane increased the concentration of GABA, but there were no significant changes with any other amino acids (Figure 5).
Figure 5.
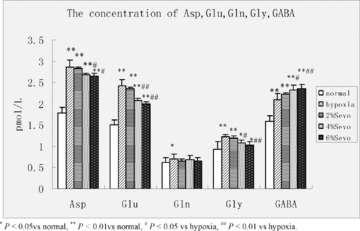
Concentration of amino acid neurotransmitters in aCSF (pmol•L−1,  ± s, n = 8).
± s, n = 8).
Discussion
Sevoflurane preconditioning is well known to provide neuroprotection [2], but its postconditioning ability in neuroprotection has not been widely studied. Deyhimy et al. [10] reported that, in myocardial protection, sevoflurane postconditioning was as effective as preconditioning, but did not address neuroprotection. Postconditioning effects of Isoflurane on ischemic neurologic injury were demonstrated by a recent study in which isoflurane administrated after OGD or brain ischemia provides neuroprotection [5]. In our study, we obtained further evidence demonstrating that sevoflurane‐induced postconditioning ameliorates oxygen–glucose—reperfusion injury in hippocampal slices, suggesting that exposure to sevoflurane may be beneficial following neurologic injury.
The portions of the brain most vulnerable to hypoxia are the pyramidal cells in the hippocampus. The in vitro model of the hippocampal slice, therefore, has been widely used in the study of hypoxic brain injury and protective effects of various interventions. This model relies on the cultivation of hippocampal slices in a temperature‐controlled environment containing controlled levels of oxygen and a continuous renewal of aCSF. Properly prepared, the model allows exchange of substances with surrounding perfusate and maintenance of normal physiologic function. Under normal study conditions, the slices can survive for 10 or more hours.
In our study, sevoflurane demonstrated anesthetic neuroprotection via postconditioning in vitro in a well‐established brain slice model [15, 16]. The in vitro model controls and excludes the confounding variables of blood pressure, temperature, electrolyte variation and the blood–brain barrier. Sevoflurane administered to hippocampal slices in vitro following OGD‐reperfusion markedly reduced injury in a dose‐dependent fashion. When neuronal morphology was observed in the CA1 sector of the hippocampus by both light and electron microscopy, exposure to sevoflurane improved preservation of normal structure in a dose‐dependent fashion at concentrations greater than 2%. Neurons exposed to 4% and 6% sevoflurane demonstrated only slight edema, fairly clear karyon and karyotheca, and greater numbers of normal neurons were seen. The same conclusions can also be drawn from the slices that underwent PI‐induced fluorescence. PI is a fluorescent dye of very strong polarity. When the cell membranes are damaged it enters the cytoplasm and combines with nucleic acids, increasing fluorescence intensity by 20–30 times. Hence, PI fluorescence staining is widely used to evaluate the integrity of cell membranes following an insult [11, 12]. Previous studies have shown a linear relationship between fluorescence intensity and the number of dead cells [11, 13]. MFI was significantly decreased in all of our study groups that were exposed to sevoflurane, relative to the OGD‐reperfusion/hypoxia group. In our study, 6% sevoflurane had the greatest effect on suppression of MFI, suggesting that higher concentrations possess a greater postconditioning neuroprotective capacity.
Hippocampal nerve loops are composed of tertiary neurons, that is, dentate gyrus granular cells, CA3 pyramidal cells and CAl pyramidal cells. CAl pyramidal cells are selectively susceptible to hypoxic injury and their electrophysiologic function recovers slowly, even after reoxygenation. [14, 15]. There are some synaptic connections between CA3 pyramidal cells and CAl cells, such that when the Schaffer collateral is electrically stimulated in area CA3, OPS can be recorded in the CAl pyramidal cell layer and the OPS changes significantly when brain is hypoxic [16]. Hence, OPS was selected as a sensitive indicator of synaptic transmission function in our study. In the OGD group, OPS disappearance time was 2.9 ± 0.7 min, but after reoxygenation, the OPS recovery degree and OPS recovery rate were still lower than in the control group. This is consistent with a significant hypoxic injury of synaptic transmission. When the neurons were exposed to sevoflurane during reoxygenation, we found OPS disappearance time was significantly increased, in a dose‐dependent fashion to a peak of 5.0 ± 1.1 min in the 6% sevoflurane group. The corresponding OPS recovery degree also significantly increased, especially in the 6% sevoflurane group, where OPS recovery degree increased to 67.9%± 21.8% from 9.5 ± 19.2% (P < 0.01) following postconditioning. Correspondingly, OPS recovery rate also increased. This demonstrated that sevoflurane‐induced postconditioning significantly improved recovery of synaptic transmission function after hypoxic injury.
There is growing evidence that excitatory amino acids (EAA) play a key role in the development of hypoxic brain injury, including Glu and Asp. EAA are the main neurotransmitters of excitatory neuronal synapses in the central nervous system, but they are also neurotoxins. Hypoxic brain injury induced by EAA (mainly as Glu) occurs when increased quantities are released into synaptic space and energy deficiency prevents the reuptake of EAA by glial cells and the presynaptic membrane. This leads to further EAA accumulation in the extra cellular spaces. Excessive EAA thus results in a cascade of cellular toxicities, such as protein degradation, enzyme degradation, progressive neuronal necrosis and induction of NOS to synthesize large amounts of NO, producing NO neurotoxicity [17, 18, 19]. Extra cellular K+ accumulates, leading to increased cell excitability [20]. Our results confirm that sevoflurane‐induced postconditioning significantly reduces EAA neurotransmitter concentrations in aCSF. Inhibition of EAA release may be one protective pathway of ischemic brain injury. Interestingly, in previous studies, glutamine concentration of corticostriatal slices was not affected [21]. Gly is not a member of the EAA, but it can amplify EAA toxicities [22]. In our study, sevoflurane also reduced Gly concentration, which may explain cerebral protection during hypoxia from another perspective. The central inhibitory neurotransmitter GABA inhibits neuronal hypoxic injury and plays an active role in neuronal growth, development, and plasticity [23, 24]. Accumulation of GABA in synapses can play a protective role in the brain [22]. Our study results show GABA concentration increases significantly after sevoflurane postconditioning, suggesting an increase in GABA concentration is also involved in the protective effect. Gln could affect the release of the neurotransmitter Glu. Its concentration, however, did not significantly increase in our study. Thus, Gln may not participate in sevoflurane‐induced postconditioning.
In summary, this study examined sevoflurane‐induced postconditioning following OGD‐reperfusion injury in rat hippocampal slices in vitro. Effects on morphology, immunofluorescence, electrophysiology, and neurotransmitter release were studied. All of our results demonstrate that postconditioning with sevoflurane ameliorates OGD‐reperfusion injury in hippocampal slices. Further studies are necessary to better define the potential clinical applications of this therapy following brain injury.
Conflict of Interest
Dr. Mychaskiw is a consultant who has received research support and is a member of the speakers bureau of Baxter Healthcare, Inc. The authors have no other conflicts of interest.
Acknowledgments
This work was completed in Jiangsu Province Key Laboratory of Anesthesiology.
References
- 1. Tomai F, Crea F, Chiariello L, Gioffre PA. Ischemic preconditioning in humans: Models, mediators, and clinical relevance. Circulation 1999;100:559–563. [DOI] [PubMed] [Google Scholar]
- 2. Ferrari R, Ceconi C, Curello S, Percoco G, Toselli T, Antonioli G. Ischemic preconditioning, myocardial stunning, and hibernation: Basic aspects. Am Heart J 1999;138:S61–S68. [DOI] [PubMed] [Google Scholar]
- 3. Payne RS, Akca O, Roewer N, Schurr A, Kehl F. Sevoflurane‐induced preconditioning protects against cerebral ischemic neuronal damage in rats. Brain Res 2005;1034:147–152. [DOI] [PubMed] [Google Scholar]
- 4. Kapinya KJ, Löwl D, Fütterer C, Maurer M, Waschke KF, Isaev NK, Dirnagl U. Tolerance against ischemic neuronal injury can be induced by volatile anesthetics and is inducible NO synthase dependent. Stroke 2002;33:1889–1898. [DOI] [PubMed] [Google Scholar]
- 5. Lee JJ, Li L, Jung HH, Zuo Z. Postconditioning with isoflurane reduced ischemia‐induced brain injury in rats. Anesthesiology 2008;108:1055–1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Li H, Wang JK, Zeng YM, et al Sevoflurane post‐conditioning protects against myocardial reperfusion injury by activation of phosphatidylinositol‐3‐kinase signal transduction. Clin Exp Pharmacol Physiol 2008;35:1043–1051. [DOI] [PubMed] [Google Scholar]
- 7. Steurer M, Schläpfer M, Steurer M, et al The volatile anaesthetic sevoflurane attenuates lipopolysaccharide‐induced injury in alveolar macrophages. Clin Exp Immunol 2009;155:224–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lipton P, Aitken PG, Dudek FE, et al Making the best of brain slices: Comparative methods. J Neurosci Methods 1995;59:151–156. [DOI] [PubMed] [Google Scholar]
- 9. Cronberg T, Rytter A, Asztély F., Söder A, Wieloch T. Glucose but not lactate in combination with acidosis aggravates ischemic neuronal death in vitro. Stroke 2004;35:753–757. [DOI] [PubMed] [Google Scholar]
- 10. Deyhimy DI, Fleming NW, Brodkin IG, Liu H. Anesthetic preconditioning combined with postconditioning offers no additional benefit over preconditioning or postconditioning alone. Anesth Analg 2007;105:1863–1864. [DOI] [PubMed] [Google Scholar]
- 11. Laake JH, Haug FM, Wieloch T, Ottersen OP. A simple in vitro model of ischemia based on hippocampal slice cultures and propidium iodide fluorescence. Brain Res Brain Res Protoc 1999;4:173–184. [DOI] [PubMed] [Google Scholar]
- 12. Noraberg J, Kristensen BW, Zimmer J. Markers for neuronal degeneration in organotypic slice cultures. Brain Res Brain Res Protoc 1999;3:278–290. [DOI] [PubMed] [Google Scholar]
- 13. Newell DW, Barth A, Papermaster V, Malouf AT. Glutamate and non‐glutamate receptor mediated toxicity caused by oxygen and glucose deprivation in organotypic hippocampal cultures. J Neuroscience 1995;15:7702–7711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Balestrino M, Aitken PG, Somjen GG. Spreading depression‐like hypoxic depolarization in CA1 and fascia dentate of hippocampal slice: Relationship to selective vulnerability. Brain Res 497:102–107. [DOI] [PubMed] [Google Scholar]
- 15. Schurr A, Teyler TJ, Tseng MT, editors. Brain slices, fundamental, applications and implications. New York : S Karge Publishers Inc, 1986:105–117. [Google Scholar]
- 16. Fairchild MD, Parsons JE, Wasterlain CG, Rinaldi PC., Wallis RA. A hypoxic injury potential in the hippocampal slice. J Brain Res 1988;453:357–361. [DOI] [PubMed] [Google Scholar]
- 17. Pellegrini‐Giampietro DE, Gorter JA, Bennett MV, Zukin RS. The GluR2 (GluR‐B) hypothesis: Ca(2+)‐permeable AMPA receptors in neurological disorders. Trends Neurosci 1997;20:464–470. [DOI] [PubMed] [Google Scholar]
- 18. Hirose K, Chan PH. Blockade of glutamate excitoxicity and its clinical applications. J Neurochem Res 1993;18:479–483. [DOI] [PubMed] [Google Scholar]
- 19. Kollegger H, McBean HG, Tipton KF. Reduction of Striatal N‐methyl‐D‐aspartate toxicity by inhibition of nitric oxide synthase. J Biochem Pharmacol 1993;45:260–264. [DOI] [PubMed] [Google Scholar]
- 20. Katsura K, Ekholm A, Siesijo BK. Coupling among changes in energy metabolism, acidbase homestasis, and ion fluxes in ischemia. Can Physiol Pharmacol 1992;70(Suppl):S170–S175. [DOI] [PubMed] [Google Scholar]
- 21. Jung HH, Lee JJ, Washington JM, Zuo Z. Inability of volatile anesthetics to inhibit oxygen‐glucose deprivation‐induced glutamate release via glutamate transporters and anion channels in rat corticostriatal slices. Brain Res 2008;1227:234–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Globus MY, Busto R, Dietnich WD, et al Comparative effect of transient global ischemia on extracellular levels glutamate, glycine and γ‐amino butyric acid in vulnerable and nonvulnerable brain regions in the rat. J Neurochem 1991;57:470–475. [DOI] [PubMed] [Google Scholar]
- 23. Han VK, Lauder JM, D'Ercole AJ. Characterization of somatomedin/insulin‐like growth factor receptors and correlation with biologic action in cultured neonatal rat astroglial cells. J Neurosci 1987;7:501–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Cobas A, Fairén A, Alvarez‐Bolado G, Sánchez MP. Prenatal development of the intrinsic neurons of the rat neocortex: A comparative study of the distribution of GABA‐immunoreactive cells and the GABAA receptor. Neuroscience 1991;40:375–397. [DOI] [PubMed] [Google Scholar]