

Abstract
Cilostazol increases intracellular cyclic adenosine monophosphate (cyclic AMP) levels by inhibiting type III phosphodiesterase. It was approved by the Food and Drug Administration for the treatment of intermittent claudication. Its principal actions include inhibition of platelet aggregation, antithrombotic action in cerebral ischemia, and vasodilation, mediated by increased cyclic AMP levels. In a multicenter, randomized, placebo‐controlled, double‐blind clinical trial, cilostazol has been shown to protect patients from recurrent cerebral infarction. It has been recently suggested that cilastozol could be useful in the treatment of transient focal cerebral ischemic injury. Beneficial effects of cilostazol in cerebral ischemic infarction and edema formation has been confirmed in rats by the magnetic resonance imaging (MRI). The preventive effect was ascribed to cAMP‐dependent protein kinase (PKA)‐coupled maxi‐K channel activation with additional antioxidant and poly(adenosine diphosphate [ADP]‐ribose) polymerase inhibitory actions. Most recently, cilostazol has been shown to prevent vacuolation and rarefaction in the white matter of the rats subjected to chronic cerebral hypoperfusion in association with suppression of astrocyte and microglial activation. Taken together, recent experimental studies with cilostazol showed promising results in cerebral ischemia and chronic cerebral hypoperfusion.
Keywords: Anti‐inflammatory drugs, Antioxidants, Apoptosis, Brain ischemia, Cilostazol, K+ channels, Neuroprotective drugs
Introduction
Cilostazol [Pletaal®] was developed as a selective inhibitor of cyclic nucleotide phosphodiesterase 3 (PDE3) (Kimura et al. 1985). Under inhibition of PDE3 activity (recombinant human PDE3A, IC50= 0.54 ± 0.06 μM; Shakur et al. 2002) and suppression of cyclic AMP degradation, intracellular cyclic AMP level was increased in platelets and blood vessels, leading to inhibition of platelet aggregation and dilation of vascular smooth muscle cells (Kimura et al. 1985). Additional reports described the pleiotropic actions of cilostazol, thereby providing a variety of clinical uses including prevention of recurrent stroke (Gotoh et al. 2000), coronary restenosis (Douglas et al. 2005), and peripheral occlusive disease. These indications were based on antiplatelet, vasodilator, and antiproliferative actions of cilastozol in preclinical studies (Kambayashi et al. 2003).
Cilostazol has been shown to inhibiting platelet aggregation induced by a variety of stimuli, including arachidonic acid, adenosine diphosphate (ADP), epinephrine, collagen, thrombin, and high shear stress (Minami et al. 1997; Matsumoto et al. 1999). Cilostazol was approved in the United States for treatment of intermittent claudication (Dawson et al. 1998).
Antiplatelet therapy modestly improved outcome in both acute stroke (aspirin) and in secondary stroke prevention (aspirin with or without dipyridamole) (Bednar 2000). This therapy is a key component of secondary preventive strategies in ischemic stroke (Majid et al. 2001). The secondary prevention strategies rely largely on the risk factor reduction, carotid endarterectomy, anticoagulation for cardioembolic stroke, and antiplatelet agents for atherothrombotic stroke (Easton 1998). In line with these reports, cilostazol was suggested for treatment of cerebral ischemic events caused by thrombus formation in the carotid artery (Kohda et al. 1999). Cilostazol has been shown in a multicenter, randomized, and double‐blind clinical trial to provide a considerable risk reduction (about 41.7%) in patients with recurrent cerebral infarction (Gotoh et al. 2000).
Our recent studies demonstrated the protective effects of cilostazol against transient focal cerebral ischemia and chronic cerebral hypoperfusion injury. Cilostazol significantly decreased ischemic brain infarction, inhibited apoptotic and oxidative cell death (Choi et al, 2002), and attenuated gray and white matter damage 24 h after focal cerebral ischemia in rats (Honda et al. 2006). Reduction of the brain ischemic infarction and edema by cilostazol was confirmed by magnetic resonance imaging (MRI) in rats (Lee et al. 2003). These results were further supported by Ye et al. (2007), in that cilostazol was effective in mice in chronic as well as in acute injury induced by focal cerebral ischemia. Additionally, cilostazol protected rats, subjected to permanent bilateral common carotid artery ligation (BCCAL), from cognitive impairment and white matter lesions induced by chronic cerebral hypoperfusion (Lee et al. 2006; Watanabe et al. 2006). The present article summarizes our recent knowledge of the neuroprotective effects of cilostazol in in vitro and in vivo studies.
Chemistry
Chemical structure of cilostazol (Pletaal®; OPC‐130136) is shown in Figure 1. Its chemical name is 6‐[4‐(1‐cyclohexyl‐1H‐tetrazol‐5‐yl)butoxy]‐3,4‐dihydro‐2(1H)‐quinolinone and its molecular weight is 369.47. It is manufactured and distributed by Otsuka Pharmaceutical Co. Ltd (Tokushima, Japan). Cilostazol has high plasma protein binding ability. In an in vitro study, 95% of [14C]cilostazol was found to bind to plasma proteins (Akiyama et al. 1985). In vivo after a single oral dose (50 mg/kg), 95.5% of cilastozol was bound to plasma proteins (Bramer et al. 1999).
Figure 1.
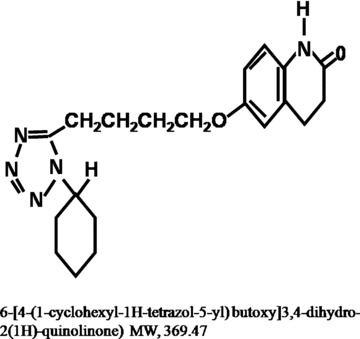
Chemical structure of cilostazol (6‐[4‐(1‐cyclohexyl‐1H‐tetrazol‐5‐yl)butoxy]3,4‐dihydro‐2(1H)‐quinolinone).
Pharmacology
In Vitro Protective Action of Cilostazol
Inhibition of lipopolysaccharide (LPS)‐induced apoptosis by cilostazol in human umbilical vein endothelial cells (HUVECs) has been reported by Kim et al. (2002). They demonstrated in vitro inhibition of LPS‐induced apoptosis in HUVECs by cilostazol and its metabolites (OPC‐13015 and OPC‐13213) and found that cilastozol can reverse the LPS‐induced decrease in Bcl‐2 protein, increase in Bax protein and cytochrome c release from mitochondria. Additionally, they documented that cilostazol and its analogs scavenge hydroxyl radicals, suppress production of the intracellular reactive oxygen species, and decrease the formation of tumor necrosis factor‐α (TNF‐α). The cell survival studies emphasized the importance of cAMP stimulation in the somatostatin transcription by cyclic AMP response element binding protein (CREB) phosphorylation at Ser‐133 (Gonzalez and Montminy 1989), and the survival‐promoting effect of brain‐derived neurotrophic factor or neurotrophin‐3 (Franke et al. 2000). Consistent with these results, TNF‐α‐induced reduced viability of SK‐N‐SH (human brain neuroblastoma cell line) and HCN‐1A cells (human brain cortical cell line) were significantly reversed by cilostazol (0.1∼100 μM) in a concentration‐dependent manner (Hong et al. 2003). These authors further elucidated the signal transduction pathway by which cilostazol recovers the cell viability, and found that TNF‐α (50 ng/mL)‐induced suppressed Akt and CREB‐phosphorylation were markedly elevated (up to 3‐ to 4‐fold) by cilostazol at 10 μM (p < 0.01). It remains to be defined in further studies whether expression of these neurotrophic factors is involved in the action of cilostazol.
Activation of maxi‐K+ channels appears to be involved in the downstream signal transduction pathway leading to the recovery of cell viability in SK‐N‐SH cells. It has been reported that maxi‐K+ channels, large conductance calcium‐activated K+ channels, are rapidly activated by depolarization and increased intracellular calcium (Latorre et al. 1989), and that the K+ channel opening reduces neurotransmitter release by suppressing accumulation of pathological levels of Ca2+, and attenuates ischemic injury (Robitaille and Charlton 1992). Activation of maxi‐K+ channels has been found to protect neurons against glutamate release and excitotoxicity, and to reduce the pathological consequences of ischemia (Lawson 2000). Recent studies have documented that the maxi‐K+ channel opener BMS 204352 [maxipost, (S)‐3‐(5‐chloro‐2‐methoxyphenyl)‐3‐fluoro‐6‐(trifluoromethyl)‐1,3‐dehydro‐2H–indole‐2‐one; Cheney et al. 2001] protects neuronal cells against acute ischemic damage by blocking Ca2+ entry and minimizing neuronal depolarization (Gribkoff et al. 2001). In the previous electrophysiological studies (Hong et al. 2003), cilostazol increased the outward K+ currents by approximately 4‐fold in SK‐N‐SH cells by activating the maxi‐K+ channels without affecting the K+ ATP channels. In line with this report, cilostazol significantly decreased the cytosolic Ca2+ level elevated by TNF‐α (50 ng/mL) in the SK‐N‐SH cells. In vitro (Kim et al., 2004) and in vivo (Lee et al. 2004) studies, cilostazol ameliorated neuronal damage by suppressing apoptotic cell death. This effect involved opening of maxi‐K+ channels associated with an increase in the Akt phosphorylation, which was suppressed by iberiotoxin. These results strongly suggest that the antiapoptotic effect of cilostazol is mediated by maxi‐K+ channel opening.
Anti‐Inflammatory Effects
Shin et al. (2004) reported that remnant lipoprotein particles (RLP)‐induced decreased cell viability of HUVECs in association with decreased oligonucleosomal DNA fragmentation was significantly reversed by pretreatment with cilostazol (0.01∼100 μmol/L). In accordance with these results, cilostazol significantly suppressed RLP‐stimulatd NAD(P)H oxidase‐dependent superoxide formation and cytokine production (TNF‐α and interleukin‐1β). Recently, Park et al. (2005) showed that cilostazol reduces expression of adhesion molecules and chemokines and monocyte adhesion to HUVECs. Cilostazol suppressed RLP (50 μg/mL)‐stimulated monocyte adhesion (3.3‐fold) in HUVECs as well as increased cell surface expression of vascular cell adhesion molecule‐1 (VCAM‐1), intercellular adhesion molecule‐1, E‐selectin, and monocyte chemoattractant protein‐1 (MCP‐1). At 10 μM BAY 11‐7085, a specific inhibitor of inhibitory kappaB (IκB) phosphorylation, had similar effect. Further study (Park et al. 2006) showed that cilostazol (1 μM) increased K+ currents in human endothelial cells by activating maxi‐K+ channels, and that this effect was abolished by iberiotoxin (100 nM), a maxi‐K+ channel blocker. In line with these findings, cilostazol significantly suppressed TNF‐α‐induced IκBα degradation, so that translocation of the nuclear factor‐κB (NF‐κB) p65 subunit into the nucleus was blocked. These effects of cilostazol were consistently antagonized by iberiotoxin (p < 0.05), KT 5720 or Rp‐cAMPs (cAMP‐dependent protein kinase [PKA] inhibitors; p < 0.05) but not by KT 5823 or Rp‐cGMPs (cGMP‐dependent protein kinase [PKG] inhibitors). These findings indicate that increased intracellular cAMP levels by cilostazol are directly coupled to its maxi‐K+ channel opening action via protein kinase A activation in human endothelial cells, thereby suppressing TNF‐α‐stimulated superoxide production and expression of adhesion molecules. These results are further supported by the reports that PKA activation by cAMP‐elevating agents, such as forskolin and dibutyryl cAMP, inhibited TNF‐α‐induced NF‐κB‐dependent reporter gene expression and reduced NF‐κB‐dependent expression of adhesion molecules and chemokines (Ollivier et al. 1996; Aizawa et al. 2003).
Based on these in vitro findings, the antiatherogenic effect of cilostazol was elucidated in the low‐density lipoprotein receptor‐null mice fed a high cholesterol diet, where cilostazol significantly suppressed multiple plaque lesions in the proximal ascending aorta including aortic sinus. This effect was accompanied by decreased macrophage accumulation with reduced superoxide and TNF‐α formation, as well as VCAM‐1 and MCP‐1 expression. Accumulating evidence suggests that the anti‐inflammatory action of cilostazol is responsible for the antiatherosclerotic effect of cilostazol (Lee et al. 2005).
Protection from Focal Cerebral Ischemic Injury
Many reports have shown that cilostazol exerts neuroprotective effects against transient focal ischemic brain injury (Table 1). Cilostazol decreased ischemic brain infarction in association with inhibition of apoptotic and oxidative cell death (Choi et al. 2002), and attenuated gray and white matter damage at 24 h after focal cerebral ischemia in rats (Honda et al. 2006). Reduction of the brain ischemic infarction by cilostazol was confirmed by MRI in rats (Lee et al. 2003). The infarct areas were reduced in accordance with the apparent diffusion coefficient (ADC) and T2 images obtained after treatment with cilostazol. Hoehn‐Berlage et al. (1995) previously suggested that the decline of ADC is related to the reduction in cerebral blood flow that causes a failure of high‐energy metabolism and leads to cytotoxic edema. The blood brain barrier (BBB) breakdown was reported to occur following postischemic reperfusion of the brain (Sage et al. 1984). The integrity of the BBB plays an important role in the pathological changes, since BBB disruption is associated with brain edema formation and cerebral infarction following postischemic reperfusion (Betz 1996). Consistent with the report of Yang et al. (1999), disruption of BBB in the ipsilateral hemisphere was observed after focal cerebral ischemia, which was significantly suppressed by cilostazol (Lee et al. 2004).
Table 1.
In vivo effects of cilostazol on animal models of cerebral ischemia and chronic cerebral hypoperfusion.
| Animal model | Dose of cilostazol | Effects | Reference |
|---|---|---|---|
| Transient MCAO in rat | 10 mg/kg, i.v. (5 min or 1 h after ischemia) | Increased the cAMP, Bcl‐2 and decreased the TNF‐α, Bax scavenged hydroxyl, and peroxyl radicals | Choi et al. 2002 |
| Transient MCAO in rat | 30 mg/kg, p.o. (5 min, 4 h after ischemia) | Reduced the ischemic edema (MRI study) | Lee et al. 2003 |
| Transient MCAO in rat | 30 mg/kg, p.o. (5 min, 4 h after ischemia) | Increased the CREB, Akt phosphorylation, and the Bcl‐2 | Lee et al. 2004 |
| Transient MCAO in rat | 30 mg/kg, p.o. (5 min, 4 h after ischemia) | PARP inhibition, decreased the TUNEL, microglia, TNF‐α positive cells, and AIF translocation | Lee et al. 2007 |
| Transient MCAO in mice | 3–10 mg/kg, i.p. (30 min before MCAO) | Attenuated ischemic neuronal injury and inhibited blood–brain barrier disruption | Ye et al. 2007 |
| 10 mg/kg, i.p. (1, 2, and 3 h after ischemia) | Inhibited astrocyte proliferation/glial scar formation | ||
| 10 mg/kg, i.p. (1, 4, and 7 h after ischemia) | Accelerated the angiogenesis | ||
| Permanent MCAO in rat | 30 or 50 mg/kg, p.o. (30 min, 4 h after MCAO) | Reduced the gray and white matter damage; improved the CBV and CBF in the peri‐infarct area; increased perfusion in the ischemic penumbra | Honda et al. 2006 |
| Permanent MCAO in mice | 30 mg/kg, i.p. (12 h, 1 h before and after MCAO) | Increased metallothionein‐1 and ‐2 in penumbra | Wakida et al. 2006 |
| BCCAL in rat | 50 mg/kg/day, p.o. | Protected against cerebral hypoperfusion‐induced cognitive impairment and white matter damage; increased p‐CREB, Bcl‐2, and COX‐2 | Watanabe et al. 2006 |
| BCCAL in rat | 60 mg/kg/day, p.o. | Suppressed the activation of microglia and astrocytes but also diminished oligodendrocyte; decreased TNF‐α and apoptosis in white matter lesion | Lee et al. 2006 |
MCAO = middle cerebral artery occlusion; BCCAL = bilateral common carotid artery ligation; CREB = cAMP‐response element binding protein; CBF = cerebral blood flow; CBV = cerebral blood volume; i.v. = intravenous; i.p. = intraperitoneal; p.o. = peroral.
In the penumbral region, blood flow is reduced to a critical level during middle cerebral artery occlusion (MCAO), but reperfusion after ischemia provides an excess of oxygen with restored blood flow, leading to not only enhancement of neuronal viability but also to catalysis of enzymatic oxidative reaction. The produced reactive oxygen species trigger apoptosis and laddered DNA fragmentation (Bredesen 1995). Recently, Lee et al. (2004) reported that cilostazol (30 mg/kg, orally) significantly suppressed the laddering feature of DNA fragmentation in the samples of 24 and 48 h reperfusion after 2 h MCAO in rats. In the process of apoptosis, accumulating evidence points to a significant role of Bcl‐2 and cytochrome c release from mitochondria to cytosol, and caspase‐3 activation in promoting cell survival and cell death (Chan 2005). Martinou et al. (1994) showed that the overexpression of Bcl‐2 in transgenic mice protected neurons from ischemia‐induced cell death. Similarly, human Bcl‐2 overexpression with herpes simplex virus vectors limits neuronal death in focal cerebral ischemia (Lawrence et al. 1997). Bcl‐2 protects the integrity of mitochondrial oxidative phosphorylation and thus limits mitochondrial dysfunction induced by several apoptosis stimuli (Kluck et al. 1997). In line with these important proteins, CREB is suggested to be a post‐translationally activated transcription factor that is involved in the numerous brain functions, including cell survival (Walton et al. 1999). CREB is a key mediator coupling neurotrophin signals to survival messages (Finkbeiner 2000). Neurotrophins, such as nerve growth factor and brain‐derived neurotrophic factor, have neuroprotective effects that are activated by CREB‐mediated transcription of various neuroprotective genes, including Bcl‐2 and BDNF (Bonni et al. 1999; Walton and Dragunow 2000). Recently, Tanaka (2001) showed that about 80% of the phosphorylated CREB‐positive neurons coexpressed Bcl‐2 in the ischemic area of the brain. The finding that cilostazol enhances CREB phosphorylation in association with increased Bcl‐2 levels, thereby reducing the caspase‐3‐positive cells in the penumbral region, provides evidence for neuroprotection from cerebral ischemia (Lee et al. 2004) (Fig. 2F).
Figure 2.
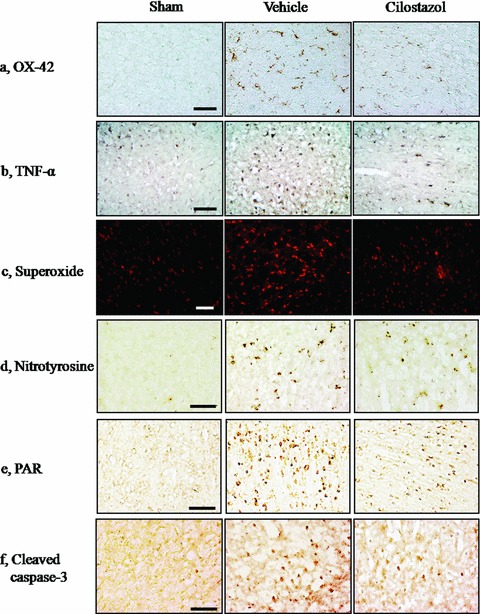
Representative immunohistochemical staining of the CD11b (OX‐42), TNF‐α, superoxide, nitrotyrosine, poly(ADP‐ribose) polymer (PAR), and cleaved caspase‐3‐positive cells in the penumbral region of the vehicle‐ and cilostazol‐treated group in comparison with sham group. Compared with the vehicle group, the OX‐42, TNF‐α, superoxide, nitrotyrosine, PAR, and cleaved caspase‐3‐positive cells in the penumbral region of the cilostazol‐treated group are less prominent at 24 h reperfusion after 2 h MCAO. Little immunoreactivity was shown in the sham groups. Each representative Figure is derived from 4∼6 rat brains of each group. Scale bar = 50 μm.
Evidence has accumulated that under moderate to severe stress conditions, activation of PARP, a nuclear protein, contributes to DNA repair in response to moderate DNA damage (de Murcia et al. 1997). The pharmacological inhibition of PARP attenuates brain injury in rodents, and mice lacking PARP gene are markedly protected from cerebral ischemic damage (Eliasson et al. 1997; Endres et al. 1997). It has been also shown that extensive DNA strand breaks that occur following ischemia/reperfusion can lead to overactivation of PARP, followed by the massive consumption of NAD+, and eventual cell death (Ducrocq et al. 2000). In the direct enzyme assay cilostazol, that has a quinolinone moiety in its structure, inhibited PARP with relatively low IC50 value (883 ± 41 nM). The 50% inhibitory concentration of cilostazol was about two orders of magnitude lower than that of 3‐aminobenzamide (IC50, 20.77 ± 5.19 μM). In vitro, in PC12 cells, cilostazol (0.1∼100 μM) concentration‐dependently inhibited H2O2 (100 μM)‐induced PARP activity. Additionally, cilostazol (30 mg/kg, two times orally) significantly reduced the elevated PARP activity and increased the density of poly(ADP‐ribose) polymer (PAR)‐positive cells (a product of activated PARP) in the penumbral zones of the rat cerebral cortex subjected to focal cerebral ischemic injury (Lee et al. 2007) (Fig. 2E). In accordance with these results, decreased NAD+ levels in in vitro and in vivo studies were ameliorated by cilostazol. This effect was associated with reduction in microglia (measured by the immunohistochemistry for OX‐42) and in TNF‐α levels (Fig. 2A and B). Additionally, at 30 mg/kg cilostazol significantly suppressed both superoxide and nitrotyrosine (a marker of peroxynitrite, interaction product of nitric oxide, and superoxide) levels in the penumbral sample of the brain subjected to 24 h reperfusion after 2 h MCAO (Fig. 2C and D).
It has been shown that overactivation of PARP can initiate a nuclear signal that propagates to mitochondria and triggers the release of apoptosis‐inducing factor (AIF), a mitochondrial protein, from mitochondria to the nucleus (Yu et al. 2002; Hong et al. 2004). In addition, AIF was implicated in the PARP‐1‐dependent cell death after focal cerebral ischemia (Hong et al. 2004; Komjati et al. 2004; Plesnila et al. 2004). In rat MCAO model, the number of AIF‐positive cells was strikingly increased in the infarcts of vehicle‐treated rats and significantly reduced by cilostazol. Simultaneous staining of AIF‐positive cells with Hoechst 33342, a marker of nucleus, revealed that increased AIF‐translocation to the nuclei was significantly decreased by cilostazol (Lee et al. 2007). These results indicated that AIF‐inhibiting and neuroprotective effects of cilostazol are closely related to its PARP inhibitory activity.
Protection from Chronic Cerebral Hypoperfusion Injury
The white matter lesions are frequently observed in aging, hypertension, and cerebrovascular disease, and are thought to be responsible for cognitive decline and gait disorders in the elderly population (Boone et al. 1992). There is ample evidence that chronic cerebral ischemia induced experimentally by the bilateral ligation of common carotid artery (BCCA) in rats leads to demyelination and axonal damage (Ihara et al. 2001). This experimental model has been proposed as a model for vascular dementia and cerebrovascular white matter lesions (Wakita et al. 1994; Tanaka et al. 1996; Tomimoto et al. 2003). Wakita et al. (1999) reported suppression of the activated microglia by anti‐inflammatory drugs with attenuation of the white matter lesions, suggestive of importance of inflammatory reaction in provoking the white matter lesion. Microglia and astrocytes are the major sources of TNF‐α (Sawada et al. 1989). TNF‐α, a proinflammatory cytokine in the central nervous system, induces various pathological effects, including edema formation after ischemia and demyelination, and these lesions are ameliorated by anti‐TNF‐α antibody (Taupin et al. 1997). On the other hand, oligodendroglial cell death has been reported following BCCA occlusion in gerbils (Kurumatani et al. 1998), and after transient global cerebral ischemia in rats (Petito et al. 1998). Apoptosis of oligodendrocytes, a major cellular component of the white matter, may, therefore, contribute directly to the white matter lesions. Microglial activation is the major source of inflammatory cytokines in the ischemic brain (Gregersen et al. 2000). Therefore, protection against the impact of oxidative stress, inflammatory damage, and apoptosis is of importance in reducing critical damage of the white matter and in preventing the cognitive impairment. Chronic cerebral hypoperfusion caused a large increase in the astrocytes and microglia with decreased oligodendrocytes in the white matter (optic tract, corpus callosum, internal capsule) of the rats subjected to BCCAL (Cho et al. 2006; Lee et al. 2006; Watanabe et al. 2006). The activation of astrocytes and microglia was significantly suppressed by cilostazol. In addition, cilostazol also prevented the loss of oligodendrocytes (Lee et al., 2006).
The evidence is accumulating that in the human brain apoptosis of the oligodendrocytes is increased in the white matter lesions; it involves myelin degeneration, astrogliosis, activation of microglia, and loss of oligodendrocytes (Akiguchi et al. 1997; Tomimoto et al. 2003). Oligodendrocytes, a major cellular component of the white matter, are more vulnerable to the oxidative stress than other glial cells such as astrocytes (Pantoni et al. 1996; Hollensworth et al. 2000). They readily undergo apoptosis in different paradigms causing ischemic and nonischemic damage with an activation of caspase pathways (Petito et al., 1998). After cerebral hypoperfusion in male Wistar rats, cilostazol reduced apoptosis of oligodendrocytes through the phosphorylation of CREB and subsequent elevation of Bcl‐2 in the white matter region (Watanabe et al. 2006). In line with these findings, cilostazol (60 mg/kg/day, orally) significantly reduced the apoptotic cell death in association with reduced caspase‐3 immunoreactivity and TUNEL‐positive cells in the white matter of rat brains subjected to BCCAL (Lee et al. 2006).
The protective effects of cilostazol against cerebral hypoperfusion‐induced cognitive impairment and white matter damage were assessed in a rat BCCA ligation model (Lee et al. 2006; Watanabe et al., 2006). In the experiment with Wistar rats in Morris water maze task, the prolonged latency induced by BCCA ligation was significantly shortened by the administration of cilostazol (50 mg/kg/day) throughout the entire period of the experiment (at 3, 7, 14, 21, and 28 days). In the vehicle group of chronic cerebral hypoperfusion model, the vacuolation and rarefactions were significantly increased in the white matter, which were accompanied by extensive activation of both microglia and astrocytes and suppression of oligodendrocytes in association with increased TNF‐α production, caspase‐3 immunoreactivity, and TUNEL‐positive cells in the white matter including optic tract. In conjunction with the improvement of the spatial learning memory, cilostazol (60 mg/kg/day, orally) prevented the occurrence of vacuolation and rarefaction of the white matter with reduced apoptosis (Lee et al. 2006). Additionally, post‐treatment with cilostazol strongly reversed not only activation of microglia and astrocytes but also diminished oligodendrocytes following chronic cerebral hypoperfusion. These results were supported by the reports of Watanabe et al. (2006), in that cilostazol (50 mg/kg/day) upregulated the expressions of Bcl‐2 and cyclooxygenase‐2, leading to a protective effect through increased CREB phosphorylation pathway. Taken together, these findings provide a novel therapeutic strategy for cilostazol treatment of cognitive impairment in poststroke patients.
Recent Clinical Trials in Stroke
A randomized, placebo‐controlled, double‐blind, comparative trial (the Cilostazol Stroke Prevention Study; CSPS), has shown that cilostazol (100 mg, twice daily) reduces the risk of secondary stroke by 41.7% in comparison with placebo, suggesting that the cilostazol can be highly effective in reducing the risk of subsequent cerebral infarction (Gotoh et al. 2000). Most recently, Huang (2007) reported that both cilostazol and aspirin prevented ischemic stroke recurrence in patients with ischemic disease, but severe cerebral hemorrhage was higher in aspirin‐treated as compared to cilostazol‐patients. Another recent report showed that cilostazol is cost‐effective in prevention of the recurrence of cerebral infarction (Inoue et al. 2006). Cilostazol also effectively arrested the progression of asymptomatic infarction areas when assessed by MRI in Japanese subjects with type II diabetes mellitus without symptomatic coronary vascular events (Shinoda‐Tagawa et al. 2002). Interestingly, Shinohara (2006) emphasized that administration of cilostazol to patients with cerebral infarction in the chronic stage reduced the recurrence of infarction and the incidence of pneumonia at least in Japanese patients.
Conclusions
Current treatment options for stroke and neurodegenerative diseases are still limited. In vitro studies and in vivo in different experimental animal models, cilostazol exerted a highly significant neuroprotection due to its pleiotropic effects. The neuroprotective potential of cilostazol are ascribed to its anti‐inflammatory and antiapoptotic effects mediated by its antioxidant, maxi‐K channel opening, and PARP inhibitory activities (Fig. 3). Taken together, these findings suggest that cilostazol is a promising candidate for pharmacological intervention in stroke and neurodegenerative diseases.
Figure 3.
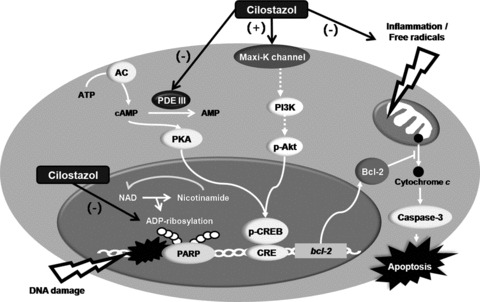
Neuroprotective mechanisms of cilostazol against focal cerebral ischemia. The neuroprotective potentials of cilostazol are ascribed to its anti‐inflammatory and antiapoptotic effects mediated by scavenging hydroxyl radicals and intracellular ROS, maxi‐K channel opening, and inhibition of PARP activity.
Conflict of Interest
The authors have no conflict of interest.
Addendum
The chemical names of drugs mentioned in the text by code number only are:
BAY 11‐7085 is (E)‐3‐(4‐t‐butylphenyl‐sulfonyl)‐2‐propenenitrile.
BMS‐204352 or maxopost is (S)‐3‐(5‐chloro‐2‐methoxyphenyl)‐3‐fluoro‐6‐(trifluoromethyl)‐1,3‐dehydro‐2H‐ indole‐2‐one.
KT 5720 is [9R,10S,12S]‐2,3,9,10,11,12‐hexahydro‐10‐hydroxy‐9‐methyl‐1‐oxo‐9,12‐epoxy‐1H‐diindolol [1,2,3‐fg; 3,2,1‐kl] pyrrolo [3,4‐1][1,6] benzodiazocine‐10 carboxylic acid hexyl ester.
KT 5823 is (9S,10R,12R)‐2,3,9,10,11,12‐hexahydro‐10‐methoxy‐2,9‐dimethyl‐1‐oxo‐9,12‐epoxy‐1H‐diindol[1,2,3‐fg:3',2',1'‐ll]pyrrolo[3,4‐i][1,6] benzoic acid methyl ester.
Acknowledgment
This work was supported by the MRC program of MOST/KOSEF (R13‐2005‐009).
References
- Aizawa T, Wei H, Miano JM, Abe J, Berk BC, Yan C (2003) Role of phosphodiesterase 3 in NO/cGMP‐mediated antiinflammatory effects in vascular smooth muscle cells. Circ Res 93: 406–413. [DOI] [PubMed] [Google Scholar]
- Akiguchi I, Tomimoto H, Suenaga T, Wakita H, Budka H (1997) Alterations in glia and axons in the brains of Binswanger's disease patients. Stroke 28: 1423–1429. [DOI] [PubMed] [Google Scholar]
- Akiyama H, Kudo S, Shimizu T (1985) The absorption, distribution and excretion of a new antithrombotic and vasodilating agent, cilostazol, in rat, rabbit, dog and man. Arzneimittelforschung 35: 1124–1132. [PubMed] [Google Scholar]
- Bednar MM (2000) Stroke: Antithrombin versus anti‐platelet therapy. Expert Opin Investig Drugs 9: 355–369. [DOI] [PubMed] [Google Scholar]
- Betz AL (1996) Alterations in cerebral endothelial cell function in ischemia. Adv Neurol 71: 301–311. [PubMed] [Google Scholar]
- Bonni A, Brunet A, West AE, Datta SR, Takasu MA, Greenberg ME (1999) Cell survival promoted by the Ras‐MAPK signaling pathway by transcription‐dependent and ‐independent mechanisms. Science 286: 1358–1362. [DOI] [PubMed] [Google Scholar]
- Boone KB, Miller BL, Lesser IM, Mehringer CM, Hill‐Gutierrez E, Goldberg MA, Berman NG (1992) Neuropsychological correlates of white‐matter lesions in healthy elderly subjects. A threshold effect. Arch Neurol 49: 549–554. [DOI] [PubMed] [Google Scholar]
- Bramer SL, Forbes WP, Mallikaarjun S (1999) Cilostazol pharmacokinetics after single and multiple oral doses in healthy males and patients with intermittent claudication resulting from peripheral arterial disease. Clin Pharmacokinet 37(Suppl. 2):1–11. [DOI] [PubMed] [Google Scholar]
- Bredesen DE (1995) Neural apoptosis. Ann Neurol 38: 839–851. [DOI] [PubMed] [Google Scholar]
- Chan PH (2005) Mitochondrial dysfunction and oxidative stress as determinants of cell death/survival in stroke. Ann N Y Acad Sci 1042: 203–209. [DOI] [PubMed] [Google Scholar]
- Cheney JA, Weisser JD, Bareyre FM, Laurer HL, Saatman KE, Raghupathi R, Gribkoff V, Starrett JE Jr., McIntosh TK (2001) The maxi‐K channel opener BMS‐204352 attenuates regional cerebral edema and neurologic motor impairment after experimental brain injury. J Cereb Blood Flow Metab 21: 396–403. [DOI] [PubMed] [Google Scholar]
- Cho KO, La HO, Cho YJ, Sung KW, Kim SY (2006) Minocycline attenuates white matter damage in a rat model of chronic cerebral hypoperfusion. J Neurosci Res 83: 285–291. [DOI] [PubMed] [Google Scholar]
- Choi JM, Shin HK, Kim KY, Lee JH, Hong KW (2002) Neuroprotective effect of cilostazol against focal cerebral ischemia via antiapoptotic action in rats. J Pharmacol Exp Ther 300: 787–793. [DOI] [PubMed] [Google Scholar]
- Dawson DL, Cutler BS, Meissner MH, Strandness DE Jr. (1998) Cilostazol has beneficial effects in treatment of intermittent claudication: Results from a multicenter, randomized, prospective, double‐blind trial. Circulation 98: 678–686. [DOI] [PubMed] [Google Scholar]
- De Murcia JM, Niedergang C, Trucco C, Ricoul M, Dutrillaux B, Mark M, Oliver FJ, Masson M, Dierich A, LeMeur M, et al. (1997) Requirement of poly(ADP‐ribose) polymerase in recovery from DNA damage in mice and in cells. Proc Natl Acad Sci USA 94: 7303–7730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douglas JS Jr., Holmes DR Jr., Kereiakes DJ, Grines CL, Block E, Ghazzal ZM, Morris DC, Liberman H, Parker K, Jurkovitz C, et al. (2005) Coronary stent restenosis in patients treated with cilostazol. Circulation 112: 2826–2832. [DOI] [PubMed] [Google Scholar]
- Ducrocq S, Benjelloun N, Plotkine M, Ben‐Ari Y, Charriaut‐Marlangue C (2000) Poly(ADP‐ribose) synthase inhibition reduces ischemic injury and inflammation in neonatal rat brain. J Neurochem 74: 2504–2511. [DOI] [PubMed] [Google Scholar]
- Easton JD (1998) What have we learned from recent antiplatelet trials? Neurology 51: S36–S38. [DOI] [PubMed] [Google Scholar]
- Eliasson MJ, Sampei K, Mandir AS, Hurn PD, Traystman RJ, Bao J, Pieper A, Wang ZQ, Dawson TM, Snyder SH, et al. (1997) Poly(ADP‐ribose) polymerase gene disruption renders mice resistant to cerebral ischemia. Nat Med 3: 1089–1095. [DOI] [PubMed] [Google Scholar]
- Endres M, Wang ZQ, Namura S, Waeber C, Moskowitz MA (1997) Ischemic brain injury is mediated by the activation of poly(ADP‐ribose) polymerase. J Cereb Blood Flow Metab 17: 1143–1151. [DOI] [PubMed] [Google Scholar]
- Finkbeiner S (2000) CREB couples neurotrophin signals to survival messages. Neuron 25: 11–14. [DOI] [PubMed] [Google Scholar]
- Franke B, Bayatti N, and Engele J (2000) Neurotrophins require distinct extracellular signals to promote the survival of CNS neurons in vitro. Exp Neurol 165: 125–135. [DOI] [PubMed] [Google Scholar]
- Gonzalez GA, Montminy MR (1989) Cyclic AMP stimulates somatostatin gene transcription by phosphorylation of CREB at serine 133. Cell 59: 675–680. [DOI] [PubMed] [Google Scholar]
- Gotoh F, Tohgi H, Hirai S, Terashi A, Fukuuchi Y, Otomo E, Shinohara Y, Itoh E, Matsuda T, Sawada T, et al. (2000) Cilostazol stroke prevention study: A placebo‐controlled double‐blind trial for secondary prevention of cerebral infarction. J Stroke Cerebrovasc Dis 9: 147–157. [DOI] [PubMed] [Google Scholar]
- Gregersen R, Lambertsen K, Finsen B (2000) Microglia and macrophages are the major source of tumor necrosis factor in permanent middle cerebral artery occlusion in mice. J Cereb Blood Flow Metab 20: 53–65. [DOI] [PubMed] [Google Scholar]
- Gribkoff VK, Starrett JE Jr., Dworetzky SI, Hewawasam P, Boissard CG, Cook DA, Frantz SW, Heman K, Hibbard JR, Huston K, et al. (2001) Targeting acute ischemic stroke with a calcium‐sensitive opener of maxi‐K potassium channels. Nat Med 7: 471–477. [DOI] [PubMed] [Google Scholar]
- Hoehn‐Berlage M, Norris DG, Kohno K, Mies G, Leibfritz D, Hossmann KA (1995) Evolution of regional changes in apparent diffusion coefficient during focal ischemia of rat brain: the relationship of quantitative diffusion NMR imaging to reduction in cerebral blood flow and metabolic disturbances. J Cereb Blood Flow Metab 15: 1002–1011. [DOI] [PubMed] [Google Scholar]
- Hollensworth SB, Shen C, Sim JE, Spitz DR, Wilsonn GL, LeDoux SP (2000) Glial cell type‐specific responses to menadione‐induced oxidative stress. Free Radic Biol Med 28: 1161–1174. [DOI] [PubMed] [Google Scholar]
- Honda F, Imai H, Ishikawa M, Kubota C, Shimizu T, Fukunaga M, Saito N (2006) Cilostazol attenuates gray and white matter damage in a rodent model of focal cerebral ischemia. Stroke 37: 223–228. [DOI] [PubMed] [Google Scholar]
- Hong KW, Kim KY, Shin HK, Lee JH, Choi JM, Kwak YG, Kim CD, Lee WS, Rhim BY (2003) Cilostazol prevents tumor necrosis factor‐alpha‐induced cell death by suppression of phosphatase and tensin homolog deleted from chromosome 10 phosphorylation and activation of Akt/cyclic AMP response element‐binding protein phosphorylation. J Pharmacol Exp Ther 306: 1182–1190. [DOI] [PubMed] [Google Scholar]
- Hong SJ, Dawson TM, Dawson VL (2004) Nuclear and mitochondrial conversations in cell death: PARP‐1 and AIF signaling. Trends Pharmacol Sci 25: 259–264. [DOI] [PubMed] [Google Scholar]
- Huang YN (2007) Cilostazol reduces cerebral hemorrhage in secondary stroke prevention: A multicenter, randomized, double‐blinded, aspirin control study. 16th Annual European Stroke Conference , Glasgow , UK .
- Ihara M, Tomimoto H, Kinoshita M, Oh J, Noda M, Wakita H, Akiguchi I, Shibasaki H (2001) Chronic cerebral hypoperfusion induces MMP‐2 but not MMP‐9 expression in the microglia and vascular endothelium of white matter. J Cereb Blood Flow Metab 21: 828–834. [DOI] [PubMed] [Google Scholar]
- Inoue T, Kobayashi M, Uetsuka Y, Uchiyama S (2006) Pharmacoeconomic analysis of cilostazol for the secondary prevention of cerebral infarction. Circ J 70: 453–458. [DOI] [PubMed] [Google Scholar]
- Kambayashi J, Liu Y, Sun B, Shakur Y, Yoshitake M, Czerwiec F (2003) Cilostazol as a unique antithrombotic agent. Curr Pharm Des 9: 2289–2302. [DOI] [PubMed] [Google Scholar]
- Kim KY, Shin HK, Choi JM, Hong KW (2002) Inhibition of lipopolysaccharide‐induced apoptosis by cilostazol in human umbilical vein endothelial cells. J Pharmacol Exp Ther 300: 709–715. [DOI] [PubMed] [Google Scholar]
- Kim KY, Shin HK, Lee JH, Kim CD, Lee WS, Rhim BY, Shin YW, Hong KW (2004) Cilostazol enhances casein kinase 2 phosphorylation and suppresses tumor necrosis factor‐alpha‐induced increased phosphatase and tensin homolog deleted from chromosome 10 phosphorylation and apoptotic cell death in SK‐N‐SH cells. J Pharmacol Exp Ther 308: 97–104. [DOI] [PubMed] [Google Scholar]
- Kimura Y, Tani T, Kanbe T, Watanabe K (1985) Effect of cilostazol on platelet aggregation and experimental thrombosis. Arzneimittelforschung 35: 1144–1149. [PubMed] [Google Scholar]
- Kluck RM, Bossy‐Wetzel E, Green DR, Newmeyer DD (1997) The release of cytochrome c from mitochondria: A primary site for Bcl‐2 regulation of apoptosis. Science 275: 1132–1136. [DOI] [PubMed] [Google Scholar]
- Kohda N, Tani T, Nakayama S, Adachi T, Marukawa K, Ito R, Ishida K, Matsumoto Y, Kimura Y (1999) Effect of cilostazol, a phosphodiesterase III inhibitor, on experimental thrombosis in the porcine carotid artery. Thromb Res 96: 261–268. [DOI] [PubMed] [Google Scholar]
- Komjati K, Mabley JG, Virag L, Southan GJ, Salzman AL, Szabo C (2004) Poly(ADP‐ribose) polymerase inhibition protect neurons and the white matter and regulates the translocation of apoptosis‐inducing factor in stroke. Int J Mol Med 13: 373–382. [PubMed] [Google Scholar]
- Kurumatani T, Kudo T, Ikura Y, Takeda M (1998) White matter changes in the gerbil brain under chronic cerebral hypoperfusion. Stroke 29: 1058–1062. [DOI] [PubMed] [Google Scholar]
- Latorre R, Oberhauser A, Labarca P, Alvarez O (1989) Varieties of calcium‐activated potassium channels. Annu Rev Physiol 51: 385–399. [DOI] [PubMed] [Google Scholar]
- Lawrence MS, McLaughlin JR, Sun GH, Ho DY, McIntosh L, Kunis DM, Sapolsky RM, Steinberg GK (1997) Herpes simplex viral vectors expressing Bcl‐2 are neuroprotective when delivered after a stroke. J Cereb Blood Flow Metab 17: 740–744. [DOI] [PubMed] [Google Scholar]
- Lawson K (2000) Potassium channel openers as potential therapeutic weapons in ion channel disease. Kidney Int 57: 838–845. [DOI] [PubMed] [Google Scholar]
- Lee JH, Lee YK, Ishikawa M, Koga K, Fukunaga M, Miyakoda G, Mori T, Hosokawa T, Hong KW (2003) Cilostazol reduces brain lesion induced by focal cerebral ischemia in rats‐an MRI study. Brain Res 994: 91–98. [DOI] [PubMed] [Google Scholar]
- Lee JH, Kim KY, Lee YK, Park SY, Kim CD, Lee WS, Rhim BY, Hong KW (2004) Cilostazol prevents focal cerebral ischemic injury by enhancing casein kinase 2 phosphorylation and suppression of phosphatase and tensin homolog deleted from chromosome 10 phosphorylation in rats. J Pharmacol Exp Ther 308: 896–903. [DOI] [PubMed] [Google Scholar]
- Lee JH, Oh GT, Park SY, Choi JH, Park JG, Kim CD, Lee WS, Rhim BY, Shin YW, Hong KW (2005) Cilostazol reduces atherosclerosis by inhibition of superoxide and tumor necrosis factor‐alpha formation in low‐density lipoprotein receptor‐null mice fed high cholesterol. J Pharmacol Exp Ther 313: 502–509. [DOI] [PubMed] [Google Scholar]
- Lee JH, Park SY, Shin YW, Hong KW, Kim CD, Sung SM, Kim KY, Lee WS (2006) Neuroprotection by cilostazol, a phosphodiesterase type 3 inhibitor, against apoptotic white matter changes in rat after chronic cerebral hypoperfusion. Brain Res 1082: 182–191. [DOI] [PubMed] [Google Scholar]
- Lee JH, Park SY, Shin HK, Kim CD, Lee WS, Hong KW (2007) Poly(ADP‐ribose) polymerase inhibition by cilostazol is implicated in the neuroprotective effect against focal cerebral ischemic infarct in rat. Brain Res 1152: 182–190. [DOI] [PubMed] [Google Scholar]
- Majid A, Delanty N, Kantor J (2001) Antiplatelet agents for secondary prevention of ischemic stroke. Ann Pharmacother 35: 1241–1247. [DOI] [PubMed] [Google Scholar]
- Martinou JC, Dubois‐Dauphin M, Staple JK, Rodriguez I, Frankowski H, Missotten M, Albertini P, Talabot D, Catsicas S, Pietra C, et al. (1994) Overexpression of BCL‐2 in transgenic mice protects neurons from naturally occurring cell death and experimental ischemia. Neuron 13: 1017–1030. [DOI] [PubMed] [Google Scholar]
- Matsumoto Y, Marukawa K, Okumura H, Adachi T, Tani T, Kimura Y (1999) Comparative study of antiplatelet drugs in vitro: Distinct effects of cAMP‐elevating drugs and GPIIb/IIIa antagonists on thrombin‐induced platelet responses. Thromb Res 95: 19–29. [DOI] [PubMed] [Google Scholar]
- Minami N, Suzuki Y, Yamamoto M, Kihira H, Imai E, Wada H, Kimura Y, Ikeda Y, Shiku H, Nishikawa M (1997) Inhibition of shear stress‐induced platelet aggregation by cilostazol, a specific inhibitor of cGMP‐inhibited phosphodiesterase, in vitro and ex vivo. Life Sci 61: 383–389. [DOI] [PubMed] [Google Scholar]
- Ollivier V, Parry GCN, Cobb RR, De Prost D, Mackman N (1996) Elevated cyclic AMP inhibits NF‐κB‐mediated transcription in human monocytic cells and endothelial cells. J Biol Chem 271: 20828–20835. [DOI] [PubMed] [Google Scholar]
- Pantoni L, Garcia JH, Gutierrez JA (1996) Cerebral white matter is highly vulnerable to ischemia. Stroke 27: 1641–1646. [DOI] [PubMed] [Google Scholar]
- Park SY, Lee JH, Kim YK, Kim CD, Rhim BY, Lee WS, Hong KW (2005) Cilostazol prevents remnant lipoprotein particle‐induced monocyte adhesion to endothelial cells by suppression of adhesion molecules and monocyte chemoattractant protein‐1 expression via lectin‐like receptor for oxidized low‐density lipoprotein receptor activation. J Pharmacol Exp Ther 312: 1241–1248. [DOI] [PubMed] [Google Scholar]
- Park SY, Lee JH, Kim CD, Lee WS, Park WS, Han J, Kwak YG, Kim KY, Hong KW (2006) Cilostazol suppresses superoxide production and expression of adhesion molecules in human endothelial cells via mediation of cAMP‐dependent protein kinase‐mediated maxi‐K channel activation. J Pharmacol Exp Ther 317: 1238–1245. [DOI] [PubMed] [Google Scholar]
- Petito CK, Olarte JP, Roberts B, Nowak TS Jr., Pulsinelli WA (1998) Selective glial vulnerability following transient global ischemia in rat brain. J Neuropathol Exp Neurol 57: 231–238. [DOI] [PubMed] [Google Scholar]
- Plesnila N, Zhu C, Culmsee C, Groger M, Moskowitz MA, Blomgren K (2004) Nuclear translocation of apoptosis‐inducing factor after focal cerebral ischemia. J Cereb Blood Flow Metab 24: 458–466. [DOI] [PubMed] [Google Scholar]
- Robitaille R, Charlton MP (1992) Presynaptic calcium signals and transmitter release are modulated by calcium‐activated potassium channels. J Neurosci 12: 297–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sage JI, Van Uitert RL, Duffy TE (1984) Early changes in blood brain barrier permeability to small molecules after transient cerebral ischemia. Stroke 15: 46–50. [DOI] [PubMed] [Google Scholar]
- Sawada M, Kondo N, Suzumura A, Marunouchi T (1989) Production of tumor necrosis factor‐α by microglia and astrocytes in culture. Brain Res 491: 394–397. [DOI] [PubMed] [Google Scholar]
- Shakur Y, Fong M, Hensley J, Cone J, Movsesian MA, Kambayashi J, Yoshitake M, Liu Y (2002) Comparison of the effects of cilostazol and milrinone on cAMP‐PDE activity, intracellular cAMP and calcium in the heart. Cardiovasc Drugs Ther 16: 417–427. [DOI] [PubMed] [Google Scholar]
- Shin HK, Kim YK, Kim KY, Lee JH, Hong KW (2004) Remnant lipoprotein particles induce apoptosis in endothelial cells by NAD(P)H oxidase‐mediated production of superoxide and cytokines via lectin‐like oxidized low‐density lipoprotein receptor‐1 activation: Prevention by cilostazol. Circulation 109: 1022–1028. [DOI] [PubMed] [Google Scholar]
- Shinoda‐Tagawa T, Yamasaki Y, Yoshida S, Kajimoto Y, Tsujino T, Hakui N, Matsumoto M, Hori M (2002) A phosphodiesterase inhibitor, cilostazol, prevents the onset of silent brain infarction in Japanese subjects with Type II diabetes. Diabetologia 45: 188–194. [DOI] [PubMed] [Google Scholar]
- Shinohara Y (2006) Antiplatelet cilostazol is effective in the prevention of pneumonia in ischemic stroke patients in the chronic stage. Cerebrovasc Dis 22: 57–60. [DOI] [PubMed] [Google Scholar]
- Tanaka K (2001) Alteration of second messengers during acute cerebral ischemia‐adenylate cyclase, cyclic AMP‐dependent protein kinase, and cyclic AMP response element binding protein. Prog Neurobiol 65: 173–207. [DOI] [PubMed] [Google Scholar]
- Tanaka K, Ogawa N, Asanuma M, Kondo Y, Nomura M (1996) Relationship between cholinergic dysfunction and discrimination learning disabilities in Wistar rats following chronic cerebral hypoperfusion. Brain Res 729: 55–65. [PubMed] [Google Scholar]
- Taupin V, Renno T, Bourbonniere L, Peterson AC, Rodriguez M, Owens T (1997) Increased severity of experimental autoimmune encephalomyelitis, chronic macrophage/microglial reactivity, and demyelination in transgenic mice producing tumor necrosis factor‐α in the central nervous system. Eur J Immunol 27: 905–913. [DOI] [PubMed] [Google Scholar]
- Tomimoto H, Ihara M, Wakita H, Ohtani R, Lin JX, Akiguchi I, Kinoshita M, Shibasaki H (2003) Chronic cerebral hypoperfusion induces white matter lesions and loss of oligodendroglia with DNA fragmentation in the rat. Acta Neuropathol 106: 527–534. [DOI] [PubMed] [Google Scholar]
- Wakida K, Morimoto N, Shimazawa M, Hozumi I, Nagase H, Inuzuka T, Hara H (2006) Cilostazol reduces ischemic brain damage partly by inducing metallothionein‐1 and ‐2. Brain Res 1116: 187–193. [DOI] [PubMed] [Google Scholar]
- Wakita H, Tomimoto H, Akiguchi I, Kimura J (1994) Glial activation and white matter changes in the rat brain induced by chronic cerebral hypoperfusion: An immunohistochemical study. Acta Neuropathol 87: 484–492. [DOI] [PubMed] [Google Scholar]
- Wakita H, Tomimoto H, Akiguchi I, Lin JX, Miyamoto K, Oka N (1999) A cyclooxygenase‐2 inhibitor attenuates white matter damage in chronic cerebral ischemia. NeuroReport 10: 1461–1465. [DOI] [PubMed] [Google Scholar]
- Walton MR, Dragunow I (2000) Is CREB a key to neuronal survival? Trends Neurosci 23: 48–53. [DOI] [PubMed] [Google Scholar]
- Walton M, Woodgate AM, Muravlev A, Xu R, During MJ, Dragunow M (1999) CREB phosphorylation promotes nerve cell survival. J Neurochem 73: 1836–1842. [PubMed] [Google Scholar]
- Watanabe T, Zhang N, Liu M, Tanaka R, Mizuno Y, Urabe T (2006) Cilostazol protects against brain white matter damage and cognitive impairment in a rat model of chronic cerebral hypoperfusion. Stroke 37: 1539–1545. [DOI] [PubMed] [Google Scholar]
- Yang GY, Gong C, Qin Z, Liu XH, Lorris Betz A (1999) Tumor necrosis factor α expression produces increased blood‐brain barrier permeability following temporary focal cerebral ischemia in mice. Brain Res Mol Brain Res 69: 135–143. [DOI] [PubMed] [Google Scholar]
- Ye YL, Shi WZ, Zhang WP, Wang ML, Zhou Y, Fang SH, Liu LY, Zhang Q, Yu YP, Wei EQ (2007) Cilostazol, a phosphodiesterase 3 inhibitor, protects mice against acute and late ischemic brain injuries. Eur J Pharmacol 557: 23–31. [DOI] [PubMed] [Google Scholar]
- Yu SW, Wang H, Poitras MF, Coombs C, Bowers WJ, Federoff HJ, Poirier GG, Ted M, Dawson TM, Dawson VL (2002) Mediation of poly(ADP‐ribose) polymerase‐1‐dependent cell death by apoptosis‐inducing factor. Science 297: 259–264. [DOI] [PubMed] [Google Scholar]