

Abstract
TPA023 and α5IA are structurally related compounds that selectively modulate certain GABAA receptor subtypes. Hence, TPA023 has weak partial agonist efficacy at the α2 and α3 subtypes whereas α5IA has inverse agonist efficacy at the α5 subtype. These efficacy characteristics translate into novel pharmacological profiles in preclinical species with TPA023 being a nonsedating anxiolytic in rats and primates whereas α5IA enhanced cognition in rats but was devoid of the proconvulsant or kindling liabilities associated with nonselective inverse agonists. In vitro and in vivo metabolic studies showed that TPA023 was metabolized via CYP3A4‐mediated t‐butyl hydroxylation and N‐deethylation whereas α5IA was metabolized to produce the hydroxymethyl isoxazole, the latter of which was highly insoluble and caused renal toxicity in preclinical species. In humans, TPA023 had a half‐life in the region of 6–7 h whereas the half‐life of α5IA was 2–2.5 h. TPA023 was clearly differentiated from the nonselective agonist lorazepam in terms of saccadic eye movement and unlike lorazepam, it did not impair either postural stability, as judged by body sway, or cognition. The occurrence of the hydroxymethyl isoxazole metabolite of α5IA in human urine precluded the use of α5IA in prolonged dosing studies. Nevertheless, α5IA was evaluated in an alcohol‐induced cognitive impairment model in healthy normal volunteers and was found to reverse the memory‐impairing effects of alcohol. To date, however, no efficacy data for either TPA023 or α5IA in patient populations has been reported, although at the very least, the preclinical and limited clinical data with TPA023 and α5IA validate the approach of targeting specific GABAA receptors through subtype‐selective efficacy.
Keywords: α5 Subtype, α2/α3 Subtype, Anxiolytics, GABAA receptor, Benzodiazepines, Cognition enhancers, Subtype‐selectivity
Introduction
The GABAA receptor is a pentameric assembly of subunits derived from the GABAA gene family (α1–6, β1–3, γ1–3, δ, ∈, θ and π; Simon et al. 2004) and is responsible for the ligand (GABA)‐gated flow of chloride ions into the neuron, which result in a hyperpolarization of the cell membrane that reduces the neuronal firing rate and therefore has an inhibitory effect on neuronal activity. The most common combination of subunits are α, β, and γ subunits arranged in a αβαβγ configuration as viewed from the synapse (Minier and Segal 2004).
The classical benzodiazepines, including diazepam, chlordiazepoxide, midazolam, lorazepam and alprazolam all exert their anxiolytic, sedative, anticonvulsant, cognition‐impairing and myorelaxant effects via the modulation of subtypes of GABAA receptor that contain β, γ2 and either an α1, α2, α3 or α5 subunit. These receptors represent around 75% of the total rat brain GABAA receptor population (McKernan and Whiting 1996) and the specific benzodiazepine recognition site occurs at the interface of the γ2 and α subunit (Sieghart 1995, 2006). The contribution of the α subunit to benzodiazepine binding is highlighted by the fact that the corresponding receptors containing either an α4 or α6 subunit (in which a histidine residue is replaced by an arginine) have essentially no affinity for classical benzodiazepines (Benson et al. 1998; Wieland et al. 1992) and represent the so‐called diazepam‐insensitive GABAA receptors.
Given that the pharmacological effects of benzodiazepines are mediated via four different subtypes, namely α1‐, α2‐, α3‐, and α5‐containing GABAA receptors, and that these various receptor populations have distinct neuroanatomical localizations (Fritschy and Möhler 1995; Pirker et al. 2000; Sperk et al. 1997)—and, therefore, presumably subserve distinct functions—it is not unreasonable to assume that individual GABAA receptor subtypes may be responsible for different aspects of the pharmacological profile of nonselective benzodiazepine site ligands. The delineation of the roles of the different GABAA receptor subtypes has been greatly facilitated by the generation of transgenic mouse containing either mutations or deletions of specific subunits (Rudolph and Möhler 2004; Vicini and Ortinski 2004) as well as the availability of novel, subtype‐selective pharmacological tools. A summary of these data, which form the basis for the development of an α2/α3‐selective partial agonist as a nonsedating anxiolytic and an α5‐selective inverse agonist as a cognition enhancer, will be presented in the following section.
Preclinical Pharmacological Proof of Concept
α2/α3 Subtype‐Selective Nonsedating Anxiolytic
The initial wave of benzodiazepines (BZs), such as chlordiazepoxide and diazepam, was superseded to a certain extent by the so‐called high‐potency BZs such as alprazolam, clonazepam, and lorazepam (Chouinard 2004; Moroz 2004), although mechanistically, the low‐, medium‐ and high‐potency benzodiazepines all share the same mode of action with clinical differences between compounds being primarily related to the pharmacokinetic and/or pharmaceutical properties of these various drugs. Nevertheless, it has long remained a goal to develop compounds that retain the anxiolytics efficacy but are devoid of sedative properties. In this regard, initial attempts to develop nonsedating anxiolytic focused on nonselective partial agonist such as bretazenil (Atack 2003). However, although these compounds showed good separation between doses required for anxiolysis and sedation in preclinical species (Haefely et al. 1990), this failed to translate into the clinic (Atack 2003).
More recently, the strategy has been to selectively modulate GABAA subtypes associated with anxiolytic‐like activity without affecting the subtype associated with sedation (Atack 2005). Evidence that α1‐containing GABAA receptors are associated with sedation includes the fact that the hypnotic zolpidem has higher affinity for α1‐ compared to α2‐ or α3‐containing GABAA receptors (Sieghart 1995). In addition, mice in which α1‐containing GABAA receptors are rendered insensitive to diazepam (α1H101R mice) show a reduced susceptibility to the sedating effects of diazepam (McKernan et al. 2000; Rudolph et al. 1999). Thus, it would appear that agonist activity at the α1 subtype should be avoided in a potential nonsedating anxiolytic. Accordingly, the prototypic efficacy‐selective compound L‐838417, which has no efficacy at the α1 subtype but is a partial agonist at the α2, α3 and α5 subtypes, is a nonsedating anxiolytic in rodents and primates (McKernan et al. 2000; Rowlett et al. 2005).
Although several converging lines of evidence point to a role of the α1 subtype in mediating sedation, the identity of the "anxiolytic" GABAA subtype appears less certain. Hence, using a molecular genetic approach (i.e., α2H101R and α3H126R mice), it was shown that the α2 subtype appears to be responsible for the anxiolytic affects of diazepam (Löw et al. 2000). On the other hand, pharmacological data with the α3 selective inverse agonist α3IA (Atack et al. 2005) and the α3 selective agonist TP003 (Dias et al. 2005) point to a role of α3‐containing GABAA receptors in mediating anxiolytic‐like effects. The reasons for the discrepancy between the transgenic mouse and pharmacological data are unclear but, from a practical point of view, a compound with selectivity for the α2 and α3 subtypes would be expected to exert anxiolytic efficacy.
α5 Subtype‐Selective Cognitive Enhancer
Whereas nonselective agonists have been shown to impair cognition in man (Ghoneim and Mewaldt 1990), nonselective inverse agonists have been shown to increase vigilance in preclinical species (Venault et al. 1986). However, they are also associated with convulsant or proconvulsant activity as well as anxiogenic properties (Venault and Chapouthier 2007). Indeed, the nonselective partial inverse agonist FG 7142 has been reported to provoke a strong anxiogenic response in human volunteers (Dorow et al. 1983) and due to these liabilities, nonselective inverse agonists are of no clinical utility. Clearly, a compound that had inverse agonist activity that specifically targeted the GABAA receptor subtype associated with cognition might possess the cognition enhancing properties in the absence of the proconvulsant and anxiogenic liabilities associated with the nonselective compounds (Maubach 2003).
Data from a variety of neuroanatomical, molecular genetic, and pharmacological studies have implicated α5‐containing GABAA receptors in cognitive processes and, more specifically, hippocampally mediated cognition. For example, the preferential expression of α5‐containing GABAA receptors in the hippocampus (Fritschy and Mohler 1995; Pirker et al. 2000; Sperk et al. 1997; Sur et al. 1999), implicates this subtype in hippocampal‐specific functions, such as learning and memory (Maubach 2003). Moreover, mice in which the expression of α5‐containing GABAA receptors had been reduced (Crestani et al. 2002) or was absent (Collinson et al. 2002) demonstrated improved performance in certain cognitive tasks (Collinson et al. 2002; Crestani et al. 2002). Additional evidence implicating the role of the α5 subtype in cognition comes from observations that these receptors play a role in the amnestic effects of the anesthetic etomidate (Cheng et al. 2006).
Although the evidence supporting the role of the α5 subtype in cognition suggests that an α5‐selective inverse agonist may enhance cognition, a critical issue is that of whether or not such a compound, which specifically activates the hippocampus, a region if the brain often associated with seizure activity, might be proconvulsant or not. Indeed, initial studies with the α5 binding‐selective compounds RY‐023, −024, and −080 showed these compounds to be either convulsant or proconvulsant (Liu et al. 1996). Subsequently, however, more detailed studies showed that the proconvulsant effects of RY‐080 could not be solely attributed to actions at the α5 subtype but were probably associated with inverse agonist activity at other subtypes (Atack et al. 2006a). Moreover, at doses that selectively occupied α5‐containing receptors, L‐655708, which is structurally related to RY‐080 (Quirk et al. 1996), was not proconvulsant, although it did improve performance in the Morris water maze (Atack et al. 2006b). Additional α5‐selective compounds reported to enhance cognition in rodents in the absence of any overt convulsant or proconvulsant activity include a thiophene, which has a degree of binding‐ and efficacy‐ selectivity (Compound 43; Chambers et al. 2003), as well as efficacy selective compounds from the pyrazolotriazine (Compound 13; Chambers et al. 2004) and triazolophthalazine series (Compound 16, Sternfeld et al. 2004), the latter of which was designated α5IA (Dawson et al. 2006). Because α5IA has progressed further than any of the other publicly disclosed α5‐selective inverse agonists, its properties will be described in more detail in the following sections.
Common Origins of TPA023 and α5IA
In the initial pursuit of a compound with higher affinity for α2/α3 compared to α1 and α5 receptors (i.e., binding selectivity), features of a screening hit, plus the known compounds CL 218872 and Compound 80 (Tarzia et al. 1988) were combined to result (Fig. 1) in Compound 62 (Carling et al. 2004; Compound 3, Carling et al. 2005; Compound 1, Russell et al. 2005). The structure‐activity relationship of this class of compounds along with 10 other structurally diverse series identified from screening of the Merck chemical collection revealed that at best only a modest (∼10‐fold) α2/α3 binding selectivity could be achieved. At this stage, the strategy was changed in order to try to identify compounds which bind with equivalent affinity at the different subtypes but only have agonist or partial agonist efficacy and only at the α2 and α3 subtypes (i.e., efficacy selectivity; Atack, 2005).
Figure 1.

Origins of TPA023 and α5IA. Both compounds were derived from a common ancestor (3‐Phenyl‐6‐(2‐pyridyl)methyloxy‐7,8,9,10‐tetrahydro‐(7,10‐ethano)‐1,2,4‐triazolo[3,4‐a]phthalazine; Compound 62, Carling et al. 2004; Compound 3, Carling et al. 2005; Compound 1, Russell et al. 2005) and entailed systematic replacements of the 2.2.2 bicyclic group (replaced with a t‐butyl group at the 7‐position in L‐838417 and TPA023 or a benzo fused ring structure in α5IA), the pendant phenyl at the 3‐position (2‐fluorophenyl in TPA023 and 3‐methyl isoxazole in α5IA), and the O‐linked group at the 6‐position (2‐ethyl‐1,2,4 triazole in TPA023 and 1‐methyl‐1,2,3 triazole in α5IA).
The replacement of the 2‐pyridyl group of Compound 62 (Carling et al. 2004) with a 1,2,4‐triazole and the [2.2.2] bicyclic ring system with a pendant phenyl group and thereafter a t‐butyl group resulted (Fig. 1) in the prototypic α2/α3 selective partial agonist L‐838417 (McKernan et al. 2000) and TPA023; compounds that differed from each other with regard to the extent and positioning of fluorination of the 3‐phenyl ring (fluorination decreased the metabolic liability associated with the 3‐phenyl group) as well as substitution on the 1,2,4 triazole group (2‐methyl and 2‐ethyl for L‐838417 and TPA023, respectively).
An analogous strategy was employed in the development of α5IA (Fig. 1). Hence, replacement of the 3‐phenyl group with an isoxazole introduced inverse agonism and changing the [2.2.2] bicyclic ring system for a benzo fused ring, which gave a 1,2,4‐triazolophthalazine core structure, imparted greater affinity at the expense of some α5 inverse agonism. However, this could be restored by the introduction at the C‐6 position of various isomeric triazoles of which the 1‐methyl 1,2,3‐triazole was of most interest as it was not proconvulsant whereas the 2‐ and 3‐methyl 1,2,3‐triazoles were (Sternfeld et al. 2004).
In vitro Properties of TPA023 and α5IA
Affinity
TPA023 has equivalent affinity for the benzodiazepine binding site of recombinant human α1‐, α2‐, α3‐ and α5‐containing GABAA receptors, with Ki values ranging from 0.19 to 0.41 nM (Fig. 2) with much lower affinity being observed at α4‐ and α6‐containing GABAA receptors (the so‐called diazepam‐insensitive GABAA receptors; Ki values of 60 and 418 nM at the α4 and α6 subtype, respectively; Atack et al. 2006c). Similarly, α5IA is also nonselective in terms of GABAA receptor affinity with Ki values at the α1, α2, α3, and α5 subtypes ranging from 0.58 to 0.88 nM and again, much lower affinity was observed at α4 and α6 subtypes (respective Ki values of 37 and >10,000 nM; Dawson et al. 2006). The affinity of TPA023 and α5IA at native rat brain GABAA receptors (∼0.3 and ∼1 nM, respectively) indicates that both compounds have similar affinity at rat and human GABAA receptors affinity. Neither compound had significant additional activities in a panel of over 100 receptor and enzyme assays and the binding of [14C]TPA023 or [14C]α5IA to rat brain sections could be blocked by the nonselective benzodiazepine flunitrazepam, indicating that both compounds had specificity for the benzodiazepine recognition site of GABAA receptors.
Figure 2.

Affinity and efficacy of TPA023 and α5IA at recombinant human GABAA receptors containing β3, γ2 and either an α1, α2, α3, or α5 subunit stably expressed in mouse fibroblast cells. Affinity was measured using a [3H]flumazenil radioligand binding assay under equilibrium conditions whereas efficacy was measured using whole‐cell patch clamp electrophysiology with the extent of the modulation of a GABA EC20‐equivalent concentration being expressed relative to that observed with the nonselective full agonist chlordiazepoxide in the case of the GABA‐induced currents being potentiated (i.e., agonism) or the non‐selective full inverse agonist DMCM when GABA‐induced currents were attenuated (i.e., inverse agonism). (Data modified from Atack et al. 2006c and Dawson et al. 2006.)
Intrinsic Efficacy
Although TPA023 and α5IA share structural similarities and have comparable affinity for the different, stably transfected human GABAA receptor subtypes, they have markedly different selective efficacy profiles, with TPA023 being an α2/α3‐selective agonist whereas α5IA is an α5‐selective inverse agonist (Fig. 2). Hence, although TPA023 is an antagonist at the α1 and α5 subtypes, it is a weak partial agonist at the α2 and α3 subtypes. On the other hand, α5IA is essentially an antagonist at the α2 and α3 subtypes, but has weak inverse agonist efficacy at the α1 subtype and pronounced inverse agonism at the α5 subtype. The agonist efficacy of TPA023 at the α3 subtype had an EC50 of 1.7 nM compared to a binding affinity of 0.19 nM. Similarly, the EC50 for the inverse agonism effects of α5IA was higher than the binding affinity with the EC50 and Ki values being 2.2 nM and 0.66 nM, respectively (Dawson et al. 2006).
As the efficacy profile of both compounds is the key determinant of their respective in vivo pharmacological profiles, additional studies were performed with human receptors in a different expression system (transient transfection in Xenopus laevis oocytes) or with recombinant rat receptors (also transiently transfected in Xenopus laevis oocytes). For both compounds, the intrinsic efficacy profiles were similar across different GABAA receptor expression systems. Hence, in each system, TPA023 was an α2/α3 weak partial agonist (unpublished data) whereas α5IA was an α5‐selective inverse agonist (Dawson et al. 2006).
The intrinsic efficacy of α5IA was further evaluated in an in vitro mouse hippocampal slice electrophysiological preparation in which long‐term potentiation (LTP) can be produced. These long‐term changes in synaptic efficacy may be a model of the changes in synaptic efficacy that presumably occur during learning and memory and in this assay, the nonselective full inverse agonist DMCM has previously been reported to enhance LTP (Seabrook et al. 1997). The selective modulation of α5‐containing GABAA receptors by α5IA was also able to enhance long‐term potentiation (Dawson et al. 2006). Importantly, the effects of α5IA were not accompanied by the paroxysmal burst discharges observed with DMCM and which may be predictive of proconvulsant or convulsant activity in vivo (Seabrook et al. 1997). Similarly, modulation of α5 receptors by the α5 binding‐selective inverse agonist L‐655708 also enhanced LTP (Atack et al. 2006b).
In vivo Properties of TPA023 and α5IA
TPA023
When dosed orally, TPA023 was readily absorbed and penetrated into the brain as judged by the fact that the dose required to give 50% inhibition of [3H]flumazenil rat in vivo binding (i.e., 50% occupancy, the Occ50) was 0.42 mg/kg p.o. (Table 1). The anxiolytic‐like properties of TPA023 were assessed in a variety of rat models; the ethological elevated plus maze, in which anxiety is measured under relatively baseline (i.e., nonconditioned) conditions as well as assays in which rats are conditioned to produce an increased level of fear or anxiety in either the fear‐potentiated startle or conditioned suppression of drinking assays (Table 1). In all three assays, TPA023 produced an anxiolytic‐like effect at doses corresponding to receptor occupancy in the region of 70–88%. More specifically, in the elevated plus maze, TPA023 significantly increased the time spent on the open arms whereas it completely attenuated the increase in startle produced by the expectation of an electric shock in the fear‐potentiated startle assay and was able to partially reverse the suppression of licking of a water spout produced by the anticipation of an electric shock (Atack et al. 2006c). TPA023 also had anxiolytic‐like activity in the squirrel monkey conditioned emotional response assay (which is analogous to the rat‐conditioned suppression of drinking assay). In this assay, the minimal effective dose (0.3 mg/kg) produced plasma drug concentrations corresponding to 65% occupancy (based upon the rat plasma‐occupancy relationship; Atack et al. 2006c).
Table 1.
Summary of preclinical invitro properties of TPA023 and α51A.
| Species | Assay | TPA023 | α5IA |
|---|---|---|---|
| Receptor occupancy | |||
| Rat | [3H]flumazenil in vivo binding | Occ50= 0.42 mg/kg p.o. | Occ50= 0.35 mg/kg p.o. |
| Anxiety assays | |||
| Rat | Elevated plus maze | Anxiolytic at 70% occ. | Not anxiogenic at 79% occ. |
| Rat | Fear potentiated startle | Anxiolytic at 70% occ. | — |
| Rat | Conditioned suppression of drinking | Anxiolytic at 88% occ. | — |
| Sq. monkey | Conditioned emotional response | Anxiolytic at 65% occ. * | — |
| Sedation/motor incoordination | |||
| Mouse | Rotarod, baseline | No effect at >99% occ. | No effect at 95% |
| Rat | Chain‐pulling | No effect at 99% occ. | — |
| Sq. monkey | Lever pressing | No effect at >99% occ.* | — |
| Seizure activity | |||
| Mouse | PTZ‐induced convulsions | Anticonvulsant, ED50∼50% occ. | Not proconvulsant at 98% occ. |
| Mouse | Kindling | — | No effect at 94% occ. |
| Ethanol interaction | |||
| Mouse | Rotarod, + ethanol | Modest potentiation at 83% occ. | No ethanol potentiation |
| Cognition | |||
| Rat | DMTP Morris water maze | — | Enhanced at 25% occ. |
| Withdrawal, drug discrimination, and self‐administration | |||
| Mouse | FG 7142‐precipitated withdrawal | No effect at 80% occ. | — |
| Baboon | Self‐administration | No effect at >95% occ | — |
| Baboon | Lorazepam drug discrimination | No effect | — |
Occ50, dose required to give 50% occupancy; Sq. monkey, squirrel monkey; PTZ, pentylenetetrazole; DMTP, delayed matching to place.
* squirrel monkey occupancy values were estimated using the squirrel monkey plasma drug concentrations and were calculated from the rat plasma concentration occupancy curve.
As regards sedation and/or motor incoordination, TPA023 did not impair performance in either the mouse rotarod, the rat chain‐pulling or squirrel monkey level‐pressing assays at doses corresponding to ≥99% occupancy. Based upon these data, there is a clear separation between the level of occupancy required for anxiolytic‐like activity (65–88%, depending on the assay) and sedation (≥99%).
In mice treated for 7 days with TPA023, and in contrast to the nonselective full agonist triazolam, there were no signs of physical dependence as demonstrated by a lack of FG 7142‐precipitated seizure activity (Atack et al. 2006c). A clear distinction between TPA023 and benzodiazepine nonselective full agonists was also observed in baboons in which TPA023 did not generalize to lorazepam in drug‐discrimination experiments (Ator 2005). In addition, baboons trained to self‐administer cocaine did not self‐administer TPA023 to a greater extent than vehicle although they did self‐administer lorazepam, indicating the TPA023 has little abuse potential (Ator 2005).
α5IA
The inhibition of rat brain [3H]flumazenil in vivo binding by α5IA was dose dependent with the dose corresponding to 50% occupancy, the Occ50, being 0.35 mg/kg p.o. (Table 1). When dosed to mice, α5IA did not produce convulsions in its own right nor was it proconvulsant in that it did not reduce the dose of pentylenetetrazole required to cause either clonic or tonic convulsions, even at a dose that occupied 98% of mouse brain GABAA receptors.
Acute, subthreshold doses of a nonselective partial inverse agonists such as FG7142 will not produce overt convulsions but over a period of time, the repeated dosing at this same dose level can sensitize the brain such that convulsions are produced; a process known as kindling. Therefore, although α5IA was not proconvulsant, it was important to establish whether it could produce kindling. Accordingly, mice were dosed for 19 days with either α5IA or FG 7142 and the number of clonic or tonic convulsions was recorded during a 45‐min period after injection. Unlike FG 7142, which produced clonic convulsions in a third of mice by day 8 that progressed to tonic convulsions later in the study, α5IA‐treated mice showed no signs of clonic convulsions, even after 19 days of dosing (Dawson et al. 2006). Similarly, an α5 efficacy‐selective pyrazolotriazine compound also showed no signs of kindling (Chambers et al. 2004). Unlike FG 7142, α5IA did not increase the % time spent in the closed arms of the elevated plus maze, suggesting that it did not have anxiogenic‐like activity (Dawson et al. 2006). Neither did α5IA affect locomotor activity measured either indirectly as the distance traveled during the elevated plus maze trial or as judged by performance in the mouse rotarod assay (Dawson et al. 2006).
Performance in the delayed matching to place version of the Morris water maze is thought to be hippocampus‐dependent (Steele and Morris 1999). Given the preferential hippocampal localization of α5‐containing GABAA receptors, this task was chosen to assess the effects of α5IA on rat cognitive performance. In this assay, rats have to “remember” the position of a hidden platform in relation to spatial cues, with the platform position remaining constant within any given day but changing from day‐to‐day. α5IA increased the speed to find the platform in the second trial, which occurred 4 h after the initial trial, suggesting that during the second trial, the rat "remembered" the position of the platform better than vehicle‐treated rats, either as a consequence of better encoding during Trial 1 or better retrieval during Trial 2. The receptor occupancy during Trials 1 and 2 was similar, being ∼25% (Dawson et al. 2006).
Hence, α5IA was able to enhance cognitive performance in the delayed matching to place version of the Morris water maze in normal rats (i.e., rats without pharmacologically‐induced or age‐ or lesion‐related deficits) but was without the anxiogenic and proconvulsant liabilities associated with nonselective inverse agonists such as FG 7142.
Metabolism of TPA023 and α5IA
In vitro incubation of TPA023 with microsomes from various species, including man, produced alcohol (M1) and free triazole (M2) as the predominant metabolites (1, 3). Both the t‐butyl hydroxylation and the N‐deethylation reactions in human liver microsomes were greatly inhibited by CYP3A‐specific antibodies and inhibitors, with CYP3A4 rather than CYP3A5 playing the major role in TPA023 metabolism (Ma et al. 2007). The M1 and M2 metabolites of TPA023 were also found in vivo in rat as well as human but are not considered to play a pharmacological role based on their poor blood‐brain barrier penetration as well as, in the case of the M2 metabolite, lower affinity for GABAA receptors than the parent. More specifically, the brain‐to‐plasma ratios of M1 and M2 are ∼0.02 compared to ∼0.4 for the parent (Polsky‐Fisher et al. 2006).
Figure 3.

The major metabolic pathways of TPA023 and α5IA. Metabolism of TPA023 proceeds via the predominantly CYP3A4‐mediated t‐butyl hydroxylation and N‐deethylation (Ma et al. 2007) whereas the single major metabolite of α5IA is the hydroxymethyl isoxazole.
The in vitro incubation of α5IA with microsome preparations from various species, including humans, produced a single metabolite (M1, Fig. 3). This hydroxymethyl isoxazole metabolite was also found in vivo, including man (Xue et al. 2004). However, based upon the very low solubility of this metabolite (0.6 μg/mL in water) plus the observations of crystalluria in preclinical species, α5IA was considered unsuitable for prolonged administration to humans (Merschman et al. 2005).
Human Pharmacokinetics of TPA023 and α5IA
The pharmacokinetics of TPA023 appear relatively dose dependent, with the Cmax values being in the region of 5, 13, and 28 ng/mL at the doses of 0.5, 1.5, and 3.0 mg, respectively (Fig. 4A) with maximum plasma concentrations being achieved approximately 2 h after dosing. Analysis of these data demonstrated a mean apparent half‐life of 7.3 h for the 0.5 and 1.5 mg doses (de Haas et al. 2007) or 6.7 h at the 3.0 mg dose (Polsky‐Fisher et al. 2006). These data compare to the half‐lives (and oral bioavailability,%F) reported in rat and dog of 1.4 and 1.5 h (35 and 53%), respectively (Carling et al. 2005). In vitro studies implicated members of the CYP3A subfamily in the metabolism of TPA023 (Ma et al. 2007) and this was confirmed experimentally in man by the fact that the potent CPY3A inhibitor itraconazole produced an approximately 5‐fold increase in the TPA023 plasma area‐under‐the‐curve (Polsky‐Fisher et al. 2006).
Figure 4.
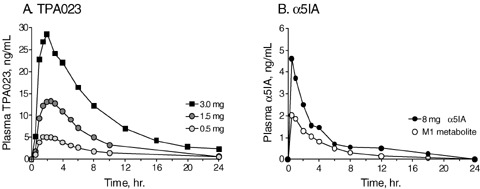
Plasma pharmacokinetics of TPA023 and α5IA in man. A. Plasma TPA023 concentrations were measured after single doses of 0.5, 1.5, or 3.0 mg of an immediate release formulation. Data were collected from two separate studies, one of which was a pharmacodynamic study (0.5 and 1.5 mg doses; de Haas et al. 2007) and the other was a metabolism and disposition study (3 mg dose; Polsky‐Fisher et al. 2006). Data represent mean (n= 12 for the 0.5 and 1.5 mg doses and n= 5 for the 3.0 mg dose). B. Plasma concentrations of α5IA and the hydroxymethyl isoxazole M1 metabolite following a single dose of 8 mg α5IA. Data are the mean (n= 12; Xue et al. 2004). (Data redrawn from Xue et al. 2004; Polsky‐Fisher et al. 2006 and de Haas et al. 2007.)
In man, α5IA was rapidly absorbed (Xue et al. 2004), with maximum plasma concentrations after an 8 mg dose (c. 4.5 ng/mL) being observed at the earliest measured time point (0.5 h). The half‐life was in the region of 2–2.5 h. The kinetics of the plasma concentrations of the hydroxymethyl isoxazole metabolite mirrored those of the parent, with maximum concentrations (c.2 ng/mL) occurring at 0.5 h postdose, with the ratio of parent to metabolite being in the region of 2:1 (Fig. 4B).
Human Pharmacodynamic Studies
TPA023
To date, no studies describing the anxiolytic efficacy of TPA023 have been described. However, the pharmacodynamic effects of TPA023 have been compared to those of lorazepam (de Haas et al. 2007). In these studies, it was shown that although lorazepam produced a significant decrease in alertness, impaired memory and increased body sway, TPA023 had no effect on any of these parameters (Table 2). Nevertheless, TPA023 clearly had a pharmacodynamic effect as it impaired saccadic eye movement peak velocity to a similar extent as lorazepam, although it did not affect either the peak latency or inaccuracy, both of which were impaired by lorazepam. Therefore, the pharmacodynamic responses of TPA023 are clearly differentiated from lorazepam and whereas it may be tempting to speculate that the similar effects of TPA023 and lorazepam on saccadic peak velocity may be a surrogate of anxiolytic efficacy (de Haas et al. 2007)—indicating that TPA023 may be a nonsedating anxiolytic in man—it remains to be determined whether TPA023 possesses anxiolytic efficacy in the clinic.
Table 2.
Comparison of pharmacodynamic effects of TPA023 and lorazepam in healthy male volunteers (de Haas et al. 2007)
| Placebo versus | |||
|---|---|---|---|
| 0.5 mg TPA023 | 1.5 mg TPA023 | 2 mg Lorazepam | |
| Saccadic eye movements | |||
| Peak velocity | 0.002 | <0.001 | <0.001 |
| Peak latency | N.S. | N.S. | <0.001 |
| Inaccuracy | N.S. | N.S. | <0.001 |
| Visual analogue scales | |||
| Alertness | N.S. | N.S. | <0.05 |
| Calmness | N.S. | 0.002 | N.S. |
| Log body sway | |||
| Eyes open | N.S. | N.S. | <0.001 |
| Eyes closed | N.S. | N.S. | <0.001 |
| Memory impairment | |||
| Performance | N.S. | N.S. | <0.05 |
| Reaction time | N.S. | N.S. | <0.05 |
N.S., not significantly different (P > 0.05)
α5IA
As mentioned above, the low solubility of the hydroxymethyl isoxazole metabolite of α5IA precluded this compound being evaluated in prolonged human dosing studies. Nevertheless, α5IA was evaluated in an experimental medicine model in which ethanol was used to produce a cognitive impairment and the ability of α5IA (administered 2 h prior to ethanol administration) to reduce the effect of ethanol on the ability to recall a list of 20 words was measured. In these studies, α5IA did not affect the breath‐alcohol levels nor did it alter the alcohol‐induced reduction in saccadic peak velocity (Nutt et al. 2007). However, α5IA did significantly improve performance on the word recall task (Fig. 5). Hence, subjects given placebo could recall on average 5.3 ± 0.9 out of the total of 20 words whereas when given α5IA, they could recall 8.4 ± 0.5 words. Furthermore, those subjects that showed the greatest alcohol‐induced impairments on placebo tended to show the greatest improvement in performance when given α5IA (Nutt et al. 2007).
Figure 5.

α5IA reduces the ethanol‐induced impairment on word recall. Twelve male volunteers (mean age 24) were given either α5IA (4 mg) or placebo 2 h prior to being given alcohol in the form of vodka at a dose of 0.8 g/kg during a 0.5‐h period. One‐hour later they were asked to memorize a list of 20 words and 0.5 h later (i.e., 4 h after placebo or α5IA administration) were asked to write down as many words from that list as they could recall. Values shown are mean ± SEM. (n= 12). ***, P < 0.001.
Summary
Nonselective benzodiazepine site agonists and inverse agonists exert pharmacological effects via modulation of GABAA receptors containing either an α1, α2, α3, or α5 subunit. In vivo the pharmacological properties of nonselective agonists include anxiolytic, sedative/hypnotic, myorelaxant and anticonvulsant properties whereas the nonselective inverse agonists are anxiogenic, increase vigilance, and are proconvulsant. The selective efficacy approach has provided the pharmacological tools to enable subtype selective modulation of GABAA receptors. Such compounds possess unique pharmacological profiles in preclinical species with, for example, the α2/α3 selective partial agonist TPA023 being a nonsedating anxiolytic in preclinical rat and primate assays whereas the α5 selective inverse agonist α5IA enhances cognitive performance in rats. Data from pharmacodynamic studies in normal volunteers suggest that, at the very least, these compounds get into the brain and exert pharmacological effects in man. The poor solubility of the hydroxymethyl isoxazole metabolite of α5IA prevented the further clinical development of this compound and the current development status of TPA023 has not been disclosed. Consequently, no efficacy data in patient populations have been described for either compound and so clear evidence that the novel pharmacological properties of these compounds translate into clinical utility is currently lacking.
Conflict of Interest
The authors have no conflict of interest.
References
- Atack JR (2003) Anxioselective compounds acting at the GABAA receptor benzodiazepine binding site. Curr Drug Targets CNS Neurol Disord 2: 213–232. [DOI] [PubMed] [Google Scholar]
- Atack JR (2005) The benzodiazepine binding site of GABAA receptors as a target for the development of novel anxiolytics. Expert Opin Investig Drugs 14: 601–618. [DOI] [PubMed] [Google Scholar]
- Atack JR, Hutson PH, Collinson N, Marshall G, Bentley G, Moyes C, Cook SM, Collins I, Wafford K, McKernan RM, et al. (2005) Anxiogenic properties of an inverse agonist selective for α3 subunit‐containing GABAA receptors. Br J Pharmacol 144: 357–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atack JR, Bayley PJ, Fletcher SR, McKernan RM, Wafford KA, Dawson GR (2006a) The proconvulsant effects of the GABAAα5 subtype‐selective compound RY‐080 may not be α5‐mediated. Eur J Pharmacol 548: 77–82. [DOI] [PubMed] [Google Scholar]
- Atack JR, Bayley PJ, Seabrook GR, Wafford KA, McKernan RM, Dawson GR (2006b) L‐655,708 enhances cognition in rats but is not proconvulsant at a dose selective for α5‐containing GABAA receptors. Neuropharmacology 51: 1023–1029. [DOI] [PubMed] [Google Scholar]
- Atack JR, Wafford KA, Tye SJ, Cook SM, Sohal S, Pike A, Sur C, Melillo D, Bristow L, Bromidge F, et al. (2006c) TPA023 [7‐(1,1‐dimethylethyl)‐6‐(2‐ethyl‐2H‐1,2,4‐triazol‐3‐ylmethoxy)‐3‐(2‐fluorophenyl)‐1,2,4‐triazolo[4,3‐b]pyridazine], an agonist selective for α2‐ and α3‐containing GABAA receptors, is a non‐sedating anxiolytic in rodents and primates. J Pharmacol Exp Therap 316: 410–422. [DOI] [PubMed] [Google Scholar]
- Ator NA (2005) Contributions of GABAA receptor subtype selectivity to abuse liability and dependence potential of pharmacological treatments for anxiety and sleep disorders. CNS Spectr 10: 31–39. [DOI] [PubMed] [Google Scholar]
- Benson JA, Löw K, Keist R, Mohler H, Rudolph U (1998) Pharmacology of recombinant γ‐aminobutyric acidA receptors rendered diazepam‐insensitive by point‐mutated α‐subunits. FEBS Lett 431: 400–404. [DOI] [PubMed] [Google Scholar]
- Carling RW, Moore KW, Street LJ, Wild D, Isted C, Leeson PD, Thomas S, O'Connor D, McKernan RM, Quirk K, et al. (2004) 3‐Phenyl‐6‐(2‐pyridyl)methyloxy‐1,2,4‐triazolo[3,4‐a]phthalazines and analogues: High‐affinity γ‐Aminobutyric acid‐A benzodiazepine receptor ligands with α2, α3, and α5‐subtype binding selectivity over α1. J Med Chem 47: 1807–1822. [DOI] [PubMed] [Google Scholar]
- Carling RW, Madin A, Guiblin A, Russell MGN, Moore KW, Mitchinson A, Sohal B, Pike A, Cook SM, Ragan CI, et al. (2005) 7‐(1,1‐Dimethylethyl)‐6‐(2‐ethyl‐2H‐1,2,4‐triazol‐3‐ylmethoxy)‐3‐(2‐fluorophenyl)‐1,2,4‐triazolo[4,3‐b]pyridazine: A functionally selective γ‐aminobutyric acidA (GABAA) α2/α3‐subtype selective agonist that exhibits potent anxiolytic activity but is not sedating in animal models. J Med Chem 48: 7089–7092. [DOI] [PubMed] [Google Scholar]
- Chambers MS, Atack JR, Broughton HB, Collinson N, Cook S, Dawson GR, Hobbs SC, Marshall G, Maubach KA, Pillai GV, et al. (2003) Identification of a novel, selective GABAAα5 receptor inverse agonist which enhances cognition. J Med Chem 46: 2227–2240. [DOI] [PubMed] [Google Scholar]
- Chambers MS, Atack JR, Carling RW, Collinson N, Cook SM, Dawson GR, Ferris P, Hobbs SC, O'Connor D, Marshall G, et al. (2004) An orally bioavailable, functionally selective inverse agonist at the benzodiazepine site of GABAAα5 receptors with cognition enhancing properties. J Med Chem 47: 5829–5832. [DOI] [PubMed] [Google Scholar]
- Cheng VY, Martin LJ, Elliott EM, Kim JH, Mount HTJ, Taverna FA, Roder JC, Macdonald JF, Bhambri A, Collinson N, et al. (2006) α5GABAA receptors mediate the amnestic but not sedative‐hypnotic effects of the general anesthetic etomidate. J Neurosci 26: 3713–3720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chouinard G (2004) Issues in the clinical use of benzodiazepines: Potency, withdrawal, and rebound. J Clin Psychiatry 65 Suppl 5:7–12. [PubMed] [Google Scholar]
- Collinson N, Kuenzi FM, Jarolimek W, Maubach KA, Cothliff R, Sur C, Smith A, Otu FM, Howell O, Atack JR, et al. (2002) Enhanced learning and memory and altered GABAergic synaptic transmission in mice lacking the α5 subunit of the GABAA receptor. J Neurosci 22: 5572–5580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crestani F, Keist R, Fritschy J‐M, Benke D, Vogt K, Prut L, Blüthmann H, Möhler H, Rudolph U (2002) Trace fear conditioning involves hippocampal α5 GABAA receptors. Proc Natl Acad Sci USA 99: 8980–8985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson GR, Maubach KA, Collinson N, Cobain M, Everitt BJ, MacLeod AM, Choudhury HI, McDonald LM, Pillai G, Rycroft W, et al. (2006) An inverse agonist selective for α5 subunit‐containing GABAA receptors enhances cognition. J Pharmacol Exp Therap 316: 1335–1345. [DOI] [PubMed] [Google Scholar]
- De Haas SL, De Visser SJ, Van Der Post JP, De Smet M, Schoemaker RC, Rijnbeek B, Cohen AF, Vega JM, Agrawal NGB, Goel TV, et al. (2007) Pharmacodynamic and pharmacokinetic effects of TPA023, a GABAAα2,3 subtype‐selective agonist, compared to lorazepam and placebo in healthy volunteers. J Psychopharmacol 21: 374–383. [DOI] [PubMed] [Google Scholar]
- Dias R, Sheppard WFA, Fradley RL, Garrett EM, Stanley JL, Tye SJ, Goodacre S, Lincoln RJ, Cook SM, Conley R, et al. (2005) Evidence for a significant role of α3‐containing GABAA receptors in mediating the anxiolytic effects of benzodiazepines. J Neurosci 25: 10682–10688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorow R, Horowski R, Paschelke G, Amin M (1983) Severe anxiety induced by FG 7142, a β‐carboline ligand for benzodiazepine receptors. Lancet 2: 98–99. [DOI] [PubMed] [Google Scholar]
- Fritschy J‐M, Möhler H (1995) GABAA receptor heterogeneity in the adult rat brain: Differential regional and cellular distribution of seven major subunits. J Comp Neurol 359: 154–194. [DOI] [PubMed] [Google Scholar]
- Ghoneim MM, Mewaldt SP (1990) Benzodiazepines and human memory: A review. Anesthesiology 72: 926–938. [PubMed] [Google Scholar]
- Haefely W, Martin JR, Schoch P (1990) Novel anxiolytics that act as partial agonists at benzodiazepine receptors. Trends Pharmacol Sci 11: 452–456. [DOI] [PubMed] [Google Scholar]
- Liu R, Hu RJ, Zhang P, Skolnick P, Cook JM (1996) Synthesis and pharmacological properties of novel 8‐substituted imidazobenzodiazepines: High‐affinity, selective probes for α5‐containing GABAA receptors. J Med Chem 39: 1928–1934. [DOI] [PubMed] [Google Scholar]
- Löw K, Crestani F, Keist R, Benke D, Brünig I, Benson JA, Fritschy J‐M, Rülicke T, Bluethmann H, Möhler H, et al. (2000) Molecular and neuronal substrate for the selective attenuation of anxiety. Science 290: 131–134. [DOI] [PubMed] [Google Scholar]
- Ma B, Polsky‐Fisher SL, Vickers S, Cui D, Rodrigues AD (2007) Cytochrome P450 3A‐dependent metabolism of a potent and selective γ‐aminobutyric acidAα2/3 receptor agonist in vitro: Involvement of cytochrome P450 3A5 displaying biphasic kinetics. Drug Metab Dispos 35: 1301–1307. [DOI] [PubMed] [Google Scholar]
- Maubach K (2003) GABAA receptor subtype selective cognition enhancers. Curr Drug Targets CNS Neurol Disord 2: 233–239. [DOI] [PubMed] [Google Scholar]
- McKernan RM, Whiting PJ (1996) Which GABAA‐receptor subtypes really occur in the brain? Trends Neurosci 19: 139–143. [DOI] [PubMed] [Google Scholar]
- McKernan RM, Rosahl TW, Reynolds DS, Sur C, Wafford KA, Atack JR, Farrar S, Myers J, Cook G, Ferris P, et al. (2000) Sedative but not anxiolytic properties of benzodiazepines are mediated by the GABAA receptor α1 subtype. Nature Neurosci 3: 587–592. [DOI] [PubMed] [Google Scholar]
- Merschman SA, Rose MJ, Pearce GES, Woolf EJ, Schaefer BH, Huber AC, Musson DG, Petty KJ, Rush DJ, Varsolona RJ, et al. (2005) Characterization of the solubility of a poorly soluble hydroxylated metabolite in human urine and its implications for potential renal toxicity. Pharmazie 60: 359–363. [PubMed] [Google Scholar]
- Minier F, Sigel E (2004) Techniques: Use of concatenated subunits for the study of ligand‐gated ion channels. Trends Pharmacol Sci 25: 499–503. [DOI] [PubMed] [Google Scholar]
- Moroz G (2004) High‐potency benzodiazepines: Recent clinical results. J Clin Psychiatry 65 Suppl 5:13–18. [PubMed] [Google Scholar]
- Nutt DJ, Besson M, Wilson SJ, Dawson GR, Lingford‐Hughes AR (2007) Blockade of alcohol's amnestic activity in humans by an α5 subtype benzodiazepine receptor inverse agonist. Neuropharmacology 53: 810–820. [DOI] [PubMed] [Google Scholar]
- Pirker S, Schwarzer C, Wieselthaler A, Sieghart W, Sperk G (2000) GABAA receptors: Immunocytochemical distribution of 13 subunits in the adult brain. Neuroscience 101: 815–850. [DOI] [PubMed] [Google Scholar]
- Polsky‐Fisher SL, Vickers S, Cui D, Subramanian R, Arison BH, Agrawal NGB, Goel TV, Vessey LK, Murphy MG, Lasseter KC, et al. (2006) Metabolism and disposition of a potent and selective GABA‐Aα2/3 receptor agonist in healthy male volunteers. Drug Metab Dispos 34: 1004–1011. [DOI] [PubMed] [Google Scholar]
- Quirk K, Blurton P, Fletcher S, Leeson P, Tang F, Mellilo D, Ragan CI, McKernan RM (1996) [3H]L‐655,708, A novel ligand selective for the benzodiazepine site of GABAA receptors which contain the α5 subunit. Neuropharmacology 35: 1331–1335. [DOI] [PubMed] [Google Scholar]
- Rowlett JK, Platt DM, Lelas S, Atack JR, Dawson GR (2005) Different GABAA receptor subtypes mediate the anxiolytic, abuse‐related, and motor effects of benzodiazepine‐like drugs in primates. Proc Natl Acad Sci USA 102: 915–920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudolph U, Möhler H (2004) Analysis of GABAA receptor function and dissection of the pharmacology of benzodiazepines and general anesthetics through mouse genetics. Annu Rev Pharmacol Toxicol 44: 475–498. [DOI] [PubMed] [Google Scholar]
- Rudolph U, Crestani F, Benke D, Brünig I, Benson JA, Fritschy J‐M, Martin JR, Bluethmann H, Möhler H (1999) Benzodiazepine actions mediated by specific γ‐aminobutyric acidA receptor subtypes. Nature 401: 796–800. [DOI] [PubMed] [Google Scholar]
- Russell MGN, Carling RW, Atack JR, Bromidge FA, Cook SM, Hunt P, Isted C, Lucas M, McKernan RM, Mitchinson A, et al. (2005) Discovery of functionally selective 7,8,9,10‐tetrahydro‐7,10‐ethano‐1,2,4‐triazolo[3,4‐a]phthalazines as GABAA receptor agonists at the α3 subunit. J Med Chem 48: 1367–1383. [DOI] [PubMed] [Google Scholar]
- Seabrook GR, Easter A, Dawson GR, Bowery BJ (1997) Modulation of long‐term potentiation in CA1 region of mouse hippocampal brain slices by GABAA receptor benzodiazepine site ligands. Neuropharmacology 36: 823–830. [DOI] [PubMed] [Google Scholar]
- Sieghart W (1995) Structure and pharmacology of γ‐aminobutyric acidA receptor subtypes. Pharmacol Rev 47: 181–234. [PubMed] [Google Scholar]
- Sieghart W (2006) Structure, pharmacology, and function of GABAA receptor subtypes. Adv Pharmacol 54: 231–263. [DOI] [PubMed] [Google Scholar]
- Simon J, Wakimoto H, Fujita N, Lalande M, Barnard EA (2004) Analysis of the set of GABAA receptor genes in the human genome. J Biol Chem 279: 41422–41435. [DOI] [PubMed] [Google Scholar]
- Sperk G, Schwarzer C, Tsunashima K, Fuchs K, Sieghart W (1997) GABAA receptor subunits in the rat hippocampus I: Immuncytochemical distribution of 13 subunits. Neuroscience 80: 987–1000. [DOI] [PubMed] [Google Scholar]
- Steele RJ, Morris RGM (1999) Delay‐dependent impairment of a matching‐to‐place task with chronic and intrahippocampal infusion of the NMDA‐antagonist D‐AP5. Hippocampus 9: 118–136. [DOI] [PubMed] [Google Scholar]
- Sternfeld F, Carling RW, Jelley RA, Ladduwahetty T, Merchant KJ, Moore KW, Reeve AJ, Street LJ, O'Connor D, Sohal B, et al. (2004) Selective, orally active γ‐aminobutyric acidAα5 receptor inverse agonists as cognition enhancers. J Med Chem 47: 2176–2179. [DOI] [PubMed] [Google Scholar]
- Street LJ, Sternfeld F, Jelley RA, Reeve AJ, Carling RW, Moore KW, McKernan RM, Sohal B, Cook S, Pike A, et al. (2004) Synthesis and biological evaluation of 3‐heterocyclyl‐7,8,9,10‐tetrahydro‐(7,10‐ethano)‐1,2,4‐triazolo[3,4‐a]phthalazines and analogues as subtype‐selective inverse agonists for the GABAAα5 benzodiazepine binding site. J Med Chem 47: 3642–3657. [DOI] [PubMed] [Google Scholar]
- Sur C, Fresu L, Howell O, McKernan RM, Atack JR (1999) autoradiographic localization of α5 subunit‐containing GABAA receptors in rat brain. Brain Res 822: 265–270. [DOI] [PubMed] [Google Scholar]
- Tarzia G, Occelli E, Toja E, Barone D, Corsico N, Gallico L, Luzzani F (1988) 6‐(Alkylamino)‐3‐aryl‐1,2,4‐triazolo [3,4‐a]phthalazines A new class of benzodiazepine receptor ligands. J Med Chem 31: 1115–1123. [DOI] [PubMed] [Google Scholar]
- Venault P, Chapouthier G (2007) From the behavioral pharmacology of beta‐carbolines to seizures, anxiety, and memory. Sci World J 7: 204–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venault P, Chapouthier G, De Carvalho LP, Simiand J, Morre M, Dodd RH, Rossier J (1986) Benzodiazepine impairs and β‐carboline enhances performance in learning and memory tasks. Nature 321: 864–866. [DOI] [PubMed] [Google Scholar]
- Vicini S, Ortinski P (2004) Genetic manipulations of GABAA receptor in mice make inhibition exciting. Pharmacol Ther 103: 109–120. [DOI] [PubMed] [Google Scholar]
- Wieland HA, Lüddens H, Seeburg PH (1992) A single histidine in GABAA receptors is essential for benzodiazepine agonist binding. J Biol Chem 267: 1426–1429. [PubMed] [Google Scholar]
- Xue L, Lin L, Hsieh JY‐K, Matuszewski BK (2004) Determination of a selective GABA‐A α5 receptor inverse agonist in human plasma by high‐performance liquid chromatography with tandem mass spectrometric detection. J Liquid Chromat Rel Tech 27: 689–704. [Google Scholar]