

Abstract
Vinpocetine (ethyl‐apovincaminate, Cavinton), a synthetic derivative of the Vinca minor alkaloid vincamine, has been used now for decades for prevention and treatment of cerebrovascular diseases predisposing to development of dementia. Both vinpocetine and its main metabolite cis‐apovincaminic acid (cAVA) exert a neuroprotective type of action. Bilateral N‐methyl‐D‐aspartate (NMDA)‐induced neurodegeneration in the entorhinal cortex of rat was used as a dementia model to confirm the neuroprotective action of these compounds in vivo. NMDA‐lesioned rats were treated 60 min before lesion and throughout 3 postoperative days with a 10 mg/kg intraperitoneal dose of vinpocetine or cAVA. Behavioral tests started after termination of drug treatment and consisted of novel object recognition, social discrimination, and spontaneous alternation in a Y‐maze, and spatial learning in the Morris water maze. At the end of behavioral testing brains were perfused with fixative and the size of the excitotoxic neuronal lesion and that of microglial activation around the lesion were assayed quantitatively on brain sections immunostained for neuron‐specific nuclear protein (NeuN) and integrin CD11b, respectively. Entorhinal NMDA lesions impaired recognition of novel objects and the new social partner, and suppressed spontaneous alternation and spatial learning performance in the Morris maze. Both vinpocetine and cAVA effectively attenuated the behavioral deficits, and significantly decreased lesion size and the region of microglia activation. Both lesion‐induced attention deficit and learning disabilities were markedly alleviated by vinpocetine and cAVA. The morphological findings corroborated the behavioral observations and indicated reduced lesion size and microglia activation especially after vinpocetine treatment which supports an in vivo neuroprotective mode of action of vinpocitine and a less potent action of cAVA.
Keywords: Vinpocetine, Dementia model, Neuroprotection, Attention and learning, Lesion size, Microglia activation
Vinpocetine (ethyl‐apovincaminate, Cavinton) a synthetic derivative of the Vinca minor alkaloid vincamine is currently prescribed for the treatment of disorders arising from cerebrovascular and cerebral neurodegenerative diseases leading ultimately to dementia in the elderly. Although the complex neuroprotective mechanism underlying the therapeutic action of vinpocetine is not yet fully clarified several reports during the last decade point to molecular targets of this drug which are associated with its effects on voltage‐dependent sodium channels, glutamate receptors, voltage‐operated calcium channels, and calcium‐ and calmodulin‐dependent cyclic guanosine monophosphate‐phosphodiesterase. [1, 2, 3, 4] Vinpocetine may also act through the peripheral benzodiazepine receptors in the brain [5, 6] which are highly expressed in microglia but have also been identified in neurons [7].
Neuroprotection in vivo by vinpocetine has been studied in a number of hypoxia‐ischemia models (reviewed by Bönöczk et al. [3]). Several mechanisms have been postulated to explain neuronal cell death in hypoxia, ischemia and chronic neurodegenerative diseases, including excessive excitotoxic neurostimulation, calcium overload, oxidative stress, programmed cell death and brain vascular deterioration [8]. However to date experimental neurodegenerative models other than hypoxia‐ischemia have not yet been applied to explore the in vivo mode of action of vinpocetine and its derivatives, but neuronal overexcitation by glutamate, formation of free radicals, chronic inflammation, or trauma may all contribute to the neurodegenerative process. The aim of the present study was to apply an in vivo glutamate‐induced overexcitation model to study the potential neuroprotective action of vinpocetine.
In humans, vinpocetine is rapidly absorbed orally and subject to an intense first pass metabolic turnover during which the larger part of the substance is hydrolyzed into its active metabolite, cis‐apovincaminic acid (cAVA) [9, 10, 11]. Therefore, the question came up whether this main metabolite plays an active role in the neuroprotective action of vinpocetine. Consequently we compared the putative neuroprotective potential of vinpocetine as well as that of cAVA.
For the current study we chose for N‐methyl‐D‐aspartate (NMDA)‐induced excitotoxic lesioning of the entorhinal cortex in rats. Applying this model a clear deficit in the Morris maze spatial learning test could be obtained as a result of hippocampal dysfunction due to the partial disruption of the massive perforant pathway connection of the entorhinal cortex to the dentate gyrus of the hippocampus [12, 13, 14]. Degeneration of the perforant path results in a shrinking of the molecular layer of the dentate gyrus [15, 16]. In essentially all cases of Alzheimer's disease the deranged connectivity between the entorhinal cortex and the hippocampus is the initial event leading to short‐term memory disturbance and attention deficit [17, 18, 19]. Therefore, in our animal model in addition to spatial learning, we also assessed attention deficits that resulted from the NMDA‐induced entorhinal lesion. Drug effects were established with a preselected optimal dose of 10 mg/kg of vinpocetine and of its metabolite cAVA applied both pre‐ and postsurgery. Behavioral analysis was followed by a postmortem neuropathology analysis in which we quantified the size of neuronal lesion in the entorhinal cortex and the accompanying microglia activation.
Experimental Procedures
Animals
Male Harlan‐Wistar rats weighing 300–400g outbred in our laboratory were group housed with three animals per cage with free access to food and water at a standard temperature of 23 ± 1°C in a light‐controlled room (light on from 07:00 a.m. to 07:00 p.m.). Behavioral tests were performed during the light phase between 9:00 a.m. and 3:00 p.m. The animal experiments were carried out in accordance with the European Community Council Directive for the care and use of laboratory animals and were approved by the Scientific Ethical Committee on Animal Experimentation, Animal Health Care Station Budapest 1135, Lehel u. 43–47 (1327/003/Fov/2005).
Drug Treatments
Vinpocetine (cis:‐ethyl‐apovincaminate) and cAVA were supplied by Gedeon Richter Company (Budapest, Hungary). The 10 mg/kg dose was selected for both compounds as optimal dose based on previous in vivo studies in our laboratories. Rats were divided into seven groups: intact (9); sham‐lesioned vehicle‐injected control (12); sham‐lesioned vinpocetine‐treated (6); sham‐lesioned cAVA‐treated (6); NMDA‐lesioned and vehicle‐treated (14); NMDA‐lesioned and vinpocetine‐treated (8); and NMDA‐lesioned and cAVA‐treated (8). One animal from the 7th group (NMDA + cAVA) was discarded because of health problems and severe loss of body weight. Drug‐treated rats received either 10 mg/kg vinpocetine or 10 mg/kg cAVA intraperitoneally 60 min before and 90 min after surgery, and two injections per day for three consecutive days (altogether 8 injections) in 1 mL/kg physiological saline solution containing 0.1% Tween 80 and ascorbic acid. Sham‐lesioned and NMDA‐lesioned controls received the vehicle solution alone.
NMDA‐lesion
Excitotoxic lesioning of the entorhinal cortex was carried out bilaterally. Rats were anesthetized with sodium pentobarbital by intraperitoneal injection and positioned in a Narishige stereotactic frame. With a Hamilton syringe of 10 μL, 30 nmol N‐methyl‐D,L‐aspartate (NMDA, Sigma, St. Louis) dissolved in phosphate‐buffered saline pH7.4 (PBS) was injected bilaterally into each of three different locations of the entorhinal cortex. The volume of each injection was 0.5μL and the infusion time was 0.1μL/min. At the end of each injection the needle was left in place for another 3min to prevent loss of the toxin in the needle track. Altogether six injections (six times 30 nmol NMDA) were applied to the entorhinal cortices. The stereotaxic coordinates measured from Bregma according to the atlas of Paxinos and Watson [20] were: craniocaudal −6.4, −6.7, −6.8; lateral 4.0, 2.4, 3.5; dorsoventral 8.2, 7.8, 7.4. The injection locations were approached at an angle of 15°. Sham‐lesioned animals received PBS solution alone. Animals were allowed to recover after surgery for 3 days during which period they were treated either with vinpocetine or with cAVA as described above before the behavioral testing was started.
Behavioral Tests
All behavioral tests were performed blindly and the individual animals from the different groups and cages were selected randomly for the testing. The experimental animals could not see the experimenter, because the test arenas were observed through an optical camera positioned 1.5 m above the arenas, which also allowed to carry out video recording. The behavioral tests started in the morning of the 5th postoperative day, that is, one and a half days after the last drug injection.
Novel Object Recognition
On postsurgery day 5 novel object recognition was tested in a habituated open field arena according to Myhrer et al. [21]. During the first session two identical objects were placed into the arena at an equal distance from the wall in an asymmetric position with respect to the center of the arena. These objects became familiar objects during the 5 min test period. After a 2 hours intersession interval spent in the home‐cage the rats were replaced into the open‐field arena for the 2nd session that lasted another 5 min. During the second session one of the familiar objects was replaced by a novel object. The time (sec) spent on visiting the objects were recorded. The total duration of visits towards both objects served as general exploratory activity, while for discriminating the novel from the familiar object the following calculation was applied:
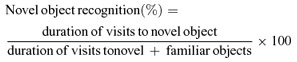 |
Fifty percent represented no discrimination or no recognition of novelty.
Social Discrimination Test
The experiment was performed 6 days after surgery according to description of Von Frijtag et al. [22] in a 70 cm rectangular arena surrounded by a 35 cm high wall. The floor was covered with sawdust, which was mixed after each test. The arena was illuminated from a height of 120 cm by a red bulb of 40W. After 2h habituation in the observation room the test animal was allowed to explore the arena for 3 min. After that a juvenile male partner rat was placed into the arena for 5 additional min (learning phase). During the inter‐trial interval, animals were replaced into their home cage for 7 min. The retrieval trial was started by replacing the test rat into the arena upon which the familiar rat was introduced together with a new juvenile male from the same cage. Social interactions (sniffing, grooming, following) towards the familiar and the novel juvenile rat, respectively, were recorded for 3 min. Data were expressed as frequency (in scores per 3 min) or time (in seconds) of social interactions towards the new partner divided by interactions towards the familiar partner. In case of attention and/or memory deficits, test rats were unable to differentiate between the familiar and the new partner, and the ratio was around 1.
Spontaneous Alternation
Attention and working memory was assessed on the seventh postoperative day in the course of spontaneous alternation in a Y‐maze described earlier [23]. The maze was made from black plastic material with sawdust on the floor. Sawdust was mixed after each test for eliminating odor prints. The arms were 50 cm long, 30 cm high, 10 cm wide and converged at a 120° angle. Each rat was placed at the center of the maze and allowed to move freely for 5 min. The number of arm entries was recorded. Alternation was defined as entering the arm different from the two previous arms entered and was expressed as percent of total entries.
Spatial Learning in a Morris Water Maze
The test procedure started at the 8th postoperative day and continued for another 4 days, that is, five daily sessions were performed. The Morris water maze test [24] was carried out in a black round water tank (diameter 153cm, height 63cm) filled to a depth of 53 cm with water of 26 ± 1°C. The tank was divided into four (imaginary) quadrants. A black hidden platform with a diameter of 8 cm was submerged in the center of one quadrant 1.5cm below the water surface. Four different starting positions were equally spaced around the perimeter of the tank. The animals had to learn to find the hidden platform orientating by different visual cues placed on the wall of the experimental chamber. The order of starting positions varied randomly between sessions, but was constant within a daily session. Four trials were given per daily session. Rats were placed into the water at a starting position facing the wall of the tank. Each trial lasted until the rats found the platform. If the platform was not found during 90 sec, the operator led the animal to it. Rats spent 30 sec on the platform at the end of each trial. Latency time to find the platform was measured during each trial. Latency to find the platform at the first trial was considered as a measure of reference memory and computed separately. Spatial learning performance was reflected by the mean escape latency calculated by averaging all four trials.
Brain Tissue Analysis
Brain Fixation and Sectioning
Brain fixation was carried out at postoperative days 13 by transcardial perfusion of heparinized PBS pH7.4 followed by 4% paraformaldehyde (Sigma‐Aldrich, Hungary) solution containing 0.05% glutaraldehyde (Sigma‐Aldrich, Hungary) in 0.1M phosphate buffer (PB, pH7.4) after adequate deep pentobarbital anesthesia. After postfixation of the brains for 48 h in the same fixative the brains were stored in 0.1M PB containing 0.1% Na‐azide until histological examinations. For histological processing the brains were cryoprotected by storage in PB containing 30% sucrose and sectioned in a Leica cryostat microtome at a thickness of 30 μm to obtain horizontal sections of the entorhinal cortex and ventral hippocampus in sham‐operated control and NMDA‐lesioned animal.
Immunocytochemistry
Identification of neurons and activated microglia. Mouse anti‐neuron‐specific nuclear protein (Neuronal Nuclei, NeuN, MAB377, Chemicon) antibody recognizes most neuronal types in the central nervous system but not glial cells. Therefore, it is an adequate tool for outlining the precise area of loss of neurons in an anatomical structure after different lesions. This staining, therefore, allows a quantitative measurement of lesion size, which reflects the area of lost neurons. The antibody against the integrin, CD11b, was selected to label activated microglia. In the control brain baseline level activity of microglia is minimal and hardly visible after immunocytochemical staining. Microglia activation becomes markedly enhanced in and around the lesion or degeneration of the nervous tissue. Mouse anti‐rat CD11b (MAB1405Z, Chemicon), as primary antibody was used in the microglia immunoassay. Both NeuN and CD11b antibodies were applied in a dilution rate of 1:1000. The biotinylated secondary horse anti‐mouse antibody and the Vectastain ABC kit were obtained from Vector Labs (CA, USA) and used in 1:500 dilution rate. The staining was completed with nickel‐enhanced diaminobenzidine (DAB, 3,3’‐diamino benzidine, Sigma) reaction in the presence of H2O2.
The extent of neuronal loss and that of microglial activation around the lesion were manually delineated and quantified with the Quantimet 600 (Leica, Cambridge, UK) image analysis system. Three brain sections were analyzed per animal and the results were averaged.
Statistical Analysis
Statistical comparisons between groups were conducted using one‐way ANOVA or two‐way ANOVA with repeated measures on one factor, where appropriate. In cases in which the overall ANOVA indicated significant effects, post hoc analysis was applied using Tukey HSD test. All statistical analyses were done applying the Statistica 8.0 program. The statistical significance was set at P < 0.05. Means and standard errors of means (SEM) are presented to demonstrate the results.
Results
Behavioral Analysis
Novel Object Recognition
NMDA‐induced excitotoxic lesioning of the entorhinal cortex markedly affected the recognition of a novel object positioned in the habituated open field (Fig. 1). One‐way ANOVA revealed a significant treatment effect: F6,55= 5.63, P < 0.001. Post hoc Tukey test showed that the NMDA‐lesioned group performed inferior to sham control (P < 0.01) and both vinpocetine and cAVA were effective in preventing the attention deficit (P < 0.05 and P < 0.01 vs. NMDA control, respectively). Performances of intact and sham‐injected groups were not different. The novel object recognition of the NMDA group was also impaired against intact and both drug‐treated sham groups (not shown in the figure to avoid overcrowded statistical presentations).
Figure 1.
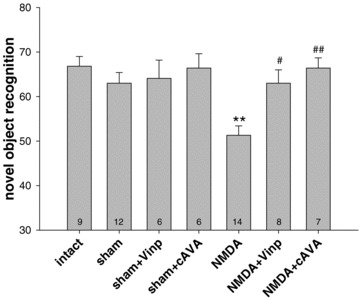
Attention ability of rats to recognize a novel object in a habituated open‐field arena tested 5 days after surgery. The time spent with exploration of the novel object in percent of time spent with exploration of both novel and familiar objects is shown. The 50% represents the chance level. Animals with bilateral entorhinal lesion (NMDA) performed worse than sham controls (**P < 0.01) and treatments with vinpocetine (Vinp) and cis‐apovincaminic acid (cAVA) in a dose of 10mg/kg i.p. prevented this deficit (#P < 0.05 and ##P < 0.01 vs. NMDA group, respectively). The numbers of animals per groups are showed inside the columns. Columns represent means ± SEM.
Social Discrimination
Similar to novel object recognition the social discrimination ability was also impaired by the lesion tested at postoperative day 6 (Fig. 2). The frequency ratio of visiting the new versus the familiar partner was different between groups: F6.55= 3.55, P < 0.01, so was the duration ratio: F6.55= 3.03, P < 0.02. Post hoc comparison of two groups showed that the NMDA lesion decreased social recognition against sham control rats as shown in figure 2 (frequency: P < 0.01, duration: P < 0.05). Furthermore the lesion decreased social interaction compared with intact animals (P < 0.01 and P < 0.05 for frequency and duration respectively); however, this statistical finding is not depicted in the figure, because the sham control group is considered as the appropriate control. Both drugs attenuated the effect of the NMDA lesion since the NMDA plus drug‐injected groups performed statistically similar as the sham controls and intact groups. With respect to the duration of interactions the NMDA+Vinp group differed significantly from the NMDA control group (P < 0.05, Fig. 2).
Figure 2.
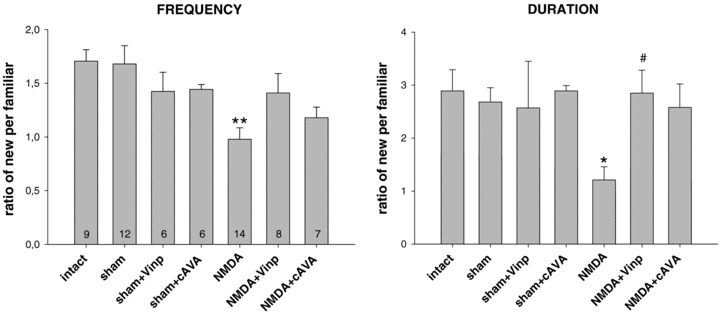
Social discrimination performance expressed as a ratio of interactions with the new vs. familiar partners tested at 6th postioperative day. Frequency and duration of social interactions after NMDA lesion against sham controls were reduced (**P < 0.01 and *P < 0.05, respectively). Drug treatments (vinpocetine and cAVA) attenuated the deficit caused by the lesion (#P < 0.05 vs. NMDA). The performances were comparable among the intact and sham‐lesioned groups with or without drug treatments. Columns represent means ± SEM.
Spontaneous Alternation
Spontaneous alternation in the Y maze was tested at postoperative day 7 (Fig. 3). The performance of different groups was evaluated by one‐way ANOVA: F6,55= 3.33, P < 0.01. Post hoc Tukey test showed that NMDA lesions significantly impaired alternation (P < 0.01) against sham controls, while both treatments (vinpocetine and cAVA) attenuated the lesion effect. Drug‐treated lesioned groups did not differ from the sham control group. Vinpocetine‐treated lesioned animals performed better compared with the NMDA‐lesioned controls (P < 0.05).
Figure 3.
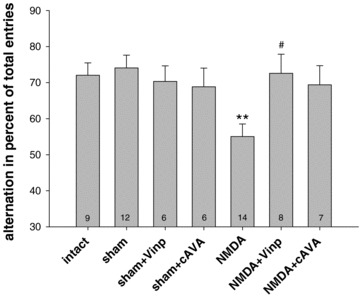
Spontaneous alternation in percent of total arm entries in the Y maze 7 days following NMDA‐induced excitotoxic lesion in the entorhinal cortex. NMDA lesion decreased alternation (**P < 0.01 vs. sham) and the two drug treatments (vinpocetine and cAVA) proved to be preventive, that is, no differences were found against the sham and intact controls. The difference between the NMDA + Vinp and NMDA groups was significant (#P < 0.05). Columns represent means ± SEM.
Spatial Memory
Acquisition of spatial memory to find the submerged platform in the Morris water maze was tested throughout five consecutive daily sessions. The spatial learning performance of the seven groups is shown in figure 4. Escape latency of the first trials of each daily session was separately evaluated and served as a measure of reference memory (RM, Fig. 4A and B). The means of all four trials per session was also calculated (Fig. 4 C and D). For a better overview of the results the seven goups were divided in two containing 4–4 groups: the sham‐operated and intact groups (two upper panels) and the NMDA‐injected groups and their sham‐operated controls (two lower panels).
Figure 4.
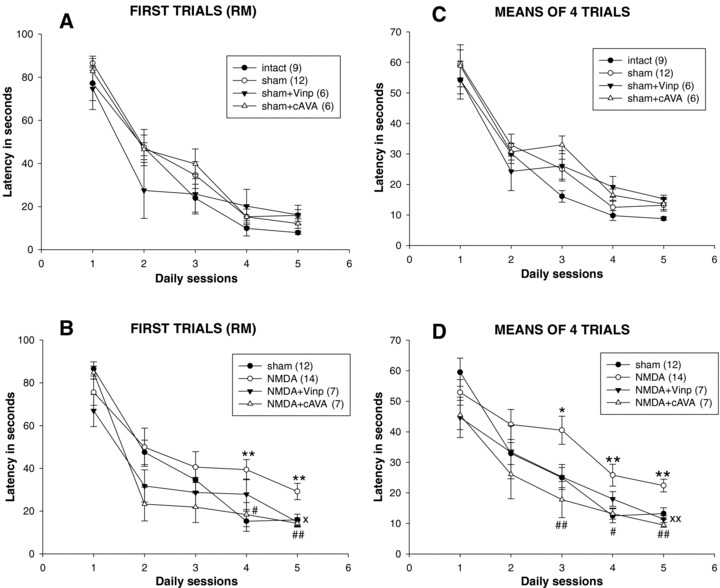
Spatial learning in the Morris water maze after entorhinal cortex lesion and drug treatments (10mg/kg i.p. vinpocetine (Vinp) or cis‐apovincaminic acid (cAVA)). Upper panels (A) and (C) show the nonlesioned control groups, while panels (B) and (D) the NMDA‐lesioned ones and their sham‐lesioned controls. Panels (A) and (B) present the escape latencies of the first trials at each sessions (reference memory, RM) and panels (C) and (D) the means of four escape latencies performed in each daily session. ANOVA analyses are in the text. Post hoc test results are the followings: *P < 0.05, **P < 0.01 between NMDA versus sham animals; ×P < 0.05, ×P < 0.01 between NMDA + Vinp and NMDA groups; #P < 0.05, ##P < 0.01 between NMDA‐cAVA and NMDA groups.
The escape latency performed at the first trial of sessions while leaving out the first session (Fig. 4A and B) showed significant effects of the group factor (F6,54= 5.33, P < 0.001) and that of session factor with repeated measures (F3,162= 16.70, P < 0.001) without a significant group–session interaction. In the statistical evaluation of the first trials (RM) the session one was not included, as mentioned above, because the very first latency to find the platform is influenced mainly by chance and clearly accidental. Evaluating means of daily trials (Fig. 4C and D), including all five sessions, resulted in a similar picture (groups: F6,54= 4.94, P < 0.001; sessions: F4,216= 101.69, P < 0.001; no groups–sessions interaction) indicating that there was a difference in the performance between groups and all groups showed learning.
In the next step of statistical analysis based on the two‐way ANOVA with repeated measures the differences between two groups was evaluated with the post hoc Tukey test. For the evaluation of the first trials (RM) in sessions 2–5, the NMDA‐lesioned group differed from sham‐controls (P < 0.05) and was also different from intact controls (P < 0.005). NMDA‐vinpocetine‐ as well as NMDA‐cAVA‐ treated groups were different from the NMDA control group (P < 0.05 and P < 0.001, respectively), but the two drug‐treated groups were not different from sham controls. No difference was found between the control groups: intact; sham‐lesioned; and sham‐lesioned treated either with vinpocetine or cAVA. Similar picture emerged in evaluating learning performance when the means of four trials were considered (panels C and D). In this case including also session 1, there was a significantly impaired performance of the NMDA group against sham control (P < 0.05) and also against normal control groups (P < 0.005). The drug treatments (vinpocetine and cAVA) prevented the deficit of lesioned animals: P < 0.05 and P < 0.001, respectively, versus NMDA control. The performance of drug‐treated groups was not different from that of sham controls. Furthermore, no difference was found between the intact and sham‐lesioned groups. Thus, either the first trials or the mean trials test evaluations clearly showed that NMDA lesions in the entorhinal cortex decreased spatial memory performance and both vinpocetine and cAVA significantly interfered with this deficit.
The daily performance could also be analyzed separately with one‐way ANOVA. There were significant differences among groups shown on figure 4(B) and (D) at session 4 and 5 testing latency of first trials (P < 0.01 both) and at sessions 3, 4, and 5 testing mean performances (P < 0.001, P < 0.02 and P < 0.001, respectively). Post hoc Tukey test revealed further group differences depicted on the figure. Both for first and mean trials, the marked distinctions between the groups was only found at the last two or three sessions.
Brain Tissue Analysis
Extent of Neuronal Lesion and Microglia Activation in the Entorhinal Cortex
The different drug treatments significantly reduced the size of entorhinal cortex lesion and that of microglia activation in the entorhinal cortex area (Fig. 5; two‐way ANOVA with treatment and lesion factors and with repeated measures on the last factor: F2,26= 7.56, P < 0.01). It was also found that the region of immunoreactive microglia was larger than that of neuronal cell death (F2,26= 11.90, P < 0.002), which means that microglia becomes activated in the areas surrounding the neuronal lesion that may be considered as a penumbra zone. There was no significant interaction between the treatment–lesion factors. One‐way ANOVA applied separately to neuronal lesion size and microglia activation area pointed to the same direction (F2,26= 7.17, P < 0.01 and F2,26= 4.71, P < 0.05 respectively), that is, treatments reduced the effect of lesion. Vinpocetine reduced the lesion size significantly assessed by post hoc Tukey test regarding both parameters (P < 0.05), while cAVA treatment effect was only marginal (P= 0.05) in reducing microglia activation if the two groups were compared. The decrements in neuronal lesion size were 23% and 16% after vinpocetine and cAVA, respectively, and those of the microglial activation area were 27% and 23%, respectively.
Figure 5.
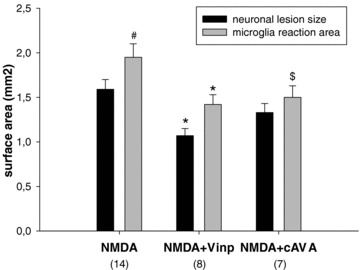
Extent of neuronal lesion and the area of microglia activation in the entorhinal cortex‐hippocampus complex following NMDA injections at the –7.6 Bregma level. Groups contained 7–14 animals. Vinpocetine (Vinp) treatment reduced the size of both parameters: *P < 0.05 versus NMDA‐vehicle‐treated control group. Treatment with cis‐apovincaminic acid (cAVA) did not diminish significantly the neuronal lesion size, while practically significantly reduced the area covered by activated microglia ($P= 0.05 vs. NMDA‐vehicle group). Area invaded by activated microglia extended the size of neuronal lesion but only in case of NMDA‐vehicle group (# P < 0.05).
Figures 6 and 7 shows representative examples of the location of neuronal lesion and the extent of immunoreactive microglia in the entorhinal cortex‐ventral hippocampus at the level of Bregma –7.6, which was the level of maximal size of the entorhinal lesion. It can be observed that the lesion (Fig. 6) and the microglia invasion (Fig. 7) cover the entire medial entorhinal cortex and the larger part of the lateral entorhinal cortex, while the subiculum has also been damaged in the NMDA‐treated animals without drug treatment (Fig. 6). Cavinton seemed to be more effective to diminish the size of the lesion compared with cAVA. Microglia activation was reduced by both compounds. Figure 8 shows with higher magnification the survived NeuN positive neurons (left panel) and the activated microglia cells (right panel). The border of lesioned and non‐lesioned tissue is rather sharp which allowed to outline the size of neuronal lesion at low magnification. The same holds true for microglia activation although some scattered groups of cells could also be found (marked by asterisk) around the border of possibly lesioned and non‐lesioned tissue. It might also be that the scattered activated microglia followed the path of degenerating axons originated from the lesion core towards the hippocampal projection area.
Figure 6.
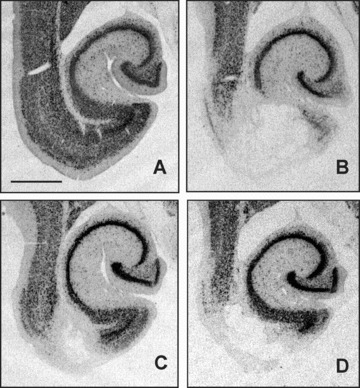
Size of neuronal lesion in the entorhinal cortex‐ventral hippocampus region outlined by the lack of neuron‐specific nuclear protein (NeuN) immunostained cells. Photomicrographs are from representative animals of sham‐lesioned vehicle‐treated (A), NMDA‐lesioned vehicle‐treated (B), NMDA‐lesioned vinpocetine‐treated (C), and NMDA‐lesioned cAVA‐treated (D) groups. Bar represents 1 mm.
Figure 7.
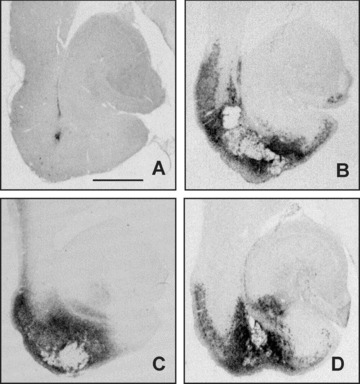
Areas of microglial activation in the entorhinal cortex‐ventral hippocampus region outlined by DC11b immunostaining. Photomicrographs are from representative animals of sham‐lesioned vehicle‐treated (A), NMDA‐lesioned vehicle‐treated (B), NMDA‐lesioned vinpocetine‐treated (C), and NMDA‐lesioned cAVA‐treated (D) groups. Bar represents 1 mm.
Figure 8.
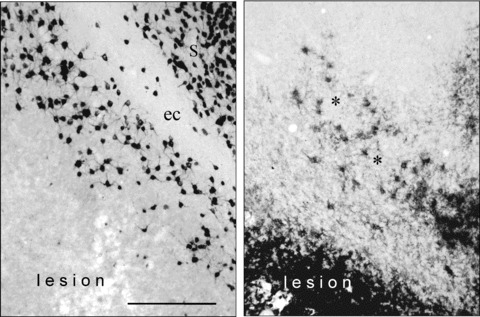
Photomicrographs with higher magnification allow to identify the stained neurons and microglia cells. The left panel shows the neuron‐specific nuclear protein (NeuN) positive neuronal staining, the right panel the CD11b positive microglia staining. It is noteworthy that the border of neuronal lesion and the microglia activation is sharp and can be clearly outlined. In some cases a scattered appearance of activated microglia could also be observed (asterisks). S, subiculum; ec, external capsule. In the external capsule no neuronal cell bodies are stained. Bar represents 100 μ.
Discussion
Both vinpocetine and its metabolite cis‐apovincaminate proved to be neuroprotective in an in vivo animal model of entorhinal cortex lesion with the glutamate analogue NMDA. The excitotoxic lesion itself markedly impaired attention in rats measured as the ability to recognize a novel object or a novel social partner. The lesioned rats also performed worse in spontaneous alternation in the Y‐maze which reflects an attention deficit associated with impaired working memory. In addition the entorhinal cortex lesioned animals showed impaired spatial learning in the Morris water maze including a deficit in reference memory performance. This entorhinal lesion model, therefore, mimics all the prominent early cognitive symptoms of Alzheimer's disease. that is, a deficient attention capability and an impaired episodic memory [17, 18].
The lesion in the entorhinal cortex was rather extensive and accompanied by a massive microglia activation covering a penumbra‐like region around the lesion core which hosts the potentially surviving neurons. Vinpocetine and cAVA in a dose of 10mg/kg given shortly before and continuously for 4 days after the excitotoxic injection of NMDA attenuated the attention deficit, and prevented the learning and memory impairment in the spontaneous alternation and Morris water maze tests. Vinpocetine significantly, and cAVA almost significantly, reduced lesion size, while both drugs reduced microglia activation.
Cis‐apovincaminic acid is the main metabolite of vinpocetine in human, thus, it became interesting to compare its action to that of vinpocetine on neurodegeneration. In the present study it turned out that cAVA has almost the same neuroprotective potency as vinpocetine itself in rats based first of all on the behavioral findings. Its effectiveness in reducing the size of the neuronal lesion and the degree of microglial activation was less pronounced compared with vinpocetine. We may conclude from the present experiments that vinpocetine and somewhat less potently cAVA rescued part of the entorhinal neurons from the effects of excitotoxicity and preserved part of the perforant pathway connection with the hippocampal formation and thereby contributed markedly to the improved behavioral performance of the lesioned rats.
Vinpocetine, based on a number of observations during the past decades, possesses a neuroprotective type of action in chronic neurodegenerative diseases that involve a pathological vascular component. In human it has been shown that vinpocetine increases cerebral blood flow and metabolism in the penumbra zone in stroke patients [25]. Animal studies have shown that vinpocetine reduces neurodegeneration initiated by decreased blood flow [26, 27, 28]. Vinpocetine in clinical studies yielded a significantly better outcome on global cognitive assessments if the memory problems were associated with poor brain circulation [29]. Both the clinical and the experimental studies focused almost exclusively on those neurodegenerative diseases that are strongly associated with vascular abnormalities. This is understandable since in about 80% of dementia cases vascular pathology is a major factor that contributes to the diseased state and promotes or helps to exacerbate pathobiochemical processes characteristic to Alzheimer's disease [30, 31, 32]. These considerations support the notion that the target structures involved in the pharmacological action of vinpocetine, indeed are the endothelial cell, the brain vasculature and the blood–brain barrier.
The immediate subsequent question in view of the results of the present study therefore is: which other neuronal and glial target structures may be involved in the neuroprotective action of vinpocetine? The neurodegeneration model used in the present study is principally based on the in vivo overexcitation of the NMDA glutamate receptor, the subsequent increased intracellular Ca2+ concentration and the onset of necrotic and apoptotic mechanisms [33, 34]. Vinpocetine and to a lesser extent also cAVA diminished glutamatergic excitotoxicity and attenuated the size of lesion in the neuronal population of entorhinal cortex. It has been documented earlier in vitro that vinpocetine is cellprotective by interfering with the NMDA receptor‐Ca2+ channel overexcitation [35], which lends further support for the NMDA receptor as target for drug development in Alzheimer's disease [34, 36, 37, 38]. According to our additional observation published recently [39] vinpocetine was found to be neuroprotective in vitro against glutamate toxicity in primary cortical neuronal culture. Since vinpocetine has a binding affinity to the peripheral‐type benzodiazepine recetors (PBRs, [6]) we investigated whether the neuroprotection by vinpocetine can be at least partially due to an effect on PBRs [39]. The answer in this study was that during in vitro glutamate excitotoxicity neuronal PBRs are involved but not likely the primary targets for the neuroprotective action exerted by vinpocetine.
Vinpocetine and its metabolite cAVA decreased microglia activation around the lesion in the entorhinal cortex in the present study. While without drug treatment the area covered by activated microglia significantly exceeded the lesioned area outlined by the loss of NeuN positive neurons in the NMDA group, treatment with either vinpocetine or cAVA reduced the extent of microglia activation relative to the extent of neuronal loss. In other words in the penumbra region 2 weeks following the NMDA lesion inflammatory processes were still active which were strongly reduced by the drug treatments.
Microglia activation is present in essentially all conditions where the vitality of the brain tissue is seriously challenged [40, 41, 42]. It is not yet fully understood whether the microglial activation is neuroprotective or neurodestructive in nature, but most likely both mechanisms may occur under specified time and stage‐dependent conditions [43, 44, 45, 46]. In this respect it is essential to obtain further information on the phase of the inflammatory process and expression level of life or death signals and their respective receptor systems [42, 47]. Interestingly, activated microglia synthetizes increased amounts of peripheral benzodizepine receptors which act as receptors for vinpocetine [7, 48] and therefore may provide an important molecular target for vinpocetine in conditions where brain tissues is challenged.
Conflict of Interest
The authors declare no conflict of interest.
Acknowledgments
This work was partly supported by the Hungarian Scientific Research Fund (OTKA) K68875 to CN.
References
- 1. Kiss B, Cai NS, Erdö SL. Vinpocetine preferentially antagonizes quisqualate/AMPA receptor responses: Evidence from release and ligand binding studies. Eur J Pharmacol 1991;209:109–112. [DOI] [PubMed] [Google Scholar]
- 2. Erdö SL, Molnár P, Lakics V, Bence J, Tömösközi Z. Vincamine and vincanol are potent blockers of voltage‐gated Na+ channels. Eur J Pharmacol 1996;314:69–73. [DOI] [PubMed] [Google Scholar]
- 3. Bönöczk P, Gulyás B, Adam‐Vizi V, et al Role of sodium channel inhibition in neuroprotection: Effect of vinpocetine. Brain Res Bull 2000;53:245–254. [DOI] [PubMed] [Google Scholar]
- 4. Vas A, Gulyás B. Eburnamine derivatives and the brain. Med Res Rev 2005;25:737–757. [DOI] [PubMed] [Google Scholar]
- 5. Braestrup C, Squires RF. Specific benzodiazepine receptors in rat brain characterized by high‐affinity (3H)diazepam binding. Proc Natl Acad Sci U S A 1977;74:3805–3809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Gulyás B, Halldin C, Vas A, et al [11C]Vinpocetine: A prospective peripheral benzodiazepine receptor ligand for primate PET studies. J Neurol Sci 2005;229–230: 219–223. [DOI] [PubMed] [Google Scholar]
- 7. Kassiou M, Meikle SR, Banati RB. Ligands for peripheral benzodiazepine binding sites in glial cells. Brain Res Brain Res Rev 2005;48:207–210. [DOI] [PubMed] [Google Scholar]
- 8. De Keyser J, Sulter G, Luiten PG. Clinical trials with neuroprotective drugs in acute ischaemic stroke: Are we doing the right thing? Trends Neurosci 1999;22:535–540. [DOI] [PubMed] [Google Scholar]
- 9. Miskolczi P, Vereczkey L, Szalay L, Göndöcs C. Effect of age on the pharmacokinetics of vinpocetine (Cavinton) and apovincaminic acid. Eur J Clin Pharmacol 1987;33:185–189. [DOI] [PubMed] [Google Scholar]
- 10. Szakács T, Veres Z, Vereczkey L. In vitro‐in vivo correlation of the pharmacokinetics of vinpocetine. Pol J Pharmacol 2001;53:623–628. [PubMed] [Google Scholar]
- 11. Chen J, Cai J, Tao W, Mei N, Cao S, Jiang X. Determination of apovincaminic acid in human plasma by high‐performance liquid chromatography using solid‐phase extraction and ultraviolet detection. J Chromatogr 2006;830:201–206. [DOI] [PubMed] [Google Scholar]
- 12. Eijkenboom M, Blokland A, Van Der Staay FJ. Modelling cognitive dysfunctions by bilateral lesions of the entorhinal cortex in the rat. Neuroscience 1999;101:27–39. [DOI] [PubMed] [Google Scholar]
- 13. Squire LR, Stark CE, Clark RE. The medial temporal lobe. Annu Rev Neurosci 2004;27:279–306. [DOI] [PubMed] [Google Scholar]
- 14. Kopniczky Z, Dochnal R, Mácsai M, Pál A, Kiss G, Mihály A, Szabó G. Alterations of behavior and spatial learning after unilateral entorhinal ablation of rats. Life Sci 2006;78:2683–2688. [DOI] [PubMed] [Google Scholar]
- 15. Nadler JV, Cotman CW, Lynch GS. Histochemical evidence of altered development of cholinergic fibers in the rat dentate gyrus following lesions. I. Time course after complete unilateral entorhinal lesion at various ages. J Comp Neurol 1977;171:561–587. [DOI] [PubMed] [Google Scholar]
- 16. Nyakas C, Luiten PGM, Balkan B, Spencer DG. Changes in septo‐hippocampal projections after lateral entorhinal or combined entorhinal‐raphé lesions as studied by anterograde tracing methods. Brain Res Bull 1988;21:285–293. [DOI] [PubMed] [Google Scholar]
- 17. Hyman BT, Van Hoesen GW, Damasio AR, Barnes CL. Alzheimer's disease: Cell‐specific pathology isolates the hippocampal formation. Science 1984;225:1168–1170. [DOI] [PubMed] [Google Scholar]
- 18. Hyman BT, Van Hoesen GW, Kromer LJ, Damasio AR. Perforant pathway chnages and the memory impairment of Alzheimer's disease. Ann Neurol 1986;20:472–481. [DOI] [PubMed] [Google Scholar]
- 19. Thal DR, Rub U, Schultz C, et al Sequence of Aβ‐protein deposition in the human medial temporal lobe. J Neuropathol Exp Neurol 2000;59:733–748. [DOI] [PubMed] [Google Scholar]
- 20. Paxinos G, Watson G. The Rat Brain in Stereotaxic Coordinates. Sydney : Academic Press, 1982. [Google Scholar]
- 21. Myhrer T, Enger S, Aas P. Cognitive side effects in rats caused by pharmacological agents used to prevent soman‐induced lethality. Eur J Pharmacol 2004;483:271–279. [DOI] [PubMed] [Google Scholar]
- 22. Von Frijtag JC, Reijmers LGJE, Van Der Harst JE, Leus IE, Van Den Bos N, Spruijt BM. Defeat followed by individual housing results in long‐term impaired reward‐ and cognition‐related behaviors in rat. Behav Brain Res 2000;117:137–146. [DOI] [PubMed] [Google Scholar]
- 23. Lalonde R. The neurobiological basis of spontaneous alternation. Neurosci Biobehav Rev 2002;26:91–104. [DOI] [PubMed] [Google Scholar]
- 24. Morris R. Developments of a water‐maze procedure for studying spatial learning in the rat. J Neurosci Methods 1984;11:47–60. [DOI] [PubMed] [Google Scholar]
- 25. Szakáll S, Boros I, Balkay L, et al Cerebral effects of a single dose of intravenous vinpocetine in chronic stroke patients: A PET study. J Neuroimaging 1998;8: 197–204. [DOI] [PubMed] [Google Scholar]
- 26. King GA. Protective effects of vinpocetine and structurally related drugs on the lethal consequences of hypoxia in mice. Arch Int Pharmacodyn Ther 1987;286:299–307. [PubMed] [Google Scholar]
- 27. Sauer D, Rischke R, Beck T, Rossberg C, Mennel HD, Bielenberg GW, Krieglstein J. Vinpocetine prevents ischemic cell damage in rat hippocampus. Life Sci 1988;43:1733–1739. [DOI] [PubMed] [Google Scholar]
- 28. Dézsi L, Kis‐Varga A, Nagy J, Komlódi Z, Kárpáti E. Neuroprotective effects of vinpocetine in vivo and in vitro. Apovincaminic acid derivatives as potential tools in ischemic stroke. Acta Pharm Hung 2002;72:84–91. [PubMed] [Google Scholar]
- 29. McDaniel MA, Maier SF, Einstein GO. Brain‐specific” nutrients: A memory cure? Nutrition 2003;19:957–975. [DOI] [PubMed] [Google Scholar]
- 30. Farkas E, Luiten PG. Cerebral microvascular pathology in aging and Alzheimer's disease. Prog Neurobiol 2001;64:575–611. [DOI] [PubMed] [Google Scholar]
- 31. Jellinger KA. Alzheimer disease and cerebrovascular pathology: An update. J Neural Transm 2002;109:813–836. [DOI] [PubMed] [Google Scholar]
- 32. Jellinger KA. The pathology of “vascular dementia”: A critical update. J Alzheimers Dis 2008;14:107–123. [DOI] [PubMed] [Google Scholar]
- 33. Meldrum B, Garthwaite J. Excitatory amino acid neurotoxicity and neurodegenerative diseases. Trends Pharmacol Sci 1990;11:379–387. [DOI] [PubMed] [Google Scholar]
- 34. Lipton SA. Pathologically‐activated therapeutics for neuroprotection: Mechanism of NMDA receptor block by memantine and S‐nitrosylation. Curr Drug Targets 2007;8:621–632. [DOI] [PubMed] [Google Scholar]
- 35. Kaneko S, Sugimura M, Inoue T, Satoh M. Effects of several cerebroprotective drugs on NMDA channel function: Evaluation using Xenopus oocytes and [3H]MK‐801 binding. Eur J Pharmacol 1991;207:119–128. [DOI] [PubMed] [Google Scholar]
- 36. Molinuevo JL, Garcia‐Gil V, Villar A. Memantine: An antiglutamatergic option for dementia. Am J Alzheimers Dis Other Demen 2004;19:10–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lipton SA. The molecular basis of memantine action in Alzheimer's disease and other neurologic disorders: Low‐affinity, uncompetitive antagonism. Curr Alzheimer Res 2005;2:155–165. [DOI] [PubMed] [Google Scholar]
- 38. Lleó A, Greenberg SM, Growdon JH. Current pharmacotherapy for Alzheimer's disease. Annu Rev Med 2006;57:513–533. [DOI] [PubMed] [Google Scholar]
- 39. Tárnok K, Kiss E, Luiten PGM, Nyakas C, Tihanyi K, Schlett K, Eisel ULM. Effects of Vinpocetine on mitochondrial function and neuroprotection in primary cortical neurons. Neurochem Int 2008;53:289–295. [DOI] [PubMed] [Google Scholar]
- 40. Streit WJ, Walter SA, Pennell NA. Reactive microgliosis. Prog Neurobiol 1999;57:563–581. [DOI] [PubMed] [Google Scholar]
- 41. Kreutzberg GW. Microglia: A sensor for pathological events in the CNS. Trends Neurosci 1996;19:312–318. [DOI] [PubMed] [Google Scholar]
- 42. Hanisch UK, Kettenmann H. Microglia: Active sensor and versatile effector cells in the normal and pathologic brain. Nat Neurosci 2007;10:1387–1394. [DOI] [PubMed] [Google Scholar]
- 43. Eisel UL. Cytokines in degenerative brain diseases: Lessons from transgenic animals. Curr Top Microbiol Immunol 2002;265:49–62. [DOI] [PubMed] [Google Scholar]
- 44. Van Rossum D, Hanisch UK. Microglia. Metab Brain Dis 2004;19:393–411. [DOI] [PubMed] [Google Scholar]
- 45. Streit WJ. Microglia and neuroprotection: Implications for Alzheimer's disease. Brain Res Brain Res Rev 2005;48:234–239. [DOI] [PubMed] [Google Scholar]
- 46. Sriram K, Matheson JM, Benkovic SA, Miller DB, Luster MI, O’Callaghan JP. Deficiency of TNF receptors suppresses microglial activation and alters the susceptibility of brain regions to MPTP‐induced neurotoxicity: Role of TNF‐alpha. FASEB J 2006;20:670–682. [DOI] [PubMed] [Google Scholar]
- 47. Eisel ULM, Biber K, Luiten PGM. Life and death of nerve cells: Therapeutic cytokine signaling pathways. Curr Signal Transd Ther 2006;1:133–146. [Google Scholar]
- 48. Stephenson DT, Schober DA, Smalstig EB, Mincy RE, Gehlert DR, Clemens JA. Peripheral benzodiazepine receptors are colocalized with activated microglia following transient global forebrain ischemia in the rat. J Neurosci 1995;15:5263–5274. [DOI] [PMC free article] [PubMed] [Google Scholar]