

Abstract
Developing effective treatments for chronic neurodegenerative disorders such as amyotrophic lateral sclerosis (ALS) has proven extremely difficult. ALS is universally fatal, characterized by progressive weakness due to the degeneration of upper and lower motor neurons, and leads eventually to respiratory failure which is the usual cause of death. Only a single treatment has been approved, the modestly effective nonspecific neuroprotectant Rilutek® (riluzole; 2‐amino‐6‐(trifluoromethoxy)benzothiazole). KNS‐760704 [(6R)‐4,5,6,7‐tetrahydro‐N6‐propyl‐2,6‐benzothiazole‐diamine dihydrochloride, RPPX], a synthetic amino‐benzothiazole with demonstrated activity in maintaining mitochondrial function, is being developed as a treatment for ALS. It has proven to be effective in multiple in vitro and in vivo assays of neuroprotection, including the G93A‐SOD1 mutant mouse model; however, its specific mechanism of action remains unknown. The potential of KNS‐760604 as a treatment for ALS was first suggested by studies showing that its optical enantiomer, Mirapex®[(6S)‐4,5,6,7‐tetrahydro‐N6‐propyl‐2,6‐benzothiazole‐diamine; pramipexole dihydrochloride; PPX], a high‐affinity agonist at dopamine D2, D3, and D4 receptors, exhibits important neuroprotective properties independent of its dopamine receptor agonism. In cell‐based assays, both RPPX and PPX reduce the production of reactive oxygen species (ROS), attenuate the activation of apoptotic pathways, and increase cell survival in response to a variety of neurotoxins. However, PPX has limited utility as a clinical neuroprotective agent because the drug concentrations required for neuroprotection would likely produce unacceptable dopaminergic side effects. RPPX, on the other hand, while possessing the same neuroprotective potential as PPX, is a much lower‐affinity dopamine receptor agonist and may therefore be more useful in the treatment of ALS. This review will examine the data supporting the hypothesis that the RPPX may have therapeutic potential for the treatment of neurodegenerative disorders including ALS. In addition, we will briefly review recent preclinical data in support of RPPX, and discuss the current status of its clinical development.
Keywords: Benzothiazole, Enantiomer, KNS‐760704, Mirapex®, Mitochondria, Neuroprotection, Pramipexole, (R)‐(+)‐pramipexole, Rilutek®
Introduction
Amyotrophic Lateral Sclerosis
Amyotrophic lateral sclerosis (ALS; Lou Gehrig's disease, Charcot's sclerosis) is a rapidly progressing neurodegenerative disorder characterized by the specific, targeted death of motor neurons, resulting in eventual paralysis and death. As the disease progresses, the patient loses upper and lower motor neuron function, accompanied by spasticity, muscle degeneration, and paralysis, including the loss of respiratory function that is the usual cause of death [1, 2, 3]. The disease does spare some motor neurons, preserving function to some degree in those muscles controlling eye movement and bladder control, although there is some debate about whether selective motor neuron sparing occurs [4]. Clinically significant dementia is uncommon, but there is increasing recognition that mild cognitive impairment frequently accompanies the disease and the recently identified TDP‐43 proteinopathy suggests a potential pathogenetic link between ALS and frontotemporal lobar degeneration [5, 6, 7]. While rare, ALS is one of the most common of the chronic neurodegenerative disorders. There are approximately 30,000 patients in the United States, with about 5000 new patients diagnosed each year. It affects adults, predominantly in the 5th to 7th decades of life, with a small preponderance in males, and the median survival time is 3–5 years from symptom onset and approximately 1.5 years from definitive diagnosis [3, 8, 9, 10].
The causes of ALS are largely unknown, with ∼90% of patients having no known hereditary disease etiology (sporadic ALS, sALS). The remaining patients demonstrate evidence of genetic linkage (familial ALS, fALS); currently four major chromosomal loci, termed ALS1, ALS3, ALS6, and ALS7, have been associated with typical disease presentations [1, 11, 12, 13, 14]. Additional loci have been associated with atypical forms of ALS, including slow‐progressing juvenile forms and ALS with concurrent dementia. ALS1 is the most frequently occurring of the known loci, accounting for about 20% of fALS patients, and reflects mutations in the gene responsible for the enzyme copper/zinc (Cu/Zn) superoxide dismutase‐1 (SOD1) [15, 16]. The other major loci associated with typical ALS code for genes with unknown function, and currently provide little in the way of insights for drug development. Recently, several studies have implicated TAR DNA binding protein 43 (TDP‐43) in both frontotemporal lobar degeneration with ubiquitinated inclusions and ALS, including sALS [6, 7, 17, 18, 19, 20]. In one of these studies it was observed that the TDP‐43 inclusions were found in sALS and in some cases of SOD1‐negative fALS, but were absent in ALS1 [17]. These results, while still preliminary (e.g., there is not yet an animal model demonstrating that mutated TDP‐43 results in ALS‐like disease), may suggest a promising future disease target.
SOD1, on the other hand, has been definitively linked to one form of fALS. SOD1 is a ubiquitous enzyme, usually found in the cytosol, that catalyzes the conversion (dismutation) of superoxide (O2 −) to water and hydrogen peroxide and is therefore a major antioxidant mechanism [21]. However, the mutation of this gene in ALS1 apparently contributes to the disease in a complex way not predicated on the loss of SOD function, but rather primarily from emergent toxicity, possibly associated with protein aggregates of the mutant enzyme [22, 23, 24, 25, 26, 27, 28, 29, 30]. These discoveries have yielded transgenic mouse models of ALS1‐linked fALS [31, 32, 33] that are currently employed in the search for new therapeutic agents in ALS; however, their utility may be limited because the mutations in this gene are responsible for only ∼2% of ALS patients. Should a potential drug be specific for some aspect of mutant SOD1 toxicity, as opposed to a final common pathway of motor neuron death common to all forms of ALS, including sALS, its utility would (by definition) be limited to that small percentage of the patient population.
Despite these limitations, the identification and characterization of the ALS1 SOD1 mutations responsible for some cases of ALS holds promise for future drug development in the disease, and may provide some insights into more general mechanisms of ALS disease etiology. In general, however, the current picture with regard to disease mechanisms remains elusive and speculative [1, 34, 35, 36]. Putative causal mechanisms must account for the pathognomonic features of ALS, including its targeting of motor neurons, its characteristic progression, and its largely sporadic etiology. One current focus in the search for common mechanisms of motor neuron death across ALS (and other forms of neurodegeneration) is the mitochondrion, the organelle responsible for supplying cellular energy [37, 38, 39, 40, 41, 42] and whose dysfunction may contribute significantly to the pathology underlying the major chronic neurodegenerative disorders.
Mitochondrial Dysfunction and Neurodegeneration
Mitochondria are membrane‐bound organelles present in eukaryotic cells, and are the major source of cellular energy. They consist of an outer mitochondrial membrane (OMM), an inner mitochondrial membrane (IMM), an intermembrane space, and a mitochondrial matrix bounded by the extensively folded IMM, which forms the characteristic mitochondrial “cristae” easily observed in electron micrographs [43, 44, 45]. As the site of oxidative phosphorylation, they funnel electrons harvested from catabolism and transferred to molecular acceptors (nicotinamide nucleotides, NADH, or flavin nucleotides, FADH2) to oxygen via an electron transport chain, eventually resulting in the reduction of molecular oxygen to water. The electron transport chain is located on the IMM and consists of four complexes (complex I–IV) responsible for the redox reactions accomplishing various steps in electron transfer [42, 43, 44, 46, 47, 48, 49]. The final component, complex IV, is a cytochrome c oxidase, which transfers electrons from cytochrome c to oxygen, and simultaneously transfers protons across the IMM. The transport of protons from the matrix to the intermembrane space creates a potential (electrical) difference and a pH gradient across the IMM. This pH and electrical gradient creates an electronegative environment in the matrix (proton motive force). The IMM is highly impermeable to protons, and they are transferred into the matrix by the action of ATP synthase, thereby coupling the production of cellular energy (ATP) to cellular catabolism. To accomplish these functions, mitochondria possess a small, maternally‐inherited genome that encodes 13 protein components of the respiratory chain, and additional tRNAs and rRNAs involved in intramitochondrial protein synthesis [50, 51]. The protein constituents of mitochondria are supplemented significantly by the specific, targeted transfer of proteins from elsewhere in the cell [52].
Due to their critical function in cell energy production, mitochondria have been implicated in a large number of diseases, including neurodegenerative disorders [37, 38, 39, 53, 54, 55, 56]. Of particular interest are the pathological production of highly reactive and toxic oxygen species, and the induction of apoptotic processes [47]. At several points in the electron transport chain, small amounts of superoxide and peroxide are produced, which can result in the production of the reactive oxygen species (ROS) hydrogen peroxide and hydroxyl radical [57, 58]. While highly damaging if left unchecked, ROS are normally eliminated by SOD and other endogenous antioxidants. Breakdown of normal mechanisms of ROS removal have been hypothesized to result in a number of diseases, including acute and chronic neurodegenerative disorders [39, 40, 59, 60].
This brief overview of mitochondrial function suggests several potential pathological mechanisms of disease. Any disruption of electron transport or its sequelae, such as reduced ATP production, will result in cellular compromise, particularly during oxidative stress. An overproduction of ROS, or a breakdown in mechanisms for their scavenging, can result in damage to cellular components. Another major potential pathway of mitochondrial disease is the activation of cell death pathways (apoptosis) via a breakdown in the permeability of the mitochondrial membrane and the release of normally impermeable or tightly regulated intramitochondrial components. This latter mechanism is generally referred to as mitochondrial permeability transition (mPT). This process is voltage‐dependent, can be elicited by oxidative stress and calcium (Ca2+) entry into the matrix, and appears to depend on the activation of an incompletely characterized protein complex involving principally the IMM, but eventually affecting the OMM as well [61, 62]. This protein complex, known as the mPT pore, permits indiscriminant proton entry into the matrix, ultimately resulting in swelling of the IMM, disruption of the OMM, and a relatively nonspecific increase in permeability that prominently includes diffusion of cytochrome c into the cytosol [61, 63, 64]. Cytosolic cytochrome c activates the caspase system leading to apoptotic cell death [42, 47, 48, 59, 64, 65]. Opening of this extremely high‐conductance mPT “pore” can be recorded using patch clamp techniques [66, 67, 68], and can be inhibited by pharmacological agents such as cyclosporin A and related compounds [67, 69]. It is currently unclear if mPT or the mPT pore has a normal, nonpathological function, but it has become an important target in the search for treatments of mitochondrial dysfunction, including the characterization of the mitochondrial effects of the (6S)‐ and (6R)‐4,5,6,7‐tetrahydro‐N6‐propyl‐2,6‐benzothiazolediamine enantiomers, Mirapex® and KNS‐760704 (see below).
Neurons are highly metabolically demanding cells, and the number, structure, and localization of mitochondria in neurons reflect these high energy requirements. In addition to “normal” cellular processes, neurons and other electrically excitable cells utilize the tightly regulated and electronegative transmembrane potential to set the stage for characteristic events (such as action potentials and neurotransmitter mobilization, release, and reception) that allow for intercellular communication. While the opening and closing of voltage‐gated ion channels underlying phenomena such as the action potential do not require energy per se, the restoration and maintenance of the ionic balances that contribute to the neuronal membrane potential and the high levels of synthesis of neurotransmitters represent major and to some degree unique neuronal energy requirements. The energy requirements of neurons are therefore high, but also quite variable, and depend both on cell type and functional requirements at any given moment. These energy requirements underlie the impressive sensitivity of neurons to acute hypoxia [70], and make them vulnerable to trauma and the acute degenerative processes following strokes, underscoring the role of mitochondria in acute neurodegeneration [71, 72]. Mitochondria were once considered highly efficient organelles unlikely to be involved in the etiology of common chronic diseases. Increasingly, however, it has been recognized that these organelles are complicated, fragile, and susceptible to many known and unknown functional disruptions. This has been particularly true in the study of neurodegenerative disorders. In addition to their response to acute hypoxia, mitochondrial dysfunction has now been suggested to underlie or contribute to essentially all of the major neurodegenerative disorders, including chronic illnesses such as Alzheimer's disease, Parkinson's disease (PD), ALS, and Huntington's disease [37, 39, 40, 54, 55].
In the specific case of ALS, several lines of evidence suggest a common mitochondrial mechanism, although as yet no single defect is proven to initiate the disease, except in the case of SOD1 fALS. Even in this case, as discussed above, it is apparently not the loss of SOD1 function that leads to the disease, but rather a gain in toxic function. Recent evidence has shown that the mutant dismutase‐negative SOD1 has a lower repulsive charge, and can form insoluble disulfide‐bond linked aggregates that correlate with the onset of a disease‐like phenotype in transgenic mice [73, 74, 75, 76]. These aggregated proteins are located at least in part in the mitochondria, while the usual localization of SOD1 is predominately cytosolic. The aggregated protein can apparently recruit wild‐type SOD1 into the aggregates in double‐transgenic (mutant SOD‐1 and wt‐ SOD‐1) mice, associating with the IMM and causing damage to mitochondrial cristae [76]. Protein aggregation in mitochondria can lead to swelling and other mitochondrial malformations, conceivably leading to opening of the mPT pore and cell death, although mutant SOD1 may also have other toxic effects, such as peroxynitrite production [77]. Some of these findings parallel some of the earliest observations concerning disease mechanisms in ALS, which arose from ultrastructural studies of mitochondria in muscle and spinal neurons in ALS patients. For example, neuronal mitochondria in these patients showed swelling and vacuolization consistent with mitochondrial injury and similar to the mitochondrial damage observed in transgenic SOD1 mutant mouse motorneurons [78, 79]. While there is evidence that certain SOD1 mutants may damage mitochondria, causing or contributing to ALS symptoms, the relationship between mitochondria and sporadic ALS (sALS) is less extensive. There have been many reports of abnormalities in mitochondria from sALS patients, including alterations in the processing and or levels of mitochondrial electron transport chain constituents [80, 81]. Other abnormalities observed in sporadic ALS patients have included mitochondrial DNA deletions and reduced ATP levels [82]. Data implicating mitochondrial mechanisms in ALS‐induced neuronal degeneration suggest a number of possible targets for the discovery of new and effective treatments for ALS.
Benzothiazoles and Neuroprotection
Benzothiazoles are structurally uncomplicated molecules with a broad range of biologic activity. Simple synthetic substitutions to the benzothiazole nucleus (Fig. 1A)have yielded compounds possessing diverse therapeutic potential as anti‐infective, anti‐inflammatory, and antitumor agents [83, 84, 85]. In the central nervous system, benzothiazoles have demonstrated neuroprotective properties in both acute and chronic models of neurodegeneration [86, 87]. They have also shown utility as in vivo labels of brain amyloid in patients with Alzheimer's disease and have been shown to prevent protein aggregation of huntingtin in models of Huntington's disease [88, 89]. Currently, the only medicine approved for use in ALS is a benzothiazole, Rilutek® (riluzole; 2‐amino‐6‐trifluoromethoxy‐benzothiazole; Fig 1B.). This compound is a low‐potency and nonspecific modulator of many pharmaceutical targets, including excitatory amino acid receptors, ion channels (sodium, calcium, and potassium), and protein aggregates [90, 91, 92, 93, 94, 95, 96, 97, 98, 99], and its mechanism‐of‐action clearly remains speculative. Nevertheless, this compound is the only clinically‐effective drug developed to date for the treatment of ALS, producing a small, but significant effect increase in survival without a significant improvement in measures of motor function. A recent review of clinical trials with Rilutek® demonstrated that the increase in mean survival time for ALS patients varied from 3 months for a pooled patient sample from trials where patients received 100 mg of the drug, to 1.7 months for a pooled sample of patients receiving various dose levels [100]. Considering the severity of the disease and the modest efficacy of Rilutek®, it is clear that ALS remains a pressing unmet medical need. The remainder of this review will focus on a drug currently entering clinical trials for the treatment of ALS, KNS‐760704.
Figure 1.
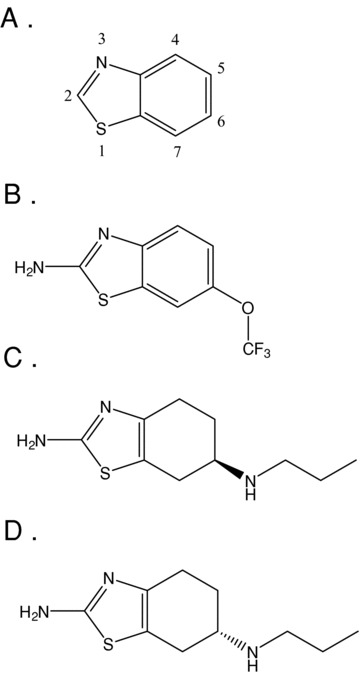
Structures of the benzothiazole core (A), Rilutek® (B), RPPX (C), and Mirapex® (D).
KNS‐760704 and Mirapex®: Evidence for Neuroprotection Independent of Dopamine Receptor Interaction
KNS‐760704 (Fig. 1C) is an amino‐benzothiazole that, like Rilutek®, has been demonstrated to be neuroprotective in multiple in vitro and in vivo assays but whose specific mechanism of action remains unknown. Its potential as a treatment for ALS was first suggested by studies of its optical enantiomer, Mirapex® (pramipexole dihydrochloride; PPX; (6S)‐4,5,6,7‐tetrahydro‐N6‐propyl‐2,6‐benzothiazole‐diamine; Fig. 1D) in a wide range of neuroprotective assays. PPX is a nonergot dopamine receptor agonist discovered in the 1980s as a synthetic analogue of the dopamine “autoreceptor” agonist (R)‐(–)‐apomorphine [101]. It was subsequently characterized as a high‐affinity agonist at dopamine D2, D3, and D4 receptors [102, 103] and approved in 1997 for the treatment of PD and for restless legs syndrome (RLS) in 2006.
Like ALS, PD is a neurodegenerative disorder that has been linked to mitochondrial dysfunction [104, 105]. Defects in the electron transport chain and associated increases in ROS contribute to the neurodegeneration characteristic of cells in the substantia nigra (SN) [106]. Hall et al. [107] evaluated the neuroprotective potential of PPX in attenuating the postischemic degeneration of dopaminergic neurons in the SN using a gerbil bilateral carotid occlusion model of brain ischemia. When dosed twice daily for 28 days, the compound reduced cell loss in the SN, but not the hippocampus, relative to control. Although PPX reduced dopamine turnover in SN cells, the reduction was insufficient to account for its observed neuroprotective effects, suggesting that a more likely mechanism of PPX neuroprotection might be through a direct antioxidant action.
In this light, further studies of PPX focused on establishing the compound's potential as a neuroprotectant in nondopaminergic neurons, in determining the degree to which dopamine receptor agonism contributed to its ability to protect neurons, and in delineating the interaction of the molecule with mitochondrial mechanisms thought to be related to neuronal degeneration. Sethy et al. [108] found that in rats treated with the nicotinamide antagonist neurotoxin 3‐acetylpyridine (3‐AP), PPX administration for up to 3 h post‐3‐AP treatment significantly reduced toxin‐related reductions in ATP levels in cerebellum, protected neurons in the inferior olivary nucleus, and improved motor coordination measures. PPX also reduced the accumulation of cytochrome c (and α‐synuclein, a presynaptic protein found in aggregated form in PD) in response to administration of the neurotoxin methylpyridinium ion (MPP+) in SH‐SY5Y cells [109], and has been shown to increase the amount of antiapoptotic protein Bcl‐2 in the dendritic processes of hippocampal and cortical neurons following multi‐day administration to rats [109, 110, 111]. Bcl‐2 has been shown to inhibit the release of cyctochrome c under a number of proapoptotic conditions, so these results were further evidence of a neuroprotective potential of the compound.
Cassarino et al. [112] examined the production of ROS in cultured SH‐SY5Y neuroblastoma cells in vitro and in rat striatum in vivo (via microdialysis) in response to treatment with MPP+. In these systems, PPX significantly reduced the concentration of oxygen radicals, albeit nonpotently. Most interesting, they also used an indirect measure of mitochondrial swelling to estimate the effects of PPX on mPT. In these experiments, using mitochondria from liver cells, PPX was able to reduce mPT induced by calcium and phosphate in a concentration‐dependent manner. Cyclosporine A, a known blocker of mPT and the mPT pore, was fully effective at a concentration of 25 μM, as was PPX at a concentration of 100 μM. In later experiments, Sayeed et al. [68] looked directly at the interaction of PPX with the mPT pore using patch clamp recording in mitoplasts prepared from liver cells. The compound blocked opening of the pore in a concentration‐dependent manner with an apparent IC50 of 500 nM (compared with an IC50 of 26 nM for cyclosporine A).
Subsequent work with PPX examined the contribution of dopamine agonism to observed effects on mitochondria and neuroprotection. Several lines of evidence have demonstrated that PPX exerts its effects, at least in part, by mechanisms that are independent of its dopamine receptor affinity. For example, in pheochromocytoma (PC12) cells, pretreatment with PPX (1–100 μM) resulted in protection against H2O2‐induced cell death [113], an effect that was not blocked by several dopamine receptor antagonists. In a similar study, PPX (5–20 μM) was shown to protect cultured dopaminergic mesencephalic‐derived (MES) 23.5 cells from dopamine, 6‐OH‐dopamine, and H2O2‐induced cytotoxicity [114], and these effects were not blocked by the dopamine D2 antagonist raclopride or the D3 antagonist U99194A. Ramirez et al. [115] reported that PPX was neuroprotective in 1‐methyl‐4‐phenyl‐1,2,3,6‐tetrahydropyridine (MPTP) treated male C57BL/6 mice in which the D3 receptor gene was deleted (D3–/– mice) although the dopamine sparing effect in the knockout mice was not as robust as in mice expressing the wildtype gene (D3+/+ mice). Similarly, the significant neuroprotective effects of PPX in this model were attenuated but not eliminated by the coadministration of the D3 receptor antagonist A‐437203.
KNS‐760704: Exploiting the Neuroprotective Potential of Pramipexole
KNS‐760704 is a >99.95% chirally pure formulation of (6R)‐4,5,6,7‐tetrahydro‐N6‐propyl‐2,6‐benzothiazolediamine dihydrochloride and, as such, is devoid of any of the dopamine agonist effects that might be attributed to trace contamination by the S(–) enantiomer, PPX. RPPX, which refers generally to the R(+)‐enantiomer of pramipexole dihydrochloride without regard to a specific formulation, had been anecdotally reported to have modestly lower affinity for D2 and D3 receptors. Our recent work demonstrates that KNS‐760704 is in fact a very low‐affinity agonist (as much as 1000‐fold lower affinity) at these receptors (Gribkoff, unpublished data). While PPX is a potent and effective treatment for PD, its utility as a clinical neuroprotective agent is limited because the drug concentrations required for neuroprotection would likely produce unacceptable dopaminergic side effects such as hypotension and seizures. KNS‐760704, with its low dopamine receptor affinity, thus has a unique and potentially more useful profile than PPX for the treatment of neurodegenerative diseases such as ALS. This potential, however, is predicated on its having neuroprotective properties equipotent with PPX.
Given the magnitude of stereo‐specific receptor affinity difference between the enantiomers, a number of studies used both PPX and RPPX to test whether dopamine receptor affinity is a critical requirement for pramipexole neuroprotection and to evaluate the relative neuroprotective potency of the enatiomers. Abramova et al. [116] examined the ability of PPX and RPPX to modulate the activation of caspases and the retention of calcein in response to MPP+ application to SH‐SY5Y cells. Both enantiomers were equipotent and effective at blocking these components of the mitochondrial‐induced cell death pathways, and in the case of calcein retention, appeared to be maximally effective at concentrations below 1 μM for both PPX and RPPX. Gu et al. [117] showed in SHSY‐5Y cells exposed to 5 mM MPP+ that PPX and RPPX in concentrations from 1 to 100 μM produced similar, dose‐dependent reductions in cytochrome c release and preserved mitochondrial membrane potential at 10 μM. Using nondopaminergic JK cells, they also showed that both PPX and RPPX (10 μM) significantly increased cell survival following exposure to 5mM MPP+.
In a recent study, Danzeisen et al. [118] examined the uptake of radiolabeled PPX ([3H]PPX) into neurons and neuronal mitochondria, the antioxidant properties of both PPX and RPPX, the effects of the compounds on glutamate‐induced cell death, and the effects of PPX and RPPX on survival time in SOD1 (G93A) mutant mice. Uptake of [3H]PPX (and presumably RPPX, although this was not measured) into both neurons and neuronal mitochondria was confirmed, and intramitochondrial uptake was shown to be energy‐dependent and reduced by a reduction in mitochondrial membrane potential. Both enantiomers were found to have equipotent (although not potent) antioxidant potential against H2O2 and superoxide, and somewhat more potent and equal effects on the release of nitric oxide in response to the nitric oxide donor (Z)‐1‐[2‐(2‐aminoethyl)‐N‐(2‐ammonioethyl)amino] diazen‐1‐ium‐1,2‐diolate (DETA). They were also protective against the cytotoxic effects of glutamate in cultured H‐22 hippocampal neuroblastoma cells. These cells are killed following application of glutamate by the generation of ROS, rather than by a receptor‐mediated excitotoxic cascade, and both enantiomers were very effective, although not potent, neuroprotectants with EC50s in the range of 370 μM for PPX to 190 μM for RPPX. In control mice, both PPX and RPPX were shown to enter brain with a brain‐to‐plasma ratio in excess of 6. Following oral dosing at 100 mg/kg commencing at 45 days of age in G93A‐SOD1 mutant mice, RPPX was protective, resulting in a modest but significant 7‐day increase in survival time. PPX was dosed at 3 mg/kg in the G93A‐SOD1 mutant mice, presumably because of intolerable dopaminergic side effects at higher doses. Even at this much lower dose, PPX administration was associated with a significant increase in wheel‐running behavior attributed to dopaminergic activation. No significant difference was observed between control (vehicle) group survival and that of the PPX group at this low dose.
Testing the Hypothesis that RPPX may Provide Benefit in ALS
This paper is not intended to comprehensively review all preclinical data associated with PPX and RPPX, but rather to present the logic for initiating the clinical development of RPPX as a treatment for ALS. The data examined strongly suggest that PPX is neuroprotective, and may be accomplishing this via one or more mitochondrial mechanisms. They also indicate that these effects do not depend on dopamine receptor interaction, and that the low‐affinity enantiomer RPPX exerts equipotent cytoprotection in every modality measured to date. Given this, and the dose‐limiting, dopamine related side‐effects associated with Mirapex®, it follows that RPPX would be the more useful member of this enantiomer pair for clinical tests of effectiveness in ALS or any neurodegenerative disorder.
In support of future clinical trials, we have examined the physicochemical, pharmacokinetic, and toxicokinetic profiles of RPPX in preclinical studies. KNS‐760704, the formulation of RPPX currently used in our clinical studies (see below), is a white crystalline solid with a molecular weight of 211.3 as the free base (formula weight of 302.3 as the dihydrochloride monohydrate actually used in the studies). It has chemical purity ≥99.99% and enantiomeric purity ≥99.95%. It has a melting point between 285 and 287°C, and is highly water soluble (>600 mg/mL). It is highly stable in solution in water and physiological buffer solutions, and is nonhygroscopic.
KNS‐760704 did not significantly affect any of the major cytochrome P450 isoenzymes and was only moderately bound to human plasma proteins (40.3%). The compound enters the CNS efficiently, with brain‐to‐plasma ratios of about 5–15, depending on species and dose. KNS‐760704 was negative for any mutagenic or genotoxic signal in a full battery of in vitro and in vivo assessments. The compound did not significantly affect the cardiac delayed rectifier (hERG) potassium current at or near concentrations of clinical relevance (IC50∼ 100 μM for hERG inhibition in vitro) and in a good laboratory practices (GLP) study of cardiovascular safety in Gottingen minipigs, there were no adverse effects observed at the highest dose tested (75 mg/kg).
Acute and chronic toxicology studies have been completed in two species, rat and minipigs, and the results from these studies strongly support the ongoing development of KNS‐760604 in ALS. The compound is rapidly and essentially completely absorbed when dosed orally and its half‐life was in the range of 3–8 h across species. Both species achieved greater‐than or equal‐to dose‐proportional peak plasma KNS‐760704 levels and exposures in single and repeat‐dose studies. The no‐observed‐adverse‐effect‐level (NOAEL) in rats of 100 mg/kg for males and ≥300 mg/kg in females after 6 months of dosing provides multiples of exposure of about 7 and 25, respectively, over the highest dose being evaluated in clinical studies (300 mg). In minipigs, the NOAEL of 50 mg/kg after 9 months of dosing provides exposure multiples of about 10 to 15 over the 300 mg dose in human subjects.
The clinical development of RPPX is being advanced on two fronts. RPPX has been evaluated in ALS subjects under research IND 60,948 held by James Bennett at the University of Virginia. In these open label studies, Bennett et al. have evaluated single and multiple doses of R‐(+)‐pramipexole in approximately 100 ALS subjects at a variety of dosages and durations of dosing up to a maximum daily dose of 100 mg TID. The drug has been safe and well‐tolerated. In a futility study of 30 subjects with early sALS treated for 6 months with open‐label RPPX at 10 mg TID following a 3‐month lead‐in phase, RPPX yielded a nonsignificant 13% reduction in the slope of decline on the ALSFRS‐R. A subset of these patients evaluated at a dose of RPPX 20 mg TID for an average of 7.9 months showed a further, but still nonsignificant, reduction of 17% in the slope of decline on the ALSFRS‐R compared with treatment at 10 mg TID [119].
Knopp Neurosciences, Inc. has licensed rights to the use of RPPX from the University of Virginia, and opened IND 75,959 for the evaluation of KNS‐760704 in ALS. Randomized, placebo‐controlled, double blind phase 1 safety studies of the drug in a total of 82 healthy adult subjects were completed in 2007. KNS‐760704 has been safe and well‐tolerated at single doses of 50, 150, and 300 mg and in twice‐daily doses of 50, 100, and 150 mg for 4 1/2 days. It is highly orally bioavailable, ∼90% renally excreted as unchanged drug and has demonstrated linear pharmacokinetics (Fig. 2). A phase 2A placebo‐controlled clinical trial in ALS patients (http://www.clintrials.gov) was initiated in March 2008.
Figure 2.
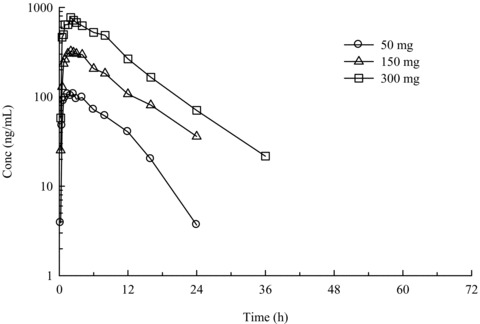
Mean human plasma concentrations of KNS‐760704 after single oral administration of a 50, 150, or 300 mg dose to adult volunteers under fasting conditions. Note semi‐logarithmic ordinate scale.
Conclusions
This review presented the logic for examining mitochondrial‐targeted neuroprotective agents in ALS and other neurodegenerative disorders, and presented evidence that the D2/D3 dopamine receptor agonist and approved PD/RLS drug Mirapex® (PPX) is neuroprotective and interacts with several potentially important mitochondrial mechanisms. However, due to its very high affinity for dopamine receptors PPX is limited to low doses which may not fully exploit neuroprotective mechanisms. RPPX, which has significantly lower dopamine receptor affinity, has been shown to be tolerated at much higher doses, and therefore may be a more useful neuroprotective agent in chronic and acute neurodegenerative disorders, including ALS. Open‐label studies in ALS patients with RPPX are being completed under IND 60,948, and placebo‐controlled clinical trials in ALS patients with enantiomerically pure KNS‐760704 are being initiated in 2008 under IND 75,959.
Conflict of Interest
The authors have no conflict of interest.
References
- 1. Boillee S, Vande VC, Cleveland DW. ALS: A disease of motor neurons and their nonneuronal neighbors. Neuron 2006;52:39–59. [DOI] [PubMed] [Google Scholar]
- 2. Brooks BR. Natural history of ALS: Symptoms, strength, pulmonary function, and disability. Neurology 1996;47:S71–S81. [DOI] [PubMed] [Google Scholar]
- 3. Jackson CE, Bryan WW. Amyotrophic lateral sclerosis. Semin Neurol 1998;18:27–39. [DOI] [PubMed] [Google Scholar]
- 4. Palmowski A, Jost WH, Prudlo J, Osterhage J, Kasmann B, Schimrigk K, Ruprecht KW. Eye movement in amyotrophic lateral sclerosis: A longitudinal study. Ger J Ophthalmol 1995;4:355–362. [PubMed] [Google Scholar]
- 5. Irwin D, Lippa CF, Swearer JM. Cognition and amyotrophic lateral sclerosis (ALS). Am J Alzheimers Dis Other Demen 2007;22:300–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Arai T, Hasegawa M, Akiyama H, Ikeda K, Nonaka T, Mori H, Mann O, Tsuchiya K, Yoshida M, Hashizame Y, et al TDP‐43 is a component of ubiquitin‐positive tau‐negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem Biophys Res Commun 2006;351:602–611. [DOI] [PubMed] [Google Scholar]
- 7. Dickson DW, Josephs KA, Mador‐Ortiz C. TDP‐43 in differential diagnosis of motor neuron disorders. Acta Neuropathol 2007;114:71–79. [DOI] [PubMed] [Google Scholar]
- 8. Beghi E, Logroscino G, Chio A, Hardiman O, Mitchell D, Swingler R, Traynor BJ. The epidemiology of ALS and the role of population‐based registries. Biochim Biophys Acta 2006;1762:1150–1157. [DOI] [PubMed] [Google Scholar]
- 9. Brooks BR. Clinical epidemiology of amyotrophic lateral sclerosis. Neurol Clin 1996;14:399–420. [DOI] [PubMed] [Google Scholar]
- 10. Strong M, Rosenfeld J. Amyotrophic lateral sclerosis: A review of current concepts. Amyotroph Lateral Scler Other Motor Neuron Disord 2003;4:136–143. [DOI] [PubMed] [Google Scholar]
- 11. Brown RH, Jr . Amyotrophic lateral sclerosis. Insights from genetics Arch Neurol 1997;54:1246–1250. [DOI] [PubMed] [Google Scholar]
- 12. Bruijn LI, Miller TM, Cleveland DW. Unraveling the mechanisms involved in motor neuron degeneration in ALS. Annu Rev Neurosci 2004;27:723–749. [DOI] [PubMed] [Google Scholar]
- 13. Cleveland DW, Rothstein JD. From Charcot to Lou Gehrig: Deciphering selective motor neuron death in ALS. Nat Rev Neurosci 2001;2:806–819. [DOI] [PubMed] [Google Scholar]
- 14. Majoor‐Krakauer D, Willems PJ, Hofman A. Genetic epidemiology of amyotrophic lateral sclerosis. Clin Genet 2003;63:83–101. [DOI] [PubMed] [Google Scholar]
- 15. Rosen DR, Bowling AC, Patterson D, Usdin TB, Sapp P, Mezey E, McKenna‐Yasek D, O'Regan J, Rahmani Z, Ferrante RJ, et al A frequent ala 4 to val superoxide dismutase‐1 mutation is associated with a rapidly progressive familial amyotrophic lateral sclerosis. Hum Mol Genet 1994;3:981–987. [DOI] [PubMed] [Google Scholar]
- 16. Wong PC, Pardo CA, Borchelt DR, Lee MK, Copeland NG, Jenkins NA, Sisodia SS, Cleveland DW, Price DL. An adverse property of a familial ALS‐linked SOD1 mutation causes motor neuron disease characterized by vacuolar degeneration of mitochondria. Neuron 1995;14:1105–1116. [DOI] [PubMed] [Google Scholar]
- 17. Mackenzie IR, Bigio EH, Ince PG, Geser F, Neumann M, Cairns NJ, Kwong LK, Forman MS, Ravits J, Stewart H, et al Pathological TDP‐43 distinguishes sporadic amyotrophic lateral sclerosis from amyotrophic lateral sclerosis with SOD1 mutations. Ann Neurol 2007;61:427–434. [DOI] [PubMed] [Google Scholar]
- 18. Sreedharan J, Blair IP, Tripathi VB, Hu X, Vance C, Rogelj B, Ackerley S, Durnall JC, William KL, Buratti E, et al TDP‐43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science 2008;319:1668–1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Tan CF, Eguchi H, Tagawa A, Onodera O, Iwasaki T, Tsujino A, Nishizawa M, Kakita A, Takahashi H. TDP‐43 immunoreactivity in neuronal inclusions in familial amyotrophic lateral sclerosis with or without SOD1 gene mutation. Acta Neuropathol 2007;113:535–542. [DOI] [PubMed] [Google Scholar]
- 20. Van Deerlin VM, Leverenz JB, Bekris LM, Bird TD, Yuan W, Elman LB, Clay D, Wood EM, Chen‐Plotkin AS, Martinez‐Lage M, et al TARDBP mutations in amyotrophic lateral sclerosis with TDP‐43 neuropathology: A genetic and histopathological analysis. Lancet Neurol 2008;7:409–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zelko IN, Mariani TJ, Folz RJ. Superoxide dismutase multigene family: A comparison of the CuZn‐SOD (SOD1), Mn‐SOD (SOD2), and EC‐SOD (SOD3) gene structures, evolution, and expression. Free Radic Biol Med 2002;33:337–349. [DOI] [PubMed] [Google Scholar]
- 22. Borchelt DR, Guarnieri M, Wong PC, Lee MK, Slunt HS, Xu ZS, Sisodia SS, Price DL, Cleveland DW. Superoxide dismutase 1 subunits with mutations linked to familial amyotrophic lateral sclerosis do not affect wild‐type subunit function. J Biol Chem 1995;270:3234–3238. [DOI] [PubMed] [Google Scholar]
- 23. Borchelt DR, Lee MK, Slunt HS, Guarnieri M, Xu ZS, Wong PC, Brown RH Jr, Prize DL, Sisodia SS, Cleveland DW. Superoxide dismutase 1 with mutations linked to familial amyotrophic lateral sclerosis possesses significant activity. Proc Natl Acad Sci U S A 1994;91:8292–8296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Bruijn LI, Houseweart MK, Kato S, Anderson KL, Anderson SD, Ohama E, et al Aggregation and motor neuron toxicity of an ALS‐linked SOD1 mutant independent from wild‐type SOD1. Science 1998;281:1851–1854. [DOI] [PubMed] [Google Scholar]
- 25. Furukawa Y, O'Halloran TV. Posttranslational modifications in Cu,Zn‐superoxide dismutase and mutations associated with amyotrophic lateral sclerosis. Antioxid Redox Signal 2006;8:847–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Potter SZ, Valentine JS. The perplexing role of copper‐zinc superoxide dismutase in amyotrophic lateral sclerosis (Lou Gehrig's disease). J Biol Inorg Chem 2003;8:373–380. [DOI] [PubMed] [Google Scholar]
- 27. Rakhit R, Chakrabartty A. Structure, folding, and misfolding of Cu, Zn superoxide dismutase in amyotrophic lateral sclerosis. Biochim Biophys Acta 2006;1762:1025–1037. [DOI] [PubMed] [Google Scholar]
- 28. Valentine JS, Hart PJ. Misfolded CuZnSOD and amyotrophic lateral sclerosis. Proc Natl Acad Sci U S A 2003;100:3617–3622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Williamson TL, Corson LB, Huang L, Burlingame A, Liu J, Bruijn LI, Cleveland DW. Toxicity of ALS‐linked SOD1 mutants. Science 2000;288:399. [DOI] [PubMed] [Google Scholar]
- 30. Wood JD, Beaujeux TP, Shaw PJ. Protein aggregation in motor neurone disorders. Neuropathol Appl Neurobiol 2003;29:529–545. [DOI] [PubMed] [Google Scholar]
- 31. Bruijn LI, Cleveland DW. Mechanisms of selective motor neuron death in ALS: Insights from transgenic mouse models of motor neuron disease. Neuropathol Appl Neurobiol 1996;22:373–387. [DOI] [PubMed] [Google Scholar]
- 32. Cleveland DW, Bruijn LI, Wong PC, Marszalek JR, Vechio JD, Lee MK, Xu XS, Borchelt DR, Sisodia SS, Price DL. Mechanisms of selective motor neuron death in transgenic mouse models of motor neuron disease. Neurology 1996;47:S54–S61. [DOI] [PubMed] [Google Scholar]
- 33. Julien JP, Kriz J. Transgenic mouse models of amyotrophic lateral sclerosis. Biochim Biophys Acta 2006;1762:1013–1024. [DOI] [PubMed] [Google Scholar]
- 34. Ludolph AC, Meyer T, Riepe MW. The role of excitotoxicity in ALS—what is the evidence? J Neurol 2000;247(Suppl. 1):I7–16. [DOI] [PubMed] [Google Scholar]
- 35. Przedborski S. Programmed cell death in amyotrophic lateral sclerosis: A mechanism of pathogenic and therapeutic importance. Neurologist 2004;10:1–7. [DOI] [PubMed] [Google Scholar]
- 36. Valentine JS. Do oxidatively modified proteins cause ALS? Free Radic Biol Med 2002;33:1314–1320. [DOI] [PubMed] [Google Scholar]
- 37. Dupuis L, Gonzalez de Aguilar JL, Oudart H, De TM, Barbeito L, Loeffler JP. Mitochondria in amyotrophic lateral sclerosis: A trigger and a target. Neurodegener Dis 2004;1:245–254. [DOI] [PubMed] [Google Scholar]
- 38. Bauer MF, Neupert W. Import of proteins into mitochondria: A novel pathomechanism for progressive neurodegeneration. J Inherit Metab Dis 2001;24:166–180. [DOI] [PubMed] [Google Scholar]
- 39. Calabrese V, Lodi R, Tonon C, D'Agata V, Sapienza M, Scapagnini G, Mangiameli A, Pennisi G, Stella AM, Butterfield DA. Oxidative stress, mitochondrial dysfunction and cellular stress response in Friedreich's ataxia. J Neurol Sci 2005;233:145–162. [DOI] [PubMed] [Google Scholar]
- 40. Emerit J, Edeas M, Bricaire F. Neurodegenerative diseases and oxidative stress. Biomed Pharmacother 2004;58:39–46. [DOI] [PubMed] [Google Scholar]
- 41. Gallaher BW, Hille R, Raile K, Kiess W. Apoptosis: Live or die—hard work either way! Horm Metab Res 2001;33:511–519. [DOI] [PubMed] [Google Scholar]
- 42. Gottlieb RA. Mitochondria: Execution central. FEBS Lett 2000;482:6–12. [DOI] [PubMed] [Google Scholar]
- 43. Frey TG, Mannella CA. The internal structure of mitochondria. Trends Biochem Sci 2000;25:319–324. [DOI] [PubMed] [Google Scholar]
- 44. Lloreta‐Trull J, Serrano S. Biology and pathology of the mitochondrion. Ultrastruct Pathol 1998;22:357–367. [DOI] [PubMed] [Google Scholar]
- 45. Mannella CA. Structure and dynamics of the mitochondrial inner membrane cristae. Biochim Biophys Acta 2006;1763:542–548. [DOI] [PubMed] [Google Scholar]
- 46. Brand MD, Affourtit C, Esteves TC, Green K, Lambert AJ, Miwa S, Pakag JL, Parker N. Mitochondrial superoxide: Production, biological effects, and activation of uncoupling proteins. Free Radic Biol Med 2004;37:755–767. [DOI] [PubMed] [Google Scholar]
- 47. Cai J, Yang J, Jones DP. Mitochondrial control of apoptosis: The role of cytochrome c. Biochim Biophys Acta 1998;1366:139–149. [DOI] [PubMed] [Google Scholar]
- 48. McBride HM, Neuspiel M, Wasiak S. Mitochondria: More than just a powerhouse. Curr Biol 2006;16:R551–R560. [DOI] [PubMed] [Google Scholar]
- 49. Sherratt HS. Mitochondria: Structure and function. Rev Neurol (Paris) 1991;147:417–430. [PubMed] [Google Scholar]
- 50. Chen XJ, Butow RA. The organization and inheritance of the mitochondrial genome. Nat Rev Genet 2005;6:815–825. [DOI] [PubMed] [Google Scholar]
- 51. Stuart JA, Brown MF. Mitochondrial DNA maintenance and bioenergetics. Biochim Biophys Acta 2006;1757:79–89. [DOI] [PubMed] [Google Scholar]
- 52. Koehler CM. New developments in mitochondrial assembly. Annu Rev Cell Dev Biol 2004;20:309–335. [DOI] [PubMed] [Google Scholar]
- 53. Howell N, Elson JL, Chinnery PF, Turnbull DM. mtDNA mutations and common neurodegenerative disorders. Trends Genet 2005;21:583–586. [DOI] [PubMed] [Google Scholar]
- 54. Onyango IG, Khan SM. Oxidative stress, mitochondrial dysfunction, and stress signaling in Alzheimer's disease. Curr Alzheimer Res 2006;3:339–349. [DOI] [PubMed] [Google Scholar]
- 55. Trushina E, McMurray CT. Oxidative stress and mitochondrial dysfunction in neurodegenerative diseases. Neuroscience 2007;145:1233–1248. [DOI] [PubMed] [Google Scholar]
- 56. Xu Z, Jung C, Higgins C, Levine J, Kong J. Mitochondrial degeneration in amyotrophic lateral sclerosis. J Bioenerg Biomembr 2004;36:395–399. [DOI] [PubMed] [Google Scholar]
- 57. Dam‐Vizi V. Production of reactive oxygen species in brain mitochondria: Contribution by electron transport chain and non‐electron transport chain sources. Antioxid Redox Signal 2005;7:1140–1149. [DOI] [PubMed] [Google Scholar]
- 58. Turrens JF. Mitochondrial formation of reactive oxygen species. J Physiol 2003;552:335–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Chan PH. Mitochondria and neuronal death/survival signaling pathways in cerebral ischemia. Neurochem Res 2004;29:1943–1949. [DOI] [PubMed] [Google Scholar]
- 60. Robberecht W. Oxidative stress in amyotrophic lateral sclerosis. J Neurol 2000;247(Suppl. 1):I1–I6. [DOI] [PubMed] [Google Scholar]
- 61. Bernardi P, Krauskopf A, Basso E, Petronilli V, Blachly‐Dyson E, Di LF, Forte MA. The mitochondrial permeability transition from in vitro artifact to disease target. FEBS J 2006;273:2077–2099. [DOI] [PubMed] [Google Scholar]
- 62. Crompton M, Virji S, Doyle V, Johnson N, Ward JM. The mitochondrial permeability transition pore. Biochem Soc Symp 1999;66:167–179. [DOI] [PubMed] [Google Scholar]
- 63. Halestrap AP, McStay GP, Clarke SJ. The permeability transition pore complex: Another view. Biochimie 2002;84:153–166. [DOI] [PubMed] [Google Scholar]
- 64. Javadov S, Karmazyn M. Mitochondrial permeability transition pore opening as an endpoint to initiate cell death and as a putative target for cardioprotection. Cell Physiol Biochem 2007;20:1–22. [DOI] [PubMed] [Google Scholar]
- 65. Halestrap AP, Doran E, Gillespie JP, O'Toole A. Mitochondria and cell death. Biochem Soc Trans 2000;28:170–177. [DOI] [PubMed] [Google Scholar]
- 66. Andrabi SA, Sayeed I, Siemen D, Wolf G, Horn TF. Direct inhibition of the mitochondrial permeability transition pore: A possible mechanism responsible for anti‐apoptotic effects of melatonin. FASEB J 2004;18:869–871. [DOI] [PubMed] [Google Scholar]
- 67. De MU, Basso E, Szabo I, Zoratti M. Electrophysiological characterization of the Cyclophilin D‐deleted mitochondrial permeability transition pore. Mol Membr Biol 2006;23:521–530. [DOI] [PubMed] [Google Scholar]
- 68. Sayeed I, Parvez S, Winkler‐Stuck K, Seitz G, Trieu I, Wallesch CW, Schönfeld P, Siemen D. Patch clamp reveals powerful blockade of the mitochondrial permeability transition pore by the D2‐receptor agonist pramipexole. FASEB J 2006;20:556–558. [DOI] [PubMed] [Google Scholar]
- 69. Bernardi P, Broekemeier KM, Pfeiffer DR. Recent progress on regulation of the mitochondrial permeability transition pore; a cyclosporin‐sensitive pore in the inner mitochondrial membrane. J Bioenerg Biomembr 1994;26:509–517. [DOI] [PubMed] [Google Scholar]
- 70. Clarkson AN, Sutherland BA, Appleton I. The biology and pathology of hypoxia‐ischemia: An update. Arch Immunol Ther Exp (Warsz) 2005;53:213–225. [PubMed] [Google Scholar]
- 71. Mattson MP, Culmsee C, Yu ZF. Apoptotic and antiapoptotic mechanisms in stroke. Cell Tissue Res 2000;301:173–187. [DOI] [PubMed] [Google Scholar]
- 72. Sullivan PG, Rabchevsky AG, Waldmeier PC, Springer JE. Mitochondrial permeability transition in CNS trauma: Cause or effect of neuronal cell death? J Neurosci Res 2005;79:231–239. [DOI] [PubMed] [Google Scholar]
- 73. Antonyuk S, Elam JS, Hough MA, Strange RW, Doucette PA, Rodriguez JA, Hayward LJ, Valentine JS, Hart PJ, Hasnain SS. Structural consequences of the familial amyotrophic lateral sclerosis SOD1 mutant His46Arg. Protein Sci 2005;14:1201–1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Furukawa Y, O'Halloran TV. Amyotrophic lateral sclerosis mutations have the greatest destabilizing effect on the apo‐ and reduced form of SOD1, leading to unfolding and oxidative aggregation. J Biol Chem 2005;280:17266–17274. [DOI] [PubMed] [Google Scholar]
- 75. Watanabe M, Dykes‐Hoberg M, Culotta VC, Price DL, Wong PC, Rothstein JD. Histological evidence of protein aggregation in mutant SOD1 transgenic mice and in amyotrophic lateral sclerosis neural tissues. Neurobiol Dis 2001;8:933–941. [DOI] [PubMed] [Google Scholar]
- 76. Deng HX, Shi Y, Furukawa Y, Zhai H, Fu R, Liu E, Gorrie GH, Khan MS, Hung WY, Bigio EH, et al Conversion to the amyotrophic lateral sclerosis phenotype is associated with intermolecular linked insoluble aggregates of SOD1 in mitochondria. Proc Natl Acad Sci U S A 2006;103:7142–7147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Martin LJ, Chen K, Liu Z. Adult motor neuron apoptosis is mediated by nitric oxide and Fas death receptor linked by DNA damage and p53 activation. J Neurosci 2005;25:6449–6459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Hirano A, Nakano I, Kurland LT, Mulder DW, Holley PW, Saccomanno G. Fine structural study of neurofibrillary changes in a family with amyotrophic lateral sclerosis. J Neuropathol Exp Neurol 1984;43:471–480. [DOI] [PubMed] [Google Scholar]
- 79. Kusaka H, Hirano A. Fine structure of anterior horns in patients without amyotrophic lateral sclerosis. J Neuropathol Exp Neurol 1985;44:430–438. [DOI] [PubMed] [Google Scholar]
- 80. Echaniz‐Laguna A, Zoll J, Ponsot E, N'guessan B, Tranchant C, Loeffler JP, Lampert E. Muscular mitochondrial function in amyotrophic lateral sclerosis is progressively altered as the disease develops: A temporal study in man. Exp Neurol 2006;198:25–30. [DOI] [PubMed] [Google Scholar]
- 81. Swerdlow RH, Parks JK, Cassarino DS, Trimmer PA, Miller SW, Maguire DJ, Sheehan JP, Maguire RS, Pattee G, Juel VC, et al Mitochondria in sporadic amyotrophic lateral sclerosis. Exp Neurol 1998;153:135–142. [DOI] [PubMed] [Google Scholar]
- 82. Dhaliwal GK, Grewal RP. Mitochondrial DNA deletion mutation levels are elevated in ALS brains. Neuroreport 2000;11:2507–2509. [DOI] [PubMed] [Google Scholar]
- 83. Spillane CB, Fletcher NC, Rountree SM, Van Den BH, Chanduloy S, Morgan JL, Keene FR. Benzothiazole bipyridine complexes of ruthenium(II) with cytotoxic activity. J Biol Inorg Chem 2007;12:797–807. [DOI] [PubMed] [Google Scholar]
- 84. Oketani K, Nagakura N, Harada K, Inoue T. In vitro effects of E3040, a dual inhibitor of 5‐lipoxygenase and thromboxane A(2) synthetase, on eicosanoid production. Eur J Pharmacol 2001;422:209–216. [DOI] [PubMed] [Google Scholar]
- 85. Yildiz‐Oren I, Yalcin I, Ki‐Sener E, Ucarturk N. Synthesis and structure‐activity relationships of new antimicrobial active multisubstituted benzazole derivatives. Eur J Med Chem 2004;39:291–298. [DOI] [PubMed] [Google Scholar]
- 86. Wahl F, Renou E, Mary V, Stutzmann JM. Riluzole reduces brain lesions and improves neurological function in rats after a traumatic brain injury. Brain Res 1997;756:247–255. [DOI] [PubMed] [Google Scholar]
- 87. Plesnila N, Von BL, Retiounskaia M, Engel D, Ardeshiri A, Zimmermann R, Hoffmann F, Landshamer S, Wagner E, Culmsee C. Delayed neuronal death after brain trauma involves p53‐dependent inhibition of NF‐kappaB transcriptional activity. Cell Death Differ 2007;14:1529–1541. [DOI] [PubMed] [Google Scholar]
- 88. Klunk WE, Engler H, Nordberg A, Wang Y, Blomqvist G, Holt DP, Bergström M, Savitcheva I, Huang GF, Estrada S, et al Imaging brain amyloid in Alzheimer's disease with Pittsburgh Compound‐B. Ann Neurol 2004;55:306–319. [DOI] [PubMed] [Google Scholar]
- 89. Heiser V, Engemann S, Brocker W, Dunkel I, Boeddrich A, Waelter S, Nordhoff E, Lurz R, Schugardt N, Rautenberg S, et al Identification of benzothiazoles as potential polyglutamine aggregation inhibitors of Huntington's disease by using an automated filter retardation assay. Proc Natl Acad Sci U S A 99 Suppl 2002;4:16400–16406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Doble A. The pharmacology and mechanism of action of riluzole. Neurology 1996;47:S233–S241. [DOI] [PubMed] [Google Scholar]
- 91. Hockly E, Tse J, Barker AL, Moolman DL, Beunard JL, Revington AP, Holt K, Sunshine S, Moffitt H, Sathasivam K, et al Evaluation of the benzothiazole aggregation inhibitors riluzole and PGL‐135 as therapeutics for Huntington's disease. Neurobiol Dis 2006;21:228–236. [DOI] [PubMed] [Google Scholar]
- 92. Iwasaki Y, Ikeda K, Kinoshita M. Molecular and cellular mechanism of glutamate receptors in relation to amyotrophic lateral sclerosis. Curr Drug Targets CNS Neurol Disord 2002;1:511–518. [DOI] [PubMed] [Google Scholar]
- 93. Hebert T, Drapeau P, Pradier L, Dunn RJ. Block of the rat brain IIA sodium channel alpha subunit by the neuroprotective drug riluzole. Mol Pharmacol 1994;45:1055–1060. [PubMed] [Google Scholar]
- 94. Huang CS, Song JH, Nagata K, Yeh JZ, Narahashi T. Effects of the neuroprotective agent riluzole on the high voltage‐activated calcium channels of rat dorsal root ganglion neurons. J Pharmacol Exp Ther 1997;282:1280–1290. [PubMed] [Google Scholar]
- 95. Mathie A, Veale EL. Therapeutic potential of neuronal two‐pore domain potassium‐channel modulators. Curr Opin Investig Drugs 2007;8:555–562. [PubMed] [Google Scholar]
- 96. Song JH, Huang CS, Nagata K, Yeh JZ, Narahashi T. Differential action of riluzole on tetrodotoxin‐sensitive and tetrodotoxin‐resistant sodium channels. J Pharmacol Exp Ther 1997;282:707–714. [PubMed] [Google Scholar]
- 97. Wang SJ, Wang KY, Wang WC. Mechanisms underlying the riluzole inhibition of glutamate release from rat cerebral cortex nerve terminals (synaptosomes). Neuroscience 2004;125:191–201. [DOI] [PubMed] [Google Scholar]
- 98. Wu SN. Large‐conductance Ca2+‐ activated K+ channels:physiological role and pharmacology. Curr Med Chem 2003;10:649–661. [DOI] [PubMed] [Google Scholar]
- 99. Xing Y, Zhang Y, Stabernack CR, Eger EI, Gray AT. The use of the potassium channel activator riluzole to test whether potassium channels mediate the capacity of isoflurane to produce immobility. Anesth Analg 2003;97:1020–4. [DOI] [PubMed] [Google Scholar]
- 100. Miller RG, Mitchell JD, Lyon M, Moore DH. Riluzole for amyotrophic lateral sclerosis (ALS)/motor neuron disease (MND). Cochrane Database Syst Rev CD00(1447), 2007. [DOI] [PubMed] [Google Scholar]
- 101. Schneider CS, Mierau J. Dopamine autoreceptor agonists: Resolution and pharmacological activity of 2,6‐diaminotetrahydrobenzothiazole and an aminothiazole analogue of apomorphine. J Med Chem 1987;30:494–498. [DOI] [PubMed] [Google Scholar]
- 102. Kreiss DS, Bergstrom DA, Gonzalez AM, Huang KX, Sibley DR, Walters JR. Dopamine receptor agonist potencies for inhibition of cell firing correlate with dopamine D3 receptor binding affinities. Eur J Pharmacol 1995;277:209–214. [DOI] [PubMed] [Google Scholar]
- 103. Mierau J, Schneider FJ, Ensinger HA, Chio CL, Lajiness ME, Huff RM. Pramipexole binding and activation of cloned and expressed dopamine D2, D3 and D4 receptors. Eur J Pharmacol 1995;290:29–36. [DOI] [PubMed] [Google Scholar]
- 104. Dodson MW, Guo M. Pink1, Parkin, DJ‐1 and mitochondrial dysfunction in Parkinson's disease. Curr Opin Neurobiol 2007;17:331–337. [DOI] [PubMed] [Google Scholar]
- 105. Schapira AH. Mitochondrial dysfunction in Parkinson's disease. Cell Death Differ 2007;14:1261–1266. [DOI] [PubMed] [Google Scholar]
- 106. Chinta SJ, Kumar MJ, Hsu M, Rajagopalan S, Kaur D, Rane A, Nicholls DG, Choi J, Anderson JK. Inducible alterations of glutathione levels in adult dopaminergic midbrain neurons result in nigrostriatal degeneration. J Neurosci 2007;27:13997–14006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Hall ED, Andrus PK, Oostveen JA, Althaus JS, VonVoigtlander PF. Neuroprotective effects of the dopamine D2/D3 agonist pramipexole against postischemic or methamphetamine‐induced degeneration of nigrostriatal neurons. Brain Res 1996;742:80–88. [DOI] [PubMed] [Google Scholar]
- 108. Sethy VH, Wu H, Oostveen JA, Hall ED. Neuroprotective effects of the dopamine agonists pramipexole and bromocriptine in 3‐acetylpyridine‐treated rats. Brain Res 1997;754:181–186. [DOI] [PubMed] [Google Scholar]
- 109. Kakimura J, Kitamura Y, Takata K, Kohno Y, Nomura Y, Taniguchi T. Release and aggregation of cytochrome c and alpha‐synuclein are inhibited by the antiparkinsonian drugs, talipexole and pramipexole. Eur J Pharmacol 2001;417:59–67. [DOI] [PubMed] [Google Scholar]
- 110. Kitamura Y, Taniguchi T, Shimohama S, Akaike A, Nomura Y. Neuroprotective mechanisms of antiparkinsonian dopamine D2‐receptor subfamily agonists. Neurochem Res 2003;28:1035–1040. [DOI] [PubMed] [Google Scholar]
- 111. Takata K, Kitamura Y, Kakimura J, Kohno Y, Taniguchi T. Increase of bcl‐2 protein in neuronal dendritic processes of cerebral cortex and hippocampus by the antiparkinsonian drugs, talipexole and pramipexole. Brain Res 2000;872:236–241. [DOI] [PubMed] [Google Scholar]
- 112. Cassarino DS, Fall CP, Smith TS, Bennett JP Jr. Pramipexole reduces reactive oxygen species production in vivo and in vitro and inhibits the mitochondrial permeability transition produced by the parkinsonian neurotoxin methylpyridinium ion. J Neurochem 1998;71:295–301. [DOI] [PubMed] [Google Scholar]
- 113. Fujita Y, Izawa Y, Ali N, Kanematsu Y, Tsuchiya K, Hamano S, Tamaki T, Yoshizumi M. Pramipexole protects against H2O2‐induced PC12 cell death. Naunyn Schmiedebergs Arch Pharmacol 2006;372:257–266. [DOI] [PubMed] [Google Scholar]
- 114. Le WD, Jankovic J, Xie W, Appel SH. Antioxidant property of pramipexole independent of dopamine receptor activation in neuroprotection. J Neural Transm 2000;107:1165–1173. [DOI] [PubMed] [Google Scholar]
- 115. Ramirez AD, Wong SK, Menniti FS. Pramipexole inhibits MPTP toxicity in mice by dopamine D3 receptor dependent and independent mechanisms. Eur J Pharmacol 2003;475:29–35. [DOI] [PubMed] [Google Scholar]
- 116. Abramova NA, Cassarino DS, Khan SM, Painter TW, Bennett JP Jr. Inhibition by R(+) or S(–) pramipexole of caspase activation and cell death induced by methylpyridinium ion or beta amyloid peptide in SH‐SY5Y neuroblastoma. J Neurosci Res 2002;67:494–500. [DOI] [PubMed] [Google Scholar]
- 117. Gu M, Iravani MM, Cooper JM, King D, Jenner P, Schapira AH. Pramipexole protects against apoptotic cell death by non‐dopaminergic mechanisms. J Neurochem 2004;91:1075–1081. [DOI] [PubMed] [Google Scholar]
- 118. Danzeisen R, Schwalenstoecker B, Gillardon F, Buerger E, Krzykalla V, Klinder K, Schild L, Hengerer B, Ludolph AC, Dorner‐Ciossek C, et al Targeted antioxidative and neuroprotective properties of the dopamine agonist pramipexole and its nondopaminergic enantiomer SND919CL2x [(+)2‐amino‐4,5,6,7‐tetrahydro‐6‐Lpropylamino‐benzathiazole dihydrochloride]. J Pharmacol Exp Ther 2006;316:189–199. [DOI] [PubMed] [Google Scholar]
- 119. Wang H, Larriviere KS, Keller KE, Ware KA, Burns TM, Conaway MA, Lacomis D, Pattee GL, Phillips LH, Solenski NJ, et al R+ pramipexole as a mitochondrially focused neuroprotectant: Initial early phase studies in ALS. Amyotroph Lateral Scler 2008;9:50–58. [DOI] [PubMed] [Google Scholar]