

Abstract
Lithium is a monovalent cation that was introduced in 1949 by John Cade for the treatment of bipolar disorder. Clinical reports and subsequent studies confirmed this application and the beneficial effects of this compound. However, over the last 15 years, various authors have also demonstrated the neuroprotective effects of lithium against several neurotoxic paradigms. Thus, experimental studies in neuronal cell cultures and animal models of Alzheimer disease and others pathologies have provided strong evidence for the potential benefits of lithium. The main mechanism underlying its neuroprotective effects is thought to be inhibition of glycogen synthase kinase‐3 (GSK‐3), although other biochemical pathways in the brain could also be affected. In this review, the main mechanisms of lithium action are summarized, including the modulation of glutamate receptors, effects on arachidonic acid metabolism, its role with respect to AKT, and other potential mechanisms. In addition, its effects on neuroprotective proteins such as Bcl‐2 and p53 are also discussed. Although the cellular and molecular biological effects of lithium may constitute an effective therapeutic strategy for Alzheimer disease, further clinical and experimental studies with this drug and specific GSK‐3 inhibitors are necessary to confirm the use of lithium in therapeutic approaches to neurodegenerative diseases.
Keywords: Alzheimer disease, Autophagy, GSK‐3β, Lithium, Oxidative stress, Parkinson disease
Introduction
The main clinical pharmacological application of lithium is currently in the treatment of manic‐depressive illness, by means of its well‐documented mood‐ stabilizing effects [1]. However, recent experimental data suggest that lithium also shows neuroprotective effects in preventing programmed neuronal cell death in different paradigms [2, 3, 4, 5, 6]. The vast majority of these studies suggest that the neuroprotective effects of lithium are provided through the modulation of multiple mechanisms; however, it seems that the enzyme glycogen synthase kinase‐3 (GSK‐3) is probably the main target involved in lithium's neuroprotective effects [4, 7]. Thus, GSK‐3 inhibition could account for the potential benefits of this ion in the treatment of chronic neurodegenerative diseases such as Alzheimer disease (AD), Parkinson disease (PD), Huntington disease (HD), and amyotrophic lateral sclerosis (ALS) [1, 4, 7]. However, a distinction needs to be made, at least in neuronal cell cultures, between the effects of acute versus chronic treatment with lithium. Thus, whereas GSK‐3 inhibition by lithium occurs rapidly and constitutes an acute effect, the long‐term ameliorative effects of lithium have been attributed to Bcl‐2, p53, and Bax regulation, events that may also be dependent on c‐Jun modulation by lithium and changes in the expression of all these proteins will be discussed below, as they may explain the neuroprotective effects of this compound in chronic treatments.
Inhibition of GSK‐3 by Lithium
Research has demonstrated that lithium at therapeutic concentrations (1–2 mM) inhibits GSK‐3 [1, 8, 9, 10]. This enzyme has two isoforms: α and β. Although it was initially identified as a regulator of glycogen synthesis, it is now known to play a key role in the central nervous system by regulating various processes or transcription factors, for example, in tau phosphorylation, regulation of c‐jun, the nuclear translocation of β‐catenin and nuclear export of the nuclear factor of activated T cells (NF‐Atc), nuclear factor kappa B (NFκB), pCreb and the myocyte enhancer factor (MEF2), a prosurvival transcription factor [1, 9, 10, 11, 12, 13]. Moreover, GSK‐3 is currently attracting widespread interest as a regulator of neuronal apoptosis [4, 5, 6]. In this regard, it has been widely demonstrated that inhibition of GSK‐3 prevents apoptosis, whereas increased activity is proapoptotic [14]. However, it has only recently been shown that GSK‐3 may represent a possible therapeutic target of lithium. In addition, a range of evidence suggests that the behavioral effects of lithium in rodent models of mania may be due to inhibition of GSK‐3 [13, 15]. One of the well‐characterized mechanisms of GSK‐3 inhibition by lithium is through direct competition for a magnesium‐binding site with GSK‐3β. Thus, studies performed in cultures of cerebellar granule neurons (CGNs) lend support to the hypothesis that acute administration of lithium in a millimolar range inhibits GSK‐3β activity [16]. Moreover, lithium indirectly regulates GSK‐3 activity, which is regulated negatively by phosphorylation of serine‐9 in the pseudosubstrate domain through the activation of Akt (also called protein kinase B). This is important because the activation of Akt suggests that this drug has additional neuroprotective effects that are probably due to modulation of targets downstream of Akt, such as FOXO, Bad, and murine double minute (MDM2) [13, 15, 17] (Fig. 1). Thus, our group demonstrated that after deprivation of serum and potassium as proapoptotic stimuli, CGNs died through an apoptotic process, which included the activation of the classical intrinsic mitochondrial route [18]. In this context, mitochondrial proteins such as cytochrome C are released as a result of these apoptotic stimuli and caspase‐3 is activated. Likewise, lithium was able to inhibit caspase‐3 activation, which is a key enzyme involved in the apoptotic process [19]. One question that remains to be clarified is whether selective GSK‐3 inhibitors such as SB‐415286, which have shown neuroprotective effects in different paradigms, are indeed useful for the treatment of neurodegenerative diseases.
Figure 1.
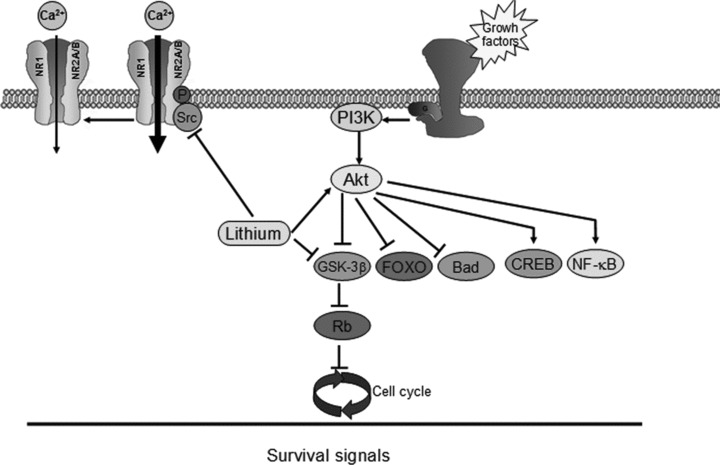
Network integrating the potential neuroprotective effects of lithium in neurons. Thus, activation of Akt by lithium suggests that neuroprotective effects of this drug are also probably due to modulation of downstream targets of Akt such as Bad, FOXO, CREB, and NF‐κβ. Furthermore, lithium inhibits directly GSK‐3β and modulates NMDA receptor.
Furthermore, with respect to AD, the amyloid cascade hypothesis suggests an explicit role for GSK‐3β through tau phosphorylation [20, 21, 22]. Thus, the inhibition of this enzyme constitutes a potential therapeutic target in this disease. Interestingly, lithium ameliorated symptoms in an AD study [23].
More recently, it was reported that a reduction in GSK‐3α levels reduced the production of amyloid beta (Aβ), whereas a reduction in GSK‐3β increased its production [24]. Different studies have postulated that lithium decreases the production of Aβ from its precursor, amyloid precursor protein (APP), and can thus be considered a powerful therapeutic agent for treating AD [20, 21, 22, 23, 24, 25, 26]. However, Feyt et al. demonstrated in two cell types expressing human APP695 that lithium at noncytotoxic concentrations increases the production of Aβ by increasing β‐secretase activity [27].
These results, coupled with the fact that lithium can inhibit both GSK‐3 isoforms, suggest that the effect of lithium on Aβ protein production could be unrelated to its inhibition of GSK‐3 [21, 24]. Clinical studies have demonstrated that lithium could be used as a preventive treatment for AD, although further clinical studies are obviously needed to confirm its action [23]. In this context, inhibition of GSK‐3 levels by lithium has been proposed as a possible therapy for various diseases, not only AD disease but also other neurodegenerative disorders [28, 29, 30] (Table 1).
Table 1.
Examples of the neuroprotective effects of lithium in neuronal cell cultures or models for studies of specific aspects of different experimental neuropathological processes
| Neuropathological process (disorder) | Neurotoxin | Cell/tissue culture/experimental model | References |
|---|---|---|---|
| Alzheimer disease | Amyloid beta‐peptide | Hippocampal neurons and PC12 cells | [4] |
| Transgenic mouse model of AD hippocampus | [7, 20, 25] | ||
| Amyotrophic lateral sclerosis | Mouse model | [3, 6] | |
| Parkinson disease | MPTP | Rat treatment | [64] |
| Huntington disease | Huntingtin overexpression | Caenorhabditis elegans | [68] |
| 3‐Nitropropionic acid | Rat treatment | [28] | |
| Cerebral ischemia | Model of ischemia | Rat | [81] |
| Epilepsy | Kainic acid | Rat | [76] |
| Aging | C. elegans | [86] | |
| SAMP‐8 | [40, 90] |
The possible involvement of cell cycle activation in neuronal apoptosis has been extensively investigated in recent years [31, 32, 33]. Studies carried out on brains of AD patients demonstrated that the expression of cell cycle proteins and DNA synthesis both increase in this pathology [32, 33]. Further, phosphorylation of the retinoblastoma protein (Rb) increases in the brains of PD patients [32]. Thus, both in vivo and in vitro data from neuronal cell cultures suggest that reentry into the cell cycle is a potential step in the neuronal apoptotic cascade. In this process of cell cycle reentry, the phosphorylation of Rb by cyclin‐dependent kinases (CDKs) is a key step, as unphosphorylated Rb can block cell cycle progression by binding to the transcription factor E2F‐1 [34, 35, 36]. However, recent studies suggest that Rb could be phosphorylated by other kinases such as CDK5 in neuronal cells and GSK‐3β in nonneuronal cells [35, 36, 37, 38]. Our group demonstrated that Rb is rapidly phosphorylated in CGNs under serum‐ and potassium‐deprivation conditions (a well‐known apoptotic model). Since pRb phosphorylation was prevented in the presence of GSK‐3β inhibitors, we suggested that targeting GSK‐3β could be involved in the inhibition of cell cycle reentry mediated by lithium. This would also explain the neuroprotective properties of this drug and its application to the treatment of neurodegenerative diseases. Lithium can inhibit two key features of AD, namely the process of cell cycle reentry and tau phosphorylation [39, 40].
Lithium Modulates Neuronal Growth Factors
A possible explanation for lithium's neuroprotective effects after chronic treatment is through an increase in the levels of neurotrophic factors [41]. Using a pharmacological approach with the compound K252a, a Trk tyrosine kinase inhibitor and a brain derived neurotrophic factor (BDNF)‐neutralizing antibody, lithium‐induced neuroprotection against excitotoxicity was also suppressed [14]. These data strongly implicate BDNF/TrkB activation in lithium's neuroprotection. Further evidence in support of this hypothesis comes from studies performed in cortical neurons, where lithium transiently increased intracellular BDNF followed by an increase in levels of phosphorylated TrkB. Therefore, long‐term lithium treatment enhances BDNF expression, secretion, and subsequent activation of TrkB receptors [2, 4, 9]. Furthermore, this hypothesis is confirmed using cortical neurons derived from either heterozygous (+/−) or homozygous (−/−) BDNF knockout mice, because lithium does not protect them from glutamate excitotoxic cell death, whereas it does protect cultures derived from wild‐type mice [1].
Modulation of the Excitotoxic Response by Lithium
Chronic treatment of CGNs with lithium modulates glutamate receptor hyperactivity, which might explain its effects in the treatment of manic depression [42]. Although the mechanism involved in this process is unknown, studies in rat cortical neurons showed that long‐term lithium treatment decreased the activation of Src kinase, which is involved in N‐methyl‐d‐aspartate (NMDA) receptor activation [15]. Interestingly, phosphorylation of NR2B tyrosine is modulated by two kinases: Src and Fyn. In the many studies reported on Src kinases, lithium has been shown to down‐regulate Src kinase activation and, therefore, inhibit NR2B tyrosine phosphorylation, thus implicating lithium in the regulation of the NMDA receptor. Another study demonstrated that long‐term lithium treatment in rats caused decreases in Tyr‐402 phosphorylation of Pyk2 and Tyr‐416 phosphorylation of Src. The inhibition of Pyk2 Tyr‐402 phosphorylation in the rat hippocampus was associated with a lithium‐induced decrease in NR2A/NR2B tyrosine phosphorylation and a loss of Src kinase activity [4, 14]. In addition, the effects of lithium on changes in the phosphorylation state of NMDAR subunits could prevent or modulate the NMDAR‐mediated intracellular calcium increase in neurons [42].
Thus, chronic lithium treatment, through the inhibition of Src tyrosine kinase‐mediated pathways, may be beneficial for the treatment of excitotoxicity‐related neurodegenerative diseases. By inhibiting NMDA receptor activation, lithium down‐regulates the brain arachidonic acid (AA) cascade [43]. Cyclooxygenase‐2 is responsible for AA production in brain phospholipids, phospholypase A2 (cPLA2) activities and an increase in the concentration of prostaglandin E2. Therefore, blocking NMDAR signaling and cPLA2 could be important for lithium's therapeutic efficacy. Moreover, changes in the expression of brain mRNA from NMDA receptor subunits have been demonstrated in patients with bipolar disorders. Moreover, lithium modulates AMPA GluR1/2 synaptic expression in the hippocampus; specifically, this expression was shown to be down‐regulated after chronic treatment with therapeutically relevant concentrations of lithium, both in vitro and in vivo[14, 17]. Furthermore, electrophysiological studies demonstrated that the AMPA/NMDA ratio is decreased in hippocampal CA1 neurons in animals treated chronically with lithium, and these findings suggest a novel therapeutic strategy for the development of new drugs to treat bipolar disorders and neurological diseases [8].
Overall, these studies indicate that the neuroprotective and clinical effects of lithium could be mediated by an interaction with NMDA and non‐NMDA receptors. These data are interesting because the clinical drugs used currently for AD therapy are mainly NMDA‐receptor antagonists, for example, memantine that blocks glutamate‐mediated excitotoxicity [17]. However, acetylcholinesterase inhibitors, which aim to stabilize acetylcholine levels in the synaptic cleft in order to maintain neurotransmission, are also used because it has been hypothesized that cholinergic dysfunction in the process of aging contributes to the development of AD [2]. The possibility that bipolar affective disorder is the result of some abnormality of glutamatergic neurotransmission thus warrants further investigation.
Lithium Is Involved in the Regulation of Autophagy
Autophagy is a physiological process involved in the degradation of proteins and also eliminates unwanted, damaged cell structures or organelles. It has recently been suggested that the potential benefits of lithium could be due to the induction of autophagy [44, 45, 46]. Interestingly, the role of lithium in autophagy was GSK‐3 independent and research showed the prominent role of inositol monophosphatase (IMPase) inhibition [45]. Moreover, carbamazepine and valproic acid drugs, which induced inositol depletion, also increase autophagy. The importance of autophagy in neurodegenerative diseases is to decrease both aggregated substrates—huntingtin and alpha‐synuclein—proteins that are associated with HD and some autosomal forms of PD, respectively. A recent clinical study in human patients found that lithium administration slows the progression of ALS. The potential mechanism involved in this beneficial effect of lithium is attributed to autophagy activation, since one of the features of ALS is a defect in this pathway [6]. Because autophagy is a major degradation route for aggregate‐prone proteins associated with neurodegenerative disorders, the induction of autophagy by lithium may also be a valuable strategy in the treatment of neurodegenerative diseases, in addition to the other beneficial effects of this drug.
Role of Lithium in the Inhibition of Oxidative Stress
It has been hypothesized that an increase in oxidative stress parameters could be associated with the pathophysiology of bipolar disorders [47, 48]. Thus, an increase in thiobarbituric acid reactive substance (TBARS) and superoxide dismutase and a decrease in catalase levels have been demonstrated in patients with bipolar disorders. Therefore, another potential mechanism underlying lithium's neuroprotection and efficacy in bipolar disorders is the previously demonstrated antioxidant properties of this drug. In neuronal cell preparations chronic treatment with lithium inhibited H2O2‐induced cell death. Furthermore, other studies indicate that chronic lithium treatment inhibited lipid peroxidation and protein oxidation in cortical cells [48, 49, 50, 51, 52]. This antioxidant property of lithium was attributed to an increase in glutathione levels in neurons and human neuroblastoma SH‐SY5Y cells [51]. This suggests that long‐term exposure to low lithium concentrations could confer some protection against oxidative stress. Consistent with this hypothesis, Frey et al. reported that in vivo pretreatment with lithium for 2 weeks inhibited amphetamine‐induced lipid peroxidation in rat hippocampus [53]. In vitro, Shao et al. also showed that treatment with lithium inhibited the increase of lipid peroxidation and protein oxidation induced by glutamate and the decrease of cell viability induced by H2O2[50]. These protective effects, apart from increasing the levels of glutathione and glutathione S‐transferase activity, could also be mediated by reducing intracellular calcium and stabilizing the mitochondrial membrane. Several studies have shown that GSK‐3 inhibition by lithium may also increase the resistance to oxidative stress [53]. Using HT22 neurons, lithium treatment increased the levels of serine‐phosphorylated (inactive) GSK‐3β, indicating that inhibition of this enzyme may play a role in oxidative stress resistance in HT22 cells after glutamate and H2O2 treatment [54]. Since oxidative stress is believed to occur in the pathogenesis of many neurodegenerative diseases, mainly AD and PD, drugs that lead to a decrease in oxidative stress may therefore provide a suitable strategy for the development of compounds to prevent and treat neurodegenerative conditions [37].
However, a study performed in B65 neuroblastoma cells and CGNs compared the antioxidant properties of lithium and SB‐415286 and found that mean acute treatment with lithium did not exert any antioxidant effects against H2O2, while SB‐415286 showed strong antioxidant properties [55]. Thus, because GSK‐3 is inhibited following acute treatment with lithium, it can be concluded that GSK‐3 is not involved in the antioxidant properties of this compound. Previous reports have shown that apoptosis inducing factor (AIF) plays an important physiological role in mitochondria by maintaining complex I function, and it also exerts a reactive oxygen species‐scavenging effect [56]. Although in our experiments, we have reported that lithium prevented the mitochondrial release of apoptosis‐inducing factor, it did not show any antioxidant effects. Therefore, the prevention of mitochondrial AIF localization is not involved in the antioxidant properties of lithium [57].
Additional Neuroprotective Pathways Involved in Lithium Neuroprotection
The neuroprotective effects of lithium could also be due to an increase in the antiapoptotic protein Bcl‐2. Manji et al. (2000) reported that lithium clearly increases Bcl‐2 levels in frontal cortex, hippocampus, and striatum in vivo, and in cultured cells of both rodent and human neuronal origin in vitro. In immunohistochemical studies, chronic lithium treatment of rats led to a marked increase in the number of Bcl‐2 immunoreactive cells in the dentate gyrus and striatum [59]. Furthermore, lithium increased Bcl‐2 levels in human neuroblastoma SH‐SY5Y cells, in C57BL/6 mice, in rat cerebellar granule cells, and in nucleus magnocellularis neurons [14, 58, 59, 60].
Moreover, lithium has been reported to reduce the levels of proapoptotic protein 53 in both cerebellar granule cells and SH‐SY5Y cells [58, 59, 60]. The role of lithium in p53 is important because there is evidence for a pivotal function of p53 in neuronal death in many neurodegenerative diseases. Data from in vitro and in vivo models document increased p53 levels in apoptotic neurons. Moreover, results obtained in p53‐deficient mice or neurons, and studies employing p53 antisense oligonucleotides or pharmacological p53 inhibitors, demonstrated a role for p53‐mediated neuronal apoptosis in experimental models that reproduce neurodegenerative diseases [61]. Several studies have implicated p53 in the neuronal death that occurs in stroke, traumatic brain injury, AD, PD, and ALS [62].
In another study, it has been demonstrated that lithium exerts neuroprotective effects on ethanol‐induced neuronal apoptosis via activation of the intrinsic pathway [63]. However, ethanol did not affect the activation of Akt, the best‐known antiapoptotic protein kinase. Likewise, ethanol did not alter the activity of GSK‐3β, a downstream substrate of Akt, and nor did SB415236, a specific GSK‐3β inhibitor, block ethanol‐induced apoptosis. These observations support the hypothesis that the Akt/GSK‐3β pathway is not involved in lithium's neuroprotection against ethanol neurotoxicity [63].
Parkinson disease is the second most common neurodegenerative disease and affects about 1% of the population aged 65 or more. Several neurotoxins, among them N‐methyl‐4‐phenyl‐1,2,3,6‐tetrahydropyridine (MPTP), are currently used to reproduce PD in animals. Thus, this neurotoxin is the most widely used in experimental models of PD, and chronic lithium treatment prevents MPTP neurotoxicity in mice [64]. The most plausible mechanism involved in this process is through the increase of Bcl‐2 (neuroprotective) expression and the decrease of Bax (apoptotic) expression.
Huntington disease is a fatal autosomal dominant neurodegenerative disorder characterized by behavioral pathologies, notably chorea, psychosis, and dementia. In rats, chronic lithium treatment at therapeutic doses for 16 days showed neuroprotective effects and decreased the size of striatal lesions induced by intrastriatally administered quinolinic acid [65]. Another neurotoxin used in experimental models of HD is 3‐nitropropionic acid (3‐NPA). This drug is an inhibitor of succinate dehydrogenase, a complex II respiratory enzyme required for mitochondrial energy production. In an attempt to elucidate the molecular mechanisms underlying the neuroprotective effects of lithium, our group focused its research on the administration of this compound in vivo and in vitro. We demonstrated that calpain is a key target activated by 3‐NP during neuronal cell loss. Likewise, activation of CDK5 by 3‐NPA could be a key target involved in the neurotoxic effects of this neurotoxin [66]. We also showed that lithium modulates CDK5 activation in neuronal cell cultures [67], and in vivo studies confirm this data; we therefore proposed that animals treated with 3‐NP and dietary lithium supplementation would be protected against 3‐NPA neurotoxicity. Accordingly, we suggest that lithium's neuroprotective effects are mediated by the inhibition of calpain activation and inhibition of CDK5 activation. Furthermore, prevention of p35 cleavage by calpain and formation of the p25 isoform, which is involved in an increase in CDK5 activity and neurodegeneration, are also attenuated [66, 67, 68, 69, 70]. Therefore, lithium has the capacity to reduce the breakdown of p35 to p25, thereby indicating inhibition of CDK5 activation. We also corroborated this observation by demonstrating an increase in the phosphorylated form of MEF2 [66, 67]. Since this transcription factor generates transcriptional signals for neuronal survival, its inhibition leads to cell death and, therefore, its rescue by lithium could be protective for the cell. In this regard, we propose that the neuroprotective action of lithium in response to 3‐NPA is exerted, in part, by an effect on calcium movements in neuronal cells. These data are in agreement with previous studies in which lithium treatment reversed the calcium increase mediated by glutamate and other neurotoxins, which are correlated with its neuroprotective action [49, 53]. These observations reinforce the hypothesis that inhibition of intracellular calcium increase could be the potential target of lithium neuroprotection.
A neuroprotective effect of lithium has also been demonstrated in murine human immunodeficiency virus‐1 encephalitis models. Thus, in laboratory tests lithium exerts protective effects in neurons from neurotoxic secretions of HIV‐1‐infected monocyte‐derived macrophages [71]. This neuroprotection was mediated, in part, through the classical phosphatidylinositol 3‐kinase/Akt and GSK‐3β pathways.
β‐Bungarotoxin is a neurotoxin purified from the venom of the elapid snake, Bungarus multicinctus. β‐Bungarotoxin was able to induce the activation of NMDA receptor and L‐type calcium channel, resulting in Ca2+ influx in cultured neurons [72]. The long‐term treatment of CGNs with lithium protects them against β‐bungarotoxin neurotoxicity. The neurotoxin increased intracellular Ca2+ levels and reactive oxygen species production, and decreased mitochondrial membrane potential. However, lithium was able to inhibit all these physiological changes. Accordingly, modulation of intracellular calcium homeostasis represents, at least in part, the molecular mechanism by which lithium exerts its neuroprotection and, perhaps, its clinical actions in the treatment of manic‐depressive illness [72, 73].
Ouabain, a potent inhibitor of Na,K‐ATPase, is a model of ion dysregulation that may occur in bipolar illness. Lithium pretreatment at therapeutic levels had a protective effect that was evident after 3 days in neuronal cell preparations. Although the mechanism by which lithium might protect against ouabain remains unclear, induction of new gene expression could be involved [74].
Lithium has also been shown to exert protective effects in age‐induced cerebellar granule cell death [75]. In addition, it protects SH‐SY5Y cells against thapsigargin and the 1‐methyl‐4‐phenylpyridinium ion, which induces cell death, and increases the expression of endoplasmic reticulum stress proteins in primary cultured rat cerebral cortical cells [1, 10, 49]. The activation of this kinase has been implicated in the neurotoxic effects of kainic acid [76, 77].
Staurosporine‐induced apoptosis in human neuroblastoma cells (SH‐SY5Y) is a well‐known apoptotic model. Recently, exposure of cells to different therapeutic concentrations of lithium increased the expression of the protein Six1. When the physiological effects of this protein were evaluated (overexpression and inhibition via siRNA), it was found that Six1 inhibits apoptosis through the blockade of caspase‐3 activation. Furthermore, the silencer of Six1 inhibits the neuroprotective effects of lithium. Accordingly, Six1 may be a new protein involved in the neuroprotective effects of lithium [78].
Others studies have investigated the neuroprotective effects of lithium in vivo. Inouye et al. (1995) observed that lithium pretreatment delayed radiation‐induced apoptosis in external granular layer cells of mice. In rats, lithium pretreatment attenuated both the behavioral deficits (passive avoidance and ambulatory behavior) and the reduction in choline acetyltransferase activity caused by forebrain cholinergic system lesions [79]. Chronic lithium treatment attenuated kainic acid‐induced reduction in glutamate decarboxylase levels in rats, and furthermore, it also appears that chronic low doses of lithium exerted considerable protective effects against middle cerebral artery occlusion [80, 81] and offered increased neuroprotection in a genetic ALS model, the G93A mouse [6]. In this mouse model, lithium increases the number of neurons in lamina VII.
Potential Therapeutic Avenues
The mechanism of action of lithium, whether as a neuroprotector or a drug for bipolar disorders, has remained unknown for many years. However, currently the role of lithium as an inhibitor of GSK‐3β has been established, and given greater knowledge of the functions of this kinase, it is possible to consider the use of this ion for therapeutic purposes other than its present ones. In addition, our group has demonstrated that lithium not only inhibits GSK‐3β but also acts on CDK5 in cellular models of neurotoxicity [67]. Therefore, the neuroprotective mechanisms of lithium both in vitro[67, 82] and in vivo[64] are related to its powerful action on GSK‐3β, Akt, and CDK5. Therefore, lithium is a promising drug not only as neuroprotective therapy but also as a pharmacological tool to study the neurodegenerative processes related to aging. An initial approach to treatment possibilities would be the inhibition of tau phosphorylation and structuring mechanisms that occur in the SAMP8 mouse model, which involves chronic lithium administration.
To our knowledge, the molecular mechanisms or cellular changes by which accelerated aging takes place in SAMP8, and which occur at the cerebral level, have yet to be described sufficiently. Furthermore, it has been suggested that this mouse model could be useful for studying the pathology of AD. Therefore, our group first characterized the model with respect to hyperphosphorylated tau forms, CDK5 and GSK‐3β activation. This work demonstrated a highly significant increase in hyperphosphorylated isoforms of tau, accompanied by an activation of CDK5/p25, in 5‐month‐old SAMP8 mice [83]. We also observed changes in the activity of proteases involved in neurodegeneration, such as caspases and calpains, as well as lipid peroxidation and protein damage. In addition, and as expected, we noted the development of oxidative stress, which was accompanied by modifications in the expression and activity of enzymes related to oxidative detoxification, such as catalase and superoxide dismutase; morphologically, neuronal loss was observed, with cerebral cortex and hippocampus destruction and gliosis [83]. Recently, we determined an increase of IgG extrusion in the hippocampus of 12‐month‐old SAMP8 mice, thus illustrating the dysfunction of their hematoencephalic barrier [84]. Gene expression studies of some of these proteins were also conducted, although no significant changes were demonstrated in any of them. Having demonstrated that this mice strain presents a characteristic morphology in terms of tau hyperphosphorylation and cellular changes, and given the neurodegenerative processes observed, the next step involved studying whether long‐term treatment with a GSK‐3β inhibitor would reduce tau hyperphosphorylation, and determining if this was accompanied by a reduction in the activity of kinases involved in this process, that is, CDK5 and GSK‐3β. We therefore treated 1‐month‐old senescence‐accelerated mice (SAMP8) and mouse controls (SAMR1) with lithium for 8 weeks. During the treatment period, lithium serum levels were monitored by mass spectrometry, reaching mean levels of 0.5 mEq/L; these values are considered therapeutic according to the literature [85]. The data indicated that the activity of GSK‐3β and CDK5, assessed by the breakdown of p35 to p25, was notably reduced, and that as a consequence the phosphorylation levels of tau in Ser199 were also diminished. The activation of proteases involved in the phosphorylation route, for example, calpains, was also studied. However, both biochemical measures and the study of α‐spectrin breakdown showed that lithium does not seem to act at this level. As regards gene expression, the preliminary results indicate that levels of mRNA for GSK‐3β, CDK5, and tau were not significantly modified, thus suggesting that lithium would be acting at the posttranscriptional level. The results obtained therefore indicate that inhibition of the GSK‐3β/CDK5 pathway is a promising therapeutic avenue as regards the prevention and treatment of the neurodegenerative changes associated with aging.
Mention should also be made of longevity studies conducted in the worm, Caenorhabditis elegans, which demonstrated that PI3K‐Akt plays a role in the process of cell survival in aging, probably via the modulation of chromatin structure [86]. The PI3K‐Akt pathway has previously been studied in aging, and its role in extending lifespan in adult worms has been demonstrated [87]. Interestingly, in other experimental models such as Drosophila melanogaster and mice, mutations in the insulin receptor, which regulates the Akt pathway, extend lifespan through FOXO regulation [87, 88, 89]. Accordingly, the insulin‐signaling pathway has also been shown in a variety of experimental models to be important as regards extending lifespan and in the regulation of cell survival. In a recent study, our group used a pharmacological approach to understand the role of lithium in a well‐characterized in vitro model of spontaneous (or age‐induced) neuronal apoptosis in CGNs [75, 90]. We also studied the effects of lithium in the SAMP8 mouse model to determine the role of AKT in aging. The results of these experiments demonstrated that the neuroprotective activity of lithium appears to be strongly correlated with Akt activation; this process could be due to the inhibition of its downstream targets such as GSK‐3β and FOXO‐1 [90]. Likewise, PI3K/Akt activation is the other major signaling pathway that directly modulates the activity of FOXO factors. The FOXO families of forkhead transcription factors are involved in the process of neuronal apoptosis and aging [87, 88, 89]. As noted above, FOXO‐1 is regulated by Akt phosphorylation, and once FOXO‐1 has been phosphorylated it is exported from the nucleus to the cytoplasm, thereby repressing FOXO transcriptional activity. Moreover, lithium treatment of rats has beneficial effects on neuroplasticity in the aged rat hippocampus.
Conclusion
Lithium is probably not an ideal drug since it has side effects. However, given that it is already approved for therapeutic purposes, understanding the mechanism of its action may benefit the development of neuroprotectants. As a result of more recent research findings, there has been a resurgence of interest in lithium neuropharmacology, mainly due to studies reporting important neurotrophic and neuroprotective effects. Recent advances in cellular and molecular biology have also shown that it may be useful in the treatment of acute brain injuries, such as ischemia, and chronic neurodegenerative disorders such as AD, PD, HD, and ALS. Lithium appears to offer neuroprotection through multiple intersecting mechanisms. For example, it reduces proapoptotic functions by directly inhibiting GSK‐3β activity and indirectly inhibiting NMDA‐receptor‐mediated calcium influx. It also increases the levels of neuroprotective proteins such as Bcl‐2 in the central nervous system (CNS). Furthermore, its effects on the cell cycle, where it inhibits expression of transcription factor E2F‐1 in neurons, indicate the potential application of lithium in all these neurodegenerative disorders. Finally, the potential effects of lithium on aging and its potential role as an antiaging drug are also of interest [91].
Conflict of Interest
The authors declare no conflict of interest.
Acknowledgments
This study was supported by grants from Spain's Ministerio de Educación y Ciencia (BFU/2006‐11981, SAF2006‐13092), Fondo de Investigación Sanitaria, Instituto de Salud Carlos PI080400. We are grateful to the Autonomous Government of Catalonia for supporting research groups (2005/SGR00893) and to the TV3 Marathon (063230). We thank the Language Assessment Service of the University of Barcelona for revising the manuscript.
References
- 1. Rowe MK, Chuang DM. Lithium neuroprotection: Molecular mechanisms and clinical implications. Expert Rev Mol Med 2004;6:1–18. [DOI] [PubMed] [Google Scholar]
- 2. Zhong J, Lee WH. Lithium: A novel treatment for Alzheimer's disease? Expert Opin Drug Saf 2007;6:375–383. [DOI] [PubMed] [Google Scholar]
- 3. Feng HL, Leng Y, Ma CH, Zhang J, Ren M, Chuang DM. Combined lithium and valproate treatment delays disease onset, reduces neurological deficits and prolongs survival in an amyotrophic lateral sclerosis mouse model. Neuroscience 2008;155:567–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Aghdam SY, Barger SW. Glycogen synthase kinase‐3 in neurodegeneration and neuroprotection: Lessons from lithium. Curr Alzheimer Res 2007;4:21–31. [DOI] [PubMed] [Google Scholar]
- 5. Dunn N, Holmes C, Mullee M. Does lithium therapy protect against the onset of dementia? Alzheimer Dis Assoc Disord 2005;19:20–22. [DOI] [PubMed] [Google Scholar]
- 6. Fornai F, Longone P, Ferrucci M, Lenzi P, Isidoro C, Ruggieri S, Paparelli A. Autophagy and amyotrophic lateral sclerosis: The multiple roles of lithium. Autophagy 2008;4:527–530. [DOI] [PubMed] [Google Scholar]
- 7. Engel T, Goñi‐Oliver P, Lucas JJ, Avila J, Hernández F. Chronic lithium administration to FTDP‐17 tau and GSK‐3beta overexpressing mice prevents tau hyperphosphorylation and neurofibrillary tangle formation, but pre‐formed neurofibrillary tangles do not revert. J Neurochem 2006;99:1445–1455. [DOI] [PubMed] [Google Scholar]
- 8. Bauer M, Alda M, Priller J, Young LT; International Group For The Study Of Lithium Treated Patients (IGSLI) . Implications of the neuroprotective effects of lithium for the treatment of bipolar and neurodegenerative disorders. Pharmacopsychiatry 2003;36(Suppl 3):S250–254. [DOI] [PubMed] [Google Scholar]
- 9. Gould TD, Manji HK. Glycogen synthase kinase‐3: A putative molecular target for lithium mimetic drugs. Neuropsychopharmacology 2005;30:1223–1237. [DOI] [PubMed] [Google Scholar]
- 10. Rowe MK, Wiest C, Chuang DM. GSK‐3 is a viable potential target for therapeutic intervention in bipolar disorder. Neurosci Biobehav Rev 2007;31:920–931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hongisto V, Smeds N, Brecht S, Herdegen T, Courtney MJ, Coffey ET. Lithium blocks the c‐Jun stress response and protects neurons via its action on glycogen synthase kinase 3. Mol Cell Biol 2003;23:6027–6036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Linseman DA, Cornejo BJ, Le SS, Meintzer MK, Laessig TA, Bouchard RJ, Heidenreich KA. A myocyte enhancer factor 2D (MEF2D) kinase activated during neuronal apoptosis is a novel target inhibited by lithium. J Neurochem 2003;85:1488–1499. [DOI] [PubMed] [Google Scholar]
- 13. Beaulieu JM, Sotnikova TD, Yao WD, Kockeritz L, Woodgett JR, Gainetdinov RR, Caron MG. Lithium antagonizes dopamine‐dependent behaviors mediated by an AKT/glycogen synthase kinase 3 signaling cascade. Proc Natl Acad Sci U S A 2004;101:5099–5104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chuang DM. The antiapoptotic actions of mood stabilizers: Molecular mechanisms and therapeutic potentials. Ann N Y Acad Sci 2005;1053:195–204. [DOI] [PubMed] [Google Scholar]
- 15. Beaulieu JM, Gainetdinov RR, Caron MG. The Akt‐GSK‐3 signaling cascade in the actions of dopamine. Trends Pharmacol Sci 2007;28:166–172. [DOI] [PubMed] [Google Scholar]
- 16. Chalecka‐Franaszek E, Chuang DM. Lithium activates the serine/threonine kinase Akt‐1 and suppresses glutamate‐induced inhibition of Akt‐1 activity in neurons. Proc Natl Acad Sci U S A 1999;96:8745–8750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Avila J, Hernández F. GSK‐3 inhibitors for Alzheimer's disease. Expert Rev Neurother 2007;7:1527–1533. [DOI] [PubMed] [Google Scholar]
- 18. Yeste M, Alvira D, Verdaguer E, Tajes M, Folch J, Rimbau V, Pallàs M, Camins A. Evaluation of acute antiapoptotic effects of Li+ in neuronal cell cultures. J Neural Transm 2007;114:405–416. [DOI] [PubMed] [Google Scholar]
- 19. Mora A, Sabio G, González‐Polo RA, Cuenda A, Alessi DR, Alonso JC, Fuentes JM, Soler G, Centeno F. Lithium inhibits caspase 3 activation and dephosphorylation of PKB and GSK3induced by K+ deprivation in cerebellar granule cells. J Neurochem 2001;78:199–206. [DOI] [PubMed] [Google Scholar]
- 20. Alvarez G, Muñoz‐Montaño JR, Satrústegui J, Avila J, Bogónez E, Díaz‐Nido J. Regulation of tau phosphorylation and protection against beta‐amyloid‐induced neurodegeneration by lithium. Possible implications for Alzheimer's disease. Bipolar Disord 2002;4:153–165. [DOI] [PubMed] [Google Scholar]
- 21. Engel T, Goñi‐Oliver P, Gómez de Barreda E, Lucas JJ, Hernández F, Avila J. Lithium, a potential protective drug in Alzheimer's disease. Neurodegener Dis 2008;5:247–249. [DOI] [PubMed] [Google Scholar]
- 22. Muyllaert D, Kremer A, Jaworski T, Borghgraef P, Devijver H, Croes S, Dewachter I, Van Leuven F. Glycogen synthase kinase‐3beta, or a link between amyloid and tau pathology? Genes Brain Behav 2008;7:57–66. [DOI] [PubMed] [Google Scholar]
- 23. Terao T, Nakano H, Inoue Y, Okamoto T, Nakamura J, Iwata N. Lithium and dementia: A preliminary study. Prog Neuropsychopharmacol Biol Psychiatry 2006;30:1125–1128. [DOI] [PubMed] [Google Scholar]
- 24. Phiel CJ, Wilson CA, Lee VM, Klein PS. GSK‐3alpha regulates production of Alzheimer's disease amyloid‐beta peptides. Nature 2003;423:435–439. [DOI] [PubMed] [Google Scholar]
- 25. Pérez M, Hernández F, Lim F, Díaz‐Nido J, Avila J. Chronic lithium treatment decreases mutant tau protein aggregation in a transgenic mouse model. J Alzheimers Dis 2003;5:301–308. [DOI] [PubMed] [Google Scholar]
- 26. Rockenstein E, Torrance M, Adame A, Mante M, Bar‐on P, Rose JB, Crews L, Masliah E. Neuroprotective effects of regulators of the glycogen synthase kinase‐3beta signaling pathway in a transgenic model of Alzheimer's disease are associated with reduced amyloid precursor protein phosphorylation. J Neurosci 2007;27:1981–1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Feyt C, Kienlen‐Campard P, Leroy K, N’Kuli F, Courtoy PJ, Brion JP, Octave JN. Lithium chloride increases the production of amyloid‐beta peptide independently from its inhibition of glycogen synthase kinase 3. J Biol Chem 2005;280:33220–33227. [DOI] [PubMed] [Google Scholar]
- 28. Wei H, Qin ZH, Senatorov VV, Wei W, Wang Y, Qian Y, Chuang DM. Lithium suppresses excitotoxicity‐induced striatal lesions in a rat model of Huntington's disease. Neuroscience 2001;106:603–612. [DOI] [PubMed] [Google Scholar]
- 29. Senatorov VV, Ren M, Kanai H, Wei H, Chuang DM. Short‐term lithium treatment promotes neuronal survival and proliferation in rat striatum infused with quinolinic acid, an excitotoxic model of Huntington's disease. Mol Psychiatry 2004;9:371–385. [DOI] [PubMed] [Google Scholar]
- 30. Chuang DM, Manji HK. In search of the Holy Grail for the treatment of neurodegenerative disorders: Has a simple cation been overlooked Biol Psychiatry 2007;62:4–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Verdaguer E, Jordà EG, Alvira D, Jiménez A, Canudas AM, Folch J, Rimbau V, Pallàs M, Camins A. Inhibition of multiple pathways accounts for the antiapoptotic effects of flavopiridol on potassium withdrawal‐induced apoptosis in neurons. J Mol Neurosci 2005;26:71–84. [DOI] [PubMed] [Google Scholar]
- 32. McShea A, Harris PL, Webster KR, Wahl AF, Smith MA. Abnormal expression of the cell cycle regulators P16 and CDK4 in Alzheimer's disease. Am J Pathol 1997;150:1933–1939. [PMC free article] [PubMed] [Google Scholar]
- 33. Thakur A, Siedlak SL, James SL, Bonda DJ, Rao A, Webber KM, Camins A, Pallàs M, Casadesus G, Lee HG, et al Retinoblastoma protein phosphorylation at multiple sites is associated with neurofibrillary pathology in Alzheimer disease. Int J Clin Exp Pathol 2008;1:134–146. [PMC free article] [PubMed] [Google Scholar]
- 34. Ahn KW, Joo Y, Choi Y, Kim M, Lee SH, Cha SH, Suh YH, Kim HS. Swedish amyloid precursor protein mutation increases cell cycle‐related proteins in vitro and in vivo. J Neurosci Res 2008;86:2476–2487. [DOI] [PubMed] [Google Scholar]
- 35. Jordan‐Sciutto KL, Malaiyandi LM, Bowser R. Altered distribution of cell cycle transcriptional regulators during Alzheimer disease. J Neuropathol Exp Neurol 2002;61:358–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. McShea A, Lee HG, Petersen RB, Casadesus G, Vincent I, Linford NJ, Funk JO, Shapiro RA, Smith MA. Neuronal cell cycle re‐entry mediates Alzheimer disease‐type changes. Biochim Biophys Acta 2007;1772:467–472. [DOI] [PubMed] [Google Scholar]
- 37. Marlatt MW, Lucassen PJ, Perry G, Smith MA, Zhu X. Alzheimer's disease: Cerebrovascular dysfunction, oxidative stress, and advanced clinical therapies. J Alzheimers Dis 2008;15:199–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hamdane M, Buée L. The complex p25/Cdk5 kinase in neurofibrillary degeneration and neuronal death: The missing link to cell cycle. Biotechnol J 2007;2:967–977. [DOI] [PubMed] [Google Scholar]
- 39. Yeste‐Velasco M, Folch J, Trullàs R, Abad MA, Enguita M, Pallàs M, Camins A. Glycogen synthase kinase‐3 is involved in the regulation of the cell cycle in cerebellar granule cells. Neuropharmacology 2007;53:295–307. [DOI] [PubMed] [Google Scholar]
- 40. Tajes M, Gutierrez‐Cuesta J, Folch J, Ferrer I, Caballero B, Smith MA, Casadesus G, Camins A, Pallás M. Lithium treatment decreases activities of tau kinases in a murine model of senescence. J Neuropathol Exp Neurol 2008;67:612–623. [DOI] [PubMed] [Google Scholar]
- 41. Walz JC, Frey BN, Andreazza AC, Ceresér KM, Cacilhas AA, Valvassori SS, Quevedo J, Kapczinski F. Effects of lithium and valproate on serum and hippocampal neurotrophin‐3 levels in an animal model of mania. J Psychiatr Res 2008;42:416–421. [DOI] [PubMed] [Google Scholar]
- 42. Nonaka S, Hough CJ, Chuang DM. Chronic lithium treatment robustly protects neurons in the central nervous system against excitotoxicity by inhibiting N‐methyl‐D‐aspartate receptor‐mediated calcium influx. Proc Natl Acad Sci U S A 1998;95:2642–2647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Basselin M, Chang L, Bell JM, Rapoport SI. Chronic lithium chloride administration attenuates brain NMDA receptor‐initiated signaling via arachidonic acid in unanesthetized rats. Neuropsychopharmacology 2006;31:1659–1674. [DOI] [PubMed] [Google Scholar]
- 44. Sarkar S, Floto RA, Berger Z, Imarisio S, Cordenier A, Pasco M, Cook LJ, Rubinsztein DC. Lithium induces autophagy by inhibiting inositol monophosphatase. J Cell Biol 2005;170:1101–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Fornai F, Longone P, Cafaro L, Kastsiuchenka O, Ferrucci M, Manca ML, Lazzeri G, Spalloni A, Bellio N, Lenzi P, et al Lithium delays progression of amyotrophic lateral sclerosis. Proc Natl Acad Sci U S A 2008;105:2052–2057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Klionsky DJ, Abeliovich H, Agostinis P, Agrawal DK, Aliev G, Askew DS, Baba M, Baehrecke EH, Bahr BA, Ballabio A, et al Guidelines for the use and interpretation of assays for monitoring autophagy in higher eukaryotes. Autophagy 2008;4:151–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Sarkar S, Rubinsztein DC. Inositol and IP3 levels regulate autophagy: Biology and therapeutic speculations. Autophagy 2006;2:132–134. [DOI] [PubMed] [Google Scholar]
- 48. Machado‐Vieira R, Andreazza AC, Viale CI, Zanatto V, Cereser V Jr., Da Silva Vargas R, Kapczinski F, Portela LV, Souza DO, Salvador M, et al Oxidative stress parameters in unmediated and treated bipolar subjects during initial manic episode: A possible role for lithium antioxidant effects. Neurosci Lett 2007;421:33–36. [DOI] [PubMed] [Google Scholar]
- 49. Frey BN, Andreazza AC, Kunz M, Gomes FA, Quevedo J, Salvador M, Gonçalves CA, Kapczinski F. Increased oxidative stress and DNA damage in bipolar disorder: A twin‐case report. Prog Neuropsychopharmacol Biol Psychiatry 2007;31:283–285. [DOI] [PubMed] [Google Scholar]
- 50. Shao L, Young LT, Wang JF. Chronic treatment with mood stabilizers lithium and valproate prevents excitotoxicity by inhibiting oxidative stress in rat cerebral cortical cells. Biol Psychiatry 2005;58:879–884. [DOI] [PubMed] [Google Scholar]
- 51. De Vasconcellos AP, Nieto FB, Crema LM, Diehl LA, De Almeida LM, Prediger ME, Da Rocha ER, Dalmaz C. Chronic lithium treatment has antioxidant properties but does not prevent oxidative damage induced by chronic variate stress. Neurochem Res 2006;31:1141–1151. [DOI] [PubMed] [Google Scholar]
- 52. Cui J, Shao L, Young LT, Wang JF. Role of glutathione in neuroprotective effects of mood stabilizing drugs lithium and valproate. Neuroscience 2007;144:1447–1453. [DOI] [PubMed] [Google Scholar]
- 53. Frey BN, Valvassori SS, Réus GZ, Martins MR, Petronilho FC, Bardini K, Dal‐Pizzol F, Kapczinski F, Quevedo J. Effects of lithium and valproate on amphetamine‐induced oxidative stress generation in an animal model of mania. J Psychiatry Neurosci 2006;31:326–332. [PMC free article] [PubMed] [Google Scholar]
- 54. King TD, Jope RS. Inhibition of glycogen synthase kinase‐3 protects cells from intrinsic but not extrinsic oxidative stress. Neuroreport 2005;16:597–601. [DOI] [PubMed] [Google Scholar]
- 55. Schäfer M, Goodenough S, Moosmann B, Behl C. Inhibition of glycogen synthase kinase 3 beta is involved in the resistance to oxidative stress in neuronal HT22 cells. Brain Res 2004;1005:84–89. [DOI] [PubMed] [Google Scholar]
- 56. Pizarro JG, Yeste‐Velasco M, Rimbau V, Casadesús G, Smith MA, Pallàs M, Folch J, Camins A. Neuroprotective effects of SB‐415286 on hydrogen peroxide‐induced cell death in B65 rat neuroblastoma cells and neurons. Int J Dev Neurosci 2008;26:269–276. [DOI] [PubMed] [Google Scholar]
- 57. Lai JS, Zhao C, Warsh JJ, Li PP. Cytoprotection by lithium and valproate varies between cell types and cellular stresses. Eur J Pharmacol 2006;539:18–26. [DOI] [PubMed] [Google Scholar]
- 58. Yeste‐Velasco M, Folch J, Jiménez A, Rimbau V, Pallàs M, Camins A. GSK‐3 beta inhibition and prevention of mitochondrial apoptosis inducing factorrelease are not involved in the antioxidant properties of SB‐415286. Eur J Pharmacol 20087;588:239–243. [DOI] [PubMed] [Google Scholar]
- 59. Manji HK, Lenox RH. Signaling: Cellular insights into the pathophysiology of bipolar disorder. Biol Psychiatry 2000;48:518–530. [DOI] [PubMed] [Google Scholar]
- 60. Manji HK, Moore GJ, Chen G. Clinical and preclinical evidence for the neurotrophic effects of mood stabilizers: Implications for the pathophysiology and treatment of manic‐depressive illness. Biol Psychiatry 1999;48:740–754. [DOI] [PubMed] [Google Scholar]
- 61. Chen RW, Chuang DM. Long term lithium treatment suppresses p53 and Bax expression but increases Bcl‐2 expression. A prominent role in neuroprotection against excitotoxicity. J Biol Chem 1999;274:6039–6042. [DOI] [PubMed] [Google Scholar]
- 62. Duan W, Zhu X, Ladenheim B, Yu QS, Guo Z, Oyler J, Cutler RG, Cadet JL, Greig NH, Mattson MP. p53 inhibitors preserve dopamine neurons and motor functio in experimental parkinsonism. Ann Neurol 2002;52:597–606. [DOI] [PubMed] [Google Scholar]
- 63. Culmsee C, Zhu X, Yu QS, Chan SL, Camandola S, Guo Z, Greig NH, Mattson MP. A synthetic inhibitor of p53 protects neurons against death induced by ischemic and excitotoxic insults, and amyloid beta‐peptide. J Neurochem 2001;77:220–228. [DOI] [PubMed] [Google Scholar]
- 64. Zhong J, Yang X, Yao W, Lee W. Lithium protects ethanol‐induced neuronal apoptosis. Biochem Biophys Res Commun 2006;350:905–910. [DOI] [PubMed] [Google Scholar]
- 65. Youdim MB, Arraf Z. Prevention of MPTP (N‐methyl‐4‐phenyl‐1,2,3,6‐tetrahydropyridine)dopaminergic neurotoxicity in mice by chronic lithium: Involvements of Bcl‐2 and Bax. Neuropharmacology 2004;46:1130–1140. [DOI] [PubMed] [Google Scholar]
- 66. Senatorov VV, Ren M, Kanai H, Wei H, Chuang DM. Short‐term lithium treatment promotes neuronal survival and proliferation in rat striatum infused with quinolinic acid, an excitotoxic model of Huntington's disease. Mol Psychiatry 2004;9:371–385. [DOI] [PubMed] [Google Scholar]
- 67. Crespo‐Biel N, Camins A, Pallàs M, Canudas AM. Evidence of calpain/cdk5 pathway inhibition by lithium in 3‐nitropropionic acid toxicity in vivo and in vitro. Neuropharmacology 2009;56:422–428. [DOI] [PubMed] [Google Scholar]
- 68. Jordà EG, Verdaguer E, Canudas AM, Jiménez A, Garcia de Arriba S, Allgaier C, Pallàs M, Camins A. Implication of cyclin‐dependent kinase 5 in the neuroprotective properties of lithium. Neuroscience 2005;134:1001–1011. [DOI] [PubMed] [Google Scholar]
- 69. Voisine C, Varma H, Walker N, Bates EA, Stockwell BR, Hart AC. Identification of potential therapeutic drugs for Huntington's disease using Caenorhabditis elegans. PLoS ONE 2007;2:e504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Hamdane M, Buée L. The complex p25/Cdk5 kinase in neurofibrillary degeneration and neuronal death: The missing link to cell cycle. Biotechnol J 2007;2:967–977. [DOI] [PubMed] [Google Scholar]
- 71. Camins A, Pallas M, Silvestre JS. Apoptotic mechanisms involved in neurodegenerative diseases: Experimental and therapeutic approaches. Methods Find Exp Clin Pharmacol 2008;30:43–65. [DOI] [PubMed] [Google Scholar]
- 72. Dou H, Ellison B, Bradley J, Kasiyanov A, Poluektova LY, Xiong H, Maggirwar S, Dewhurst S, Gelbard HA, Gendelman HE. Neuroprotective mechanisms of lithium in murine human immunodeficiency virus‐1 encephalitis. J Neurosci 2005;25:8375–8385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Tseng WP, Lin‐Shiau SY. Long‐term lithium treatment prevents neurotoxic effects of beta‐bungarotoxin in primary cultured neurons. J Neurosci Res 2002;69:633–641. [DOI] [PubMed] [Google Scholar]
- 74. Huang X, Lei Z, El‐Mallakh RS. Lithium normalizes elevated intracellular sodium. Bipolar Disord 2007;9:298–300. [DOI] [PubMed] [Google Scholar]
- 75. Hennion JP, El‐Masri MA, Huff MO, El‐Mailakh RS. Evaluation of neuroprotection by lithium and valproic acid against ouabain‐induced cell damage. Bipolar Disord 2002;4:201–206. [DOI] [PubMed] [Google Scholar]
- 76. Mason RP, Leeds PR, Jacob RF, Hough CJ, Zhang KG, Mason PE, Chuang DM. Inhibition of excessive neuronal apoptosis by the calcium antagonist amlodipine and antioxidants in cerebellar granule cells. J Neurochem 1999;72:1448–1456. [DOI] [PubMed] [Google Scholar]
- 77. Goodenough S, Conrad S, Skutella T, Behl C. Inactivation of glycogen synthase kinase‐3beta protects against kainic acid‐induced neurotoxicity in vivo. Brain Res 2004;1026:116–125. [DOI] [PubMed] [Google Scholar]
- 78. Plant KE, Anderson E, Simecek N, Brown R, Forster S, Spinks J, Toms N, Gibson GG, Lyon J, Plant N. The neuroprotective action of the mood stabilizing drugs lithium chloride and sodium valproate is mediated through the up‐regulation of the homeodomain protein Six1. Toxicol Appl Pharmacol 2009;235:124–134. [DOI] [PubMed] [Google Scholar]
- 79. Inouye M, Yamamura H, Nakano A. Lithium delays the radiation‐induced apoptotic process in external granule cells of mouse cerebellum. J Radiat Res 1995;36:203–208. [DOI] [PubMed] [Google Scholar]
- 80. Pascual T, Gonzalez JL. A protective effect of lithium on rat behaviour altered by ibotenic acid lesions of the basal forebrain cholinergic system. Brain Res 1995;695:289–292. [DOI] [PubMed] [Google Scholar]
- 81. Sparapani M, Virgili M, Ortali F, Contestabile A. Effects of chronic lithium treatment on ornithine decarboxylase induction and excitotoxic neuropathology in the rat. Brain Res 1997;765:164–168. [DOI] [PubMed] [Google Scholar]
- 82. Kim YR, Van Meer MP, Tejima E, Murata Y, Mandeville JB, Dai G, Chuang DM, Rosen BR, Lo EH. Functional MRI of delayed chronic lithium treatment in rat focal cerebral ischemia. Stroke 2008;39:439–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Jordà EG, Verdaguer E, Morano A, Jiménez A, Canudas AM, Camins A, Pallàs M. Lithium prevents colchicine‐induced apoptosis in rat cerebellar granule neurons. Bipolar Disord 2004;6:144–149. [DOI] [PubMed] [Google Scholar]
- 84. Sureda FX, Gutierrez‐Cuesta J, Romeu M, Mulero M, Camins A, Pallàs M. Changes in oxidative stress parameters and neurodegeneration markers in the brain of the senescence‐accelerated mice SAMP‐8. Exp Gerontol 2006;41:360–367. [DOI] [PubMed] [Google Scholar]
- 85. Pelegrí C, Canudas AM, Del Valle J, Casadesus G, Smith MA, Camins A, Pallàs M, Vilaplana J. Increased permeability of blood‐brain barrier on the hippocampus of a murine model of senescence. Mech Ageing Dev 2007;128:522–528. [DOI] [PubMed] [Google Scholar]
- 86. Pérez M, Rojo AI, Wandosell F, Díaz‐Nido J, Avila J. Prion peptide induces neuronal cell death through a pathway involving glycogen synthase kinase 3. Biochem J 2003;372:129–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. McColl G, Killilea DW, Hubbard AE, Vantipalli MC, Melov S, Lithgow GJ. Pharmacogenetic analysis of lithium‐induced delayed aging in Caenorhabditis elegans. J Biol Chem 2008;283:350–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Greer EL, Brunet A. FOXO transcription factors in ageing and cancer. Acta Physiol (Oxf) 2008;192:19–28. [DOI] [PubMed] [Google Scholar]
- 89. Hwangbo DS, Gershman B, Tu MP, Palmer M, Tatar M. Drosophila dFOXO controls lifespan and regulates insulin signalling in brain and fat body. Nature 2004;429:562–566. [DOI] [PubMed] [Google Scholar]
- 90. Greer EL, Brunet A. FOXO transcription factors at the interface between longevity and tumor suppression. Oncogene 2005;24:7410–7425. [DOI] [PubMed] [Google Scholar]
- 91. Tajes M, Yeste‐Velasco M, Zhu X, Chou SP, Smith MA, Pallàs M, Camins A, Casadesús G. Activation of Akt by lithium: Pro‐survival pathways in aging. Mech Ageing Dev 2009;130;253–261. [DOI] [PubMed] [Google Scholar]