

Abstract
The natural product caffeic acid is a specific inhibitor of 5‐lipoxygenase (5‐LOX); it also possesses antioxidant and antiinflammatory properties. The current study was designed to determine whether the neuroprotective properties of caffeic acid are due to inhibition of 5‐LOX. Cerebral damage was induced in mice by intracerebroventricular microinjection of aluminum (5.0 μg aluminum in 2.0 μL, once a day, for 5 days). Caffeic acid was administered intragastrically at 30 min prior to aluminum and repeated daily for an additional 10 days. The brain injury was determined by observation of behavioral changes in mice, as well as by measuring biochemical and pathological changes in the cerebral tissue. The levels of 5‐LOX proteins and 5‐LOX mRNA expression were measured in brain tissue. Aluminum impaired learning and memory in mice produced neuronal death in hippocampi, elevated brain malondialdehyde levels, increased protein expression of amyloid precursor protein (APP), amyloid beta, and 5‐LOX. It also increased 5‐LOX mRNA expression and decreased choline acetyl transferase (ChAT) protein expression in the brain tissue of mice. Caffeic acid prevented brain damage as well as behavioral and biochemical changes caused by aluminum overload. The results of this study suggest that overexpression of 5‐LOX accompanies the cerebral injury induced by aluminum overload in mice, and that selective inhibitors of 5‐LOX may have potential value in the treatment of aluminum neurotoxicity and conceivably of diseases associated with neuronal injury.
Keywords: Aluminum neurotoxicity, Amyloid beta, Caffeic acid, Choline acetyltransferase, Learning, Malondialdehyde, Memory, Neuronal death, 5‐Lipoxygenase
Introduction
Air, soil, and water are rich in aluminum. It is, therefore, unavoidable that we absorb this metal by various routes. Although all the physiological effects of aluminum are not clearly established, it is well known that aluminum overload can induce severe cerebral injury and neurodegeneration. The mechanism of aluminum neurotoxicity is not well understood.
Arachidonic acid is released or mobilized from membrane phospholipids by lipase pathways (the phospholipase A2 and a combination of phospholipase C and diglyceride lipase). Following mobilization, arachidonic acid is oxygenated by cyclooxygenase (COX), 5‐lipoxygenase (5‐LOX), and other enzymes. COX isozymes convert arachidonic acid into prostaglandin endoperoxides. 5‐LOX converts arachidonic acid to 5‐HPETE, which can form various leukotrienes, including leukotriene B4 (LTB4) and cysteinyl leukotrienes (LTC4, D4 and E4). The COX‐2 pathway has been shown to be a valuable target for drugs to treat brain injury.
We have shown previously that there is a close relationship between overexpression of COX‐2 and aluminum overload‐induced neuronal damage. Meloxicam, a selective inhibitor of COX‐2, protected mice from the brain damage caused by aluminum overload (Yang et al. 2006). However, it has been also recognized that inhibition of the COX‐2 pathway produces an imbalance in eicosanoid synthesis that causes undesirable effects. Inhibition of the COX‐2 pathway could lead to increased availability of PLA2‐derived polyunsaturated fatty acid and consequently to excessive production of leukotrienes and the generation of reactive oxygen species (ROS). Furthermore, increased leukotriene production leads to depletion of GSH, thus making cells vulnerable to ROS injury. It is, therefore, necessary to investigate the relationship between expression of COX‐2 and neuronal damage in mice subjected to aluminum overload.
In the periphery, 5‐LOX expression is mainly observed in granulocytes, monocytes, macrophages, B‐lymphocytes, mast cells, and Langerhans cells in skin. It was reported that 5‐LOX (−/−) mice grew normally and were fertile. Researchers considered 5‐LOX gene expression not essential for fetal development or survival. However, the available evidence indicates that 5‐LOX plays an important role in either the acute or chronic inflammatory responses (Steinhilber 1999).
5‐LOX is highly expressed in the central nervous system (CNS), particularly in thalamus, hypothalamus, brainstem, cerebellum, and hippocampus (Lammers et al. 1996). Although the physiological role of 5‐LOX in neurons is not well established, studies suggest that 5‐LOX is involved in the pathophysiology of some diseases of the CNS. 5‐LOX‐deficient mice exhibit a reduced anxiety‐like behavior (Uz et al. 2002). Qu et al. (2000) reported that the expression of 5‐LOX mRNA and protein in cerebellum and hippocampus was higher in old as compared to young rats. The 5‐LOX pathway was activated in acute cerebral damage induced by ischemia‐reperfusion or kainate, and inhibitors of 5‐LOX protect animals from brain injury (Shishido et al. 2001; Baran et al. 1994). Polymorphism of 5‐LOX promoter has been found in patients with early‐ or late‐onset Alzheimer's disease, and the onset of Alzheimer's disease was delayed in subjects with 5‐LOX promoter mutations (Qu et al. 2001).
Accordingly, we hypothesize that 5‐LOX expression participates in the pathological process of brain injury. In experiments described below we studied the relationship between 5‐LOX expression and cerebral damage induced by aluminum overload, as well as the cerebroprotective effect of caffeic acid, a selective inhibitor of 5‐LOX.
Materials and Methods
Chemicals and Antibodies
Rabbit antihuman choline acetyltransferase (ChAT) (1:200), amyloid beta (1:100) and amyloid precursor protein (APP) (1:100) polyclonal antibodies were purchased from Boster Bioteck Co. Goat antihuman 5‐LOX polyclonal antibodies (1:100) are from Santa Cruz Biotechnology Co. RNA Solv reagents were purchased from Omega Biotek Co and access RT‐PCR system from Promega Co. Caffeic acid was purchased from Sigma Chemical Co. and AlCl3·6H2O from Dongfang Reagents Co., China.
Animals
Male mice of KM strain, weighing 24–28 g, age 8 weeks, were maintained at the Laboratory Animal Care Center of Chongqing Medical University. They were kept in a regulated environment (25°C ± 1°C, 50%± 2% humidity), with 12 light/dark cycles (light from 8:00 to 20:00). All experimental procedures were approved by the Chongqing Medical University Institutional Animal Ethics Committee.
Establishment of Aluminum‐Overload Mouse Model
A stainless steel cylindrical cannula (outside diameter, 0.6 mm; inside diameter, 0.4 mm) with stopper was implanted into the left cerebral ventricle of mice (sagittal suture 1.3 mm, posterior from the bregma 0.3 mm, ventral from the dura 2.0 mm) under sodium pentobarbital anesthesia (0.5% pentobarbital solution, 50 mg/kg i.p.). The animals were allowed to recover over a 5 days period.
On the first experimental day mice were divided into four groups: (1) Aluminum‐treated group (n = 15); these animals were treated intragastrically (i.g.) with vehicle at 30 min before intracerebroventricular (i.c.v.) microinjection with 1.25% AlCl3 solution (5 μg element aluminum in 2 μL, pH 6.8) once daily for 5 successive days and for another 10 days without aluminum treatment. (2) Vehicle‐treated group (control group, n = 15); mice were given i.g. the vehicle at 30 min prior to i.c.v. microinjection with 2 μL of modified artificial cerebrospinal fluid, once daily for 5 successive days and for another 10 days after aluminum administration. The modified artificial cerebrospinal fluid with 2 ng/mL aluminum had osmotic pressure similar to that of 1.25% AlCl3 solution. (3) and (4) groups were treated with caffeic acid and aluminum (each group, n = 15), caffeic acid was administered at 30 or 10 mg/kg at 30 min prior to the administration of aluminum and for an additional 10 days after aluminum administration.
Passive Avoidance Task
On day 15 after caffeic acid administration, the step‐down type passive avoidance task was utilized as previously described (Yang et al. 2001). Briefly, mice were trained to learn to escape electric stimuli (36V). Each test mouse was placed on the grid floor with back against the platform and the intermittent electric stimuli were delivered to the grid floor for 5 min. If the mouse could immediately jump on the platform to avoid the electric stimuli, it was considered that the mouse had learned avoiding the electric stimuli. The number of trainings for mice to learn avoiding electric stimuli was recorded as a standard to evaluate its ability of study. The retention test was carried out 24 h after training. Each mouse was placed on the platform without delivery of intermittent electric stimuli to the grid floor; the time for mice to stay at the platform is recorded as the step‐down latency. An upper cut‐off time of 300 seconds was set. The step‐down latency was used as the measurement of memory retention.
Spatial Learning
The ZIL‐2 water maze was a double‐layer opaque black plastic box (80 cm × 50 cm × 20 cm) including a start point, a terminal platform, and a dead end. Near the platform was the safe region where a ladder was located for rest. The maze was filled with water to a depth of 12 cm and maintained at a temperature of 20 ± 1°C. The apparatus was used to evaluate spatial learning and memory function of mice as described by Luo et al. (2003) On the 17th day after caffeic acid administration, mice were placed at the start point nearest to the platform. If mice failed to climb the ladder within 2 min, they were removed from the water and placed on the ladder for 15 seconds. On the 18–20th day, mice were placed at the dead end farthest to the platform. On the 21st day, the latency (the time for mice to find the terminal platform) was recorded to evaluate the spatial learning performance of mice.
Malondialdehyde Levels
Cerebral tissue malondialdehyde content was determined using the thiobarbituric acid (TBA) method according to the manual (Jiancheng Bioengineering Ltd, Nanjing, China). Briefly, fresh brain tissue (100 mg) was homogenized with normal saline. 0.1 mL homogenate (10%) was used for determination of malondialdehyde content. The absorbance of samples was detected at 533 nm with a spectrophotometer. The protein content of samples was measured using Coomassie brilliant blue.
Cerebral Histological Sections and Immunohistochemistry Analysis
After behavioral evaluation, mice were anesthetized with sodium pentobarbital, 50 mg/kg i.p. and transcardially perfused with 100 mL of 0.9% saline containing heparin (250 U) followed by 100 mL of fixing solution containing 3.5% formaldehyde and 0.1 mol/L phosphate buffer (pH 7.2). The brain was removed and cut into coronal sections of 4 μm thickness. Some of the sections were stained with hematoxylin and eosin. The remaining sections were fixed with acetone for 20 min, and then kept in an ultra low temperature freezer (−70°C) for immunohistochemistry analysis.
Immunohistochemical staining for ChAT, amyloid beta, APP, and 5‐LOX protein was performed by the streptomycin‐biotin peroxidase complex (SABC) method using a Histostain™‐Plus‐ Kit (Beijing Zhongshan Reagents Co.). Briefly, the endogenous peroxidase activity of frozen cerebral sections was blocked by incubation in a hydrogen peroxide solution (3%) for l0 min. After being rinsed with distilled water, the sections were incubated with phosphate buffered saline (PBS) for 5 min, followed by incubation with nonimmune serum (l0%) to block the nonspecific binding with the second antibody. Subsequently the tissue sections were incubated at 4°C overnight with rabbit anti‐ChAT, amyloid beta, APP, and 5‐LOX antibodies. After being washed for 3 min, the sections were incubated with goat antirabbit IgG at 37°C for 20 min, and then with biotin peroxidase complex for another 20 min. This was followed by 3,3′‐diaminobenzidine (DAB) staining for 30 min. The sections were then rinsed with distilled water for 5 min and counterstained with hematoxylin. The CM‐2000B biomedicine image analysis system (Beihang) was used to measure the content of ChAT, myloid beta, APP, and 5‐LOX protein expression.
RT‐PCR Assay of 5‐LOX mRNA Content
To study the 5‐LOX mRNA expression, the reverse transcription polymerase chain reaction (RT‐PCR) was used. Total RNA was extracted from cerebral tissue of mice using RNA Solv reagents according to the manufacturer's directions. The yield of total RNA was determined by measuring the absorbance of an aliquot of precipitated stock at 260 nm and 280 nm. Sequences of the primers used for RT‐PCR amplification were as follows: 5‐LOX upstream primer was 5′‐CCTGGAGAGAGTAACCCAATCTT‐3′, while the downstream primer was 5′‐TAGTAGAGTCTCACCACCTCCAT‐3′. The length of amplification fraction was 488bp. β‐Actin upstream primer was 5′‐GTGGGGCGCCCCAGGCACCA‐3′, and downstream primer was 5′‐CTTCCTTAATGTCACGCACGATTTC‐3′. The length of amplification fraction was 540 bp. The RT‐PCR procedure was carried out according to the Access RT‐PCR system manual. Total reaction volume was 50 μL. Final reactive concentrations were 0.2 mM for dNTPmix, 1 μM for upsteam /downstream primer, 1 mM for MgSO4, 0.1 U/μL for AMV reverse transcriptase and Tfl DNA polymerase, and 4 μL for Total RNA template.
First strand cDNA was synthesized by one cycle at 48°C for 45 min followed by one cycle at 94°C for 2 min. The second step was performed by 40 cycles at 94°C for 30 seconds, at 60°C for 1 min and 68°C at 2 min. The final extension step was carried out by one cycle at 68°C for 7 min. The reaction products were visualized on the plate electrophoresis with 1% low melt point agarose gel containing ethidium bromide. Amplification was quantified using gel imaging and analysis system (BIORAD) to calculate the integrated density of products band. The amounts of 5‐LOX mRNA were calculated as ratios of 5‐ LOX mRNA amounts to the corresponding amounts of β‐Actin mRNA (5‐LOX/β‐actin).
Statistical Analysis
The data were expressed as means ± standard deviation, significances between groups were evaluated using one‐way ANOVA, followed by Bonferroni's statistic analysis to compare the model group to the control group and then the 5‐LOX group to the model group.
Results
Behavioral Changes Caused By Aluminum in Mice
Compared with the control group, the number of training runs for aluminum‐treated mice needed to avoid electric stimuli was significantly increased and step‐down latency was significantly shortened. Caffeic acid antagonized these effects (Table 1).
Table 1.
Effects of caffeic acid on aluminum‐induced impairment of learning and memory functions in mice (Mean ± S.D., n = 10)
| Number of training | Step‐down latencies (s) | Time for exploring platform(s) | |
|---|---|---|---|
| Control | 3 ± 1.41 | 246.85 ± 78.03 | 17.89 ± 10.31 |
| Model | 6.4 ± 2.70a | 46.33 ± 33.18a | 134.0 ± 74.02a |
| Caffeic acid | |||
| 10 mg/kg | 2.8 ± 1.48b | 228 ± 101.31b | 83.29 ± 80.42 |
| Caffeic acid | |||
| 30 mg/kg | 2.2 ± 1.30b | 220.4 ± 84.02b | 7.8 ± 2.6c |
a P < 0.01 versus control group; band c P < 0.05 and 0.01, versus model group, respectively. Mice in control groups were administrated intragastrically (i.g.) with vehicle 30 min before microinjection i.c.v. of 2 μL of modified artificial cerebrospinal fluid. Mice in model groups were administrated i.g. with vehicle 30 min before microinjection i.c.v. 2 μL of 1.25% AlCl3 solution (5 μg element aluminum in 2 μL, pH 6.8). Mice in caffeic acid‐treated groups were given i.g. with caffeic acid 10.0 or 30.0 mg/kg 30 min before aluminum administration.
The latencies for aluminum‐treated mice to find the terminal platform were remarkably prolonged in comparison with the control group. This finding suggests that aluminum overload causes brain damage that leads to a decline of spatial learning and memory function. Caffeic acid prevented the impairment of spatial discriminative functions in a dose‐dependent manner (Table 1).
Pathomorphological Changes of Hippocampal Neurons Induced by Aluminum
Aluminum overload produced obvious karyopyknosis of neurons in the hippocampal CA1 subfield of mouse brain. Caffeic acid antagonized this effect in a dose‐dependent manner (Fig. 1).
Figure 1.
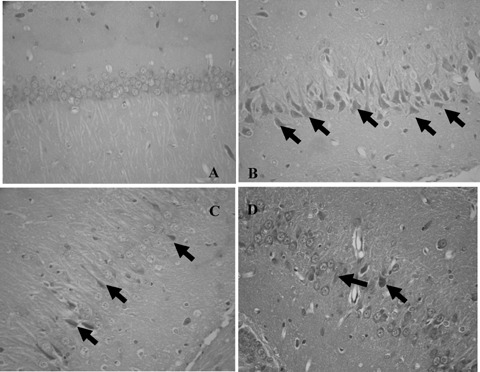
Protection of hippocampal neurons from aluminum‐induced damage by caffeic acid in mice (HE, ×400). (A) Vehicle‐treated group; (B) Vehicle plus aluminum‐treated group; (C) Caffeic acid (10 mg/kg) plus aluminum‐treated group; (D) Caffeic acid (30 mg/kg) plus aluminum‐treated group. (↑ Note karyopyknosis of cells and loss of neurons)
Changes of Malondialdehyde Content Caused by Aluminum Overload
Malondialdehyde content of brain was increased in aluminum‐overloaded mice. Caffeic acid distinctly blunted this effect (Fig. 2).
Figure 2.
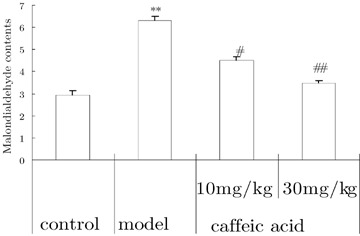
Effects of caffeic acid on changes of malondialdehyde content induced by aluminum overload in mice. (n = 5). **P < 0.01, versus control group; ## P < 0.01, versus model group. Mice in control groups were administrated intragastrically (i.g.) with vehicle 30 min before intracerebroventricular (i.c.v.) microinjection of 2 μL of modified artificial cerebrospinal fluid. Mice in model groups were administrated i.g with vehicle 30 min before microinjection i.c.v. 2 μL of 1.25% AlCl3 solution (5 μg aluminum in 2 μL, pH 6.8). Mice in caffeic acid‐treated groups were given i.g. caffeic acid 10 or 30 mg/kg 30 min before aluminum administration.
Immunohistochemsitry
In comparison to the control group, hippocampal neuronal ChAT protein expression was decreased in aluminum‐treated mice (analyzed using CM‐2000B biomedicine image analysis system). Caffeic acid prevented the decrease in ChAT protein expression (Fig. 3).
Figure 3.
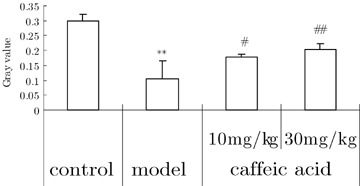
Effects of caffeic acid on changes of ChAT protein expression of hippocampi in aluminium overload mice (n = 3). **P < 0.01, versus control group; ## P < 0.01, versus model group. Mice in control groups were administrated i.g. with vehicle 30 min before i.c.v. microinjection 2 μL of modified artificial cerebrospinal fluid. Mice in model groups were administrated i.g with vehicle 30 min before i.c.v. microinjection of 2 μL of 1.25% AlCl3 solution (5 μg element aluminum in 2 μL, pH 6.8). Mice in caffeic acid‐treated groups were given i.g. caffeic acid 10 or 30 mg/kg 30 min before aluminum administration.
The APP and amyloid beta expressions were detected in hippocampal neurons of normal mice. Aluminum by i.c.v. injection distinctly increased the expression of APP and amyloid beta protein. Caffeic acid downregulated the increase of APP and amyloid beta expression (Fig. 4).
Figure 4.
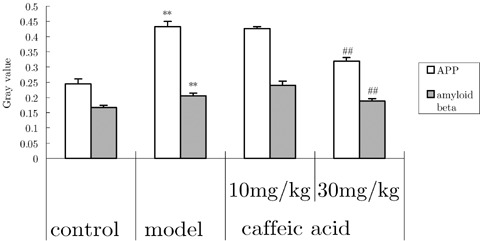
Effects of caffeic acid on changes of amyloid beta and amyloid precursor protein (APP) protein expression in hippocampi induced by aluminum overload in mice (n = 3). **P < 0.01, versus control group; ## P < 0.01, versus model group. Mice in control groups were administrated i.g. with vehicle 30 min before i.c.v. microinjection of 2 μL of modified artificial cerebrospinal fluid. Mice in model group were treated i.g. with vehicle 30 min before i.c.v. microinjection of 2 μL of 1.25% AlCl3 solution (5 μg aluminum in 2 μL, pH 6.8). Mice in caffeic acid‐treated group were given caffeic acid 10 or 30 mg/kg i.g. at 30 min before administration of aluminum.
The staining of 5‐LOX was clearly seen in perinuclei and cytoplasm of hippocampal neurons of normal mice. In aluminum‐treated mice the expression of 5‐LOX protein was more obvious. Caffeic acid significantly decreased the expression of 5‐LOX compared with that in aluminum overload mice (Fig. 5).
Figure 5.
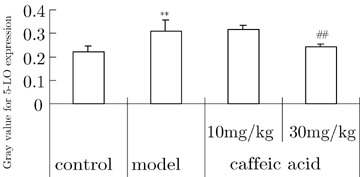
Effects of caffeic acid on changes of 5‐LOX protein expression in hippocampi induced by aluminum overload in mice (n = 3).**P < 0.01, versus control group; ## P < 0.01, versus model group. Mice in control group were treated with vehicle i.g. at 30 min prior to i.c.v. microinjection of 2 μL of modified artificial cerebrospinal fluid. Model groups received vehicle i.g. at 30 min prior to i.c.v. microinjection of 2 μL of 1.25% AlCl3 solution (5 μg element aluminum in 2 μL, pH 6.8). Mice in caffeic acid‐treated groups were given caffeic acid i.g., 10 or 30 mg/kg at 30 min prior to the administration of aluminum.
Expression of 5‐LOX mRNA in the Brain
The results of RT‐PCR analysis showed that 5‐LOX transcription (5‐LOX/β‐actin) remained at a low level in normal controls, whereas aluminum overload significantly upregulated the expression of 5‐LOX mRNA in the cerebral tissue. Caffeic acid dose‐dependently downregulated the expression of 5‐LOX mRNA (Fig. 6).
Figure 6.
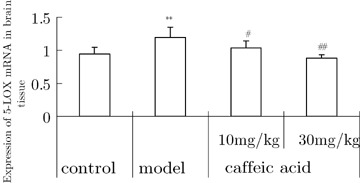
Effects of caffeic acid on changes of 5‐LOX mRNA expression in cortical brain tissue induced by aluminum overload in mice (n = 3). **P < 0.05, versus control group; # P and ## P < 0.05 and 0.01, versus model group. Mice in control groups were treated i.g. with vehicle at 30 min prior to i.c.v. microinjection of 2 μL of modified artificial cerebrospinal fluid. Mice in model groups were treated with i.g. vehicle at 30 min prior to i.c.v. microinjection of 2 μL of 1.25% AlCl3 solution (5 μg element aluminum in 2 μL, pH 6.8). Mice in caffeic acid‐treated groups were given caffeic acid 10 or 30 mg/kg i.g. at 30 min prior to administration of aluminum.
Discussion
Acute and chronic brain injury, especially that caused by neurodegenerative diseases, seriously affect the quality of human life. However, the etiology and pathogenesis of brain injury are not well understood. Thus, it is necessary to further clarify the mechanisms of cerebral damage and to develop effective cerebroprotective agents.
Our experimental results showed that aluminum impairs learning and memory functions, including spatial orientational ability, passive avoidance learning and memory in mice. Aluminum causes also neuronal death and loss of cholinergic neurons in CA1 subfield of hippocampi. It also elevates brain malondialdehyde content, APP, amyloid beta protein, 5‐LOX mRNA, and 5‐LOX protein expression. Similar results were reported by Ohtsuki et al. (1995) who found that ischemia‐reperfusion injury increases 5‐LOX mRNA expression, production of LTC4, translocation of cytosolic 5‐LOX to membranes, and generation of oxygen radicals. Uz et al. (1998) found also that the expression of 5‐LOX mRNA and protein was higher in the brain of old than young rats. Overexpression of 5‐LOX promoted senescence‐like growth arrest. Production of oxygen radicals increased in 5‐LOX‐arrested cells. Antioxidants and a low oxygen environment prevented 5‐LOX‐induced growth arrest (Catalano et al. 2004). Ikonomovic et al. (2004) reported that 5‐LOX protein is elevated in the hippocampus of Alzheimer's disease patients. Taken together, these results and our findings suggest that there may be a close relationship between overexpression of 5‐LOX and neuronal damage caused by aluminum overload, and that neuroinflammation augmented by 5‐LOX and oxidative stress may be involved in the pathogenesis of neuronal damage and neurodegeneration.
Our results also revealed that caffeic acid, a selective 5‐LOX inhibitor, protected mice from the aluminum‐induced neuronal damage that leads to deficits in learning and memory and eventual neuronal death. Caffeic acid antagonized the aluminum‐induced increase in brain malondialdehyde levels and the decrease in ChAT expression; it downregulated the overexpression of APP and of amyloid beta protein, as well as 5‐LOX mRNA and protein expression in a dose‐dependent manner. Similarly, a study with the G93A‐SOD1 mouse, a model for amyotrophic lateral sclerosis, showed increasing of 5‐LOX mRNA and protein levels on 120th day of age (West et al. 2004). Nordihydroguaiaretic acid, a 5‐LOX inhibitor, significantly extended lifespan and slowed motor dysfunction of mice, when its administration began relatively late in life (90th day). Nordihydroguaiaretic acid extended median total lifespan of G93A‐SOD1 mice by 10%, and the life expectancy following start of treatment was extended by 32% (West et al. 2004). These results further confirm our conclusion that 5‐LOX may play an important role in some neurodegenerative diseases and other neuronal injury‐related diseases, and suggest that the 5‐LOX pathway could be a candidate target for neuroprotective therapy for the cerebral damage and neurodegenerative process.
Nevertheless, further evidence should be obtained from experimental and clinical studies to confirm our hypothesis. Considering that COX‐2 is a similar inflammatory mediator derived from arachidonic acid, it should be of interest to investigate whether a combination of 5‐LOX and COX‐2 inhibitors would exert a more pronounced effect on acute and chronic cerebral damage than either inhibitor alone.
Conflict of Interest
The authors have no conflict of interest.
Acknowledgments
These studies were supported by the National Natural Science Foundation of China (No 30672211). The authors would like to thank Dr. TC He, The University of Chicago Molecular Oncology Laboratory, for his contribution and critical comments to the paper. The authors also thank Assistant Professor Q Li at UTMB, USA, for her critical comments.
References
- Baran H, Vass K, Lassmann H, Hornykiewicz O (1994) The cyclooxygenase and lipoxygenase inhibitor BW755C protects rats against kainic acid‐induced seizures and neurotoxicity. Brain Res 646: 201–206. [DOI] [PubMed] [Google Scholar]
- Catalano A, Caprari P, Soddu S, Procopio A, Romano M (2004) 5‐lipoxygenase antagonizes genotoxic stress‐induced apoptosis by altering p53 nuclear trafficking. FASEB J 18: 1740–1742. [DOI] [PubMed] [Google Scholar]
- Ikonomovic MD, Abrahamson EE, Manev H, Uz T, Isanski B, Paljug W (2004) 5‐Lipoxygenase protein is elevated in the hippocampus of Alzheimer's disease patients. Neurobiol Aging 25: S435. [Google Scholar]
- Lammers CH, Schweitzer P, Facchinetti P, Arrang JM, Madamba SG, Siggins GR, Piomelli D (1996) Arachidonate 5‐lipoxygenase and its activating protein: Prominent hippocampal expression and role in somatostatin signaling. J Neurochem 66: 147–152. [DOI] [PubMed] [Google Scholar]
- Luo J, Yin JH, Wu HZ, Wei Q (2003) Extract from Fructus cannabis activating calcineurin improved learning and memory in mice with chemical drug‐induced dysmnesia. Acta Pharmacol Sin 24: 1137–1142. [PubMed] [Google Scholar]
- Ohtsuki T, Matsumoto M, Hayashi Y, Yamamoto K, Kitagawa K, Ogawa S, Yamamoto S, Kamada T (1995) Reperfusion induces 5‐lipoxygenase translocation and leukotriene C4 production in ischemic brain. Am J Physiol 268: H1249–1257. [DOI] [PubMed] [Google Scholar]
- Qu T, Manev R, Manev H (2001) 5‐Lipoxygenase (5‐LOX) promoter polymorphism in patients with early‐onset and late‐onset Alzheimer's disease. J Neuropsychiatry Clin Neurosci 13: 304–305. [DOI] [PubMed] [Google Scholar]
- Qu T, Uz T, Manev H (2000) Inflammatory 5‐LOX mRNA and protein are increased in brain of aging rats. Neurobiol Aging 21: 647–652. [DOI] [PubMed] [Google Scholar]
- Shishido Y, Furushiro M, Hashimoto S, Yokokura T (2001) Effect of nordihydroguaiaretic acid on behavioral impairment and neuronal cell death after forebrain ischemia. Pharmacol Biochem Behav 69: 469–474. [DOI] [PubMed] [Google Scholar]
- Steinhilber D (1999) 5‐Lipoxygenase: a target for antiinflammatory drugs revisited. Curr Med Chem 6: 71–85. [PubMed] [Google Scholar]
- Uz T, Dimitrijevic N, Tueting P, Manev H (2002) 5‐lipoxygenase (5‐LOX)‐deficient mice express reduced anxiety‐like behavior. Restor Neurol Neurosci 20: 15–20. [PubMed] [Google Scholar]
- Uz T, Pesold C, Longone P, Manev H (1998) Aging‐associated up‐regulation of neuronal 5‐lipoxygenase expression: Putative role in neuronal vulnerability. FASEB J 12: 439–449. [DOI] [PubMed] [Google Scholar]
- West M, Mhatre M, Ceballos A, Floyd RA, Grammas P, Gabbita SP, Hamdheydari L, Mai T, Mou S, Pye QN et al (2004) The arachidonic acid 5‐lipoxygenase inhibitor nordihydroguaiaretic acid inhibits tumor necrosis factor alpha activation of microglia and extends survival of G93A‐SOD1 transgenic mice. J Neurochem 91: 133–143. [DOI] [PubMed] [Google Scholar]
- Yang JQ, Liu BZ, Zhou QX, He BC (2006) Protective effects of meloxicam on aluminum overload‐induced cerebral damage in mice. Eur J Pharmacol 54: 752–758. [DOI] [PubMed] [Google Scholar]
- Yang JQ, Zhou QX (2001) Protective effects of nimodipine on delayed cerebral injury induced by carbon monoxide intoxication in mice. Acta Pharmacol Sin 22: 423–426. [PubMed] [Google Scholar]