

Abstract
Cationic antimicrobial peptides (AMPs) are essential components of the innate immune system. They have attracted interest as novel compounds with the potential to treat infections with multi-drug resistant bacteria. In this study, we investigate piscidin 3 (P3), an AMP that was first discovered in the mast cells of hybrid striped bass. Prior studies have shown that P3 is less active than its homolog piscidin 1 (P1) against planktonic bacteria. However, it has the advantage of being less toxic to mammalian cells and more active on biofilms and persister cells. Both peptides cross bacterial membranes and co-localize with intracellular DNA but P3 is more condensing to DNA while P1 is more membrane active. Recently, we showed that both peptides coordinate Cu2+ through an amino-terminal copper and nickel (ATCUN) motif. We also demonstrated that the bactericidal effects of P3 are linked to its ability to form radicals that nick DNA in the presence of Cu2+. Since metal binding and membrane crossing by P3 is biologically important, we apply solid-state NMR spectroscopy to uniformly 13C-15N-labeled peptide samples to structurally characterize the ATCUN motif of P3 bound to bilayer and coordinated to Ni2+ and Cu2+. These experiments were supplemented with density functional theory calculations. Taken together these results refine the arrangement of not only the backbone but also side chain atoms of an AMP simultaneously bound to metal ions and phospholipid bilayers.
Keywords: antimicrobial peptide, magic angle spinning NMR, ATCUN motif, metallopeptide, membranes
Graphical Abstract
Mighty antimicrobial peptide: the binding of Cu2+ to antimicrobial peptide piscidin 3 enhances its potency through the formation of radicals that covalently damage neighboring molecules. Solid-state NMR and density functional theory calculations were used to study the structural effects of metal binding to the peptide. Major changes were detected at the amino end of piscidin-Cu2+ bound to bacterial cell membrane mimics.
Introduction
The study of amphipathic α-helical peptides as components of the vertebrate immune system was initiated by Zasloff’s discovery of the antibiotic activity of 23-residue magainins in frog skin.1 This was preceded by the identification of 37-residue cecropin peptides in insects, which have similar structural features and antibiotic activities.2 With obvious relevance to the treatment of infectious diseases, many natural and synthetic variants of these antimicrobial peptides (AMPs) have been characterized.3, 4 Much of the interest in these peptides results from their potential to address the increase in antibiotic resistance by human pathogens.5 In contrast to conventional antibiotics that act through specific interactions with well-defined, high-affinity targets, AMPs rely on low-affinity interactions with bacterial cell membranes.6–10 As the archetypical AMP, magainin has been subjected to extensive NMR studies in a variety of membrane mimetic environments including near-native phospholipid bilayers.11–19 Complementary results from other biophysical approaches have led to models for its mechanism of action, most of which focus on its ability to form ion channels in membranes. In recent years, it has been recognized that following their initial interactions with bacterial cell membranes, some AMPs may translocate across the membrane and affect intracellular targets, such as DNA.10, 20 The membrane activity of such AMPs arises from their cell penetrating properties rather than their ability to permeabilize and lyse bacterial membranes. In addition to their direct attack of bacteria, some AMPs have immunomodulatory effects that involve activating host cells to boost the immune response.21–24
Piscidins were the first antimicrobial cationic peptides isolated from the mast cells of hybrid striped bass.25 These 22-residue peptides have broad-spectrum activity against many pathogens, including multidrug-resistant bacteria, fungi, and viruses.26 Piscidin 1 (P1, FFHHIFRGIVHVGKTIHRLVTG) and Piscidin 3 (P3, FIHHIFRGIVHAGRSIGRFLTG) have similar sequences but are differentially expressed; although they are highly potent, their biological activities differ for unknown reasons.27 While P1 has stronger antimicrobial activity than P3 against some planktonic bacteria, P3 has higher specificity.26 Both peptides translocate across the membranes of cells, leading to co-localization with intracellular DNA.27
We previously used oriented sample solid-state NMR to solve the structures and orientations of P1 and P3 bound to bacterial cell mimics.28 These structures, which were deposited in the protein data bank (PDB), indicated that membrane-bound P1 and P3 are structurally similar. Both peptides form α-helical structures that are kinked at a central glycine (Gly13) and oriented parallel to the bilayer surface. Notably, the kink at Gly13 allows both amphipathic peptides to maximize their hydrophobic moment, as needed to optimize their hydrophobic interactions with the nonpolar core of the membrane. While the first three residues were noted to have α-helical character, this part of the α-helix was noted to be frayed due to weaker hydrogen bonding.
Recently, we demonstrated that the N-termini of both peptides coordinate Cu2+ through their amino-terminal copper and nickel (ATCUN) motif of consensus sequence XXH, where X can be any amino acid. This motif, which is FFH- for P1 and FIH- for P3, binds to Cu2+ and Ni2+ ions with high affinity. The N-terminal amino group, the two subsequent backbone amide nitrogens, and the δ-nitrogen of His3 form the metal-binding site, which has a square planar coordination geometry.29 Upon metal binding, reactive oxygen species (ROS) are formed through Fenton-like chemical reactions.30 By applying solution NMR to 15N3-H3 labelled P3, we were able to demonstrate that the side chain of H3 is involved in metal coordination.29
Our studies have shown that the antimicrobial effects of P1 are instantaneous; by contrast, it requires about one hour for those of P3 to become noticeable. These findings correlate with P1 being more membrane permeabilizing than P3 and P3 being more effective at condensing DNA and faster at cleaving isolated DNA.26 Furthermore, in living cells P3 has stronger copper-scavenging nuclease activities than P1.26 Further, P3 differs from P1 by having arginine instead of lysine at position 14. Molecular dynamics (MD) simulations show that this arginine mediates stronger interactions with DNA than the lysine in P1.26The emerging picture is that the bactericidal activities of P3 are linked to its ability to translocate across bacterial cell membranes in order to access intracellular DNA and affect it physically through aggregation and chemically through bond cleavage.
Here, we extend the characterization of the metal binding site to additional residues of P3 using solid-state NMR and gain new insights into the structure of P3 bound to not only a metal ion but also bilayers. We apply solid-state NMR spectroscopy to samples of uniformly 13C/15N-labeled peptides. The experimental results describe the effects of binding metal ions on the structure of P3 associated with phospholipid bilayers. The bilayers contained a mixture of lipids similar to those found in the bacterial cell membranes that P3 interacts with in the initial stages of its mechanism of action. We also present density functional theory (DFT) calculations to add to insights gained from the structural changes taking place at the amino terminus of the peptide upon metal coordination. Significantly, Ni2+ and Cu2+ are known to colocalize with AMPs on the surface of bacterial cells as part of the immune response.27 Therefore, it may be possible to deduce from the NMR studies the roles of metal-peptide complexes in the mechanism of antibiotic activities.
Results and Discussion
It is well established that chemical shift frequencies of individual resonances are affected by the local electronic environment, and provide local structural and dynamic information.31–34 In particular, the binding of metal ions to specific residues affects the NMR resonances of proximate residues. Since Cu2+ has unpaired electrons, the associated paramagnetic effects strongly influence both the chemical shifts and the relaxation rates of resonances from nearby sites. The shortening of relaxation rates by paramagnetic effects leads to the loss of NMR signals from sites located less than about 10 Å from the metal. Here, we take advantage of the paramagnetic effects of Cu2+ to characterize the folding of the amino terminal residues of P3 upon metal binding. We also performed experiments on peptides bound to diamagnetic Ni2+, which has previously been shown to bind the ATCUN motif in a manner similar to that of Cu2+. This enabled measurement of the paramagnetic effects of bound Cu2+.35
The photographs of the peptide-containing proteoliposome pellets in Figure 1 vividly display the colors associated with the binding of different metals to P3 in lipid bilayers constituted of 3:1 1,2-dimyristoyl-sn-glycero-3-phosphocholine (DMPC)/ 1,2-dimyristoyl-sn-glycero-3-phosphoglycerol (DMPG). The apo-P3 sample is white; in contrast, the metal-containing P3 samples are yellow and pink, reflecting absorbance of light at 420 and 525 nm by the nickel- and copper-coordinated peptide, respectively.36, 37 The solid-state NMR spectra of all three P3 samples are well-resolved. This is demonstrated by the two-dimensional 13C/13C correlation spectrum of Ni2+-bound P3 shown in Figure 2A. The 13C resonances have linewidths at half height of ~0.6 ppm. With a 10 ms mixing time, primarily cross-peaks from one-bond correlations are observed, although some signals from weak two-bond correlations are present. With a 50 ms mixing time, the signals from two- and three-bond correlations are more clearly observable in the spectra. A two-dimensional N-CA spectrum is shown in Figure 3A. A detailed analysis of 13C/13C correlation spectra obtained with 10 ms, 20 ms, 50 ms, 100 ms mixing times for proton driven spin diffusion (PDSP) as well as NCA and NCO spectra provided the resonance assignments marked in the spectra in Figures 2A and 3A.
Figure 1:
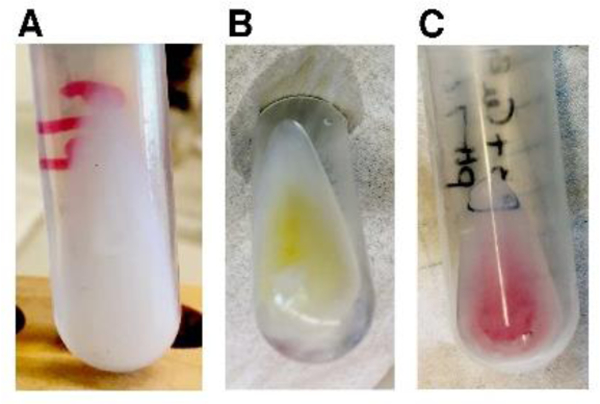
DMPC/DMPG proteoliposome samples for a 20:1 molar ratio of lipid:P3 at pH 7.4 (A) apo- (B) Ni2+ - bound (C) Cu2+-bound.
Figure 2.
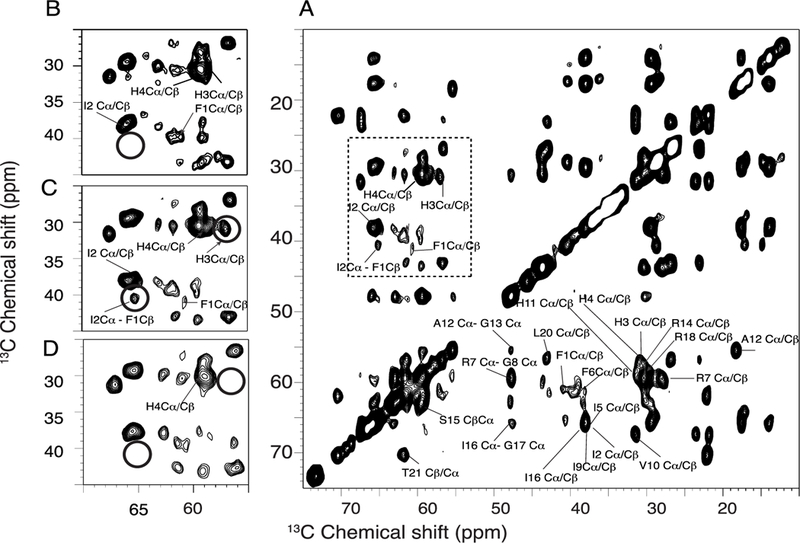
Two-dimensional 13C-13C MAS spectra of uniformly 13C/15N labeled piscidin in DMPC proteoliposomes at 7°C. All these spectra were acquired with 256 scans and 50 ms mixing time. (A) 2D homonuclear 13C/13C spin-exchange correlation spectrum of P3 bound to Ni2+. Zoomed regions for spectra of apo, Ni2+- and Cu2--bound P3 are represented in (B), (C) and (D). The experiments were performed at 750 MHz under 11.11 kHz MAS.
Figure 3.
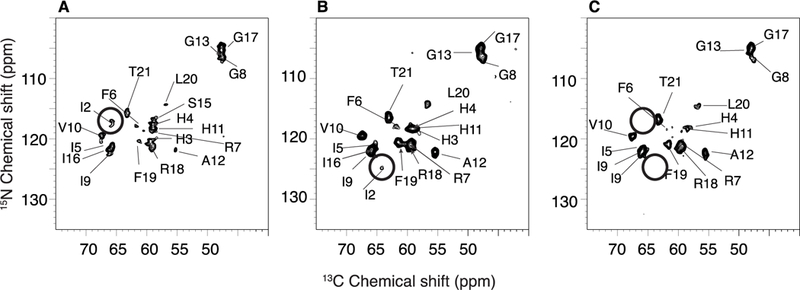
Two-dimensional heteronuclear 13C/15N correlation spectrum of P3. (A) apo-P3 (B) Ni2+-bound P3; (C) Cu2+-bound P3.metal ion.
Paramagnetic Cu2+, strongly enhances the relaxation of proximal nuclei. This identifies the location of the metal binding site. As shown in Figure 3 A and Figure 4, signals from residues 1, 2, and 3 are broadened beyond detection; as monitored by the signal intensities of other resonances, the metal ion has little or no effect on other residues in the peptide, including residue 4. In contrast to the dramatic effects of Cu2+ at the amino terminus of P3, the binding of diamagnetic Ni2+ to P3 does not abrogate the signals from residues 1, 2 and 3. However, some minor changes in linewidths (and signal intensities) are detected for the first seven residues, F1, I2, H3, H4, I5, F6, and R7. Interestingly, metal binding affects the signals of several residues near the C-terminal end of P3. For instance, the crosspeak for F19 becomes more intense upon metallation (Figure 3) while its linewidth is unaffected (Figure 4). In addition, the linewidths increase for L19 and T21 (Figure 4). These effects suggest that metal binding is sensed at the C-terminal end of P3, possibly due to intermolecular interactions mediated by hydrophobic residues present at the two extremeties of P3, such as F1, I2, I5, F6, F19 and L20.
Figure 4.
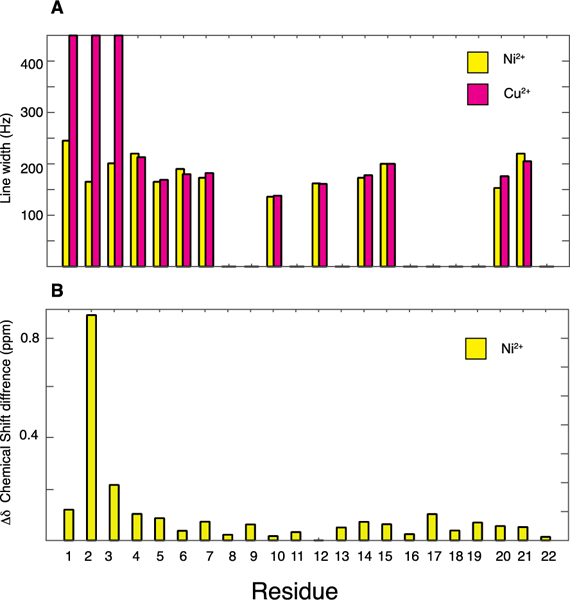
(A) Comparison of line widths between Ni2+-bound (yellow) and Cu2+-bound P3 (pink). The line widths were measured from two-dimensional 13C/13C spectra obtained with a 10 ms mixing time. (B) Chemical shift perturbations (i.e. CSDs) for 13C and 15N resonances upon Ni2+ binding to P3.
Inspection of the expanded regions of the spectra in Figures 2 and 3 demonstrate differences in the signals from the first three residues of P3. The resonances from I2 are marked by a circle. The magnitudes of metal-induced chemical shift-perturbations are quantified with the well-established equation for chemical shift deviations (CSDs): where ΔδC and ΔδN are the changes in carbon and nitrogen chemical shifts, respectively.38, 39 As shown in Figure 4B, there are significant chemical shift changes in both 13C and 15N resonances upon Ni2+ coordination at the amino end of P3.
The ATCUN motif is found in many peptides and proteins.29 The metal-bound ATCUN complexes have been shown to cleave DNA and RNA and inactivate enzymes.40, 41 The stability of the complex is governed by the basicity of the N-terminal amino group because of the competition between 1H and Cu2+ coordination. The Cu–ATCUN complex stability is significantly enhanced by bulky and hydrophobic amino acid residues at positions 1 and 2.42 Notably, P3 has an isoleucine at position 2, whose resonances are strongly affected by metal ion binding as shown in Figure 4B.
To further investigate the structure of the metallated ATCUN motif of P3, we performed density functional theory (DFT) chemical shielding calculations on the first six residues of P3 in 3:1 DMPC:DMPG28 and a model of P3-Ni2+ based on the structure of the hexamer. In this Ni2+-bound structure, the first three residues were modeled to coordinate Ni2+ with their deprotonated backbone nitrogens (N-terminal amine and two amides), with the imidazole sidechain of H3 providing a fourth coordinating ligand and generating a square planar coordination geometry (Figure 5). Residues 4-onward were modeled as an α-helix, as in the apo structure.28 The results of DFT calculations are compared to experimental shifts in Figure 6. In both the metal-bound and apo- cases, the calculated 13C chemical shieldings are consistent with the observed shifts, showing a linear trend with a slope close to −1, as expected (Figure 6A, top). This leads to a reasonable correspondence when the experimental and DFT-derived 13C CSDs are plotted for the metal-bound and apo-complexes (Figure 6B, top). This shows that the observed alpha carbon CSDs for the first three residues are consistent with the different dihedral angles adopted in the α-helix of the apo state and the square-planar geometry of the Ni2+-bound ATCUN motif. Although 15N shieldings, which are considerably more challenging to calculate accurately,43 were not well-correlated to the experimental shifts (Figure 6A, bottom), the large shift difference for the amide nitrogen of I2 was observed (Figure 6B, bottom). This CSD is consistent with the nitrogen of I2 participating in the coordination of the Ni2+ ion. Taken together, the DFT calculations provide a basis for interpreting the Ni2+-induced chemical shift changes and support a model in which the ATCUN motif of P3 binds Ni2+ in a square-planar geometry, and the bulky I2 and H4 side chains surround the metal, while residues 4–22 do not experience signficant conformational changes.
Figure 5.
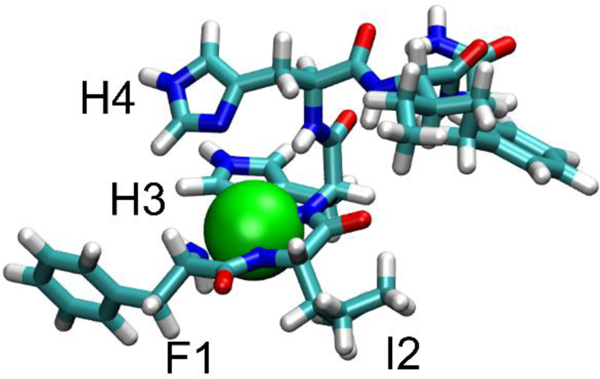
Structure of the P3 - Ni2+ complex used in the DFT chemical shift calculations. The Ni2+ ion (green sphere) is coordinated to the backbone nitrogens of F1, I2, and H3 as well as the imidazole sidechain of H3 in a square-planar geometry. The structure is consistent with bulky side chains (I2, H4) surrounding the metal ion.
Figure 6.
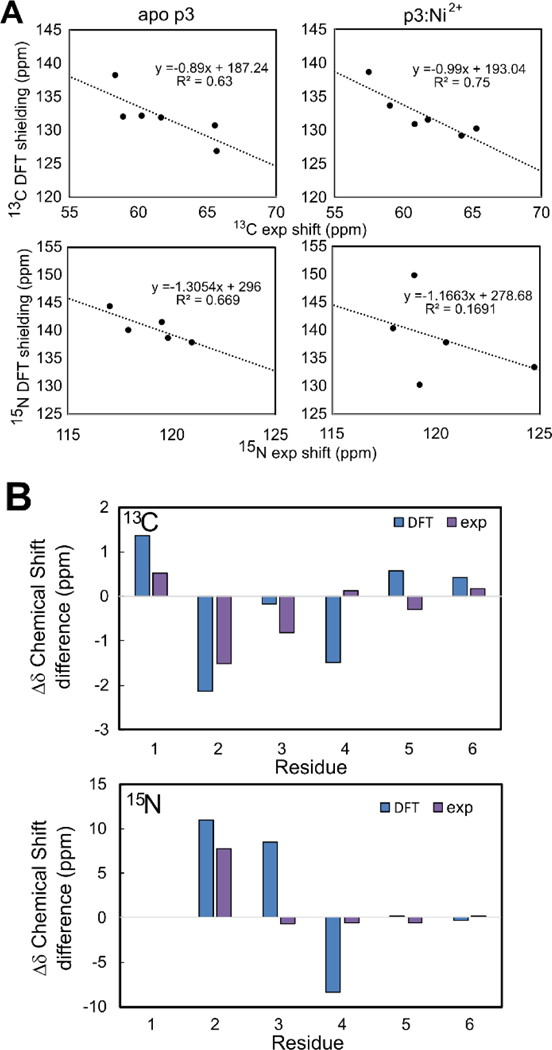
(A) Comparison of experimental chemical shifts and calculated chemical shieldings for alpha carbons (top) and amide nitrogens (bottom). Plots for apo P3 (left) and Ni2+-bound p3 (right) are shown. Symbols reflect data points and lines are linear regressions, with slopes/intercepts and R2 values displayed above the plot. (B) Comparison of experimental (purple) and DFT-derived (blue) chemical shift differences between apo-P3 and Ni2+-bound p3. Data for alpha carbons (top) and amide nitrogens (bottom) are shown.
In the apo-state, the amino terminus of P3 was previously shown to be slightly more inserted in the bilayer than the C-terminal end.28 In the halo-state, metal coordination through the ATCUN-motif results in the net loss of one positive charge. Indeed, the amine at position 1 and the amide nitrogens at positions 2 and 3 drop one proton each, resulting in a neutral amine and two negatively charged amide nitrogens.44, 45 Since the metal brings two positive charges, the peptide experiences the loss of one positive charge. This decrease in charge toghether with the bulky I2 and H4 side chains surrounding the metal ion may facilitate the insertion of the peptide into the hydrophobic environment of the bilayer and enhance its ability to cross the membrane and reach the cytoplasm.
Conclusions
The holo-state of P3, as studied here, is relevant to its natural environment and biological function. First, since AMPs such as P3 undergo post-translational modifications, including carboxyamidation, in several organelles (e.g., trans-Golgi apparatus) where copper ions are ubiquitous,46 it is highly probable that piscidins exist in the metallated state before being secreted. As a pro-peptide, the sequence of piscidin is followed by the sequence Gly-Lys.47 This dipeptide is recognized by carboxypeptidase E and the α-amidating monooxygenase (PAM) complex that remove Gly-Lys sequences at the end of pro-peptides and amidate their C-termini, as needed to generate mature peptides.48, 49 Since these post-translational modifications take place in organelles (e.g., trans-Golgi apparatus, secretory granules) where Cu2+ is present and the PAM complex needs Cu2+ for its function,46, 48 piscidins very likely bind Cu2+ during these processing steps. Second, Cu2+ ions play important roles as part of host defense and co-localize with AMPs such as P3 in cellular compartments, such as the phagosomes of immune cells.50 Third, the bactericidal effects of P3 are enhanced by copper and synergistic effects exist between Cu2+ and P3 on biofilms and persister cells.26
This study presents the first structural characterization of P3 simultaneously bound to a metal ion and bacterial cell mimics. Through a combination of solid-state NMR and DFT calculations, new insights is gained about the structure of the metal binding site, as shown in Figure 5. The first three residues form a square planar coordination geometry where the bulky side chains I2 and H4 surround the metal ion. This structural arrangement together with the net loss of one charge at the amino end of the peptide very likely play an important role in stabilizing the insertion of the N-terminus and metal ion in the hydrophobic core of the membrane it needs to cross in order to reach the cytoplasm. Our results also capture that the C-terminal end of P3 is sensitive to metal binding at the N-terminal end, possibly because intermolecular interactions taking place between multiple P3 molecules place the amino end of one peptide in proximity to the carboxylic end of another peptide.
Membrane binding is the first step in the mechanism of antimicrobial action by P3. It is followed by membrane crossing into the cytoplasm and DNA disruption. Given that P3 is expressed in vascularized tissues and exhibits low toxicity to host cells, its mechanism of action through membrane crossing followed by DNA cleavage catalyzed by metal binding may represent a novel form of specificity. The results described here provide insight into structural changes induced by metal coordination and serve to identify specific structural features of an AMP that discriminates between host and invasive cells. The results may aid in the design of anti-infective agents that demonstrate low toxicity to mammalian cells and high potency against bacterial pathogens, including those that are resistant to other classes of antibiotics.
Experimental Section
Peptide synthesis, purification, and characterization
Chemicals were purchased from Sigma-Aldrich (Saint Louis, MO) unless otherwise indicated. 13C/15N-labeled P3 (MW 2645 with labels) was synthesized from uniformly 13C/15N-labeled amino acids using Fmoc-solid-phase peptide synthesis at the University of Texas Southwestern Medical Center. The peptide was purified by reverse phase HPLC as previously described.28 Following HPLC, the peptide was washed with dilute HCl, lyophilized, and dialyzed to remove residual trifluoroacetic acid (TFA). The peptide was assessed as 98% pure by mass spectrometry.
Proteoliposome Samples
For all samples the molar peptide-to-lipid ratio (P/L) was 1:20 and the molar PC (Phosphatidylcholine) to PG (Phosphoglycerate) ratio was 3:1. The lipids were obtained from Avanti Polar Lipids (Alabaster, Alabama). The samples were prepared by co-dissolving DMPC and DMPG in a mixture of 2,2,2-trifluorethanol (TFE) and chloroform (50/50, v/v). Each sample contained approximately 4 mg of P3, after excluding an estimated 25% weight to account for the counter ions as measured by mass spectroscopy.
For apo-P3 samples ions (Figure 1A) the lipids were added to a round bottom glass tube containing the peptide dissolved in TFE. The solution was dried by passage of N2 gas. After lyophilization, the film of peptide and lipids was rehydrated with 6 mL of 20 mM HEPES at pH 7.3, warmed to about 40°C and gently mixed by vortexing and pipetting. The pH was adjusted to 7.4 with NaOH. The sample was swirled/vortexed gently to enable complete mixing of the peptide, lipids, and buffer. Next, the sample was ultra-centrifuged in a Beckman Optima-90K centrifuge at 90,000 rpm (644,000 g, Beckman NVT90 rotor) at 10°C for 17 hours. Following centrifugation, the supernatant was removed and the pellet (Figure 1A) was recovered for the NMR experiments. Pellet hydration was found to be over 10-fold in weight, therefore the pellet size was reduced by lyophilizing for three hours and then re-hydrated with water to yield a 200 μL-sample. The final HEPES buffer concentration in the NMR sample was estimated to be at or below 80 mM, and the final water content was about 1.5 times the lipid weight.
For proteoliposome samples containing metal ions (Figure 1B and 1C), the dry P3 peptide was first dissolved in ~2 mL of water, then mixed with an equimolar amount of either NiCl2 or CuSO4 in standardized aqueous solutions (Hampton Research, Aliso Viejo, CA for NiCl2 and Ricca Chemicals, Pocomoke City, MD for CuSO4), resulting in clear colorless solutions at pH 5.5 (NiCl2) and 3.5 (CuSO4). The pH was adjusted to 7.4 using NaOH in both cases. As metal ions bind to P3 and displace protons, the solution pH decreases, with characteristically slow kinetics,51 therefore this process takes several minutes. Above pH 7, metal-containing piscidin solutions display a typical yellow (nickel) or pink (copper) coloration, which provided an immediate visual test for metal binding. Indeed, ATCUN peptides bound to Ni2+ and Cu2+ absorb near 420 and 525 nm, respectively.36, 37, 52 The distinctive colors were retained in the proteoliposome pellets (Figure 1). The solutions were then diluted to a volume of 6 mL before being added to the lipids, following the same sequence of steps as for the apo-P3 sample. The final pH was 7.4. The samples were then ultracentrifuged as described above, yielding the pellets shown in Figure 1B and 1C, which were lyophilized and rehydrated to prepare the samples used for MAS solid-state NMR spectroscopy.
Solid-state NMR experiments
All of the solid-state NMR experiments were performed on a Bruker AVANCE-III HD 750 MHz spectrometer with a 4 mm H/C/N triple-resonance Bruker E-free MAS probe (Bruker https://www.bruker.com). The spinning rate was maintained at 11.11 kHz ± 2 Hz and the temperature controller was set to 7 ± 2 °C; taking into account frictional heating and calibration with the 1H chemical shift of the water resonance the actual sample temperature is estimated to be 10 ± 2 °C. The applied radiofrequency (rf) fields corresponded to π/2 pulse lengths of 2.5 μs, 3.2 μs and 5.1 μs for 1H, 13C and 15N, respectively. The peptide-containing proteoliposome pellets were packed into 4 mm zirconia rotors containing teflon inserts and sealed with Vespel drive caps.
During direct 13C detection, 100 kHz rf irradiation was used for 1H decoupling with swept frequency two-pulse phase modulation (SW-TPPM).53 Cross-polarization (CP)54 from 1H to 15N was optimized using amplitude-modulated rf irradiation (100%–70%) applied on the 1H channel; 50 kHz rf irradiation on the 1H and ∼27 kHz rf irradiation on the 15N channels were applied during the 1 ms contact time. Two-dimensional PDSD pulse sequence with 10 ms, 50 ms, and 100 ms mixing times.55–57 Double cross-polarization from 15N amide to 13CA sites using spectrally induced filtering in combination with cross-polarization (SPECIFIC-CP).58 Adiabatic tangential pulses were applied on the 13C channel. For two-dimensional N-C correlation, rf irradiations of ∼27 kHz were applied on the 15N channel and ∼16 kHz on the 13C channel for band-selective polarization transfer and continuous wave (CW) 1H irradiation (100 kHz rf field strength) was applied for decoupling during the 4.5ms contact time for N-CA and N-CO transfers. The chemical shift frequencies were referenced externally to the adamantane methylene 13C resonance at 38.48 ppm and the ammonium sulfate 15N resonance at 26.8 ppm.
Density functional theory (DFT) calculations
Two structures of the first 6 residues of p3 were used for DFT calculations: an apo structure based on the structure of p3 in 3:1 DMPC:DMPG28 (PDB ID 2mcw), and a Ni2+-bound structure also derived from it. The first six residues of 2mcw were extracted and the C-terminus was amidated in Avogadro.59 The sidechain torsion angles of I5 were adjusted to better match the experimental 13C shift of this residue. This structure was subjected to geometry optimizations in Gaussian 09,60 keeping the phi and psi torsion angles for all residues fixed using the ModRedundant option. To generate the Ni2+-bound structure, the first four residues of the resulting structure were then extracted and protons were removed from the F1 amine and I2 and H3 backbone amides in Avogadro. A Ni2+ ion was then coordinated to these nitrogens as well as the H3 sidechain δ-nitrogen. These residues were then subjected to a geometry optimization using the Universal Force Field (UFF), resulting in a square-planar geometry. Next, this structure was subjected to a second geometry optimization in Gaussian 09,60 using the B3LYP/6–31+G(d) level of theory. The first three residues of this optimized structure were then re-attached to residues 4–6 of the optimized structure of apo p3 and subjected to a third geometry optimization, with residues 4–6 held fixed with the ModRedundant option. 13C and 15N chemical shieldings were then calculated for each structure in Gaussian using the B3PW91/6–31+G(d,p) level of theory and the SCRF/PCM solvent method with water as the solvent. The resultant shieldings were then related to experimental 13C and 15N chemical shifts by linear regression in Microsoft Excel 2016. The chemical shift differences between the metallated and unmetallated structures were calculated using a slope of −1 to relate shifts to shieldings, and compared to experimental shift differences. Unless otherwise stated Gaussian geometry optimizations used the B3PW91/6–31+G(d) level of theory.
Acknowledgements
MLC acknowledges funding from the National Science Foundation (MCB 1716608). This work was also supported by grants R35GM122501 and P41EB002031 from the National Institutes of Health and utilized the Biomedical Technology Resource Center for NMR Molecular Imaging of Proteins at the University of California, San Diego. This work was performed in part using the College of William and Mary computing facilities, which were established through by contributions from the National Science Foundation, the Commonwealth of Virginia Equipment Trust Fund and the Office of Naval Research.
References
- [1].Zasloff M, Proc. Natl. Acad. Sci. U. S. A, 1987, 84, 5449–5453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Steiner H, Hultmark D, Engström Å, Bennich H, Boman HG, Nature, 1981, 292, 246–248. [DOI] [PubMed] [Google Scholar]
- [3].Fjell CD, Hiss JA, Hancock REW, Schneider G, Nat. Rev. Drug Disc, 2011, 11, 37–51. [DOI] [PubMed] [Google Scholar]
- [4].Zasloff M, Nature, 2002, 415, 389–395. [DOI] [PubMed] [Google Scholar]
- [5].Mahlapuu M, Håkansson J, Ringstad L, Björn C, Front. Cell. Infect. Microbiol, 2016, 6, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Epand RM, Vogel HJ, Biochim. Biophys. Acta, 1999, 1462, 11–28. [DOI] [PubMed] [Google Scholar]
- [7].Glukhov E, Stark M, Burrows LL, Deber CM, J. Biol. Chem, 2005, 280, 33960–33967. [DOI] [PubMed] [Google Scholar]
- [8].Wimley WC, ACS Chem. Bio, 2010, 5, 905–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Sochacki KA, Barns KJ, Bucki R, Weisshaar JC, Proc. Ntl. Acad. Sci. U.S.A, 2011, 108, E77–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Brogden KA, Nat. Rev. Microbiol, 2005, 3, 239–250. [DOI] [PubMed] [Google Scholar]
- [11].Marion D, Zasloff M, Bax A, FEBS Lett, 1988, 227, 21–26. [DOI] [PubMed] [Google Scholar]
- [12].Bechinger B, Zasloff M, Opella SJ, Protein Sci, 1993, 2, 2077–2084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Gesell J, Zasloff M, Opella SJ, Biomol J. NMR, 1997, 9, 127–135. [DOI] [PubMed] [Google Scholar]
- [14].Porcelli F, Buck-Koehntop BA, Thennarasu S, Ramamoorthy A, Veglia G, Biochemistry, 2006, 45, 5793–5799. [DOI] [PubMed] [Google Scholar]
- [15].Toke O, Cegelski L, Schaefer J, Biochim. Biophys. Acta - Biomembranes, 2006, 1758, 1314–1329. [DOI] [PubMed] [Google Scholar]
- [16].Haney EF, Hunter HN, Matsuzaki K, Vogel HJ, Biochim Biophys Acta - Biomembranes, 2009, 1788, 1639–1655. [DOI] [PubMed] [Google Scholar]
- [17].Strandberg E, Tremouilhac P, Wadhwani P, Ulrich AS, Biochim. Biophys. Acta - Biomembranes, 2009, 1788, 1667–1679. [DOI] [PubMed] [Google Scholar]
- [18].Strandberg E, Horn D, Reißer S, Zerweck J, Wadhwani P, Anne S. Ulrich, Biophys. J, 2016, 111, 2149–2161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Wakamatsu K, Takeda A, Tachi T, Matsuzaki K, Biopolymers, 2002, 64, 314–327. [DOI] [PubMed] [Google Scholar]
- [20].Nicolas P, FEBS J, 2009, 276, 6483–6496. [DOI] [PubMed] [Google Scholar]
- [21].Hancock REW, Sahl HG, Nat. Biotechnol, 2006, 24, 1551–1557. [DOI] [PubMed] [Google Scholar]
- [22].Mookherjee N, Hancock REW, Cell Mol. Life Sci, 2007, 64, 922–933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Haney EF, Hancock REW, Biopolymers, 2013, 6, 572–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Mansour SC, de la Fuente-Nunez C, Hancock RE, J. Pept. Sci, 2015, 21, 323–329. [DOI] [PubMed] [Google Scholar]
- [25].Silphaduang U, Noga EJ, Nature, 2001, 414, 268–269. [DOI] [PubMed] [Google Scholar]
- [26].Libardo MDJ, Bahar AA, Ma B, Fu R, McCormick LE, Zhao J, McCallum SA, Nussinov R, Ren D, Angeles-Boza AM, Cotten ML, FEBS J, 2017, 284, 3662–3683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Hayden RM, Goldberg GK, Ferguson BM, Schoeneck MW, Libardo MDJ, Mayeux SE, Shrestha A, Bogardus KA, Hammer J, Pryshchep S, Lehman HK, McCormick ML, Blazyk J, Angeles-Boza AM, Fu R, Cotten ML, J. Phys. Chem. B, 2015, 119, 15235–15246. [DOI] [PubMed] [Google Scholar]
- [28].Perrin BS Jr., Tian Y, Fu R, Grant CV, Chekmenev EY, Wieczorek WE, Dao AE, Hayden RM, Burzynski CM, Venable RM, Sharma M, Opella SJ, Pastor RW, Cotten ML, J. Am. Chem. Soc, 2014, 136, 3491–3504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Harford C, Sarkar B, Acc. Chem. Res, 1997, 30, 123–130. [Google Scholar]
- [30].Joyner JC, Reichfield J, Cowan JA, J. Am. Chem. Soc, 2011, 133, 15613–15626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Clore GM, Methods Enzymol, 2015, 564, 485–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Carlon A, Ravera E, Andralojc W, Parigi G, Murshudov GN, Luchinat C, Prog. Nucl. Magn. Reson. Spectrosc, 2016, 92–93, 54–70. [DOI] [PubMed] [Google Scholar]
- [33].Jaroniec CP, Solid State Nucl. Magn. Reson, 2012, 43–44, 1–13. [DOI] [PubMed] [Google Scholar]
- [34].Cheng H, Markley JL, Annu. Rev. Biophys. Biomol. Struct, 1995, 24, 209–237. [DOI] [PubMed] [Google Scholar]
- [35].Laussac JP, Sarkar B, Biochemistry, 1984, 23, 2832–2838. [DOI] [PubMed] [Google Scholar]
- [36].Melino S, Gallo M, Trotta E, Mondello F, Paci M, Petruzzelli R, Biochemistry, 2006, 45, 15373–15383. [DOI] [PubMed] [Google Scholar]
- [37].Gusman H, Lendenmann U, Grogan J, Troxler RF, Oppenheim FG, Biochim. Biophys. Acta, 2001, 1545, 86–95. [DOI] [PubMed] [Google Scholar]
- [38].Grzesiek S, Bax A, Clore GM, Gronenborn AM, Hu JS, Kaufman J, Palmer I, Stahl SJ, Wingfield PT, Nat. Struct. Biol, 1996, 3, 340–345. [DOI] [PubMed] [Google Scholar]
- [39].Cerofolini L, Giuntini S, Louka A, Ravera E, Fragai M, Luchinat C, J. Phys. Chem. B, 2017, 121, 8094–8101. [DOI] [PubMed] [Google Scholar]
- [40].Libardo MDJ, Gorbatyuk VY, Angeles-Boza AM, ACS Infect. Dis. , 2016, 2, 71–81. [DOI] [PubMed] [Google Scholar]
- [41].Libardo MD, Cervantes JL, Salazar JC, Angeles-Boza AM, ChemMedChem, 2014, 9, 1892–1901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Miyamoto T, Fukino Y, Kamino S, Ueda M, Enomoto S, Dalton transactions (Cambridge, England: : 2003), 2016, 45, 9436–9445. [DOI] [PubMed] [Google Scholar]
- [43].Fritz M, Quinn CM, Wang MZ, Hou GJ, Lu XG, Koharudin LMI, Polenova T, Gronenborn AM, J. Phys. Chem. B. , 2017, 121, 3574–3585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Vincent P, Alina Jurca S, Peter F, New J Chem, 2008, 32, 1189–1194. [Google Scholar]
- [45].McDonald MR, Fredericks FC, Margerum DW, Inorg. Chem, 1997, 36, 3119–3124. [DOI] [PubMed] [Google Scholar]
- [46].El Meskini R, Culotta VC, Mains RE, Eipper BA, J. Biol. Chem, 2003, 278, 12278–12284. [DOI] [PubMed] [Google Scholar]
- [47].Lauth X, Shike H, Burns JC, Westerman ME, Ostland VE, Carlberg JM, Van Olst JC, Nizet V, Taylor SW, Shimizu C, Bulet P, J. Biol. Chem, 2002, 277, 5030–5039. [DOI] [PubMed] [Google Scholar]
- [48].Eipper BA, Stoffers a. D A, Mains RE, Annu. Rev. Neurosci, 1992, 15, 57–85. [DOI] [PubMed] [Google Scholar]
- [49].Kreil G, Methods Enzymol, 1984, 106, 218–223. [DOI] [PubMed] [Google Scholar]
- [50].Mulero I, Noga EJ, Meseguer J, Garcia-Ayala A, Mulero V, Dev. Comp. Immunol, 2008, 32, 1531–1538. [DOI] [PubMed] [Google Scholar]
- [51].Gasmi G, Singer A, Forman-Kay J, Sarkar B, J. Pept. Res, 1997, 49, 500–509. [DOI] [PubMed] [Google Scholar]
- [52].Melino S, Santone C, Di Nardo P, Sarkar B, Febs J, 2014, 281, 657–672. [DOI] [PubMed] [Google Scholar]
- [53].Thakur RS, Kurur ND, Madhu PK, Chem. Phys. Lett, 2006, 426, 459–463. [Google Scholar]
- [54].Pines A, Gibby MG, Waugh JS, J. Chem. Phys, 1972, 56, 1776–1777. [Google Scholar]
- [55].Bloembergen N, Physica, 1949, 15, 386–426. [Google Scholar]
- [56].Frey MH, Opella SJ, J. Am. Chem. Soc, 1984, 106, 4942–4945. [Google Scholar]
- [57].Szeverenyi NM, Sullivan MJ, Maciel GE, J. Mag. Reson, 1982, 47, 462–475. [Google Scholar]
- [58].Baldus M, Petkova AT, Herzfeld J, Griffin RG, Mol. Phys, 1998, 95, 1197–1207. [Google Scholar]
- [59].Hanwell MD, Curtis DE, Lonie DC, Vandermeersch T, Zurek E, Hutchison GR, J Cheminformatics, 2012, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, Nakatsuji H, Caricato M, Li X, Hratchian HP, Izmaylov AF, Bloino J, Zheng G, Sonnenberg JL, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery JJA, Peralta JE, Ogliaro F, Bearpark M, Heyd JJ, Brothers E, Kudin KN, Staroverov VN, Keith T, Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant JC, Iyengar SS, Tomasi J, Cossi M, Rega N, Millam JM, Klene M, Knox JE, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Martin RL, Morokuma K, Zakrzewski VG, Voth GA, Salvador P, Dannenberg JJ, Dapprich S, Daniels AD, Farkas O, Foresman JB, Ortiz JV, Cioslowski J, Fox DJ in Gaussian 09 Rev. E.01, Vol. (Ed.Êds.: Editor), City, 2009. [Google Scholar]