

Abstract
Practice Gap
Pediatricians must be aware of screening indications and the evaluation
and management of a child with hematuria and/or proteinuria.
Objectives
After completing this article, readers should be able to:
1. Understand the common causes of proteinuria and hematuria and be able to differentiate between benign and serious causes.
2. Describe screening techniques for initial evaluation of hematuria and proteinuria.
3. Recognize the criteria for diagnosis of proteinuria and hematuria.
4. Plan the appropriate initial evaluation for hematuria and proteinuria and interpret laboratory findings essential for diagnosis.
5. Recognize serious causes of hematuria and proteinuria that warrant immediate referral.
INTRODUCTION
Hematuria and proteinuria are common findings in primary care practice. Although the American Academy of Pediatrics eliminated routine urine screening from its preventive care guidelines a decade ago, many pediatricians continue to use screening urinalysis (UA) as part of their health supervision visits. Most pediatric patients who are diagnosed as having hematuria or proteinuria through screening UA do not have renal disease, and abnormal findings usually resolve on repeated testing. However, hematuria or proteinuria that persists on repeated testing warrants additional evaluation, and, depending on history along with initial evaluation in the primary care office, may warrant referral to a pediatric nephrologist for further management. Although guidelines put forth by the American Academy of Pediatrics do not recommend yearly evaluation of urine by dipstick analysis for children, regular routine screening of pediatric populations has been established in Japan, Taiwan, and Korea. (1)(2)(3)(4) Our practice recommends screening of certain patient populations at increased risk for renal disease over a lifetime (Table 1).
TABLE 1.
Conditions Under Which Children Should Have a Yearly Urinalysis Performed
| History of prematurity (<32 weeks’ gestational age), very low birthweight, other neonatal complications requiring intensive care, umbilical artery line |
| Congenital heart disease (repaired or unrepaired) |
| Recurrent urinary tract infections, hematuria, or proteinuria |
| Known renal disease or urologic malformations |
| Solid organ transplant |
| Malignancy or bone marrow transplant |
| History of or prolonged treatment with drugs known to be nephrotoxic |
| History of recurrent episodes of acute kidney injury |
| Family history of inherited renal disease |
The 2 most common tests used by clinicians for initial assessment of renal function or renal injury are the urine dipstick test and UA with microscopy, where urine is centrifuged, supernatant is removed, and urine sediment is examined under a microscope. Whereas UA with microscopy is laboratory dependent, the dipstick test can be performed quickly in the provider’s office and can guide the clinician to evaluate the patient further. Urine dipstick testing should be performed on a freshly voided urine sample, within 2 hours of collection; if a specimen needs to be refrigerated, it should be allowed to return to room temperature before testing. All pads of the dipstick are fully immersed in the urine and then immediately removed from the specimen cup and placed on a horizontal surface; results should be read within 1 minute, either manually or by automated analyzer. Providers should be aware that there are a variety of urine reagent strips on the market, with a range of semiquantitative concentration results that are not interchangeable between manufacturers.
The urine dipstick tests for the peroxidase activity of hemoglobin (or myoglobin); thus, a dipstick that is positive for blood is actually positive for the detection of heme pigment, which may reflect red blood cells (RBCs) in the urine or other causes, such as hemoglobinuria or myoglobinuria. A colorimetric test is used for detection of urine protein. The dipstick measures albumin concentration as a surrogate for protein, turning different shades of green, and, ultimately, blue, according to the concentration of albumin that reacts with tetrabromophenol. Whereas the urine dipstick is usually the initial screening test for both hematuria and proteinuria, additional studies are used to better quantify and characterize abnormal dipstick test findings. These additional studies are discussed further in this review.
HEMATURIA
Prevalence
Hematuria, a finding not uncommon to pediatricians, can be benign or can be a sign of a serious underlying condition. Population studies from the 1970s to the 1990s of school-aged children suggest that approximately 1% of them have 2 or more urine dipstick tests positive for microscopic hematuria, with persistence of hematuria at 6 months in one-third of this population. (2)(3)(4)
Definitions and Measurement Methods
Macroscopic hematuria is characterized by the presence of blood in the urine in sufficient quantity to be visible to the naked eye. Grossly bloody urine may appear pink or red but may also be tea-colored or dark cola-colored with glomerular etiologies. As little as 1 mL of blood per liter of urine can induce a visible color change, and as little as 2 to 3 RBCs per high-power field (HPF) can make the urine dipstick positive. (7)(8) To our knowledge, there are no published data that correlate the number of urine sediment RBCs with the dipstick result 0–3+.
On the other hand, microscopic hematuria is hematuria in the absence of visible color change in the urine, detected only by urine dipstick and confirmed by microscopic examination of the spun urine sediment. The definition of microscopic hematuria varies from 1 to 10 RBC/HPF; however, based on previous studies in schoolaged children, we define microscopic hematuria as greater than 5 RBC/mm3 in uncentrifuged urine or greater than 5 RBCs/HPF in centrifuged urine on at least 2 to 3 different occasions. Microscopic hematuria that might undergo further evaluation should be independent of trauma, exercise, menstruation, or sexual activity (Fig 1). (9)(10)(11)(12)(13)
Figure 1.
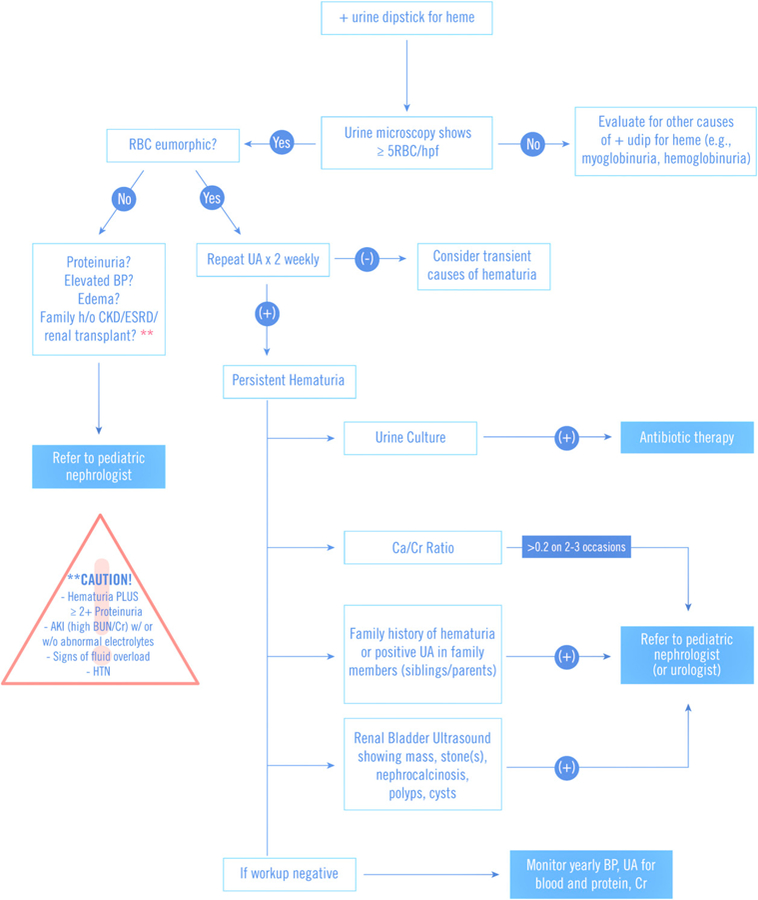
Approach to a child with microscopic hematuria. Caution items warrant urgent consultation with a nephrologist. AKI=acute kidney injury; BP=blood pressure; BUN=blood urea nitrogen; CKD= chronic kidney disease; Cr=creatinine; ESRD=end-stage renal disease; HTN=hypertension; UA=urinalysis. (Adapted from refs 14, 15, 16, and 17)
When gross hematuria is suspected due to a change in urine color, the first step in evaluation is centrifuging a fresh sample of the discolored urine. A positive urine dipstick that results from myoglobinuria, as in the case of increased skeletal muscle breakdown (eg, rhabdomyolysis, extreme exercise, or myopathies) or hemoglobinuria (from rapid hemolysis), is typically associated with discolored urine (red supernatant after centrifugation) without RBCs noted on microscopic evaluation. On the other hand, discolored urine with a negative urine dipstick should prompt questioning for and/or inclusion of other pigments (food coloring, beets, blackberries, rhubarb, paprika); drugs (sulfonamides, nitrofurantoin, salicylates, pyridium, phenytoin, rifampin, chloroquine, defuroxamine, iron sorbitol); toxins (lead, benzene); and metabolites (homogenistic acid, tyrosinosis, urates) in the differential diagnosis. (14) One of the most common of these is the pink/orange discoloration of infant diapers as a result of urate crystal precipitation. (13)
Classification
Hematuria can be transient or persistent. In obtaining a comprehensive clinical history one should determine change in color of urine, timing of color change related to urinary stream, the pattern of the hematuria (transient or persistent), and associated signs, symptoms, illness, or activity. Transient hematuria has been found in association with fever, exercise, urinary tract infections (which usually also present with dysuria and pyuria), and trauma. Passage of fresh blood with or without clots at the beginning or end of the urinary stream should prompt consideration of lower urinary tract origin, with urethritis, trauma, bladder calculus or mass, and schistosomiasis as possible etiologies. (13) A detailed history of presentation should prompt a search for associated findings (Table 2).
TABLE 2.
Causes of Hematuria in Children
| CONDITION | HEMATURIA/MICRO OR MACRO | PROTEINURIA | HISTORY | EXAMINATION | DIAGNOSTIC EVALUATION |
|---|---|---|---|---|---|
| Transient | |||||
| Exercise | Micro | + | Hematuria and/or proteinuria after exercise | Noncontributory | Absence of hematuria or proteinuria without exercise |
| Fever | Micro | + | Hematuria and/or proteinuria with febrile episode | Focal or systemic illness associated | Absence of hematuria or proteinuria after illness resolves |
| Urinary tract infection or pyelonephritis | Micro or macro | + | Dysuria, urgency, frequency, fever, flank pain, sterile pyuria | Fever, flank or lower abdominal tenderness | Urine dipstick, urine microscopy, urine culture |
| Trauma/instrumentation | Macro | − | Recent trauma or procedure | Noncontributory or findings related to trauma | Imaging studies |
| Urethritis | Micro | − | Sexually active | Normal/penile discharge | NAAT for gonorrhea/chlamydia |
| Bladder calculus/mass | Macro | − | History of nephrolithiasis | +/− Abdominal tenderness | Imaging, 24-h urine metabolic evaluation |
| Schistosomiasis | Terminal macro | − | Recent travel and contact with fresh water; previous serum sickness–like illness; dysuria, frequency | Decreased urine output | Imaging, PCR of urine or stool |
| Adenovirus hemorrhagic cystitis | Macro | − | Recent URI signs and symptoms | Active URI signs | Respiratory viral screen including adenovirus |
| Persistent | |||||
| Nonglomerular | |||||
| Hypercalciuria | Micro | − | Family history of nephrolithiasis | Noncontributory | High Ca/Cr |
| Nephrolithiasis | Micro or macro | +/− | Poor daily oral fluid intake, flank pain | Normal/CVA or flank tenderness | High Ca/Cr, imaging, crystals on UA |
| Nutcracker syndrome | Micro or macro | Up to 15% | Flank pain | Noncontributory | Imaging |
| Sickle cell trait | Micro | − | Sickle cell trait or anemia | Noncontributory | Hemoglobin electrophoresis |
| Coagulation disorders | Micro or macro | − | Known disease or family history, bruising, bleeding, petechiae, lethargy | Petechiae, ecchymosis, bleeding | Abnormal clotting profile, CBC count |
| Polycystic kidney disease | Micro or macro | + | Family history of polycystic kidney disease | Abdominal mass | Ultrasonography, genetic evaluation |
| Wilms tumor | Micro or macro | +/− | Incidental finding, gross hematuria, abdominal pain, hypertension, fever | Abdominal mass | Ultrasonography |
| Structural abnormalities of kidney, ureter, bladder (eg, UPJO, UVJO) | Micro | − | Noncontributory, recurrent or first-time UTI, family history of CAKUT | Abdominal mass | Ultrasonography, radioisotope renography |
| Glomerular | |||||
| IgA nephropathya | Micro or macro | +/− | Concomitant recurrent hematuria with infectious illness, flank pain (rare) | Noncontributory | Normal complements, kidney biopsy |
| Henoch-Schönlein purpuraa | Micro/macro | +/− | Nonblanching rash, joint swelling, abdominal pain, and intussusception | Petechial/purpuric rash, joint swelling, hypertension | Normal C3/C4 |
| Alport syndromea | Micro or macro | +/− | Family history, persistent microscopic or episodic macroscopic hematuria; poor school performance | Hearing/visual abnormalities | Genetic evaluation, kidney or skin biopsy |
| Thin basement membrane diseasea | Micro or macro | − | Family history, persistent microscopic or episodic macroscopic hematuria | Noncontributory | Nil |
| Postinfectious glomerulonephritisa | Micro or macro | +/− | Recent history of URI or skin infection | Red posterior oropharynx, hypertension, fluid overload | Antistreptolysin O titer, anti-DNase B, throat culture |
| Hemolytic uremic syndromea | Micro | +/− | Bloody diarrhea, oliguria, history of sick contact or ingestion of uncooked meat | Edema, intravascular fluid depletion, pallor, petechiae | Stool culture for Escherichia coli O157:H7 |
BUN=blood urea nitrogen; Ca/Cr=calcium/creatinine; CAKUT=congenital anomalies of the kidney and urinary tract; CBC=complete blood cell; CVA=costovertebral angle; IgA=immunoglobulin A; NAAT=nucleic acid amplification test; PCR=polymerase chain reaction; UA=urinalysis; UPJO=ureteropelvic junction obstruction; URI=upper respiratory infection; UTI=urinary tract infection; UVJO=ureterovesicular junction obstruction.
Considered to be a serious condition requiring prompt evaluation by/discussion with a pediatric nephrologist.
Persistent hematuria is defined as more than 4 to 6 weeks of positive UA results showing more than 5 RBC/ HPF in the absence of exercise activity, menses, or trauma and can be categorized based on whether the hematuria is glomerular or nonglomerular in origin (Table 2).
Nonglomerular Causes of Hematuria
Hypercalciuria, defined in children older than 2 years of age as a urine calcium/creatinine ratio greater than 0.2 (mg/mg), has been associated with persistent asymptomatic microscopic hematuria. Because infants are known to have higher calcium excretion combined with a lower urine creatinine level, a urine calcium/creatinine ratio less than 0.8 is deemed acceptable for infants younger than 6 months and less than 0.6 for infants 6 to 12 months old. Hypercalciuria may be an isolated finding or may be associated with nephrocalcinosis or frank stone disease. A recent retrospective cohort from the United Kingdom noted hypercalciuria in up to 15% of 511 children with nephrolithiasis, although other groups have described hypercalciuria in patients with urolithiasis to varying degrees, from 7% to 34%. (18)
Nephrolithiasis may be associated with microscopic or macroscopic hematuria, and depending on the location of the stone may also be associated with moderate to severe abdominal and flank pain. Nephrocalcinosis is usually asymptomatic and discovered as an incidental finding on imaging tests, or otherwise discovered on imaging during an evaluation for microscopic hematuria.
Nutcracker syndrome is a vascular disorder characterized by compression of the left renal vein between the aorta and proximal superior mesenteric artery. Although Nutcracker syndrome is not common in the United States, studies in Japanese and Korean children have found up to 30% to 45% of Doppler ultrasonographic findings consistent with Nutcracker syndrome in the setting of unexplained hematuria. (19)(20) Although most often asymptomatic, this phenomenon can present with left flank pain or abdominal pain, hematuria (which is more often microscopic than macroscopic), and varicocele. The venous compression in Nutcracker syndrome may be detected by Doppler ultrasonography or by computed tomography/magnetic resonance angiography. (21)
Glomerular Causes of Hematuria
In immunoglobulin (Ig) A nephropathy, macroscopic hematuria appears concomitantly with infectious illness, most commonly viral respiratory or gastrointestinal in origin. Often, patients with IgA nephropathy have persistent microscopic hematuria between episodes of illness. Some degree of accompanying proteinuria, at least at times of intercurrent illness, is also common. Usually no family history of renal disease is found in these patients. The most common systemic vasculitis in childhood, IgA vasculitis, more widely known as Henoch-Schönlein purpura (HSP), is characterized by palpable purpuric lesions (in the setting of neither thrombocytopenia nor coagulopathy), oligoarticular and transient nondeforming arthritis/arthralgia, abdominal pain, and renal disease. Renal involvement has been reported in up to half of children with HSP, with a tendency toward an older subset that most commonly develops hematuria with or without RBC casts and no or mild proteinuria. (22)(23)(24) Less than 5% of patients with HSP with this mild renal presentation develop chronic kidney disease (CKD) compared with ≥50% of patients with HSP with a more serious initial presentation that is nephritic/ nephrotic in nature. (22)(25) Patients with repeated episodes of isolated macroscopic hematuria are also at risk for CKD in the long term. (26) Both IgA nephropathy and IgA vasculitis are considered related diseases and display similar histologic features and IgA deposits.
Alport syndrome (AS) is a hereditary disease with both gross and microscopic hematuria that is associated with high risk of progression to end-stage renal disease (ESRD) even before the fourth decade of life. The inheritance pattern is most commonly X-linked (80% of patients with AS) but may also be autosomal recessive or dominant. The genetic abnormality that characterizes X-linked AS involves the α−5 chain of type IV collagen (COL4A5), and mutations in the COL4A3 and COL4A4 genes are responsible for the recessive and dominant forms of AS. Because type IV collagen is found in the ear and eye in addition to the glomeruli, AS is often associated with high-frequency sensorineural hearing loss and ocular abnormalities, including anterior lenticonus. (12) The rate of progression of renal disease depends on the nature of COL4A mutations. (12)
Thin basement membrane disease, also known as benign familial hematuria, is an autosomal dominant condition in which patients demonstrate persistent microscopic hematuria but no apparent CKD progression over a lifetime, hence the “benign” designation to the disease. Kidney biopsy may reveal isolated thinning of the glomerular basement membrane on electron microscopy. Although, traditionally, thin basement membrane disease is thought to be benign, the condition is not a homogenous entity and rather has been associated with various genetic mutations. It has been described as the heterozygous form of autosomal recessive AS involving COL4A3 and COL4A4 (Alport being the homozygous phenotype); and as giant fibronectin glomerulopathy, C3/CFHR5 glomerulonephritis (GN), immunotactoid GN, and fibrillary GN based on new genetic information. These rare glomerulopathies have been noted to have a not so benign renal outcome, with approximately a 20% to 40% risk of progressing to some stage of CKD. (12)(28)(29)(30)
Postinfectious GN is the most common cause of acute nephritis in children around the world. Children aged 5 to 12 years are at greatest risk. Postinfectious GN has a wide range of presentations, from asymptomatic, microscopic hematuria to full-blown acute nephritic syndrome (redbrown urine, proteinuria, edema, hypertension, and acute kidney injury). Because asymptomatic microscopic hematuria is the most common presentation, obtaining a recent history of group A streptococcal skin (2–6 weeks earlier) or throat (1–2 weeks earlier) infection becomes critical on the initial evaluation to make this diagnosis. When diagnosis is delayed or if disease is more severe, presentation may be characterized by fluid overload status (hypertension, edema, pulmonary edema). Laboratory investigation may reveal low complement protein C3 but normal C4 levels; a low C3 level in postinfectious GN usually resolves by 4 to 6 weeks (31); a low C3 level that persists beyond this time frame may warrant a renal biopsy, especially if there is continued hematuria and/or proteinuria. Children tend to have complete clinical recovery and show resolution of the disease within 1 to 2 weeks. Microscopic hematuria can persist up to 6 months.
Evaluation of Hematuria
Children with 1+ blood on urine dipstick should have a UA with microscopic evaluation of the urine to verify the presence of urinary RBCs and to assess RBC quantity and shape. If RBCs are eumorphic one should consider urinary tract infection, hypercalciuria, genitourinary malformation, and/or familial causes of hematuria (Fig 1).
Isolated microscopic hematuria has a good renal outcome in general, but the lifetime risk of CKD may be higher in certain patients, depending on the specific underlying disease. Microscopic hematuria is generally monitored on a yearly basis but may require more frequent monitoring if associated with macroscopic hematuria and/or proteinuria because these can be associated with worse renal outcome. Furthermore, genetic analysis in the setting of a positive family history of hematuria and/or proteinuria or ESRD has been recommended by several authors for early detection of rare progressive hereditary renal diseases such as giant fibronectin glomerulopathy, C3/CFHR5 GN, immunotactoid GN, and fibrillary GN, which were previously classified under the umbrella of benign familial hematuria. Therefore, these cases of persistent microscopic hematuria might also prompt consideration of subspecialty referral, especially if there is any family history of CKD. (12)
PROTEINURIA
Prevalence
Although not always pathologic, proteinuria is recognized as a marker of kidney damage and is a well-known risk factor for progression to CKD in adults and children. The prevalence of proteinuria on a random urine specimen in otherwise asymptomatic school-aged children and adolescents is approximately 5% to 15% based on multiple largescale studies. (32) This finding decreases substantially with repeated urine samples. One study examined 4 repeated urine specimens from each of approximately 9,000 children (8–15 years old); 1 of 4 specimens was positive for protein in 10.7% of patients, but only 0.1% had 4 of 4 specimens positive with persistent proteinuria. (33) The specific type of protein excreted in the urine, such as albumin or low-molecular-weight (LMW) proteins, depends on the type of kidney disease. Albuminuria is more strongly associated with CKD as a marker of glomerular disease and is a long-term complication of diabetes and hypertension. In contrast, urinary loss of LMW proteins is more reflective of tubulointerstitial disease. (32) For the purpose of this review, the term proteinuria refers to increased urinary excretion of albumin and/or other specific proteins, such as immunoglobulins or LMW proteins.
Definition
Although a small amount of protein in the urine is considered acceptable, proteinuria is defined as protein excretion greater than 100 mg/m2 per day or more than 0.2 mg protein/mg creatinine (also known as a urine protein/creatinine ratio ([U p/c] >0.2) on a single spot urine collection; in neonates and infants, a higher amount of protein excretion, up to 300 mg/m2, is allowed. Nephrotic-range proteinuria is defined as greater than 1,000 mg/m2 per day or greater than 50 mg/kg per day, or a U p/c greater than 2 on a single spot urine collection.
Measurement Methods
Different methods are used to quantify the amount of protein excreted in the urine, with the urine dipstick being the most frequently used by primary care physicians. The dipstick largely detects albumin and does not tend to detect LMW proteins. Ranges can vary depending on the manufacturer; for the purpose of this article, a negative urine dipstick for protein corresponds to a concentration of less than 0.015 g/dL (0.15 g/L) of albumin in the urine, trace corresponds to 0.015 to 0.030 g/dL (0.15–0.30 g/L) of albumin in the urine; 1+ corresponds to 0.030 to 0.100 g/dL (0.30–1.00 g/L) of albumin in the urine; 2+ corresponds to 0.100 to 0.300 g/dL (1.00–3.00 g/L) of albumin in the urine; 3+ corresponds to 0.300 to 1.000 g/dL (3.00–10.00 g/L) of albumin in the urine; and 4+ corresponds to greater than 1.000 g/dL (>10.00 g/L) of albumin in the urine. A false-positive urine dipstick for protein can occur when the urine sample has a high specific gravity (ie, a concentrated urine) or is very alkaline. Contamination with antiseptic agents or iodinated radiocontrast agents can also produce a false-positive result for protein, and as such it is recommended to wait at least 24 hours after a contrast study to test for protein in the urine. (34)
A more accurate method of measuring protein in the urine is by 24-hour urine collection. Adequacy of a 24-hour urine collection may be verified by measurement of urine creatinine, which is approximately 15 to 20 mg/kg ideal body weight in females and 20 to 25 mg/kg ideal body weight in males. (35) However, in infants and children, especially in those who are not toilet trained, this method tends to be difficult to perform accurately. As such, calculation of a U p/c in a random or spot urine sample has become recognized as an acceptable alternative to a 24-hour urine collection for protein, especially in the pediatric population. The U p/c has been shown to be a fairly reliable surrogate for a 24-hour urine collection, especially when tested in the first morning urine specimen. (36)(37) A normal U p/c is less than 0.2 mg protein/mg creatinine in children older than 2 years and less than 0.5 mg protein/mg creatinine in infants and children 6 to 24 months old. (38) Points to consider when measuring protein in this manner include a falsely elevated U p/c when there is not enough creatinine excreted or underestimation of the ratio when there is a very concentrated sample with a high creatinine level in the urine. (39)(40) Our practice is to send a urine sample for U p/c as well as UA, with the expectation that significant proteinuria will be evident on both examinations.
Testing for microalbuminuria is valuable for screening for diabetic nephropathy in the pediatric population. It is highly sensitive to detect very small quantities of albumin in the urine; this test has grown in importance with the epidemic of obesity in the pediatric population. (32)
Qualitative assessment of proteinuria to differentiate glomerular from tubular proteinuria can be performed by measuring β−2 microglobulin, α−1-macroglobulin, lysozyme, and retinol-binding protein. These levels will be 10 to 100 times higher than normal in tubular proteinuria (eg, proximal tubular dysfunction seen in Fanconi syndrome).
Classification
Proteinuria can be classified as transient, orthostatic, and persistent. Transient proteinuria, which can be defined as proteinuria noted on 1 or 2 occasions but not present on subsequent testing, is often seen in the context of fever, exercise, stress, seizures, and hypovolemic/dehydration status. (41) Orthostatic proteinuria is characterized by increased protein excretion in the upright position, which returns to normal when the patient is recumbent. On average, these patients excrete less than 1 g of protein in 24 hours in the upright position, and this normalizes to less than 50 mg in 8 hours of supine position. Orthostatic proteinuria is one of the most common causes of proteinuria in adolescents. (41) It is diagnosed when a first morning urine sample is less than 0.2 mg protein/mg creatinine in the setting of a U p/c greater than 0.2, or positive urine dipstick for proteinuria, in a random urine sample (Fig 2). The pathophysiology underlying orthostatic proteinuria remains poorly understood, but the prognosis has traditionally been thought to be good. Studies from the 1960s to the 1990s on up to 40 to 50 years after the diagnosis of orthostatic proteinuria have reported a benign course for this condition, where mortality is not shown to be greater than the average healthy population with similar demographic characteristics in the absence of other clinical evidence of renal disease. (42)(43)(44)
Figure 2.
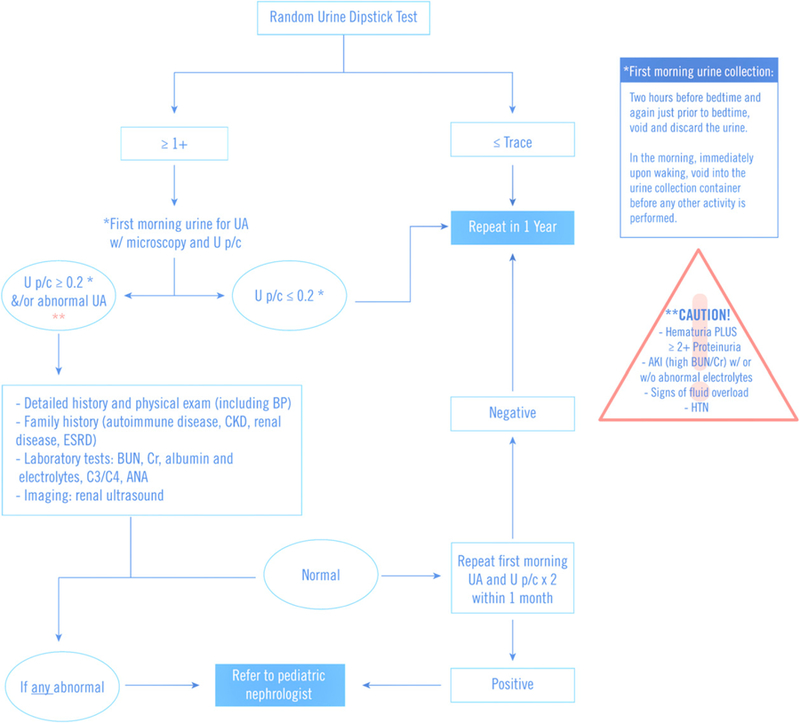
Approach to a child with asymptomatic proteinuria. Caution items warrant urgent consultation with a nephrologist. ANA=antinuclear antibody; AKI=acute kidney injury; BP=blood pressure; BUN=blood urea nitrogen; C3=complement component 3; C4=complement component 4; Ca/Cr=calcium/creatinine; CKD=chronic kidney disease; ESRD=end-stage renal disease; HPF=high-power field; HTN=hypertension; RBC=red blood cell; UA=urinalysis; U p/c=urine protein/creatinine ratio. (Adapted from refs 14, 41, 48, and 49.)
Based on the mechanism of the proteinuria, persistent proteinuria may be subclassified as glomerular, tubular, or overflow. Glomerular proteinuria refers to an anatomical or functional lesion in the glomeruli that results in an increased filtration of protein across the glomerular capillary wall. Tubular proteinuria is seen when there is an increased excretion of LMW proteins due to interference with proximal tubular reabsorption. Overflow proteinuria refers to an increased excretion of LMW proteins that results from marked overproduction of LMW proteins, leading to a level that exceeds tubular reabsorptive capacity; overflow proteinuria is very rarely seen in children and is not discussed in this review.
Among the most common causes of primary glomerular proteinuria seen in children (Table 3), minimal change disease (MCD) represents one of the most common presentations of idiopathic nephrotic syndrome. It classically presents in children (most between 3 and 9 years of age) as edema, a low albumin level (<2.5 g/dL [25.0 g/L]), proteinuria, and hyperlipidemia in the setting of normal renal function and complement levels, and absence of hypertension and/or gross hematuria. (45) Based on this clinical diagnosis, corticosteroid therapy is recommended without a confirmatory diagnosis by renal biopsy as more than 90% of cases will respond within 4 weeks. (46) Based on the response and frequency of relapses, MCD can be further subclassified. In a case where a patient is found to be corticosteroid resistant or if the clinical/laboratory presentation is different from that described previously herein, a renal biopsy should be considered. On histopathology, MCD glomeruli appear normal under light microscopy but show characteristic effacement of foot processes on electron microscopy. (47)
TABLE 3.
Persistent Proteinuria
| CONDITION | HEMATURIA/MICRO OR MACRO | PROTEINURIA | HISTORY | EXAMINATION | INVESTIGATIONS TO CONFIRM DIAGNOSIS |
|---|---|---|---|---|---|
| Glomerular | |||||
| Primary | |||||
| Minimal change disease | −/+/Micro | 4+ | Age 2–10 y, recent viral infection, nephrotic features | Anasarca | U p/c >2.0, low albumin level, hyperlipidemia, thrombocytosis, normal C3/C4 |
| Chronic kidney disease/adaptation due to nephron loss | −/+/Micro | 1–4+ | History of vesicoureteral reflux or recurrent UTIs; history of urologic abnormalities, prematurity, AKI (Table 1) | Noncontributory | Depending on history, but will likely include chem-10 and cystatin C |
| Congenital nephrotic syndrome | −/+/Micro | 4+ | Age <3 mo, prematurity, large placenta, edema at birth or first week of age | Anasarca | Elevated a-fetoprotein in amniotic fluid, U p/c >2.0, low albumin level, hyperlipidemia, normal C3/C4 |
| Focal segmental glomerular sclerosis | −/+/Micro | 1–4+ | History of corticosteroid-resistant nephrotic syndrome, history of HIV infection | Nephrotic features, hypertension | U p/c >2.0, normal to low renal function, hyperlipidemia, thrombocytosis, normal C3/C4 |
| Immune complex–mediated membranoproliferative glomerulonephritis | −/+/Micro or macro | 1–4+ | Hepatitis B or C infection, rheumatologic disease, malignancy | Nephrotic or nephritic features | Low C3, low or normal C4, hepatitis serology; depends on underlying cause |
| C3 glomerulopathy | −/+/Micro | 1–4+ | History of renal disease in family members | Nephrotic or nephritic features | Low C3, normal C4; U p/c >2.0; normal to low renal function; mutations or antibodies to complement components |
| MN | −/+/Micro | 2–4+ | History of corticosteroid resistant NS | Nephrotic features | Primary MN: + antiphospholipase A2 receptor; rule out causes for secondary MN such as hepatitis B or C, HIV, or SLE |
| Secondary | |||||
| Diabetes mellitus | + | 1–3+ | Polyuria, polydipsia, polyphagia, weight loss | Intravascular depletion | Elevated blood glucose and hemoglobin A1C levels, glucosuria |
| SLE | +/Micro or macro | 1–4+ | Fatigue, weakness, multisystem symptoms | Malar rash, joint swelling, fluid overload, hypertension | Pancytopenia, anti-dsDNA, + ANA, high ESR, low C3 and C4 |
| Tubular | |||||
| Primary | |||||
| Cystinosis | +/Micro | + (LMW) | Depends on age at onset, visual impairment, delayed puberty, Fanconi syndrome | Hepatosplenomegaly, cystine corneal crystals | Elevated cysteine level, concentrating defect, type 2 RTA,a CTNS gene mutation |
| Wilson disease | +/Micro | + (LMW) | Neurologic and behavioral symptoms, liver dysfunction, Fanconi syndrome | Kayser-Fleischer rings, hepatosplenomegaly | Low ceruloplasmin level, elevated liver enzyme levels, type 2 RTAa |
| Lowe syndrome | +/Micro | + (LMW) | Fanconi syndrome | Cataracts, cognitive impairment, hypotonia | Type 2 RTA,a OCRL 1 mutations |
| Secondary | |||||
| Acute interstitial nephritis | −/+/Micro | + | Recent use of NSAIDs, penicillin, quinolones, sulfonamides, cimetidine, cephalosporins, allopurinol | Subclinical, late in disease may have typical features of renal failure; tubulointerstitial nephritis and uveitis, red, painful eye | Urinary WBCs or WBC casts, urine eosinophils, concentrating defect |
| Acute tubular necrosis | −/+/Micro | + | Recent use of aminoglycosides, cisplatin, NSAIDs, radiocontrast media, amphotericin B. history of circulatory impairment or hypoxia | AKI, oliguria | Elevated BUN and creatinine levels; granular or muddy brown casts on microurinalysis, fraction excretion of sodium >2% |
| Heavy metal poisoning | −/+/Micro | + | Recent exposure to copper, lead, or mercury | Cognitive or behavioral impairment lead, lead lines along gum margin | Elevated level of toxin |
| Obstructive uropathy | −/+/Micro | + | Flank or abdominal pain, decreased urine output | Suprapubic mass, oliguria or anuria | Ultrasonography, VCUG, elevated BUN and creatinine levels |
AKI=acute kidney injury; ANA=antinuclear antibody; BUN=blood urea nitrogen; ESR=erythrocyte sedimentation rate; HIV=human immunodeficiency virus; LMW=low molecular weight; MN=membranous nephropathy; NSAID=nonsteroidal anti-inflammatory drug; SLE=systemic lupus erythematosus; U p/c=urine protein/creatinine ratio; UTI=urinary tract infection; VCUG=voiding cystourethrogram; WBC=white blood cell.
Type 2 RTA (renal tubular acidosis, proximal type) often accompanied by glycosuria, aminoaciduria, and LMW proteinuria.
In teenagers who present with massive proteinuria, or in children with proteinuria who are found to be corticosteroid resistant, one should consider focal segmental glomerular sclerosis (FSGS) in the differential diagnosis. This disorder is named after the typical histologic lesion characterized by some (focal) glomeruli with areas (segmental) of sclerosis or scarring, alongside areas of normal glomeruli. A recent study describes FSGS in up to 56% of children younger than 20 years of age (most of the cohort aged 1–11 years) with initial presentation of corticosteroid-resistant nephrotic syndrome. (50) The histopathologic diagnosis of FSGS is notably more prevalent in black patients compared with white patients, which may be related to the higher incidence of apolipoprotein L1 gene in this population. (51)(52)(53) Primary/idiopathic FSGS, where circulating permeability factors are thought to be involved in the pathogenesis, often presents with the classic nephrotic syndrome triad of proteinuria, hypoalbuminemia, and edema in the absence of identifiable risk factors (eg, severe obesity, decreased renal mass [as may be seen in prematurity], viral infection, drugs). Secondary FSGS, on the other hand, usually presents with subnephrotic-range proteinuria in the presence of identifiable risk factors. Several genetic forms of FSGS have been described. Mutations are most commonly described in the nephrin gene (NPHS1, which is also responsible for congenital nephrotic syndrome [see later herein]) and the podocin gene (NPHS2); both follow an autosomal recessive pattern and usually present in the first year of life. (50)(54)(55) In contrast, autosomal dominant forms of FSGS (such as mutations in alpha-actin-4 or TRPC6) tend to present in adolescence or later in adulthood.
Congenital nephrotic syndrome (CNS) refers to nephrotic syndrome that appears early in infancy, generally presenting with heavy proteinuria and marked ascites within the first 3 months after birth. In contrast to nephrotic syndrome occurring later in childhood or adolescence, CNS is more commonly associated with genetic mutations, namely, NPHS1, followed by NPSH2, encoding key components of the slit diaphragm, nephrin and podocin. Congenital nephrotic syndrome can be associated with a history of prematurity (35–38 weeks), low birthweight for gestational age, and large placenta (>25% of birthweight). The significant urinary protein losses result in hypoalbuminemia, hypogammaglobulinemia, and dysregulation of the clotting cascade, which make these children prone to poor nutritional status and growth, as well as a heightened incidence of bacterial infections and thromboembolic events. (58) Genetic cause has been described in up to 70% of infants presenting in the first 3 months of age and up to 50% of those aged 4 to 12 months. (50)(59) Children with CNS, with or without identified genetic mutations, are usually corticosteroid resistant and have a poorer prognosis. As part of nongenetic causes of CNS, one should consider prenatal and perinatal infections (TORCH infections such as rubella, toxoplasmosis, congenital syphilis, human immunodeficiency virus, cytomegalogvirus, etc), mercury intoxication, maternal systemic lupus, and neonatal alloimmunization antineutral endopeptidase; most of these causes have a specific treatment. (52)(60) Although care remains complex and the prognosis remains guarded for infants with nephrotic syndrome, improved protein supplementation, adequate nutritional support, and renal replacement therapy (if needed) may allow the child with CNS to grow big enough to become a candidate for renal transplant. (58) These advances in medical care offer the patient with CNS an overall improved prognosis.
Previously discussed acute postinfectious GN and HSP (see previously herein) are known to be secondary causes of glomerular proteinuria (Table 3). This category of disease also includes lupus nephritis, which is the term used to describe the renal (usually glomerular) involvement of systemic lupus erythematosus (SLE). Systemic lupus erythematosus is a chronic autoimmune disease that can involve any organ system, and its childhood onset is known to have a more severe course compared with adult onset. (61) Renal disease is present in 50% to 75% of children with SLE and is one of the leading causes of morbidity and mortality. (61) According to current histopathologic classification of SLE, most would consider class I (minimal mesangial) and class II (mesangial proliferative) mild lesions. Class III (focal proliferative) and class IV (diffuse proliferative), in contrast, are more severe lesions with high rates of progression to ESRD; unfortunately, class III and IV are also the most common lesions found in children. (62) Aggressive immunotherapy in these cases is recommended to mitigate the associated inflammatory damage. (63)(64) Class V (membranous lupus nephritis) on its own is considered less severe than class III and IV, although it most commonly presents in conjunction with class III or IV. The clinical presentation of SLE does not always correlate well with the severity of the histopathologic findings, and, because there is no reliable biomarker available that correlates well with disease activity, a renal biopsy should be considered when GN is suspected, including in the case of persistent mild proteinuria. (62)(65) All children with SLE should have close monitoring of blood pressure, serum creatinine level, proteinuria, and hematuria because renal disease can also be representative of disease flares, even after remission. (62)(65) For patients with previous active SLE nephritis, follow-up is recommended every 3 months. (66)
In children, as in adults, nephrotic-range proteinuria is a serious condition on its own that can be associated with myriad complications: increased risk of infections are seen as defects in humoral immunity, making these patients more prone to encapsulated bacterial infections; thromboembolic events from decreased levels of protein S, plasminogen, and antithrombin III and increased levels of fibrinogen and factors V and VIII; renal failure from recurrent episodes of acute kidney injury and hypovolemia; and anasarca, the most extreme form of fluid maldistribution, which presents with massive generalized edema, large pleural effusions, and ascites. (47) Any or all of these complications can negatively impact the clinical course.
Although proximal tubular loss of LMW proteins may be significant, isolated urine protein loss of this kind is not associated with the body swelling noted in nephrotic syndrome. Tubular proteinuria most often appears as a result of injury to the proximal tubule and in the pediatric population is more commonly secondary rather than primary in nature. Tubular damage, often induced by various drug exposures or circulatory compromise, results in impaired ability to reabsorb the LMW proteins, which are normally filtered by the glomerulus and reabsorbed by the proximal tubule. Although secondary rather than primary causes of tubular proteinuria are more likely to be encountered in children (such as acute tubular necrosis and acute interstitial nephritis), one should take note of a few primary causes that may be considered, including but not limited to cystinosis, polycystic kidney disease, Wilson disease, and mitochondrial disorders (Table 3).
Note that persistent proteinuria is associated with CKD. Proteinuria that is persistent may be the first sign of glomerular damage or loss of renal function. It has long been established that the degree of proteinuria is associated with progression of CKD. (35) Not only does protein serve as an indicator of renal damage, but it is also recognized as a perpetrator of ongoing renal damage. (35)(67)(68) As such, children who present with persistent proteinuria should undergo evaluation of renal function, and a thorough history detailing any significant illnesses or prenatal or perinatal events is essential (including but not limited to items listed in Table 1) to help determine possible causes.
Evaluation of Proteinuria
Patients with a positive urine dipstick (‡1+) of protein should have a complete UA and quantification of proteinuria with a spot U p/c, preferably in a first morning urine sample. This sample is best obtained by completely emptying the bladder before going to sleep (discarding that urine) and collecting the urine on awakening, before any other activity is performed. (48)(49) No further evaluation is necessary if the first morning urine sample has a normal U p/c of 0.2 or less (Fig 2) because the most likely diagnosis is orthostatic proteinuria and historically is not associated with long-term sequelae. Still, some pediatric nephrologists would advise repeating a first morning void on a yearly basis in patients who continue to demonstrate proteinuria. (48) Further evaluation is warranted if the first morning U p/c is greater than 0.2; these patients should undergo renal ultrasonography, serum creatinine, albumin, cholesterol and electrolytes, and C3/C4 and antinuclear antibodies, especially in the setting of a positive family history of autoimmune disorders or renal disease (Fig 2). (35)(48)(49) At this point, referral to a pediatric nephrologist may be indicated.
Being able to differentiate between temporary or benign proteinuria and proteinuria associated with a more serious condition can be challenging. Persistent proteinuria should not be overlooked because it is well-known to be associated with CKD. At the same time, the primary care provider should be aware that most adolescents who are found to have proteinuria on a screening UA do not have true renal disease and the proteinuria will resolve on repeated testing. (33) Confirming proteinuria that is or is not orthostatic in nature can and should be determined in a timely manner, as prompt referral to a pediatric nephrologist may be needed for those in whom a more serious condition is being considered. By the same token, declining further evaluation of transient and orthostatic proteinuria in an asymptomatic patient can potentially lead to avoidable family and patient anxiety, as well as unnecessary investigations and expenses.
CONCLUSION
Hematuria and proteinuria are findings that can be of concern to both clinicians and families. Fortunately, in the great majority of cases, repeated studies or further evaluation will reveal no abnormalities and little or no need for further evaluation. If hematuria and/or proteinuria is confirmed, a detailed history along with investigation for extrarenal symptoms, high blood pressure, and abnormal renal chemistries will be helpful when deciding who may be followed in the primary care office versus who to refer to nephrology for further evaluation, and how soon to refer. Further studies, including additional laboratory work, radiologic imaging, and percutaneous renal biopsy, may be undertaken by the consulting nephrologist to elucidate the cause of the urinary abnormality and to guide management.
Summary.
Based on some research evidence as well as consensus, hematuria or proteinuria that persists on repeated testing warrants additional evaluation, and, depending on history along with initial evaluation in the primary care office, may warrant referral to a pediatric nephrologist for further management. (12)(15)(35)(48)
Based on some research evidence and expert opinion, microscopic hematuria that is associated with macroscopic hematuria and/or proteinuria warrants more urgent referral to a pediatric nephrologist because these signs can be associated with worsened renal outcome. (11)(12)
Based on some research evidence as well as consensus, the urine protein/creatinine ratio has been shown to be a fairly reliable surrogate for a 24-hour urine collection, especially when tested in the first morning urine specimen. (35)(36)(37)
Based on observational studies, the long-term prognosis of orthostatic proteinuria is generally benign. (42)(43)(68)
Based on some research evidence as well as consensus, persistent proteinuria is an indicator of renal damage and is also recognized as a perpetrator of ongoing renal damage. (35)(67)(69)
PIR Quiz.
There are two ways to access the journal CME quizzes:
Individual CME quizzes are available via the blue CME link under the article title in the Table of Contents of any issue.
To access all CME articles, click “Journal CME” from Gateway’s orangemainmenu or go directly to: http://www.aappublications.org/content/journal-cme.
To learn how to claim MOC points, go to: http://www.aappublications.org/content/moc-credit.
- A 16-year-old boy is brought to your office by his parents on a Monday morning because he noted his urine appeared red in color. He is known to you. He has a history of participating in numerous high-risk behaviors in the previous year, including binge drinking and ingestion of various drugs as well as accepting eating challenges. He discloses to you that he had some recent upper respiratory infection symptoms and that he had been partying with his friends over the weekend. His physical examination does not reveal any specific abnormalities. His blood pressure is 125/65 mm Hg. A urinalysis (UA) shows reddish urine. The urine dipstick is 3+ positive for blood but negative for protein. The spun sediment performed by the laboratory shows only some granular casts but no red blood cells (RBCs). Which of the following diagnoses most explains these findings in this patient?
- Acute postinfectious nephritis.
- Acute liver injury.
- Acute muscle injury.
- Immunoglobulin A nephritis.
- Paprika ingestion.
- A 7-year-old boy is brought to your office by his parents with acute onset of brown-colored urine, vague malaise, and mild periorbital edema. He is otherwise healthy, with a negative medical history. His family history is negative for renal disease. His blood pressure is 130/75 mm Hg. His physical examination shows no abnormalities except for mild periorbital edema. Results of his laboratory studies immediately available include a serum sodium level of 140 mEq/L (140 mmol/L), potassium level of 4.0 mEq/L (4.0 mmol/L), and creatinine level of 0.4 mg/dL (35.4 µmol/L). His UA shows a specific gravity of 1.010, pH 6, 3+ blood, 1+ protein, and more than 100 RBCs per high-power field with few RBC casts. Which of the following is the most appropriate next step in the diagnosis of this patient?
- A 24-hour urine collection for protein.
- A renal biopsy.
- C3 and C4 complement levels.
- Genetic testing for renal disease.
- Serum immunoglobulin A level.
- A 10-year-old girl undergoes routine periodic screening for proteinuria because she was born at 28 weeks’ gestation and had umbilical catheters placed. She has subsequently grown and developed well and is currently healthy. Which of the following would be a reassuring screening result (that would indicate no further testing at this time)?
- A UA that shows specific gravity less than 1.005, pH 6, and 1+ protein.
- A UA that shows specific gravity 1.030, pH 8, and 2+ protein.
- A urine protein/creatinine ratio (U p/c) of 0.1.
- A U p/c of 1.0.
- A U p/c of 2.5.
- A 15-year-old girl is brought to the clinic by her parents for a routine school physical. A routine UA, performed only because her school physical required it, shows a specific gravity 1.010, pH 6, 2+ protein, no blood, and no white blood cells (leukocyte esterase). She is a healthy athlete with a normal medical history and a negative family history for renal disease. She returns after soccer practice the following day and her repeated UA shows very similar results. Which of the following is the best next step in the evaluation of the proteinuria in this patient?
- Do a 24-hour urine collection for protein quantitation.
- Obtain a first morning urine sample and send for U p/c quantitation.
- Order no further studies, sign her form, and reassure her that she’s fine.
- Refer to pediatric nephrology for renal biopsy.
- Send today’s specimen for U p/c quantitation.
- A 3-year-old boy is brought to the pediatrician’s office by his parents with a 2-day history of increasingly noticeable facial and pedal edema. He is otherwise well and playing actively in the office. He is fully immunized and has a completely negative medical history and a family history that is negative for renal disease. His physical examination shows blood pressure 88/56 mm Hg, mild periorbital edema, and edema of his feet and legs. The remainder of the examination is otherwise normal. A purified protein derivative (PPD) skin test is placed and an initial laboratory evaluation is performed, and the results of both are pending. The pediatrician’s office is located in a rural area where access to a pediatric nephrologist is not easily available. Which of the following clinical and laboratory findings in this patient would prompt an immediate referral to a pediatric nephrologist rather than initiating treatment by his pediatrician?
- Mild ascites on physical examination.
- Serum albumin level of 1.2 g/dL (12.0 g/L) (reference range, 3.5–4.5 g/dL [35.0–45.0 g/L]).
- Serum cholesterol level of 350 mg/dL (9 mmol/L) (reference range, <190 mg/dL [<5 mmol/L]).
- Serum C3 complement level of 24 mg/dL (reference range, 93–120 mg/dL).
- U p/c of 3.0.
REQUIREMENTS:
Learners can take Pediatrics in Review quizzes and claim credit online only at: http://pedsinreview.org.
To successfully complete 2018 Pediatrics in Review articles for AMA PRA Category 1 CreditTM, learners must demonstrate a minimum performance level of 60% or higher on this assessment. If you score less than 60% on the assessment, you will be given additional opportunities to answer questions until an overall 60% or greater score is achieved.
This journal-based CME activity is available through Dec. 31, 2020, however, credit will be recorded in the year in which the learner completes the quiz.
2018 Pediatrics in Review now is approved for a total of 30 Maintenance of Certification (MOC) Part 2 credits by the American Board of Pediatrics through the AAP MOC Portfolio Program. Complete the first 10 issues or a total of 30 quizzes of journal CME credits, achieve a 60% passing score on each, and start claiming MOC credits as early as October 2018. To learn how to claim MOC points, go to: http://www.aappublications.org/content/moc-credit.
ABBREVIATIONS
- AS
Alport syndrome
- CKD
chronic kidney disease
- CNS
congenital nephrotic syndrome
- ESRD
end-stage renal disease
- FSGS
focal segmental glomerular sclerosis
- GN
glomerulonephritis
- HSP
Henoch-Schönlein purpura
- HPF
high-power field
- Ig
immunoglobulin
- LMW
low molecular weight
- MCD
minimal change disease
- RBC
red blood cell
- SLE
systemic lupus erythematosus
- UA
urinalysis
- U p/c
urine protein/creatinine ratio
Footnotes
AUTHOR DISCLOSURE Drs Viteri and Reid-Adam have disclosed no financial relationships relevant to this article. Dr Viteri’s current affiliation is Children’s Hospital of Philadelphia, Philadelphia, PA. This commentary does not contain a discussion of an unapproved/investigative use of a commercial product/device.
References
- 1.Committee on Practice and Ambulatory Medicine; Bright Futures Periodicity Schedule Workgroup. 2017 recommendations for preventive pediatric health care. Pediatrics 2017;139(4):e20170254. [DOI] [PubMed] [Google Scholar]
- 2.Kitagawa T Lessons learned from the Japanese nephritis screening study. Pediatr Nephrol 1988;2(2):256–263 [DOI] [PubMed] [Google Scholar]
- 3.Dodge WF, West EF, Smith EH, Harvey Bruce III. Proteinuria and hematuria in schoolchildren: epidemiology and early natural history. J Pediatr 1976;88(2):327–347 [DOI] [PubMed] [Google Scholar]
- 4.Vehaskari VM, Rapola J, Koskimies O, Savilahti E, Vilska J, Hallman N. Microscopic hematuria in school children: epidemiology and clinicopathologic evaluation. J Pediatr 1979;95(5)(pt 1):676–684 [DOI] [PubMed] [Google Scholar]
- 5.Flynn JT, Kaelber DC, Baker-Smith CM, et al. ; Subcommittee on Screening and Management of High Blood Pressure in Children. Clinical Practice Guideline for Screening and Management of High Blood Pressure in Children and Adolescents. Pediatrics 2017;140(3):e20171904. [DOI] [PubMed] [Google Scholar]
- 6.Carmody JB, Charlton JR. Short-term gestation, long-term risk: prematurity and chronic kidney disease. Pediatrics 2013;131 (6):1168–1179 [DOI] [PubMed] [Google Scholar]
- 7.Rose BD. Pathophysiology of Renal Disease 2nd ed. New York, NY: McGraw-Hill; 1987 [Google Scholar]
- 8.Moinuddin IK, Leehey DJ. Handbook of Nephrology 1st ed. Baltimore, MD: Lippincott Williams & Wilkins; 2014:1–3 [Google Scholar]
- 9.Sutton JM. Evaluation of hematuria in adults. JAMA 1990;263(18):2475–2480 [PubMed] [Google Scholar]
- 10.Mariani AJ, Mariani MC, Macchioni C, Stams UK, Hariharan A, Moriera A. The significance of adult hematuria: 1,000 hematuria evaluations including a risk-benefit and cost-effectiveness analysis. J Urol 1989;141(2):350–355 [DOI] [PubMed] [Google Scholar]
- 11.Cohen RA, Brown RS. Clinical practice: microscopic hematuria. N Engl J Med 2003;348(23):2330–2338 [DOI] [PubMed] [Google Scholar]
- 12.Moreno JA, Yuste C, Gutiérrez E, et al. Haematuria as a risk factor for chronic kidney disease progression in glomerular diseases: a review. Pediatr Nephrol 2016;31(4):523–533 [DOI] [PubMed] [Google Scholar]
- 13.Avner ED, Harmon WE, Niaudet P, Yoshikawa N, Emma F, Goldstein SL, eds. Pediatric Nephrology 7th ed. Berlin, Germany: Springer-Verlag; 2016 [Google Scholar]
- 14.Dalrymple RA, Ramage IJ. Fifteen-minute consultation: the management of microscopic haematuria. Arch Dis Child Educ Pract Ed 2017;102(5):230–234 [DOI] [PubMed] [Google Scholar]
- 15.Meyers KE. Evaluation of hematuria in children. Urol Clin North Am 2004;31(3):559–573, x [DOI] [PubMed] [Google Scholar]
- 16.Fiore DC, Fox CL. Urology and nephrology update: proteinuria and hematuria. FP Essent 2014;416:11–21 [PubMed] [Google Scholar]
- 17.Reidy K, Del Rio M. Hematuria. In: Adam HM, Meschan Foy J, eds. Signs and Symptoms in Pediatrics Elk Grove Village, IL: American Academy of Pediatrics; 2015:471–477 [Google Scholar]
- 18.Issler N, Dufek S, Kleta R, Bockenhauer D, Smeulders N, Van’t Hoff W. Epidemiology of paediatric renal stone disease: a 22- year single centre experience in the UK. BMC Nephrol 2017;18(1):136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Okada M, Tsuzuki K, Ito S. Diagnosis of the nutcracker phenomenon using two-dimensional ultrasonography. Clin Nephrol 1998;49(1):35–40 [PubMed] [Google Scholar]
- 20.Shin JI, Park JM, Lee JS, Kim MJ. Effect of renal Doppler ultrasound on the detection of nutcracker syndrome in children with hematuria. Eur J Pediatr 2007;166(5):399–404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ahmed K, Sampath R, Khan MS. Current trends in the diagnosis and management of renal nutcracker syndrome: a review. Eur J Vasc Endovasc Surg 2006;31(4):410–416 [DOI] [PubMed] [Google Scholar]
- 22.Ghrahani R, Ledika MA, Sapartini G, Setiabudiawan B. Age of onset as a risk factor of renal involvement in Henoch- Schönlein purpura. Asia Pac Allergy 2014;4(1):42–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Trapani S, Micheli A, Grisolia F, et al. Henoch Schonlein purpura in childhood: epidemiological and clinical analysis of 150 cases over a 5-year period and review of literature. Semin Arthritis Rheum 2005;35(3):143–153 [DOI] [PubMed] [Google Scholar]
- 24.Peru H, Soylemezoglu O, Bakkaloglu SA, et al. Henoch Schonlein purpura in childhood: clinical analysis of 254 cases over a 3-year period. Clin Rheumatol 2008;27(9):1087–1092 [DOI] [PubMed] [Google Scholar]
- 25.Goldstein AR, White RH, Akuse R, Chantler C. Long-term follow-up of childhood Henoch-Schönlein nephritis. Lancet 1992;339(8788):280–282 [DOI] [PubMed] [Google Scholar]
- 26.Davin JC. Henoch-Schonlein purpura nephritis: pathophysiology, treatment, and future strategy. Clin J Am Soc Nephrol 2011;6(3):679–689 [DOI] [PubMed] [Google Scholar]
- 27.Massengill SF. Hematuria. Pediatr Rev 2008;29(10):342–348 [DOI] [PubMed] [Google Scholar]
- 28.Voskarides K, Damianou L, Neocleous V, et al. COL4A3/COL4A4 mutations producing focal segmental glomerulosclerosis and renal failure in thin basement membrane nephropathy. J Am Soc Nephrol 2007;18(11):3004–3016 [DOI] [PubMed] [Google Scholar]
- 29.Carasi C, Van’t Hoff WG, Rees L, Risdon RA, Trompeter RS, Dillon MJ. Childhood thin GBM disease: review of 22 children with family studies and long-term follow-up. Pediatr Nephrol 2005;20(8):1098–1105 [DOI] [PubMed] [Google Scholar]
- 30.Szeto CC, Mac-Moune Lai F, Kwan BC, et al. The width of the basement membrane does not influence clinical presentation or outcome of thin glomerular basement membrane disease with persistent hematuria. Kidney Int 2010;78(10):1041–1046 [DOI] [PubMed] [Google Scholar]
- 31.Eison TM, Ault BH, Jones DP, Chesney RW, Wyatt RJ. Post-streptococcal acute glomerulonephritis in children: clinical features and pathogenesis. Pediatr Nephrol 2011;26(2):165–180 [DOI] [PubMed] [Google Scholar]
- 32.Hogg RJ, Furth S, Lemley KV, et al. ; National Kidney Foundation’s Kidney Disease Outcomes Quality Initiative. National Kidney Foundation’s Kidney Disease Outcomes Quality Initiative clinical practice guidelines for chronic kidney disease in children and adolescents: evaluation, classification, and stratification. Pediatrics 2003;111(6)(pt 1):1416–1421 [DOI] [PubMed] [Google Scholar]
- 33.Vehaskari VM, Rapola J. Isolated proteinuria: analysis of a school-age population. J Pediatr 1982;101(5):661–668 [DOI] [PubMed] [Google Scholar]
- 34.Morcos SK, el-Nahas AM, Brown P, Haylor J. Effect of iodinated water soluble contrast media on urinary protein assays. BMJ 1992;305(6844):29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hogg RJ, Portman RJ, Milliner D, Lemley KV, Eddy A, Ingelfinger J. Evaluation and management of proteinuria and nephrotic syndrome in children: recommendations from a pediatric nephrology panel established at the National Kidney Foundation conference on proteinuria, albuminuria, risk, assessment, detection, and elimination (PARADE). Pediatrics 2000;105(6):1242–1249 [DOI] [PubMed] [Google Scholar]
- 36.Houser M Assessment of proteinuria using random urine samples. J Pediatr 1984;104(6):845–848 [DOI] [PubMed] [Google Scholar]
- 37.Houser MT, Jahn MF, Kobayashi A, Walburn J. Assessment of urinary protein excretion in the adolescent: effect of body position and exercise. J Pediatr 1986;109(3):556–561 [DOI] [PubMed] [Google Scholar]
- 38.Woroniecki R, Singer P. Proteinuria. In: Adam HH, Meschan Foy J, eds. Signs and Symptoms in Pediatrics , Elk Grove Village, IL: American Academy of Pediatrics; 2015:709–717 [Google Scholar]
- 39.Yang CY, Chen FA, Chen CF, et al. Diagnostic accuracy of urine protein/creatinine ratio is influenced by urine concentration. PLoS One 2015;10(9):e0137460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Naderi AS, Reilly RF. Primary care approach to proteinuria. J Am Board Fam Med 2008;21(6):569–574 [DOI] [PubMed] [Google Scholar]
- 41.Hogg RJ. Adolescents with proteinuria and/or the nephrotic syndrome. Adolesc Med Clin 2005;16(1):163–172 [DOI] [PubMed] [Google Scholar]
- 42.Robinson RR. Isolated proteinuria in asymptomatic patients. Kidney Int 1980;18(3):395–406 [DOI] [PubMed] [Google Scholar]
- 43.Levitt JI. The prognostic significance of proteinuria in young college students. Ann Intern Med 1967;66(4):685–696 [DOI] [PubMed] [Google Scholar]
- 44.Gillespie RS. Robert’s Review of Pediatric Nephrology Vol 2.1 2nd ed. Arlington, TX: KidneyWed Media; 2013 [Google Scholar]
- 45.Nephrotic syndrome in children: prediction of histopathology from clinical and laboratory characteristics at time of diagnosis: a report of the International Study of Kidney Disease in Children. Kidney Int 1978;13(2):159–165 [DOI] [PubMed] [Google Scholar]
- 46.The primary nephrotic syndrome in children: identification of patients with minimal change nephrotic syndrome from initial response to prednisone: a report of the International Study of Kidney Disease in Children. J Pediatr 1981;98(4):561–564 [DOI] [PubMed] [Google Scholar]
- 47.Andolino TP, Reid-Adam J. Nephrotic syndrome. Pediatr Rev 2015;36(3):117–125; quiz 126, 129 [DOI] [PubMed] [Google Scholar]
- 48.Sebestyen JF, Alon US. The teenager with asymptomatic proteinuria: think orthostatic first. Clin Pediatr (Phila) 2011;50 (3):179–182 [DOI] [PubMed] [Google Scholar]
- 49.Leung AK, Wong AH, Barg SS. Proteinuria in children: evaluation and differential diagnosis. Am Fam Physician 2017;95 (4):248–254 [PubMed] [Google Scholar]
- 50.Trautmann A, Bodria M, Ozaltin F, et al. ; PodoNet Consortium. Spectrum of steroid-resistant and congenital nephrotic syndrome in children: the PodoNet registry cohort. Clin J Am Soc Nephrol 2015;10(4):592–600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Freedman BI, Kopp JB, Langefeld CD, et al. The apolipoprotein L1 (APOL1) gene and nondiabetic nephropathy in African Americans. J Am Soc Nephrol 2010;21(9):1422–1426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Genovese G, Friedman DJ, Ross MD, et al. Association of trypanolytic ApoL1 variants with kidney disease in African Americans. Science 2010;329(5993):841–845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kopp JB, Nelson GW, Sampath K, et al. APOL1 genetic variants in focal segmental glomerulosclerosis and HIV-associated nephropathy. J Am Soc Nephrol 2011;22(11):2129–2137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Santín S, García-Maset R, Ruíz P, et al. ; FSGS Spanish Study Group. Nephrin mutations cause childhood- and adult-onset focal segmental glomerulosclerosis. Kidney Int 2009;76(12):1268–1276 [DOI] [PubMed] [Google Scholar]
- 55.Boute N, Gribouval O, Roselli S, et al. NPHS2, encoding the glomerular protein podocin, is mutated in autosomal recessive steroid-resistant nephrotic syndrome. Nat Genet 2000;24(4):349–354 [DOI] [PubMed] [Google Scholar]
- 56.Iatropoulos P, Noris M, Mele C, et al. Complement gene variants determine the risk of immunoglobulin-associated MPGN and C3 glomerulopathy and predict long-term renal outcome. Mol Immunol 2016;71:131–142 [DOI] [PubMed] [Google Scholar]
- 57.Licht C, et al. Membranoproliferative and C3-mediated GN in children. In: Avner ED, Harmon W, Niaudet P, Yoshikawa N, eds. Pediatric Nephrology Springer-Verlag Berlin Heidelberg; 2016:1035 [Google Scholar]
- 58.Lau KK, Chan HH, Massicotte P, Chan AK. Thrombotic complications of neonates and children with congenital nephrotic syndrome. Curr Pediatr Rev 2014;10(3):169–176 [PubMed] [Google Scholar]
- 59.Rheault MN, Gbadegesin RA. The genetics of nephrotic syndrome. J Pediatr Genet 2016;5(1):15–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Rheault MN. Nephrotic and nephritic syndrome in the newborn. Clin Perinatol 2014;41(3):605–618 [DOI] [PubMed] [Google Scholar]
- 61.Couture J, Silverman ED. Update on the pathogenesis and treatment of childhood-onset systemic lupus erythematosus. Curr Opin Rheumatol 2016;28(5):488–496 [DOI] [PubMed] [Google Scholar]
- 62.Levy DM, Kamphuis S. Systemic lupus erythematosus in children and adolescents. Pediatr Clin North Am 2012;59(2):345–364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Mina R, von Scheven E, Ardoin SP, et al. ; Carra SLE Subcommittee. Consensus treatment plans for induction therapy of newly diagnosed proliferative lupus nephritis in juvenile systemic lupus erythematosus. Arthritis Care Res (Hoboken) 2012;64(3):375–383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Schwartz N, Goilav B, Putterman C. The pathogenesis, diagnosis and treatment of lupus nephritis. Curr Opin Rheumatol 2014;26(5):502–509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Goilav B, Putterman C, Rubinstein TB. Biomarkers for kidney involvement in pediatric lupus. Biomarkers Med 2015;9 (6):529–543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sinha R, Raut S. Pediatric lupus nephritis: management update. World J Nephrol 2014;3(2):16–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Erkan E Proteinuria and progression of glomerular diseases. Pediatr Nephrol 2013;28(7):1049–1058 [DOI] [PubMed] [Google Scholar]
- 68.Rytand DA, Spreiter S. Prognosis in postural (orthostatic) proteinuria: forty to fifty-year follow-up of six patients after diagnosis by Thomas Addis. N Engl J Med 1981;305(11):618–621 [DOI] [PubMed] [Google Scholar]
- 69.Noone D, Licht C. Chronic kidney disease: a new look at pathogenetic mechanisms and treatment options. Pediatr Nephrol 2014;29(5):779–792 [DOI] [PubMed] [Google Scholar]