

Abstract
Background
Tardive dyskinesia (TD) remains a troublesome adverse effect of conventional antipsychotic (neuroleptic) medication. It has been proposed that TD could have a component of central cholinergic deficiency. Cholinergic drugs have been used to treat TD.
Objectives
To determine the effects of cholinergic drugs (arecoline, choline, deanol, lecithin, meclofenoxate, physostigmine, RS 86, tacrine, metoxytacrine, galantamine, ipidacrine, donepezil, rivastigmine, eptastigmine, metrifonate, xanomeline, cevimeline) for treating antipsychotic‐induced TD in people with schizophrenia or other chronic mental illness.
Search methods
An electronic search of the Cochrane Schizophrenia Group's Study‐Based Register of Trials (16 July 2015 and April 2017) was undertaken. This register is assembled by extensive searches for randomised controlled trials in many electronic databases, registers of trials, conference proceedings and dissertations. References of all identified studies were searched for further trial citations.
Selection criteria
We included reports identified by the search if they were of controlled trials involving people with antipsychotic‐induced TD and chronic mental illness, who had been randomly allocated to either a cholinergic agent or to a placebo or no intervention. Two review authors independently assessed the methodological quality of the trials.
Data collection and analysis
Two review authors extracted data and, where possible, estimated risk ratios (RR) or mean differences (MD), with 95% confidence intervals (CI). We analysed data on an intention‐to‐treat basis, with the assumption that people who left early had no improvement. We assessed risk of bias and created a 'Summary of findings' table using GRADE.
Main results
We included 14 studies investigating the use of cholinergic drugs compared with placebo published between 1976 and 2014. All studies involved small numbers of participants (five to 60 people). Three studies that investigated the new cholinergic Alzheimer drugs for the treatment of TD are new to this update. Overall, the risk of bias in the included studies was unclear, mainly due to poor reporting; allocation concealment was not described, generation of the sequence was not explicit, studies were not clearly blinded, we are unsure if data are incomplete, and data were often poorly or selectively reported.
We are uncertain about the effect of new or old cholinergic drugs on no clinically important improvement in TD symptoms when compared with placebo; the quality of evidence was very low (RR 0.89, 95% CI 0.65 to 1.23; 27 people, 4 RCTs). Eight trials found that cholinergic drugs may make little or no difference to deterioration of TD symptoms (low‐quality evidence, RR 1.11, 95% CI 0.55 to 2.24; 147 people). Again, due to very low‐quality evidence, we are uncertain about the effects on mental state (RR 0.50, 95% CI 0.10 to 2.61; 77 people, 5 RCTs), adverse events (RR 0.56, 95% CI 0.15 to 2.14; 106 people, 4 RCTs), and leaving the study early (RR 1.09,95% CI 0.56 to 2.10; 288 people 12 RCTs). No study reported on social confidence, social inclusion, social networks, or personalised quality of life.
Authors' conclusions
TD remains a major public health problem. The clinical effects of both older cholinergic drugs and new cholinergic agents, now used for treating Alzheimer's disease, are unclear, as too few, too small studies leave many questions unanswered. Cholinergic drugs should remain of interest to researchers and currently have little place in routine clinical work. However, with the advent of new cholinergic agents now used for treating Alzheimer's disease, scope exists for more informative trials. If these new cholinergic agents are to be investigated for treating people with TD, their effects should be demonstrated in large well‐designed, conducted and reported randomised trials.
Plain language summary
Cholinergic medication for antipsychotic‐induced tardive dyskinesia
Review question.
Are cholinergic drugs useful for treating the unpleasant side‐effect ‐ tardive dyskinesia ‐ in people with schizophrenia or other similar mental health problems who are taking antipsychotics.
Background.
People with schizophrenia often hear voices and see things (hallucinations) and have strange beliefs (delusions). These symptoms are usually treated with antipsychotic drugs. However, these drugs can have debilitating side‐effects. Tardive dyskinesia is an involuntary movement that causes the face, mouth, tongue and jaw to convulse, spasm and grimace. It is caused by long‐term or high dose of antipsychotic drugs, is difficult to treat and can be incurable. It has been proposed that tardive dyskinesia could be due to cholinergic deficiency. Older cholinergic drugs, such as deanol, lecithin and meclofenoxate, have been used to treat tardive dyskinesia. New cholinergic drugs, such as donepezil, galantamine and rivastigmine, have been developed for the treatment of Alzheimer's disease and may also be promising in the treatment for tardive dyskinesia.
Study characteristics.
We searched for trials in July 2015 and April 2017, using the Cochrane Schizophrenia Group's register of trials. The review includes 14 studies investigating the use of cholinergic drugs compared with placebo. All studies randomised small numbers of participants (five to 60 people) with schizophrenia or other chronic mental illnesses who had also developed antipsychotic‐induced tardive dyskinesia.
Key results.
We found the effects of both older and newer cholinergic drugs to be unclear as too few and too small studies are available and do not yield great evidence and leave many questions unanswered.
Quality of the evidence.
The available evidence is weak, limited, and small scale. It is not possible to recommend these drugs as a treatment for tardive dyskinesia based on our findings. To fully investigate whether the use of cholinergic drugs have any positive effects for people with tardive dyskinesia, there would have to be well‐designed, larger, longer‐term studies, particularly on new cholinergic drugs currently being used for treating Alzheimer’s disease.
Ben Gray, Senior Peer Researcher, McPin Foundation.
Summary of findings
Summary of findings for the main comparison. CHOLINERGIC DRUGS versus PLACEBO for antipsychotic‐induced tardive dyskinesia.
| CHOLINERGIC DRUGS versus PLACEBO for antipsychotic‐induced tardive dyskinesia | ||||||
| Patient or population: people with various psychiatric disorders (mainly schizophrenia) and antipsychotic‐induced tardive dyskinesia Settings: mostly inpatients in Australia, Canada, Germany, Japan, Switzerland and the USA. Intervention: CHOLINERGIC DRUGS (deanol, donepezil, galantamine, meclofenoxate, lecithin) versus PLACEBO | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of Participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| PLACEBO | CHOLINERGIC DRUGS | |||||
|
Tardive dyskinesia: No clinically important improvement follow‐up: 2 to 12 weeks |
1000 per 1000 | 890 per 1000 (650 to 1000) | RR 0.89 (0.65 to 1.23) | 27 (4 studies) | ⊕⊝⊝⊝ very low1,2 | None of the subgroups that reported on this outcome (deanol, donepezil, lecithin) found a significant difference between cholinergic drug and placebo. |
|
Tardive dyskinesia: Deterioration follow‐up: 9 days to 12 weeks |
116 per 1000 | 129 per 1000 (64 to 260) | RR 1.11 (0.55 to 2.24) | 147 (8 studies) | ⊕⊕⊝⊝ low1,3 | None of the subgroups that reported on this outcome (deanol, donepezil, lecithin, meclofenoxate) found a significant difference between cholinergic drug and placebo. |
|
Mental state: Deterioration follow‐up: 11 days to 12 weeks |
56 per 1000 | 28 per 1000 (6 to 145) | RR 0.50 (0.10 to 2.61) | 77 (5 studies) | ⊕⊝⊝⊝ very low1,2 | None of the subgroups that reported on this outcome (deanol, lecithin) found a significant difference between cholinergic drug and placebo. |
|
Adverse effects: Any adverse events follow‐up: 9 days to 8 weeks |
98 per 1000 | 55 per 1000 (15 to 210) | RR 0.56 (0.15 to 2.14) | 106 (4 studies) | ⊕⊝⊝⊝ very low1,2 | None of the subgroups that reported on this outcome (donepezil, lecithin, meclofenoxate) found a significant difference between cholinergic drug and placebo. |
|
Acceptability of treatment: Leaving the study early follow‐up: 9 days to 12 weeks |
90 per 1000 | 98 per 1000 (50 to 188) | RR 1.09 (0.56 to 2.10) | 288 (12 studies) | ⊕⊝⊝⊝ very low1,3,4 | None of the subgroups that reported on this outcome (deanol, donepezil, galantamine, meclofenoxate, lecithin) found a significant difference between cholinergic drug and placebo. |
| Social confidence, social inclusion, social networks, or personalised quality of life ‐ not reported | None of the included studies reported on this outcome. | |||||
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its CI). CI: Confidence interval; RR: Risk ratio; | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
1 Downgraded one step for risk of bias: for many studies it was unclear whether randomisation procedure and allocation concealment were carried out adequately. 2 Downgraded two steps for imprecision: very few events and participants, wide CIs that include both no effect and appreciable benefit for the intervention. 3 Downgraded one step for imprecision: wide CIs that include appreciable benefit for both the intervention and the control group, as well as no effect. 4 Downgraded one step for indirectness: leaving the study early is not a direct measure of acceptability of the intervention.
Background
Description of the condition
Since the 1950s antipsychotic (neuroleptic) medication has been used extensively to treat people with chronic mental illnesses such as schizophrenia. These drugs can effectively control symptoms such as abnormal perceptions (hallucinations), disordered thinking and fixed false beliefs (delusions). In addition, maintenance therapy with antipsychotics is associated with a reduced risk of relapse (Schooler 1993). However, antipsychotic medication has also been associated with a wide range of adverse effects, including movement disorders. The appearance of these disorders can be extremely disfiguring, compounds stigma, and is associated with poor compliance to antipsychotic treatment (Barnes 1993; Tarsy 2011).
Tardive dyskinesia (TD) is one such movement disorder and is characterised by abnormal, repetitive and involuntary movements. The clinical features include: tongue protrusion, side‐to‐side or rotatory movement of the jaw, lip smacking, puckering and pursing, and rapid eye blinking (Casey 1999). In some people rapid movements of the arms, legs, and trunk may also occur. TD is a chronic condition of insidious onset, the severity of which fluctuates spontaneously (APA 1992). Although the most frequent cause of TD is the use of antipsychotic medication, it is striking that dose reduction can lead to a temporary exacerbation in symptoms. Conversely, increasing the dose is often associated with a temporary remission (Cavallaro 1993; Smith 1980). Studies on the natural history of TD have reported widely variable remission rates (1% to 62%) depending on the patient's age, psychiatric diagnosis, course of the psychiatric disorder, and duration of therapy (Bergen 1989; Fernandez 2001; Glazer 1990).
The exact mechanisms of the pathophysiology of TD are unknown. Antipsychotic drugs block certain chemical receptor sites in the brain ‐ one of these is specific for dopamine (Casey 1995). One hypothesis explaining the cause of antipsychotic‐induced TD is that long‐term blockade of the dopamine receptors may lead to an imbalance between the activity of dopamine cells and others that employ choline (Alphs 1983; Casey 1995). This theory implies that there is an over activity of dopaminergic transmission in relation to cholinergic transmission in the striatum. Another hypothesis suggests that the chronic use of antipsychotics may also cause an abnormal production of highly active atoms and chemical groups (cytotoxic free radicals), which may damage specific cells in the brain. This, in turn, could contribute to the pathophysiology of TD (Andreassen 2000; Andreassen 2001; Cadet 1989). The latter theory is supported by the persistent nature of the syndrome, once established.
TD occurs in more than 20% of people that use first‐generation antipsychotic drugs continually for longer than three months (Kane 1982; Glazer 2000; Tarsy 2011). Every year 4% to 5% of adults and 25% to 30% of elderly persons who continually use these drugs begin to show signs of TD (APA 1992; Correll 2004). Advancing age is a risk factor for both TD's prevalence and severity, with those who are < 60 years of age being three times more likely to spontaneously remit (Jeste 2000; Smith 1980).
When the second‐generation antipsychotic drugs were introduced in the 1990’s many hoped that they would not cause TD (Miller 2007; Rosenheck 2007). Although the risk of developing TD with second‐generation antipsychotic drugs does seem to be reduced, TD risks have not been eliminated (Miller 2007; Tarsy 2011). There is even some evidence to indicate that rates of TD do not differ at all between first‐ and second‐generation antipsychotic drugs (Leucht 2009; Rosenheck 2007; Woods 2010). The large, definitive, US randomised trial of antipsychotic treatments for schizophrenia (CATIE), with a four‐year period of follow‐up, obtained an incidence rate of TD of around 17% (Miller 2008). Due to widespread use of second‐generation antipsychotic drugs, increased off‐label use, and an ageing population, the frequency of TD is likely to be higher than thought (Cloud 2014; Maher 2012), and increasing. The problem will be considerably greater for people in countries where use of newer drugs is less prevalent (Ballesteros 2000; Martins 2011).
Description of the intervention
Medications affecting the cholinergic pathway have been tested for the treatment of TD due to the hypothesis that TD is correlated with damage in cholinergic cells in striatal subregions. This damage might be caused by changes in dopaminergic transmission, as a result of prolonged antipsychotic treatment (Grimm 2001). A variety of cholinergic medications have been used in TD, starting from the 1970's. The cholinergic compounds included in this review have various functions in relation to acetylcholine, the main neurotransmitter and neuromodulator in the cholinergic pathway.
How the intervention might work
This review looked at compounds such as physostigmine, choline, and lecithin (containing phosphatidyl choline), which were some of the early cholinergic medications used for TD, as they were thought to be of potential help in treating TD by enhancing acetylcholine synthesis (Wurtman 1978). It was thought that high levels of choline in the blood (which, for instance, phosphatidyl choline converts into after digestion) would enhance acetylcholine synthesis in neurons by making the precursor more available. Other treatments included, such as deanol and meclofenoxate, have been tested due to their cholinomimetic actions (Casey 1975; Izumi 1986). Deanol, a synthetic substance, is also thought to be a precursor of acetylcholine, but this is unproven, and deanol might be an acetylcholine agonist or even a suppressor of acetylcholine synthesis. During the last 20 years many new central nervous system (CNS)‐active cholinergic compounds, predominantly acetylcholinesterase‐inhibitors such as galantamine (Loy 2006), rivastigmine (Birks 2015) and donepezil (Birks 2006b), have emerged for the treatment of Alzheimer's disease and dementia. These compounds were also included in this review, due to their potential for treating TD symptoms (Caroff 2001).
Why it is important to do this review
Several atypical antipsychotic drugs have been produced in the last decades that claim to cause less or no TD (Lieberman 1996). These claims may or may not be true, and certainly evidence does point to the fact that thoughtful use of older generation drugs is not associated with any more problems of TD than with newer treatments (Chouinard 2008). However, in a global context, it is likely that the less expensive and more familiar drugs ‐ such as chlorpromazine or haloperidol ‐ will continue to be the mainstay of treatment of people with schizophrenia (WHO Essential List 2010). Use of drugs such as these is associated with the emergence of TD and, therefore, this condition will remain a problem for years to come.
TD can result in considerable social and physical disability (Barnes 1993) and symptoms are often irreversible (Bergen 1989; Fernandez 2001; Gerlach 1988; Glazer 1990). Additionally, TD is frequently associated with lower quality of life (Ascher‐Svanum 2008) and a greater mortality rate (Chong 2009). Given the high incidence and prevalence of TD among people taking antipsychotic medication, the need for prevention or treatment is clear. Unfortunately, there has been sparse evidence to guide clinicians (NICE 2014; Taylor 2009). Although many treatments have been tested, no one intervention has been shown clearly to be effective. Cessation or reduction of the dose of antipsychotic medication would be the ideal management for TD. In clinical practice this is not always possible, not least because in many individuals such a reduction would lead to relapse. This review focuses on whether the addition of different types of cholinergic medications to those already receiving antipsychotic medication is likely to help TD.
This review is one in a series of Cochrane reviews (see Table 6) evaluating treatments for antipsychotic‐induced TD, and is an update of a Cochrane review first published in 1997 (McGrath 1997), and previously updated in 2002 (Tammenmaa 2002).
1. Other reviews in the series.
| Interventions | Reference |
| Anticholinergic medication | Soares‐Weiser 1997 |
| Benzodiazepines | Bhoopathi 2006 |
| Calcium channel blockers | Essali 2011 |
| Cholinergic medication | This review |
| Gamma‐aminobutyric acid agonists | Alabed 2011 |
| Miscellaneous treatments | Soares‐Weiser 2003 |
| Neuroleptic reduction and/or cessation and neuroleptics | Soares‐Weiser 2006 |
| Non‐neuroleptic catecholaminergic drugs | El‐Sayeh 2006 |
| Vitamin E | Soares‐Weiser 2011 |
Objectives
The primary objective of this review is to determine the effects of cholinergic drugs (arecoline, choline, deanol, lecithin, meclofenoxate, physostigmine, RS 86, tacrine, metoxytacrine, galantamine, ipidacrine, donepezil, rivastigmine, eptastigmine, metrifonate, xanomeline, cevimeline) for treating antipsychotic‐induced TD in people with schizophrenia or other chronic mental illness.
The secondary objectives are: 1. to examine whether duration of treatment has an effect on treatment response; 2. to examine whether there is difference in treatment effect for the various cholinergic compounds; 3. to examine whether treatment response differs in people with schizophrenia who are older (above 65 years) and for whom the prevalence of spontaneous dyskinesias is estimated to be higher. (This secondary objective was added in response to a comment from peer review in the first substantial up‐date of the review).
Methods
Criteria for considering studies for this review
Types of studies
All relevant randomised controlled trials. Where a trial was described as 'double‐blind' but it implied that the study was randomised and the demographic details of each group were similar, it was included and we conducted a sensitivity analysis to evaluate the effect of the presence or absence of these data. We excluded quasi‐randomised studies, such as those allocated by using alternate days of the week.
Types of participants
People with schizophrenia or any other serious mental illness, diagnosed by any criteria, irrespective of gender, age or nationality who: 1. required the use of antipsychotics for more than three months; 2. developed TD during antipsychotic treatment (diagnosed by any criteria at baseline of the trial and at least one other occasion); 3. for whom the dose of antipsychotic medication had been stable for one month or more before the trial and during the trial (the same applies for those free of antipsychotics).
Types of interventions
1. The cholinergic drugs
Arecoline, choline, deanol, lecithin, meclofenoxate, physostigmine, RS 86.
In the first substantial update of the review the following cholinergic compounds were assessed to be relevant and added to the scope of the review: 7‐methoxytacrine, cevimeline, donepezil, eptastigmine, galantamine, ipidacrine, metrifonate, rivastigmine, tacrine, xanomeline, .
2. Control condition
Placebo or no intervention.
For the 2017 update a post hoc decision was made to also include studies evaluating the above mentioned cholinergic drugs compared to any other intervention for the treatment of TD.
Types of outcome measures
When appropriate, the we grouped outcomes into time periods ‐ short term (less than six weeks), medium term (between six weeks and six months) and long term (over six months).
Primary outcomes
1. Tardive dyskinesia
No clinically important improvement in the symptoms of individuals, defined as more than 50% improvement on any TD scale ‐ any time period.
2. Adverse effects
No clinically significant extrapyramidal adverse effects ‐ any time period.
Secondary outcomes
1. Tardive dyskinesia
1.1 Any improvement in the symptoms of individuals on any TD scale, as opposed to no improvement. 1.2 Deterioration in the symptoms of individuals, defined as any deleterious change on any TD scale. 1.3 Average change in severity of TD during the trial period. 1.4 Average difference in severity of TD at the end of the trial.
2. Global outcome measures (this category of outcome measures was added in the first substantial update of the review).
2.1 The number of people per treatment group who died for any reason. 2.2 Treatment group mean and standard deviation of endpoint score on any scale of quality of life. 2.3 Treatment group mean and standard deviation of endpoint score on any scale of level of functioning.
3. General mental state changes
3.1 Deterioration in general psychiatric symptoms (such as delusions and hallucinations) defined as any deleterious change on any scale. 3.2 Average difference in severity of psychiatric symptoms at the end of the trial.
4. Acceptability of the treatment
4.1 Acceptability of the intervention to the participant group as measured by numbers of people dropping out during the trial.
5. Adverse effects
5.1 Use of any anti‐parkinsonism drugs. 5.2 Average score/change in extrapyramidal adverse effects. 5.3 Acute dystonia.
6. Other adverse effects, general and specific
7. Hospital and service utilisation outcomes
7.1 Hospital admission. 7.2 Average change in days in hospital. 7.3 Improvement in hospital status (for example: change from formal to informal admission status, use of seclusion, level of observation).
8. Economic outcomes
8.1 Average change in total cost of medical and mental health care. 8.2 Total indirect and direct costs.
9. Social confidence, social inclusion, social networks, or personalised quality of life measures
9.1. No significant change in social confidence, social inclusion, social networks, or personalised quality of life measures. 9.2 Average score/change in social confidence, social inclusion, social networks, or personalised quality of life measures.
10. Behaviour
10.1 Clinically significant agitation. 10.2 Use of adjunctive medication for sedation. 10.3 Aggression to self or others.
11. Cognitive state
11.1 No clinically important change. 11.2 No change, general and specific.
'Summary of findings' table
We used the GRADE approach to interpret findings (Schünemann 2008) and used GRADEpro to export data from this review to create a 'Summary of findings' table. This table provides outcome‐specific information concerning the overall quality of evidence from each included study in the comparison, the magnitude of effect of the interventions examined, and the sum of the available data on all outcomes we rated as important to patient care and decision making. We selected the following main outcomes for inclusion in the 'Summary of findings' table.
1. Tardive dyskinesia 1.1 Improved to a clinically important extent 1.2 Deteriorated
2. Mental state 2.1 Deteriorated
3. Adverse effect 3.1 Any adverse event 3.2 Adverse effects: no clinically significant extrapyramidal adverse effects
4. Acceptability of treatment 4.1 Leaving the study early
5. Social confidence, social inclusion, social networks, or personalised quality of life measures* 5.1 No significant change in social confidence, social inclusion, social networks, or personalised quality of life measures for either recipients of care or caregivers.
* Outcome designated important to patients. We wished to add perspectives from people’s personal experience with TD to the research agenda. A consultation with service users was planned where a previously published version of a review in the Cochrane TD series (Soares‐Weiser 2011; see Table 6) and a lay overview of that review gave the foundation for the discussions. The session was planned to provide time to reflect on current research on TD and consider gaps in knowledge. The report is published in the Health Technology Assessment (HTA) report for the UK National Institute of Health Research (Bergman 2017). We have added one figure showing one service user's expression of frustration concerning this neglected area of research (Figure 1). Informed by the results of the consultation, for this review, we updated outcomes for the 'Summary of findings' table.
1.

Message from one of the participants of the Public and patient involvement consultation of service user perspectives on tardive dyskinesia research.
Search methods for identification of studies
Electronic searches
The 2017 review update was carried out in parallel with updating eight other TD reviews, see Table 6 for details. The search for trials covered all nine TD reviews.
1. Cochrane Schizophrenia Group’s Study‐Based Register of Trials
We searched the register on 16 July, 2015 and 26 April 2017 using the following string:
*Tardive Dyskinesia* in Healthcare Condition Field of Study
In such a study‐based register, searching the major concept retrieves all the synonym keywords and relevant studies because all the studies have already been organised based on their interventions and linked to the relevant topics.
The Cochrane Schizophrenia Group’s Register of Trials is compiled by systematic searches of major resources (including AMED, BIOSIS, CINAHL, Embase, MEDLINE, PsycINFO, PubMed, and registries of clinical trials) and their monthly updates, handsearches, grey literature, and conference proceedings (see Group’s Module). There is no language, date, document type, or publication status limitations for inclusion of records into the register.
2. Details of previous electronic searches
See Appendix 1.
Searching other resources
1. Reference searching
We inspected references of all included studies for further relevant studies.
2. Personal contact
Where necessary, we contacted the first author of each included study for information regarding unpublished trials. We noted the outcome of this contact in the included studies table.
Data collection and analysis
Selection of studies
For the 2017 update, review author RA and Antonio Grande (AG) (see Acknowledgements) inspected all abstracts of studies identified as above and identified potentially relevant reports. We resolved disagreements by discussion, or where there was still doubt, we acquired the full‐text article for further inspection. We acquired the full‐text articles of all relevant reports/abstracts meeting the initial criteria for reassessment and carefully inspected for a final decision on inclusion (see Criteria for considering studies for this review). RA and AG were not blinded to the names of the authors, institutions or journal of publication. Where difficulties or disputes arose, we asked review author HB for assistance and had it not been possible to decide, or if adequate information was not available to make a decision, we planned to add these studies to those awaiting assessment and contact the authors of the papers for clarification.
Data extraction and management
1. Extraction
For the 2017 update, review authors RA and HB independently extracted data from all included studies. Again, we discussed any disagreement and documented decisions. With remaining problems KSW helped clarify issues and we documented these final decisions. We extracted data presented only in graphs and figures whenever possible, but included only if two review authors independently had the same result. We attempted to contact authors through an open‐ended request in order to obtain missing information or for clarification whenever necessary. If studies were multicentre, where possible, we extracted data relevant to each component centre separately.
2. Management
2.1 Forms
For the 2017 update we extracted data to simple forms. Extracted data are available here with a link to the original source PDF for each item.
2.2 Scale‐derived data
We included continuous data from rating scales only if: a) the psychometric properties of the measuring instrument have been described in a peer‐reviewed journal (Marshall 2000); and b) the measuring instrument has not been written or modified by one of the trialists for that particular trial. Ideally, the measuring instrument should either be i. a self‐report or ii. completed by an independent rater or relative (not the therapist). We realise that this is not often reported clearly; we noted in Description of studies if this was the case or not.
2.3 Endpoint versus change data
There are advantages of both endpoint and change data. Change data can remove a component of between‐person variability from the analysis. On the other hand, calculation of change needs two assessments (baseline and endpoint), which can be difficult in unstable and difficult to measure conditions such as schizophrenia. We decided primarily to use endpoint data, and only use change data if the former were not available. We combined endpoint and change data in the analysis as we preferred to use mean differences (MD) rather than standardised mean differences (SMD) throughout (Higgins 2011).
2.4 Skewed data
Continuous data on clinical and social outcomes are often not normally distributed. To avoid the pitfall of applying parametric tests to non‐parametric data, we applied the following standards to relevant data before inclusion.
We entered data from studies of at least 200 participants in the analysis, because skewed data pose less of a problem in large studies. We also entered all relevant change data as when continuous data are presented on a scale that includes a possibility of negative values (such as change data), it is difficult to tell whether data are skewed or not.
For endpoint data from studies < 200 participants:
(a) when a scale started from the finite number zero, we subtracted the lowest possible value from the mean, and divided this by the standard deviation (SD). If this value was lower than 1, it strongly suggests a skew and we excluded these data. If this ratio was higher than 1 but below 2, there is suggestion of skew. We entered these data and tested whether its inclusion or exclusion changed the results substantially. Finally, if the ratio was larger than 2, we included these data, because skew is less likely (Altman 1996; Higgins 2011).
(b) if a scale starts from a positive value (such as the Positive and Negative Syndrome Scale (PANSS), (Kay 1986)), which can have values from 30 to 210), we modified the calculation described above to take the scale starting point into account. In these cases skew is present if 2 SD > (S‐S min), where S is the mean score and 'S min' is the minimum score.
2.5 Common measure
Where relevant, to facilitate comparison between trials, we converted variables that can be reported in different metrics, such as days in hospital (mean days per year, per week or per month) to a common metric (e.g. mean days per month).
2.6 Conversion of continuous to binary
Where possible, we converted continuous outcome measures to dichotomous data. This can be done by identifying cut‐off points on rating scales and dividing participants accordingly into 'clinically improved' or 'not clinically improved'. It is generally assumed that if there is a 50% reduction in a scale‐derived score such as the Brief Psychiatric Rating Scale (BPRS, Overall 1962) or the Positive and Negative Syndrome Scale (PANSS, Kay 1986), this can be considered as a clinically significant response (Leucht 2005; Leucht 2005a). If data based on these thresholds were not available, we used the primary cut‐off presented by the original authors.
2.7 Direction of graphs
Where possible, we entered data in such a way that the area to the left of the line of no effect indicated a favourable outcome for cholinergic medication. Where keeping to this made it impossible to avoid outcome titles with clumsy double‐negatives (e.g. 'Not un‐improved'), we presented data where the left of the line indicates an unfavourable outcome and noted this in the relevant graphs.
Assessment of risk of bias in included studies
Review authors RA and HB independently assessed risk of bias within the included studies by using criteria described in the Cochrane Handbook for Systematic Reviews of Interventions to assess trial quality (Higgins 2011). This set of criteria is based on evidence of associations between overestimate of effect and high risk of bias of the article such as sequence generation, allocation concealment, blinding, incomplete outcome data and selective reporting.
If the raters disagreed, we made the final rating by consensus, with the involvement of another member of the review group. Where inadequate details of randomisation and other characteristics of trials were provided, we contacted authors of the studies in order to obtain further information. If non‐concurrence occurred, we reported this.
Measures of treatment effect
1. Binary data
For binary outcomes we calculated a standard estimation of the risk ratio (RR) and its 95% confidence interval (CI). It has been shown that RR is more intuitive (Boissel 1999) than odds ratios as odds ratios tend to be interpreted as RR by clinicians (Deeks 2000).
2. Continuous data
For continuous outcomes we estimated mean difference (MD) between groups. We preferred not to calculate effect size measures (standardised mean difference SMD). However, if scales of very considerable similarity were used, we presumed there was a small difference in measurement, and calculated effect size and transformed the effect back to the units of one or more of the specific instruments.
Unit of analysis issues
1. Cluster trials
Studies increasingly employ 'cluster randomisation' (such as randomisation by clinician or practice), but analysis and pooling of clustered data poses problems. Firstly, authors often fail to account for intra‐class correlation in clustered studies, leading to a 'unit of analysis' error (Divine 1992) whereby P values are spuriously low, confidence intervals unduly narrow and statistical significance overestimated. This causes type I errors (Bland 1997; Gulliford 1999).
If any of the included trials had randomised participants by clusters, and where clustering had not been accounted for in primary studies, we would have presented such data in a table, with a (*) symbol to indicate the presence of a probable unit of analysis error. In subsequent versions of this review, we will seek to contact first authors of studies to obtain intra‐class correlation coefficients (ICCs) for their clustered data and adjust for this by using accepted methods (Gulliford 1999). Where clustering has been incorporated into the analysis of primary studies, we will present these data as if from a non‐cluster randomised study, but adjust for the clustering effect.
We have sought statistical advice and have been advised that the binary data as presented in a report should be divided by a 'design effect'. This is calculated using the mean number of participants per cluster (m) and the ICC [Design effect = 1+(m‐1)*ICC] (Donner 2002). If the ICC is not reported it will be assumed to be 0.1 (Ukoumunne 1999).
If cluster studies have been appropriately analysed taking into account ICCs and relevant data documented in the report, synthesis with other studies would be possible using the generic inverse variance technique.
2. Cross‐over trials
A major concern of cross‐over trials is the carry‐over effect. It occurs if an effect (e.g. pharmacological, physiological or psychological) of the treatment in the first phase is carried over to the second phase. As a consequence, on entry to the second phase the participants can differ systematically from their initial state despite a wash‐out phase. For the same reason cross‐over trials are not appropriate if the condition of interest is unstable (Elbourne 2002). As both effects are very likely in severe mental illness, we only used data of the first phase of cross‐over studies.
3. Studies with multiple treatment groups
Where a study involved more than two treatment arms, if relevant, we presented the additional treatment arms in comparisons. If data were binary we simply added and combined within the two‐by‐two table. If data were continuous, we combined data following the formula in section 7.7.3.8 (Combining groups) of the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011). We did not use data where the additional treatment arms were not relevant.
Dealing with missing data
1. Overall loss of credibility
At some degree of loss of follow‐up, data must lose credibility (Xia 2009). We chose that, for any particular outcome, should more than 50% of data be unaccounted for, we would not reproduce these data or use them within analyses. If, however, more than 50% of those in one arm of a study were lost, but the total loss was less than 50%, we addressed this within the 'Summary of findings' table by down‐rating quality. We also planned to downgrade quality within the 'Summary of findings' table should loss be 25% to 50% in total.
2. Binary
In the case where attrition for a binary outcome was between 0% and 50% and where these data were not clearly described, we presented data on a 'once‐randomised‐always‐analyse' basis (an intention‐to‐treat (ITT) analysis). We assumed all those leaving the study early had no improvement. We undertook a sensitivity analysis to test how prone the primary outcomes were to change by comparing data only from people who completed the study to that point to the ITT analysis using the above assumptions.
3. Continuous
3.1 Attrition
We reported and used data where attrition for a continuous outcome was between 0% and 50%, and data only from people who completed the study to that point were reported.
3.2 Standard deviations
If standard deviations (SDs) were not reported, we first tried to obtain the missing values from the authors. If not available, where there were missing measures of variance for continuous data, but an exact standard error (SE) and confidence intervals available for group means, and either 'P' value or 't' value available for differences in mean, we calculated them according to the rules described in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011): When only the SE is reported, SDs are calculated by the formula SD = SE * square root (n). Chapters 7.7.3 and 16.1.3 of the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011) present detailed formulae for estimating SDs from P values, t or F values, confidence intervals, ranges or other statistics. If these formulae did not apply, we calculated the SDs according to a validated imputation method which is based on the SDs of the other included studies (Furukawa 2006). Although some of these imputation strategies can introduce error, the alternative would be to exclude a given study’s outcome and thus to lose information. We nevertheless examined the validity of the imputations in a sensitivity analysis excluding imputed values.
3.3 Assumptions about participants who left the trials early or were lost to follow‐up
Various methods are available to account for participants who left the trials early or were lost to follow‐up. Some trials just present the results of study completers, others use the method of last observation carried forward (LOCF), while more recently methods such as multiple imputation or mixed‐effects models for repeated measurements (MMRM) have become more of a standard. While the latter methods seem to be somewhat better than LOCF (Leon 2006), we feel that the high percentage of participants leaving the studies early and differences in the reasons for leaving the studies early between groups is often the core problem in randomised schizophrenia trials. We therefore did not exclude studies based on the statistical approach used. However, we preferred to use the more sophisticated approaches. (e.g. MMRM or multiple‐imputation) and only presented completer analyses if some kind of ITT data were not available at all. Moreover, we addressed this issue in the item "incomplete outcome data" of the 'Risk of bias' tool.
Assessment of heterogeneity
1. Clinical heterogeneity
We considered all included studies initially, without seeing comparison data, to judge clinical heterogeneity. We simply inspected all studies for clearly outlying people or situations which we had not predicted would arise and discussed them in the text if they arose.
2. Methodological heterogeneity
We considered all included studies initially, without seeing comparison data, to judge methodological heterogeneity. We simply inspected all studies for clearly outlying methods which we had not predicted would arise and discussed them in the text if they arose.
3. Statistical heterogeneity
3.1 Visual inspection
We visually inspected graphs to investigate the possibility of statistical heterogeneity.
3.2 Employing the I2 statistic
We investigated heterogeneity between studies by considering the I2 method alongside the Chi2 'P' value. The I2 provides an estimate of the percentage of inconsistency thought to be due to chance (Higgins 2003). The importance of the observed value of I2 depends on i. magnitude and direction of effects and ii. strength of evidence for heterogeneity (e.g. 'P' value from Chi2 test, or a confidence interval for I2). An I2 estimate greater than or equal to around 50% accompanied by a statistically significant Chi2 statistic, can be interpreted as evidence of substantial levels of heterogeneity (Section 9.5.2 Cochrane Handbook for Systematic Reviews of InterventionsHiggins 2011). We explored and discussed in the text potential reasons for substantial levels of heterogeneity (Subgroup analysis and investigation of heterogeneity).
Assessment of reporting biases
Reporting biases arise when the dissemination of research findings is influenced by the nature and direction of results (Egger 1997). These are described in Section 10 of the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011). We are aware that funnel plots may be useful in investigating reporting biases but are of limited power to detect small‐study effects. We did not use funnel plots for outcomes where there were 10 or fewer studies, or where all studies were of similar sizes. In future versions of this review, if funnel plots are possible, we will seek statistical advice in their interpretation.
Data synthesis
We understand that there is no closed argument for preference for use of fixed‐effect or random‐effects models. The random‐effects method incorporates an assumption that the different studies are estimating different, yet related, intervention effects. This often seems to be true to us and the random‐effects model takes into account differences between studies even if there is no statistically significant heterogeneity. There is, however, a disadvantage to the random‐effects model. It puts added weight onto small studies, which often are the most biased ones. Depending on the direction of effect, these studies can either inflate or deflate the effect size. We chose the fixed‐effect model for all analyses.
Subgroup analysis and investigation of heterogeneity
1. Subgroup analyses
1.1 Duration of treatment
We anticipated a subgroup analysis to examine whether any improvement occurred with short periods of intervention (less than six weeks) and, if this did occur, whether this effect was maintained at longer periods of follow‐up.
1.2 Cholinergic compound
As different cholinergic drug compounds may have differential effects on antipsychotic‐induced TD, we performed a subgroup analysis to compare the effects of different cholinergic drugs. We proposed to undertake comparisons only for primary outcomes to minimise the risk of multiple comparisons.
1.3 Older participants
We also wanted to examine whether the treatment response differs in people with schizophrenia who are older (above 65 years) and for whom the prevalence of spontaneous dyskinesias is estimated to be higher. We had hoped to present data for this subgroup for the primary outcomes.
2. Investigation of heterogeneity
We reported inconsistency if it was high. First, we investigated whether data were entered correctly. Second, if data were correct, we visually inspected the graph and successively removed outlying studies to see if homogeneity was restored. For this review we decided that should this occur with data contributing to the summary finding of no more than around 10% of the total weighting, we would present data. If not, we would not pool such data but discuss relevant issues. We know of no supporting research for this 10% cut‐off but are investigating use of prediction intervals as an alternative to this unsatisfactory state.
When unanticipated clinical or methodological heterogeneity were obvious, we simply discussed these. We did not undertake sensitivity analyses relating to these.
Sensitivity analysis
1. Implication of randomisation
If trials were described in some way as to imply randomisation we undertook a sensitivity analyses for the primary outcomes. We included these studies in the analyses and if there was no substantive difference when the implied randomised studies were added to those with better description of randomisation, then we used relevant data from these studies.
2. Assumptions for lost binary data
Where assumptions had to be made regarding people lost to follow‐up (see Dealing with missing data), we compared the findings of the primary outcomes when we used our assumption compared with completer data only. If there was a substantial difference, we reported and discussed these results, but continued to employ our assumption.
Where assumptions had to be made regarding missing SDs data (see Dealing with missing data), we compared the findings on primary outcomes when we used our assumption compared with completer data only. We undertook a sensitivity analysis to test how prone results were to change when 'completer' data only were compared to the imputed data using the above assumption. If there was a substantial difference, we reported and discussed these results, but continued to employ our assumption.
3. Risk of bias
We analysed the effects of excluding trials that we judged to be at high risk of bias across one or more of the domains of randomisation (implied as randomised with no further details available) allocation concealment, blinding and outcome reporting for the meta‐analysis of the primary outcome. If the exclusion of trials at high risk of bias did not substantially alter the direction of effect or the precision of the effect estimates, we included data from these trials in the analysis
4. Imputed values
Had we included cluster trials, we would have undertaken a sensitivity analysis to assess the effects of including data from trials where we used imputed values for ICC in calculating the design effect.
If we found substantial differences in the direction or precision of effect estimates in any of the sensitivity analyses listed above, we did not pool data from the excluded trials with the other trials contributing to the outcome, but presented them separately.
5. Fixed and random effects
We synthesised data using a fixed‐effect model, however, we also synthesised data for the primary outcome using a random‐effects model to evaluate whether this altered the significance of the results.
Results
Description of studies
Please see Characteristics of included studies and Characteristics of excluded studies.
Results of the search
The 2015 and 2017 update searches were part of an update of nine Cochrane reviews, see Table 6. The 2015 search retrieved 704 references for 344 studies, see Figure 2 for study flow diagram. After having excluded irrelevant references at title and abstract screening, we screened full texts of 72 references (63 studies). Three of these reports were new included studies to this review (Caroff 2007; Jahanian 2014; Ogunmefun 2009), added to the 11 already included studies. One of these studies (Caroff 2007) was an ongoing study in the previously published version of this review. We were able to exclude four studies that were awaiting assessment in the previously published version of this review (Gelenberg 1989; Joe 1985; Marsalek 1994; Perez Cruet 1981), and we excluded one more study for this update (Bartels 1981), added to the 44 already excluded studies from the previously published version of this review. No studies await assessment, and as far as we are aware there are no ongoing studies that would be relevant to this review.
2.
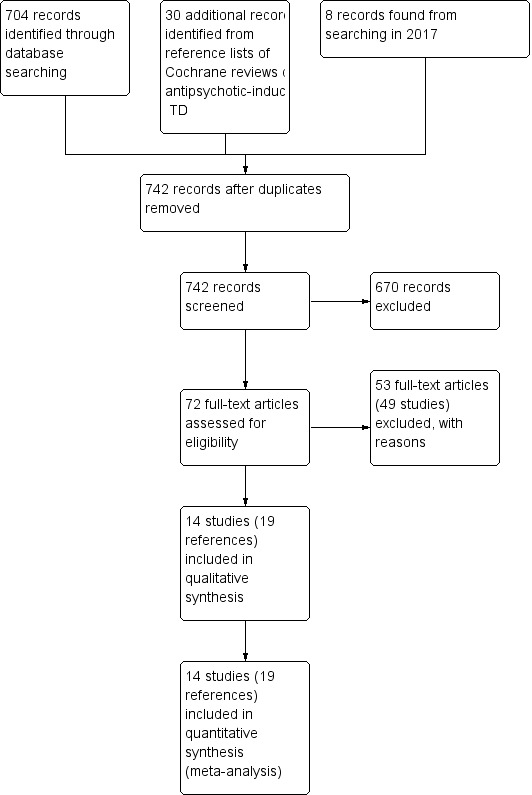
Study flow diagram for 2015 and 2017 searches.
The 2017 search found eight records (five studies). The editorial base of the Cochrane Schizophrenia Group screened these records and no new studies were relevant to this review. They could be relevant to the other reviews in this series of TD reviews (see Table 6), and were put into awaiting assessment of the miscellaneous treatments review (Soares‐Weiser 2003).
Included studies
Overall, the review now includes 14 studies with 364 participants published between 1976 and 2014. Three included studies are new to this update (Caroff 2007; Jahanian 2014; Ogunmefun 2009).
1. Methods
All studies were stated to be randomised and double‐blinded. For further details, please see sections below on Allocation (selection bias) and Blinding (performance bias and detection bias).
2. Design
All included studies presented a parallel longitudinal design. Eight of the 14 studies used a cross‐over design with two periods (Caroff 2007; Gelenberg 1990; Jackson 1978; Jackson 1979; Kocher 1980; Lucius 1976; Ogunmefun 2009; Tarsy 1977). We had considered this as likely when embarking on the review and have used only the data from before the first cross‐over for the reasons outlined above (Unit of analysis issues).
3. Duration
TD is often a chronic condition and symptoms tend to fluctuate and show considerable variability across time. Only four studies, however, all cross‐over trials, had a duration longer than six weeks (Caroff 2007 (12 weeks first treatment phase, 30 weeks total duration); Gelenberg 1990 (eight weeks first treatment phase, 18 to 20 weeks total duration); Jackson 1978 (12 weeks first treatment phase, 32 weeks total duration); Tarsy 1977 (eight weeks first treatment phase, 16 weeks total duration)). All the other included studies were of short duration (nine days to six weeks).
4. Participants
Participants now total 364 people. Both sexes were recruited and the age range was wide, though most people were men in their 50s. Diagnoses included various chronic psychiatric disorders, but mainly schizophrenia. All had antipsychotic‐induced TD diagnosed using Schooler and Kane’s research diagnostic criteria, or by a clinical psychiatrist. The number of participants ranged from five to 60 (median 21).
5. Setting
Most trials were conducted in hospital. Only two studies specifically recruited outpatients (Gelenberg 1990; Tarsy 1977). The studies themselves were from around the world, with eight conducted in the USA (Beckham 1981; Caroff 2007; Gelenberg 1990; Jackson 1978; Jackson 1979; Ogunmefun 2009; Price 1982; Tarsy 1977), and one each in Japan (Yagi 1990), Canada (de Montigny 1979), Australia (George 1981), Iran (Jahanian 2014), Switzerland (Kocher 1980) and Germany (Lucius 1976).
6. Interventions
6.1 Cholinergic drugs
6.1.1 Deanol
Six studies (de Montigny 1979; George 1981; Jackson 1978; Kocher 1980; Lucius 1976; Tarsy 1977) used deanol in doses ranging from 600 mg/day to 2000 mg/day.
6.1.2 Lecithin
Four studies (Beckham 1981; Gelenberg 1990; Jackson 1979; Price 1982) used lecithin in doses ranging from 50 g/day to 60 g/day which contained 20 g/day to 35g/day phosphatidyl choline (a theoretical precursor of acetylcholine).
6.1.3 Meclofenoxate hydrochloride
Yagi 1990 applied meclofenoxate hydrochloride in a dose of 900 mg/day.
6.1.4 Galantamine
Caroff 2007 used galantamine in doses increasing to 12 mg twice per day over 12 weeks.
6.1.5 Rivastigmine
Jahanian 2014 used rivastigmine in a dose of 1.5 mg twice per day.
6.1.6 Donepezil
Ogunmefun 2009 used donepezil in doses ranging from 5 mg to 10 mg per day.
6.2 Comparison group
In all studies placebo was used as a comparison group, with no further details given. In one study a comparison between two doses of deanol was also made (George 1981). None of the included studies compared cholinergic drugs with another active intervention.
Participants remained on schizophrenia treatment antipsychotic medication during the trials.
7. Outcomes
7.1 General
Some outcomes were presented in graphs, with inexact P values of differences, or a statement of significant or non‐significant difference. This made it impossible to acquire raw data for synthesis. Some continuous outcomes could not be extracted due to missing number of participants or missing means, standard deviations, or standard errors. We have shown details of the scales that provided usable data below. We have provided reasons for exclusions of data under 'Outcomes' in the Characteristics of included studies table.
7.2 Scales used to measure TD symptoms
7.2.1 Abnormal Involuntary Movement Scale (AIMS)
The AIMS (Guy 1976a) is a 12‐item scale consisting of a standardised examination followed by questions rating the orofacial, extremity and trunk movements, as well as three global measurements. Each of these 10 items can be scored from zero (none) to four (severe). Two additional items assess the dental status. The AIMS ranges from zero to 40, with higher scores indicating greater severity.
7.2.2 Tardive Dyskinesia Rating Scale (TDRS)
The TDRS (Simpson 1979) is a 34‐item scale consisting of measurement of the movements around the orofacial region, neck, trunk and extremities. Each of these items can be scored from zero (absent) to five (severe). This scale ranges from 10 to 102, with higher scores indicating greater severity.
7.3 Scales used to measure global state
7.3.1 Clinical Global Impression
The CGI is a three‐item scale commonly used in studies on schizophrenia to enable clinicians to quantify severity of illness and overall clinical improvement (Guy 1976b). The items are: severity of illness; global improvement and efficacy index. A seven‐point scoring system is usually employed with low scores indicating decreased severity and/or greater recovery.
7.4 Mental state
Many trials recorded changes in general mental state, and many different ways were employed to rate these changes. No scores, however, were reported in sufficient detail (number of people, mean and SD) to be used in analysis. Only the outcome of 'deterioration of mental state' could be found and used.
7.5 Adverse effects
Possible worsening of acute extrapyramidal symptoms due to cholinergic medication was assessed in some trials, however, trialists did not report scores in detail. All trials assessed general adverse effects, but, again, we often found it impossible to extract useful data (see Characteristics of included studies table); in many cases it was reported that no events occurred. Eight trials assessed specific adverse events, such as gastric adverse events.
Excluded studies
There are 49 excluded studies (53 references). Thirty‐eight of these studies were not randomised and thus excluded. One RCT was excluded because participants were not on a stable dose of antipsychotics before the trial (Simpson 1977). One RCT provided no usable data (Marsalek 1997), and nor did another nine cross‐over RCTs that did not report outcome data for the first phase before crossing over to the next treatment (Chien 1978; Domino 1985; Gelenberg 1989; Joe 1985; Jus 1978; Lieberman 1988; Nasrallah 1986; Penovich 1978; Perez Cruet 1981). We contacted the authors of eight of these studies but received no more details on outcome data. We could not identify up‐to‐date contact details for authors of two of these studies published 35 to 20 years ago (Chien 1978; Marsalek 1997); they were also excluded as we assume it very unlikely to receive data so many years later.
Studies awaiting assessment
There are no studies awaiting assessment.
Ongoing studies
We know of no ongoing studies.
Risk of bias in included studies
Please refer to Figure 3 and Figure 4 for graphical overviews of the risk of bias in the included studies.
3.
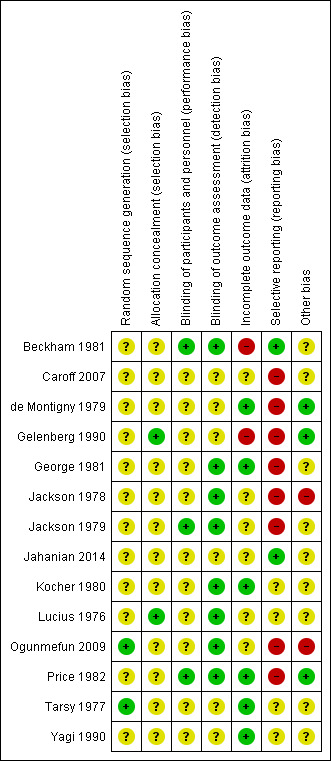
'Risk of bias, summary: review authors' judgements about each risk of bias item for each included study.
4.
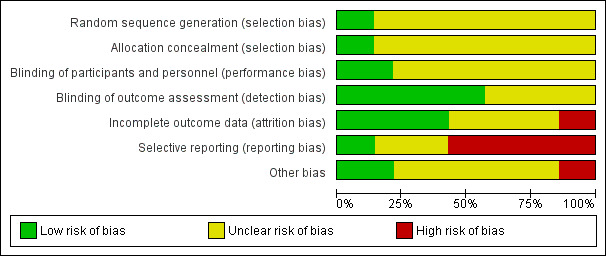
'Risk of bias' graph: review authors' judgements about each risk of bias item presented as percentages across all included studies.
Allocation
Ogunmefun 2009 and Tarsy 1977 reported using random number tables, though did not describe allocation concealment. Gelenberg 1990 and Lucius 1976 explicitly described an external allocation procedure. All other studies were not explicit about how the randomisation sequence was generated or how allocation was achieved other than using the word "randomized".
Blinding
Although all studies were conducted on a double‐blind basis, only three (Beckham 1981; Jackson 1979; Price 1982) explicitly described how this was undertaken and no study tested the blindness of raters, clinicians and trial participants. Eight trials (Beckham 1981; George 1981; Jackson 1978; Jackson 1979; Kocher 1980; Lucius 1976; Ogunmefun 2009; Price 1982) adequately described blinding of outcome assessors.
Incomplete outcome data
Two studies (Beckham 1981; Gelenberg 1990) had a drop‐out rate of greater than 30%. These studies were rated as having a high risk of attrition bias. In all cases, however, we tried to ensure that every person randomised was analysed.
Selective reporting
The majority of data in this review originates from published reports. Expected outcomes (impact on TD symptoms) were reported for most of the trials. Only Beckham 1981 and Jahanian 2014 were considered to have a low risk of reporting bias as they reported all outcomes that were stated in their published protocols (Beckham 1981 was a thesis which contained all detailed methods applied). All other studies did not fully report data for the stated outcome measures.
Other potential sources of bias
All studies had small or very small sample sizes (five to 60 participants per study). Eight of the 13 studies used a cross‐over design, and in most studies it was unclear whether other sources of bias may exist.
Effects of interventions
See: Table 1
1. Comparison 1. Cholinergic drugs versus placebo
1.1 TD symptoms
We had chosen 'any improvement in TD symptoms of more than 50% on any TD scale ‐ any time period' as a primary outcome. Although the data we found in trials did not fit this exactly, we feel that the outcome 'not improved to a clinically important extent' fits best with what we had hoped to find.
1.1.1 Not improved to a clinically important extent
There was no significant difference for cholinergic drugs over placebo on clinically important improvement after two to 12 weeks treatment (very low‐quality evidence, 4 trials, 27 people, risk ratio (RR) 0.89, 95% confidence interval (CI) 0.65 to 1.23, I2 = 0%, Analysis 1.1).
1.1. Analysis.
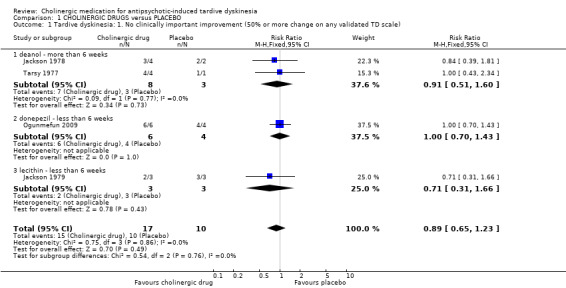
Comparison 1 CHOLINERGIC DRUGS versus PLACEBO, Outcome 1 Tardive dyskinesia: 1. No clinically important improvement (50% or more change on any validated TD scale).
1.1.2 Not any improvement
For the outcome of 'not any improvement in TD symptoms', we found no difference between cholinergic drugs and placebo after nine days to 12 weeks treatment (low‐quality evidence, 9 trials, 180 people, RR 0.87, 95% CI 0.71 to 1.07, I2=1%, Analysis 1.2). One study also provided self‐reported incidence of not any improvement in TD symptoms and found no difference between cholinergic drugs and placebo after nine to 11 days treatment (1 trial, 30 people, RR 0.92, 95% CI 0.62 to 1.36, Analysis 1.3).
1.2. Analysis.
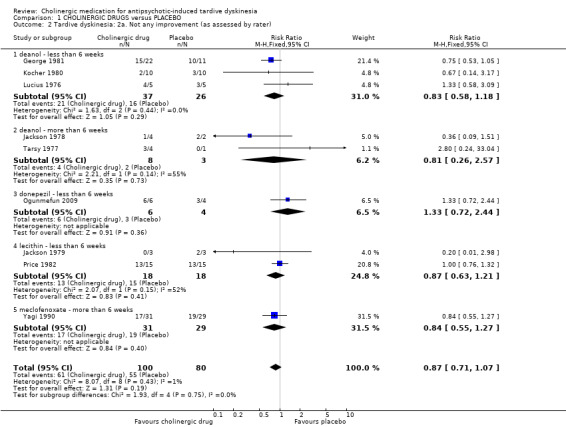
Comparison 1 CHOLINERGIC DRUGS versus PLACEBO, Outcome 2 Tardive dyskinesia: 2a. Not any improvement (as assessed by rater).
1.3. Analysis.
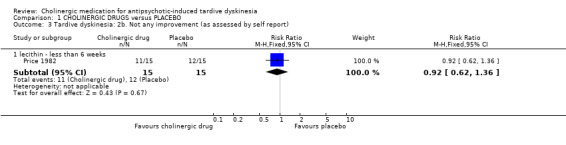
Comparison 1 CHOLINERGIC DRUGS versus PLACEBO, Outcome 3 Tardive dyskinesia: 2b. Not any improvement (as assessed by self report).
1.1.3 Average endpoint scores
TD symptoms were also measured on the continuous AIMS and TDRS scales (see Description of studies above). No difference was found between cholinergic drugs and placebo on either average AIMS endpoint scores after two to 12 weeks treatment (low‐quality evidence, 7 studies, 171 people, mean difference (MD) ‐0.12, 95% CI ‐0.44 to 0.21, I2 = 44%, Analysis 1.4) or average TDRS endpoint scores after eight weeks treatment (5 participants, 1 study, not estimable, Analysis 1.5).
1.4. Analysis.
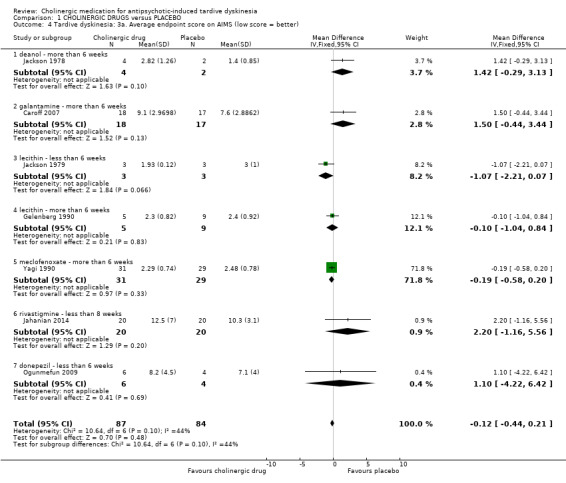
Comparison 1 CHOLINERGIC DRUGS versus PLACEBO, Outcome 4 Tardive dyskinesia: 3a. Average endpoint score on AIMS (low score = better).
1.5. Analysis.
Comparison 1 CHOLINERGIC DRUGS versus PLACEBO, Outcome 5 Tardive dyskinesia: 3b. Average endpoint score on modified Simpson TDRS (low score = better).
| Tardive dyskinesia: 3b. Average endpoint score on modified Simpson TDRS (low score = better) | |||||
|---|---|---|---|---|---|
| Study | Intervention | Mean | SD | N | Comments |
| deanol ‐ more than 6 weeks | |||||
| Tarsy 1977 | Deanol | 10 | 5.48 | 4 | |
| Tarsy 1977 | Placebo | 10 | 0 | 1 | The confidence interval of mean difference was not estimable because the placebo group only had one participant. |
1.1.4 Deterioration of symptoms
There was also no difference between cholinergic drugs and placebo with regard to deterioration of TD symptoms after nine days to 12 weeks treatment (low‐quality evidence, 8 trials, 147 people, RR 1.11, 95% CI 0.55 to 2.24, I2=0%, Analysis 1.6). One study also provided self‐reported incidence of deterioration in TD symptoms and found no difference between cholinergic drugs and placebo after nine to 11 days treatment (1 trial, 30 people, RR 3.00, 95% CI 0.13 to 68.26, Analysis 1.7).
1.6. Analysis.
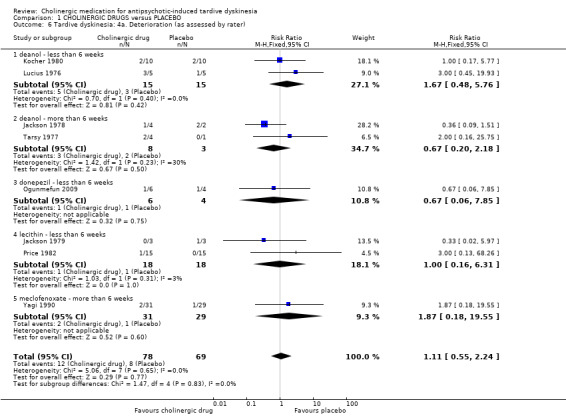
Comparison 1 CHOLINERGIC DRUGS versus PLACEBO, Outcome 6 Tardive dyskinesia: 4a. Deterioration (as assessed by rater).
1.7. Analysis.
Comparison 1 CHOLINERGIC DRUGS versus PLACEBO, Outcome 7 Tardive dyskinesia: 4b. Deterioration (as assessed by self report).
1.2 Global outcome measures
1.2.1 Death for any reason
No deaths were reported in any of the studies where this information could be extracted (11 trials, 278 people, Analysis 1.8). For cross‐over trials we counted only the first period before the cross‐over. However, in one cross‐over study (Tarsy 1977) one person died suddenly at home due to acute aspiration in the second cross‐over period, his placebo period. By this time he had completed eight weeks of deanol treatment and four weeks of placebo treatment.
1.8. Analysis.
Comparison 1 CHOLINERGIC DRUGS versus PLACEBO, Outcome 8 Global outcome: Death for any reason.
1.2.2 Global outcomes
One study assessed global usefulness of meclofenoxate on the categorical Global Usefulness Rating. This study showed no difference between meclofenoxate and placebo after eight weeks treatment (60 people, RR 0.89, 95% CI 0.59 to 1.32, Analysis 1.9).
1.9. Analysis.
Comparison 1 CHOLINERGIC DRUGS versus PLACEBO, Outcome 9 Global outcome: Intervention not useful as assessed by Global Usefulness Rating (GUR).
1.2.3 Global state
Another study assessed global state with the CGI and showed no difference between lecithin and placebo after 11 days treatment (31 people, RR ‐0.43, 95% CI ‐1.36 to 0.50, Analysis 1.10).
1.10. Analysis.
Comparison 1 CHOLINERGIC DRUGS versus PLACEBO, Outcome 10 Global state: Average endpoint score on CGI (low score = better).
1.3 Mental state
1.3.1 Deterioration
There was no significant difference in deterioration of mental state between cholinergic drugs and placebo after 11 days to 12 weeks treatment (very low‐quality evidence, RR 0.50, 95% CI 0.10 to 2.61; 77 participants, 5 studies; I2 = 0%, Analysis 1.11).
1.11. Analysis.
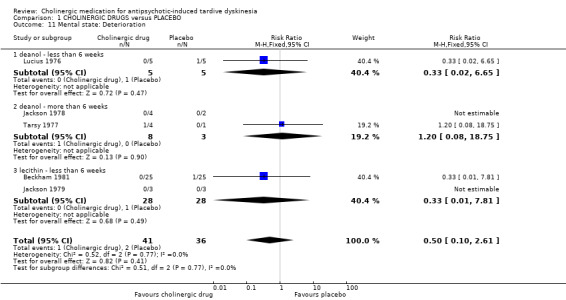
Comparison 1 CHOLINERGIC DRUGS versus PLACEBO, Outcome 11 Mental state: Deterioration.
1.4 Adverse effects
1.4.1 Any adverse effects
Although four studies (106 participants) reported on any adverse events, only one study reported that any events occurred, 3/31 in the meclofenoxate group compared with 5/29 in the placebo group after eight weeks treatment (very low‐quality evidence, RR 0.56, 95% CI 0.15 to 2.14; 60 participants; Analysis 1.12). The three studies reporting on donepezil or lecithin reported no events.
1.12. Analysis.
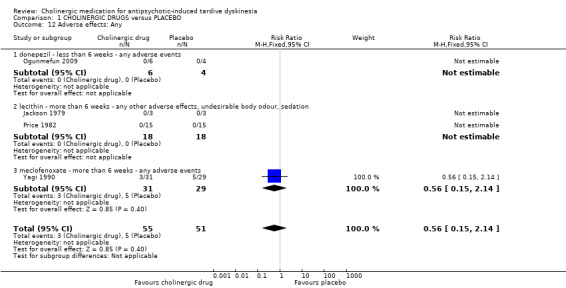
Comparison 1 CHOLINERGIC DRUGS versus PLACEBO, Outcome 12 Adverse effects: Any.
1.4.2 Various specific adverse effects
Out of eight studies (130 participants) that reported on various specific adverse events, only two studies reported that specific adverse events occurred. For deanol, one study reported that 4/10 participants had gastric adverse events versus 0/10 participants on placebo after three weeks treatment (RR 9.00, 95% CI 0.55 to 147.95; 5 studies, 61 participants; Analysis 1.13), and two studies reported that 8/32 participants had sedation, peripheral cholinergic effects, and undesirable body odour events, versus 0/22 participants on placebo after three to four weeks treatment (RR 6.83, 95% CI 0.99 to 47.25; 6 studies, 94 participants; I2 = 0%; Analysis 1.13). The four other studies reporting on deanol reported that no specific adverse events occurred, and two studies evaluating lecithin reported that no specific adverse events occurred (see Analysis 1.13).
1.13. Analysis.
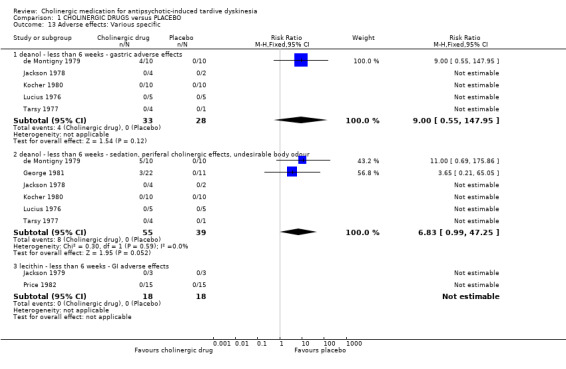
Comparison 1 CHOLINERGIC DRUGS versus PLACEBO, Outcome 13 Adverse effects: Various specific.
1.5 Leaving the study early
Using cholinergic drugs did not significantly increase the risk of a person leaving the study early after nine days to 12 weeks treatment (very low‐quality evidence, 12 studies, 288 people, RR 1.09, 95% CI 0.56 to 2.10, I2 = 47%, Analysis 1.14).
1.14. Analysis.
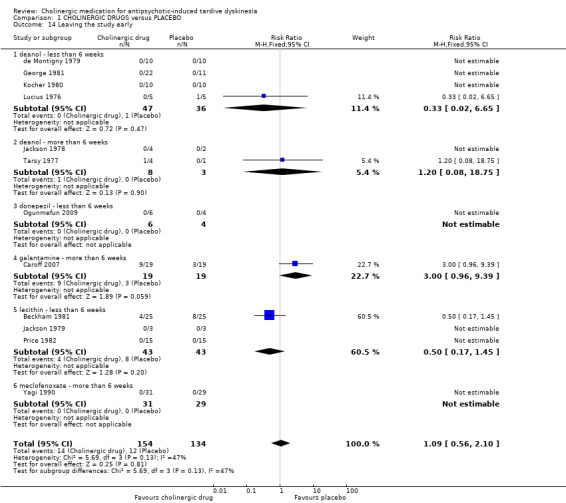
Comparison 1 CHOLINERGIC DRUGS versus PLACEBO, Outcome 14 Leaving the study early.
We did not identify any studies that reported on hospital and service utilisation outcomes, economic outcomes, social confidence, social inclusion, social networks, personalised quality of life, behaviour, or cognitive state.
1.6 Subgroup analysis
1.6.1 Duration of treatment
We stratified analyses by duration. Any effects that cholinergic drugs may have had did not clearly change in relation to duration of treatment.
1.6.2 Cholinergic compound
We stratified analyses by cholinergic compound. Any effects that cholinergic drugs may have did not clearly change in relation to type of compound.
1.6.3 Older participants
It was not possible to evaluate whether participants aged > 65 years responded differently to younger participants, since no trial reported data for different age groups that could be extracted for separate analyses.
1.7 Heterogeneity
Data were mostly homogeneous. We detected statistical heterogeneity as described in Assessment of heterogeneity for two of the outcomes.
-
TD: average endpoint score on the AIMS (I2 = 44%, P = 0.10): seven studies reported on this outcome, but there is no clear outlier.
Three studies reported an effect estimate favouring cholinergic drug over placebo, but none of the studies reported statistically significant differences between groups (see Analysis 1.4): lecithin versus placebo after two weeks treatment (short term) (MD ‐1.07, 95% CI ‐2.21 to 0.07; 6 participants, 1 RCT), lecithin versus placebo after eight weeks treatment (medium term) (MD ‐0.10, 95% CI ‐1.04 to 0.84; 14 participants, 1 RCT), and meclofenoxate versus placebo after eight weeks treatment (medium term) (MD ‐0.19, 95% CI ‐0.58 to 0.20; 60 participants, 1 RCT).
Four studies reported an effect estimate favouring placebo over cholinergic drug, but again, none of the studies reported statistically significant differences between groups (see Analysis 1.4): rivastigmine versus placebo after eight weeks treatment (short term) (MD 2.20, 95% CI ‐1.16 to 5.56; 40 participants, 1 RCT), deanol versus placebo after 12 weeks treatment (medium term) (MD 1.42, 95% CI ‐0.29 to 3.13; 6 participants, 1 RCT), galantamine versus placebo after 12 weeks treatment (medium term) (MD 1.50, 95% CI ‐0.44 to 3.44; 35 participants, 1 RCT), donepezil versus placebo after four to six weeks treatment (short term) (MD 1.10, 95% CI ‐4.22 to 6.42; 10 participants, 1 RCT).
Acceptability of treatment: leaving the study early (I2 = 47%, P = 0.13): 12 studies reported on this outcome, but only four reported that any events occurred (= that any participants left the study early), and none of these found a statistically significant difference between groups. When removing from the analysis a study that from visual inspection of the graph appears to be an outlier, the I2 statistic goes down to 0% (analysis not shown). This study reported an effect estimate in the direction favouring placebo over galantamine after 12 weeks treatment (RR 3.00, 95% CI 0.96 to 9.39; 38 participants, 1 RCT, Analysis 1.14); the other studies reported effect estimates in the direction favouring lecithin or deanol over placebo after 11 days to eight weeks treatment (see Analysis 1.14).
1.8 Sensitivity analyses
1.8.1 Implication of randomisation
We aimed to include trials in a sensitivity analysis if they were described in some way as to imply randomisation. As all studies were stated to be randomised we have not undertaken this sensitivity analysis.
1.8.2 Assumptions for lost binary data
We would have undertaken a sensitivity analysis assessing where assumptions had to be made regarding people lost to follow‐up for the primary outcome (see Dealing with missing data). We intended to compare the findings when we used our assumption compared with completer data only. No assumptions had to be made, studies that reported data for the primary outcome reported for all randomised participants.
1.8.3 Risk of bias
When excluding three trials from the primary outcome that we judged to be at high risk of bias across one or more of the domains, there was no substantial alteration to the direction of effect or the precision of the effect estimates (RR 1.00, 95% CI 0.43 to 2.34; 1 study, 5 people, analysis not shown).
1.8.4 Imputed values
We would have undertaken a sensitivity analysis to assess the effects of including data from cluster‐randomised trials where we used imputed values for ICC in calculating the design effect. No cluster‐randomised trials were included.
1.8.5 Fixed and random effects
We also synthesised data for the primary outcome using a random‐effects model. This did not alter the significance of the results (RR 0.94, 95% CI 0.71 to 1.25; 4 studies, 27 people, analysis not shown).
2. Comparison 2. Cholinergic drugs versus other cholinergic drugs
Only one study (George 1981) was identified that reported on this comparison. When comparing a higher dose of deanol (2000 mg/day) to a lower dose (1000 mg/day) there was a slight improvement in TD scores for the group with the higher dose (1 trial, 22 people, RR 0.50, 95% CI 0.25 to 0.98, Analysis 2.1). No events were reported between the different doses for death or leaving the study early (Analysis 2.2; Analysis 2.3).
2.1. Analysis.
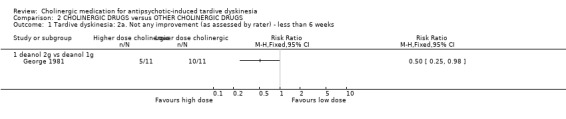
Comparison 2 CHOLINERGIC DRUGS versus OTHER CHOLINERGIC DRUGS, Outcome 1 Tardive dyskinesia: 2a. Not any improvement (as assessed by rater) ‐ less than 6 weeks.
2.2. Analysis.
Comparison 2 CHOLINERGIC DRUGS versus OTHER CHOLINERGIC DRUGS, Outcome 2 Global outcome: Death for any reason ‐ less than 6 weeks.
2.3. Analysis.
Comparison 2 CHOLINERGIC DRUGS versus OTHER CHOLINERGIC DRUGS, Outcome 3 Leaving the study early ‐ less than 6 weeks.
We did not identify any studies that reported on adverse events, mental state, hospital and service utilisation outcomes, economic outcomes, social confidence, social inclusion, social networks, personalised quality of life, behaviour, or cognitive state.
Discussion
Summary of main results
1. The search
This 2017 update has identified additional data from three studies (Caroff 2007; Jahanian 2014; Ogunmefun 2009) of new CNS‐active cholinergic agents mainly used for the treatment of Alzheimer's disease (donezepil, galantamine, and rivastigmine). It is probable, however, that the effect of these modern cholinergic agents for TD has not been comprehensively investigated within randomised trials. This update did not identify any new trials of the old cholinergic drugs (deanol, lecithin, meclofenoxate); trials of these drugs were published from 1976 to 1990.
2. Few data
Only a little over 350 people have been included in this review. It is possible that real, and important, effects have not been highlighted because of the necessarily wide CIs of the findings. Many outcomes were not measured at all (see Overall completeness and applicability of evidence), including one of our pre‐stated outcome measures. We may have been overambitious in hoping for some of these outcomes in TD trials, but simple reporting of quality of life still does not seem too demanding and does remain of interest.
3. Comparison 1. Cholinergic drugs versus placebo
3.1 TD symptoms
We are uncertain about the evidence on no clinically important improvement in TD symptoms comparing cholinergic drugs and placebo as the evidence was of very low quality (RR 0.89, 95% CI 0.65 to 1.23, 4 trials, 27 people). There may be little or no difference in deterioration of TD symptoms between cholinergic drugs and placebo, evidence was of low quality (RR 1.11, 95% CI 0.55 to 2.24, 8 trials, 147 people).
3.2 Mental state
We are uncertain about the evidence on deterioration of mental state comparing cholinergic drugs and placebo as the evidence was of very low quality (RR 0.50, 95% CI 0.10 to 2.61, 77 people, 5 trials).
3.3 Adverse events
We are uncertain about the evidence on adverse events comparing cholinergic drugs and placebo as the evidence was of very low quality (RR 0.56, 95% CI 0.15 to 2.14, 106 people, 4 trials).
3.4 Acceptability of treatment
It is always unclear what leaving the study early means. It could be to do with the participant not accepting treatment for a series of reasons, or of participants finding the trial intolerable. It also could be a function of a trial design in which willing participants are still asked to leave because of some degree of protocol violation. In any event, four studies reported that 14/53 people left the cholinergics group compared with a not significantly different 12/50 people in the placebo group. Eight studies reported that no adverse events occurred.
3.5 Social confidence, social inclusion, social networks, or personalised quality of life
This group of outcomes was selected as being of importance to patients for the 2017 review update following a service user consultation. None of the included studies reported on this outcome.
See Table 1 for a summary of the evidence.
4. Cholinergic drugs versus other cholinergic drugs
One study (George 1981) included study arms investigating two doses of deanol, 1 g and 2 g per dose, and found that twice as many participants experienced any improvement in TD symptoms with the higher dose (RR 0.50, 95% CI 0.25 to 0.98, 22 people). Very few participants were randomised, so the evidence is uncertain. Mental state, adverse events, or social confidence, social inclusion, social networks, or personalised quality of life were not reported. No events were reported for leaving the study early.
Overall completeness and applicability of evidence
1. Completeness
The most important finding of this review is that a systematic search of the literature still results in a review that is considerably underpowered to really investigate the clinical efficacy and safety of cholinergic agents in TD. All data are inconclusive. We were unable to detect any effect, good or bad of these drugs for TD. With the advent of new CNS‐active cholinergic agents for treatment of Alzheimer's disease and dementia (e.g., Birks 2006a; Birks 2006b; Birks 2015; Li 2015; Loy 2006; Maidment 2006; Rolinski 2012), the theoretical base for this review is strengthened. However, the actual central cholinergic transmission ‐enhancing effect of old cholinergic drugs, such as lecithin and especially deanol, remains unclear. Even if any of these old compounds had an effect on cholinergic transmission, the impact would probably be limited. Modern cholinergic drugs use a different mode of action and are worthy of investigation, although they are associated with gastrointestinal adverse events (Birks 2006a; Birks 2006b; Birks 2015; Li 2015; Loy 2006; Maidment 2006; Rolinski 2012). However, to date, these newer drugs have not been fully investigated in RCTs in the treatment of TD.
2. Applicability
All but two trials were hospital‐based but were nevertheless on people who would be recognisable in everyday care. Trials were set in high‐income countries in Asia, Australia, Europe and North America. Cholinergic drugs are readily accessible and most outcomes understandable in terms of clinical practice. Should cholinergic drugs have had important effects the findings may well have been applicable.
Quality of the evidence
Overall, the certainty of the evidence in this review is low to very low. This means that we have very little confidence in the effect estimates, and the true effects are likely to be substantially different from the estimates of the effect. The following are the main reasons for our low confidence in the evidence.
Poor study methodology and reporting of methods and data (see Figure 3 and Figure 4) resulting in downgrading evidence for risk of bias. Allocation concealment was not described, generation of the sequence was not explicit, studies were not clearly blinded, we are unsure if data are incomplete, and data were often poorly or selectively reported.
Very small sample sizes resulting in downgrading evidence for imprecision. The largest trial in this review randomised only 24 people. A trial of this size is unable to detect subtle, yet important differences due to cholinergic drugs with any confidence. In order to detect a 20% difference between groups, probably about 150 people are needed in each arm of the study (alpha 0.05, beta 0.8).
Wide CIs (often due to low event rates) that included appreciable benefit or harm for the intervention as well as no effect, resulting in downgrading evidence for imprecision.
Please see Table 1 for full details.
Potential biases in the review process
1. Missing studies
Every effort was made to identify relevant trials. However, these studies are all small and it is likely that we have failed to identify other studies of limited power. It is likely that such studies would also not be in favour of the cholinergics group. If they had been so, it is more likely that they would have been published in accessible literature. We do not, however, think it likely that we have failed to identify large relevant studies.
2. Introducing bias
We have tried to be balanced in our appraisal of the evidence but could have inadvertently introduced bias. We welcome comments or criticisms. New methods and innovations now make it possible to report data where, in the past, we could not report data at all or had to report data in a different way. We believe that 'Summary of findings' tables are a valuable innovation – but problematic to those not ‘blind’ to the outcome data. It is possible to ‘cherry pick’ significant findings for presentation in this table. We have tried to decrease the chance of doing this by asking a new review author (HB) to select outcomes relevant for this table before becoming familiar with the data.
Agreements and disagreements with other studies or reviews
The only other relevant quantitative reviews we know of is the previous Cochrane review (Tammenmaa 2002) and another systematic review that included the same RCTs as the Cochrane review (Tammenmaa 2004). These reviews found no evidence to support administration of the old cholinergic agents lecithin, deanol, and meclofenoxate to patients with TD, and concluded that further investigation of the clinical effects of novel cholinergic agents in TD is warranted. This update identified three new studies to include (as discussed in Results of the search), but does not substantially change these findings or conclusions.
Authors' conclusions
Implications for practice.
1. For the person with tardive dyskinesia (TD)
Given the absence of evidence for efficacy, and given that cholinergic medication is associated with adverse effects, it would be understandable if a person with TD would rather avoid these additional treatments. Should the TD be serious and warrant intervention, the person with the movement disorder or their family may wish to ask for use of one of the more modern cholinergics within a well‐designed, conducted and reported randomised trial.
2. For clinicians
The available data on the efficacy of cholinergic medications for the treatment of antipsychotic‐induced TD are insufficient to recommend their use. Adverse effects associated with some of these compounds are burdensome. Nevertheless, there is a real place for clinician‐driven research, with prescription within the context of a randomised trial and routine data collection on outcomes of relevance to people with TD and their clinicians.
3. For policy makers and funders of studies
This is one of the reviews in the series of Cochrane reviews on treatments for antipsychotic‐induced TD (see Table 6). No evidence is convincing that addition or withdrawal of another drug helps with the symptoms of TD. There are, however, many unanswered questions in this area. This unattractive adverse effect is caused, to a greater or lesser extent, by antipsychotic drugs. Clinicians and researchers should feel responsible enough to continue to try to help it. Those compiling guidance could encourage supportive activity and more research into this neglected area.
Implications for research.
1. General
If cholinergic agents are to be investigated, their effects should be demonstrated in well‐designed, conducted and reported randomised controlled clinical trials (Moher 2001).
2. Specific
2.1 Reviews
The excluded studies do contain studies which may be of value in other related reviews (Table 2).
2. Excluded studies which are relevant to other reviews.
| Excluded study | Comparison | Treatment category | Relevant review | ||||
| #1 | #2 | ||||||
| Nasrallah 1986 | Alpha‐methyl‐p‐tyrosine (AMPT) | versus choline chloride | Amino acid | Organic salt | ‐ | ||
| versus hydroxytryptophan | Amino acid (serotonin precursor) | ‐ | |||||
| versus valproic acid | Mood stabilisers | ‐ | |||||
| versus L‐DOPA | Amino acid | ‐ | |||||
| Lieberman 1988 | Benztropine versus bromocriptine | Anticholinergic | Dopamine agonist | ‐ | |||
| Bromocriptine versus haloperidol | Dopamine agonist | Antipsychotic | ‐ | ||||
| Nasrallah 1986 | Choline chloride | versus L‐DOPA | Organic salt | Amino acid | ‐ | ||
| versus hydroxytryptophan | ‐ | ||||||
| versus valproic | Anticonvulsant | ‐ | |||||
| Jus 1978 | Deanol | versus lithium carbonate | Antidepressant | Organic salt | ‐ | ||
| versus placebo | Placebo | ‐ | |||||
| Chien 1978 | versus sodium valproate | Anticonvulsant | ‐ | ||||
| versus oxpertine | Antipsychotic | ‐ | |||||
| Nasrallah 1986 | Hydroxytryptophan versus L‐DOPA | Amino acid | Amino acid | ‐ | |||
| L‐DOPA versus valproic acid | Anticonvulsant | ‐ | |||||
| Jus 1978 | Lithium carbonate versus placebo | Mood stabiliser | Placebo | ‐ | |||
| Chien 1978 | Oxypertine versus sodium valproate | Antipsychotic | Anticonvulsant | ‐ | |||
2.2 Trials
From the theoretical background, there is some evidence to suggest that cholinergics may benefit people with antipsychotic‐induced TD. It is possible that cholinergic agents have a small‐to‐moderate effect (positive or negative) that has not, as yet, been detected. The results of this review do not support further investigation of the effects of lecithin and deanol. However, there is a need for well‐designed, conducted and reported randomised trials to evaluate the effects of modern cholinergic agents for the treatment of antipsychotic‐induced TD. Such studies are of importance to people with the problem (see Figure 1) and have long been ignored.
2.2.1 Trial design
a. Use of cross‐over design
Trialists find it difficult to identify people with both TD and schizophrenia to participate in trials. Randomised cross‐over design is used in the hope of improving the power of the study to find outcomes of interest. This design initially asks participants to be randomised to one of the experimental interventions, and then, at a pre‐specified time, to be crossed over to the treatment that they did not at first receive. Conditions with a more stable time course than TD are better suited for cross‐over studies (Fleiss 1984). Further difficulties are the carry‐over effect. Unless cross‐over studies include a mid‐study washout period (where the person is free of treatment before starting the next arm of the study), any effect of cholinergic drugs may continue into the second half placebo arm of the trial – the 'carry‐over effect'. Also, carry‐over may involve the re‐growth or retreat of neuroreceptors. This slow re‐balancing, if started, could continue long after all traces of intervention drugs are gone, so the physiological half life of the experimental treatment may not be the only variable to consider when thinking though the issues of carry‐over. TD is also an unstable condition and people with TD may not remain compliant with medication. All these factors make the arguments for not using cross‐over methodology strong, despite the initial attraction (Armitage 1991; Fleiss 1984; Pocock 1983).
b. Sample size calculation
All studies included in this review had a small sample size (five to 60). However, the results suggest that a larger sample size should be used to provide more precise estimates of effect.
c. Outcomes
Many of the outcomes initially desired when this review was started have not been investigated. In addition to efficacy, it is important to assess the influence on mental state and adverse effects. Finally, a service user consultation informed the addition of outcomes of special importance to patients. We have reconsidered all these outcomes in case they were too ambitious and tried to tailor them to a real‐world pragmatic trial design (see Table 3).
3. Suggestions for design of future study.
| Methods | Allocation: randomised, with sequence generation and concealment of allocation clearly described. Blindness: double, tested. Duration: 12 months beyond end of intervention at least. Raters: independent. |
| Participants | People with antipsychotic‐induced tardive dyskinesia.* Age: any. Sex: both. History: any. N = 300.** |
| Interventions | Specific cholinergic drug (N = 150) versus placebo (N = 150) |
| Outcomes | Tardive dyskinesia: any clinically important improvement in TD, any improvement, deterioration.*** Adverse effects: no clinically significant extrapyramidal adverse effects ‐ any time period***, use of any antiparkinsonism drugs, other important adverse events. Leaving the study early. Service outcomes: admitted, number of admissions, length of hospitalisation, contacts with psychiatric services. Compliance with drugs. Economic evaluations: cost‐effectiveness, cost‐benefit. General state: relapse, frequency and intensity of minor and major exacerbations. Social confidence, social inclusion, social networks, or personalised quality of life: binary measure Distress among relatives: binary measure. Burden on family: binary measure. |
| Notes | * This could be diagnosed by clinical decision. If funds were permitting all participants could be screened using operational criteria, otherwise a random sample should suffice. ** Size of study with sufficient power to highlight about a 10% difference between groups for primary outcome. *** Primary outcome. The same applies to the measure of primary outcome as for diagnosis. Not everyone may need to have operational criteria applied if clinical impression is proved to be accurate. |
What's new
| Date | Event | Description |
|---|---|---|
| 4 October 2017 | New citation required but conclusions have not changed | Results from the latest searches did not change the overall conclusions of this review, but the results are more informative. |
| 26 April 2017 | New search has been performed | Update search run 26 April, 2017. Eight records found and assessed by editorial base Cochrane Schizophrenia, no new studies relevant to this review found. The eight records were added to Miscellaneous treatments for antipsychotic‐induced tardive dyskinesia (Table 6). |
| 31 October 2016 | Amended | Title changed from 'Cholinergic medication for neuroleptic‐induced tardive dyskinesia'. Three new trials added (Caroff 2007; Jahanian 2014; Ogunmefun 2009), analyses and text updated, outcomes list updated due to patient consultation, 'Summary of findings' table added, conclusions not substantially changed. |
History
Protocol first published: Issue 2, 1996 Review first published: Issue 2, 1997
| Date | Event | Description |
|---|---|---|
| 16 July 2015 | Amended | Update search run July 16, 2015. 704 records found and assessed by review authors |
| 18 January 2012 | Amended | Contact details updated. |
| 19 January 2011 | Amended | Contact details updated. |
| 10 November 2010 | Amended | Contact details updated. |
| 14 April 2010 | Amended | Contact details updated. |
| 16 January 2009 | Amended | Author correction. |
| 25 April 2008 | Amended | Converted to new review format. |
| 17 May 2002 | New citation required and conclusions have changed | Substantive amendment. |
| 21 February 1997 | Amended | First version of review published. |
Acknowledgements
The review authors wish to thank Prof Kristian Wahlbeck, who acted as an advisor throughout the first version of the review process, for his constant help. The review authors also wish to acknowledge Prof Toshiaki Furukawa for great help with translating and extracting data from the Japanese articles. We wish to thank the authors and conductors of trials, S Bockenheimer, Joanne Doller‐Wojcik, Edward F Domino, Alan J Gelenberg, John H Growdon, Gabriele Lucius‐Hoene, David B Newgreen, Patricia E Penovich, Jorge Perez Cruet, Lynn A Price, Andre' Villeneuve and Jan Volavka for their kind replies to our inquiries about their studies and Stanley N Caroff who also acted as peer reviewer.
For the 2017 update, we wish to thank Antonio Grande for screening literature and helping with data extraction, Farhad Sokraneh for carrying out the trial search, helping to find full‐text papers, and assessing and extracting data from a study in Persian, Ben Gray for writing the Plain language summary, and Nicholas Henschke, Linda Levi, and Loukia Spineli for assistance with preparing the report. We are also grateful to Dawn‐Marie Walker, Ruth Sayers, Megan Lees, and Vanessa Pinfold from McPin Foundation for organising and holding the public and patient involvement consultation with TD service users that contributed to selecting outcomes for the 'Summary of findings' table and to guide future research. We wish to thank the author and trial conductor William T Regenold for his kind reply to our queries about his study. We would also like to thank John McGrath for his advice and support writing the first published version of this review and Dr Eila Sailas for contributing to earlier versions of this review.
Appendices
Appendix 1. Previous methods and searches
Methods
Criteria for considering studies for this review
Types of studies
Randomised controlled trials.
Types of participants
People with schizophrenia or any other serious mental illness, diagnosed by any criteria, irrespective of gender, age or nationality who: 1. Required the use of neuroleptics for more than three months. 2. Developed tardive dyskinesia during neuroleptic treatment (diagnosed by any criteria at baseline of the trial and at least one other occasion). 3. For whom the dose of neuroleptic medication had been stable for one month or more before the trial and during the trial.
Types of interventions
1. The cholinergic drugs arecoline, choline, deanol, lecithin, meclofenoxate, physostigmine, RS 86. In the first substantial update of the review the following cholinergic compounds were assessed to be relevant and added to the scope of the review: tacrine, 7‐methoxytacrine, ipidacrine, galantamine, donepezil, rivastigmine, eptastigmine, metrifonate, xanomeline and cevimeline. 2. Control condition: Placebo or no intervention.
Types of outcome measures
Clinical efficacy (clinically relevant improvement of tardive dyskinesia symptoms) is defined in this review as an improvement in tardive dyskinesia symptoms of more than 50% on any validated tardive dyskinesia scale.
The outcomes of interest were: 1. Global outcome measures (this category of outcome measures was added in the first substantial up‐date of the review). 1.1 The number of people per treatment group who died for any reason. 1.2 Treatment group mean and standard deviation of endpoint score on any scale of quality of life. 1.3 Treatment group mean and standard deviation of endpoint score on any scale of level of functioning.
2. Tardive dyskinesia changes 2.1 The number of people per treatment group who did not show a clinically relevant improvement (improvement of more than 50% on any validated tardive dyskinesia scale) as assessed by the rater. 2.2 The number of people per treatment group who did not show any improvement by any means of tardive dyskinesia assessment as assessed by the rater. 2.3 The number of people per treatment group who deteriorated by any means of tardive dyskinesia assessment as assessed by the rater. 2.4 The number of people per treatment group who did not experience any improvement in tardive dyskinesia symptoms as rated by self‐assessment (this outcome was added in the first substantial up‐date of the review). 2.5 The number of people per treatment group who deteriorated in tardive dyskinesia symptoms as rated by self‐assessment (this outcome was added in the first substantial up‐date of the review). 2.6 Treatment group mean and standard deviation of endpoint score on any validated tardive dyskinesia scale. 2.7 Treatment group mean and standard deviation of change (baseline minus endpoint) in score on any validated tardive dyskinesia scale.
3. General mental state changes 3.1 The number of people per treatment group who deteriorated in psychiatric symptoms (such as delusions and hallucinations) by any means of assessment of psychiatric symptoms or mental state. 3.2 Treatment group mean and standard deviation of endpoint score on any scale of psychiatric symptoms.
4. Acceptability of the treatment 4.1 The number of people per treatment group who had any adverse effect (other than deterioration of tardive dyskinesia symptoms or change in mental state). 4.2 The number of people per treatment group who dropped out during the trial. When appropriate, the outcomes were grouped into time periods ‐ short term (less than 6 weeks), medium term (between 6 weeks and 6 months) and long term (over 6 months).
Search methods for identification of studies
1. Electronic searching
1.1 In the original version of the review relevant randomised trials were identified by searching the following electronic databases:
1.1.1 Biological Abstracts (January 1982 to May 1995) was searched using the CSG's phrase for randomised controlled trials (see Group search strategy) combined with the phrase: [and ((tardive near (dyskine* or diskine*) or (abnormal near movement* near disorder*) or (involuntar* near movement*))] The set of reports that resulted from this was hand searched for possible trials and researched, within the bibliographic package ProCite, with the phrase [cholinergic* or arecoline or choline or deanol or lecithin or meclofenoxate or physostigmine or RS?86]
1.1.2 The Cochrane Schizophrenia Group's Register was searched using the phrase: [cholinergic* or arecoline or choline or (#42 = 12) or deanol or (#42 = 353) or (#42 = 355) or lecithin or (#42 = 151) or meclofenoxate or physostigmine or RS?86]
1.1.3 EMBASE (January 1980 to May 1995) was searched using the CSG's phrase for randomised controlled trials (see Group search strategy) combined with the phrase: [and ((tardive dyskinesia in thesaurus ‐subheadings, prevention, drug therapy, side effect and therapy) or (neuroleptic dyskinesia in thesaurus ‐all subheadings) or (tardive or dyskines*) or (movement* or disorder*) or (abnormal or movement* or disorder*))] The set of reports that resulted from this was hand searched for possible trials and researched, within the bibliographic package ProCite, with the phrase [cholinergic* or arecoline or choline or deanol or lecithin or meclofenoxate or physostigmine or RS?86]
1.1.4 LILACS (January 1982 to September 1996) was searched using the CSG's phrase for randomised controlled trials (see Group search strategy) combined with the phrase: [and (tardive or (dyskinesia* or diskinesia*)) or (drug induced movement disorders in thesaurus))] This downloaded set of reports was hand searched for possible trials and researched, within the bibliographic package ProCite, with the phrase [cholinergic* or arecoline or choline or deanol or lecithin or meclofenoxate or physostigmine or RS?86]
1.1.5 MEDLINE (January 1966 to May 1995) was searched using the CSG's phrase for randomised controlled trials (see Group search strategy) combined with the phrase: [and (movement‐disorders in MeSH / explode all subheadings) or (anti‐dyskinesia‐agents in MeSH / explode all subheadings) or (dyskinesia‐drug‐induced in MeSH / explode all subheadings) and (psychosis in MeSH / explode all subheadings) or (schizophrenic disorders in MeSH / explode all subheadings) or (tardive near (dyskine* or diskine*)) or (abnormal* near movement* near disorder*) or (involuntar* near movement*))] The set of reports that resulted from was hand searched for possible trials and researched, within the bibliographic package ProCite, with the phrase [cholinergic* or arecoline or choline or deanol or lecithin or meclofenoxate or physostigmine or RS?86]
1.1.6 PsycLIT (January 1974 to May 1995) was searched using the CSG's phrase for randomised controlled trials (see Group search strategy) combined with the phrase: [and (explode movement‐disorders in DE) or (explode tardive‐dyskinesia in DE) or (tardive near (dyskine* or diskine*) or (abnormal* near movement* near disorder*) or (involuntar* near movement*))] The set of reports that resulted from this was hand searched for possible trials and researched, within the bibliographic package ProCite, with the phrase [cholinergic* or arecoline or choline or deanol or lecithin or meclofenoxate or physostigmine or RS?86]
1.1.7 SCISEARCH ‐ Science Citation Index. Each of the included studies was sought as a citation on the SCISEARCH database. Reports of articles that had cited these studies were inspected so that further trials could be identified.
1.2 In the first substantial up‐date of the review the Cochrane Schizophrenia Group's Register was searched (October 2001) using the phrase: [cholinergic* OR arecolin* OR arecholin* OR meclofenoxat* OR meclophenoxat* OR centrofenoxin* OR centrophenoxin* OR 'ANP 235' OR 'EN 1627' OR deanol* OR demanol* OR 'CR 121' OR 'RS 86' OR physostigmin* OR fysostigmin* OR lecithin* OR lecitin* OR choline OR cholin OR coline OR tacrin* OR takrin* OR tetrahydroaminoacridin* OR tetrahydroaminacrin* OR 'CI 970' OR THA OR THAA OR 7‐methoxyacridin* OR 7‐metoxyacridin* OR methoxytacrin* OR metoxytacrin* OR metoxytakrin* OR methoxycrin* OR metoxycrin* OR MEOTA OR ipidacrin* OR amiridin* OR NIK247 OR 'NIK 247' OR donepezil* OR E2020 OR 'E 2020' OR galanthamin* OR galantamin* OR 'CGP 37267' OR rivastigmin* OR ENA713 OR 'ENA 713' OR '212 713' OR eptastigmin* OR heptylstigmin* OR heptylphysostigmin* OR heptylfysostigmin* OR 'L 693 487' OR MF201 OR 'MF 201' OR metrifonat* OR metriphonat* OR trichlorfon* OR trichlorphon* OR trichlorfen* OR trichlorphen* OR 'L 1359' OR 'Bay a 9826' OR 'Bay 1 1359' OR xanomelin* OR 'LY 246708' OR 'FG 10232' OR cevimelin* OR AF102B OR 'AF 102B' OR 'FKS 508' OR 'SND 5008' OR SNK508 OR SNI2011]
The Cochrane Schizophrenia Group's Register is assembled by extensive searches of randomised controlled trials in electronic databases, registers for conference proceedings and dissertations etc. The search strategy of the CSG's Register contains a search strategy for trials on tardive dyskinesia. Please see search strategy in CSG module in Cochrane Library.
2. Reference searching The references of all identified studies were inspected for more studies.
3. Personal contact The first author of each included study was contacted for information regarding unpublished trials.
Data collection and analysis
[For definitions of terms used in this, and other sections, please refer to the Glossary.]
1. Selection of trials The title or abstract of each reference identified by the search was inspected independently by two reviewers (IT and ES) to assess relevance. For articles that could possibly have been RCTs, or in cases of disagreement, the full article was obtained. In turn these articles were independently inspected. There was no disagreement between the two reviewers regarding which trials were relevant.
2. Assessment of methodological quality The methodological quality of each included trial was assessed independently by two reviewers (IT and ES). Quality was evaluated using criteria described in the Cochrane Reviewers' Handbook (Clarke 2001) and the Jadad Scale (Jadad 1996). The former is based on evidence of a strong relationship between allocation concealment (blinding of random assignment of participants to intervention groups) and the potential for bias in the results (Schulz 1995), i.e. lack of adequate allocation concealment is associated with selection bias (systematic differences in comparison groups). Thus trials can, to a certain extent, be evaluated by their method of allocation concealment. The method for assigning participants to interventions undergoing comparison should be robust against selection bias (i.e. the trialist should not be able to influence which intervention the participant will receive nor should any foreknowledge of treatment assignment influence recruitment) and its description should be clear (Clarke 2001). The risk for bias in the results of a study is defined as below (Clarke 2001):
A. Low risk of bias (adequate allocation concealment) B. Moderate risk of bias (some doubt about the results) C. High risk of bias (inadequate allocation concealment)
The Jadad Scale measures a wider range of factors that impact on the quality of a trial. The scale includes three items: 1. Was the study described as randomised? 2. Was the study described as double blind? 3. Was there a description of withdrawals and drop‐outs? Each item receives 1 point if the answer is positive. In addition: 1 additional point each is given, if randomisation and/or blinding procedures are described and adequate. 1 point each can be deducted if either the randomisation or the blinding/masking procedures described were inadequate. Thus the maximum score is 5 points. (Jadad 1996).
Only trials described as randomised (category A or B) were included in the statistical analysis. The Jadad scale was used as an extra assessment of quality, however Jadad points were not used to exclude trials. If there was not enough information in the publication to assess adequate randomisation and methodological quality, or there was disagreement between the two reviewers, the article was added to those awaiting assessment and authors of the study were contacted for clarification. Justification for excluding trials from the analysis was documented.
3. Data extraction Data from the included trials were extracted by two researchers. Two kinds of measures were extracted: dichotomous (binary, yes/no) data and continuous (scale) data.
Trials in which a crossover design was used included the risk of carry over effects of a medication in the second or more stages of the trial (after crossover). To exclude potential carry over effects, only data from the first stage of the trial (before crossover) were used in the analysis. In the case of incomplete data, the article was added to those awaiting assessment and authors were contacted for clarification. If a trial met the criteria for methodological quality, but it was impossible to extract any data or collect unpublished data from the authors, the study had to be excluded from statistical analysis.
4. Data analysis 4.1 Intention to treat analysis Where possible, all data were analysed using the intention‐to‐treat principle (once randomised, always analyse). Data were excluded from studies where more than 50% of participants in any group were lost to follow up. (This did not include the outcome of 'drop‐outs'.) In studies with less than a 50% drop‐out rate, people leaving early were considered to have had the negative outcome, except for the event of death. When possible and appropriate, the 'Last Observation Carried Forward' technique was used in analysis of continuous outcomes.
4.2 Dichotomous data For dichotomous outcomes a standard estimation of the risk ratio (RR) and its 95% confidence interval (CI) was calculated. If overall results were significant, the number needed to treat (NNT) was calculated from the absolute risk difference between treatment and control groups. If heterogeneity was found it was investigated and a random effects model used.
4.3 Continuous data 4.3.1 Normally distributed versus skewed data Continuous data on clinical and social outcomes are often not normally distributed. A reliable statistical analysis of groups undergoing comparison requires that samples tested attain a normal distribution. To avoid including non‐normally distributed samples in the statistical analysis, the following standards were applied to all continuous data before inclusion: 1) Standard deviations and means were reported or derivable from data in the publication, or were obtainable from authors. 2) When a scale started from a finite number (such as zero), the standard deviation, when multiplied by two, was less than the mean (as otherwise the mean was unlikely to be an appropriate measure of the centre of the distribution ‐ Altman 1996). 3) If a scale started from a positive value (such as PANSS which can have values from 30 to 210) the calculation described above was modified to take the scale starting point into account. In these cases skewness is present if 2SD>(S‐Smin), where S is the mean score and Smin is the minimum score of the scale. Endpoint scores on scales used in clinical practice often have a definite minimum and maximum on the scale, and so these rules can be applied to them.
4.3.2 Summary statistic For continuous outcomes a pooled weighted mean difference (WMD) between groups was calculated. If heterogeneity was found it was investigated and a random effects model used.
4.3.3 Valid scales A wide range of instruments are available to measure mental health outcomes. These instruments vary in quality and many are not valid. Unpublished instruments are more likely to report statistically significant findings than those that have been published (Marshall 2000). The following minimum standards were set for valid scales: 1. The instrument had to have been described in a peer reviewed journal. 2. The instrument had to be either a self report scale or completed by an independent rater.
4.3.4 Endpoint versus change data When continuous data are presented on a scale which includes the possibility of negative values (such as change on a scale), it is impossible to tell whether data is non‐normally distributed (skewed) or not. It is thus preferable to use end point data of a scale (participants' total scores at the end of study ‐ not change in score from baseline), which typically cannot have negative values. Where possible endpoint data were presented, and if both endpoint and change (from baseline) data were available for the same outcome, then only the former were used.
5. Test for heterogeneity It is important not to pool heterogeneous studies together, as heterogeneity might reflect differences in study design or sample population, rather than true variation in results of the outcome measured. To investigate the possibility of heterogeneity of trial results, a Mantel‐Haenszel Chi‐square test was used, as well as visual inspection of graphs. A significance level less than 0.10 was pre‐defined as evidence of heterogeneity. If heterogeneity was found, the reasons for it were explored. If no study‐related explaining factor was found, data were tested pooled using the random effects model which takes into account the variation between studies. (The random effects model is more conservative in estimating treatment effect, and takes into account that some trials will produce odd results by chance.) If using the random effects model did not change the statistical significance level of the results, the results remained pooled. If the random effects model did change the statistical significance of the result, studies responsible for heterogeneity were not added to the main body of homogeneous trials, but summated and presented separately and reasons for heterogeneity investigated.
6. Addressing publication bias Data from all included studies were entered into a funnel graph (trial effect against trial size) in an attempt to investigate the likelihood of overt publication bias.
7. Sensitivity analyses Four sensitivity analyses were prespecified: 1.Treatment effect differs according to difference in the quality of trials, 2. Treatment effect differs according to different lengths of treatment, 3. Treatment effect differs for the various drugs and 4. Treatment effect differs according to the age of the participants (this sensitivity analysis was added in the first substantial up‐date of the review). These analyses were evaluated by looking at separate subgroups of trials.
8. General Data were entered into Revman in such a way that the area to the left of the line of no effect in the graph indicated a favourable outcome for cholinergic agents.
Data and analyses
Comparison 1. CHOLINERGIC DRUGS versus PLACEBO.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Tardive dyskinesia: 1. No clinically important improvement (50% or more change on any validated TD scale) | 4 | 27 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.89 [0.65, 1.23] |
| 1.1 deanol ‐ more than 6 weeks | 2 | 11 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.91 [0.51, 1.60] |
| 1.2 donepezil ‐ less than 6 weeks | 1 | 10 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.0 [0.70, 1.43] |
| 1.3 lecithin ‐ less than 6 weeks | 1 | 6 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.71 [0.31, 1.66] |
| 2 Tardive dyskinesia: 2a. Not any improvement (as assessed by rater) | 9 | 180 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.87 [0.71, 1.07] |
| 2.1 deanol ‐ less than 6 weeks | 3 | 63 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.83 [0.58, 1.18] |
| 2.2 deanol ‐ more than 6 weeks | 2 | 11 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.81 [0.26, 2.57] |
| 2.3 donepezil ‐ less than 6 weeks | 1 | 10 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.33 [0.72, 2.44] |
| 2.4 lecithin ‐ less than 6 weeks | 2 | 36 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.87 [0.63, 1.21] |
| 2.5 meclofenoxate ‐ more than 6 weeks | 1 | 60 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.84 [0.55, 1.27] |
| 3 Tardive dyskinesia: 2b. Not any improvement (as assessed by self report) | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 3.1 lecithin ‐ less than 6 weeks | 1 | 30 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.92 [0.62, 1.36] |
| 4 Tardive dyskinesia: 3a. Average endpoint score on AIMS (low score = better) | 7 | 171 | Mean Difference (IV, Fixed, 95% CI) | ‐0.12 [‐0.44, 0.21] |
| 4.1 deanol ‐ more than 6 weeks | 1 | 6 | Mean Difference (IV, Fixed, 95% CI) | 1.42 [‐0.29, 3.13] |
| 4.2 galantamine ‐ more than 6 weeks | 1 | 35 | Mean Difference (IV, Fixed, 95% CI) | 1.5 [‐0.44, 3.44] |
| 4.3 lecithin ‐ less than 6 weeks | 1 | 6 | Mean Difference (IV, Fixed, 95% CI) | ‐1.07 [‐2.21, 0.07] |
| 4.4 lecithin ‐ more than 6 weeks | 1 | 14 | Mean Difference (IV, Fixed, 95% CI) | ‐0.10 [‐1.04, 0.84] |
| 4.5 meclofenoxate ‐ more than 6 weeks | 1 | 60 | Mean Difference (IV, Fixed, 95% CI) | ‐0.19 [‐0.58, 0.20] |
| 4.6 rivastigmine ‐ less than 8 weeks | 1 | 40 | Mean Difference (IV, Fixed, 95% CI) | 2.20 [‐1.16, 5.56] |
| 4.7 donepezil ‐ less than 6 weeks | 1 | 10 | Mean Difference (IV, Fixed, 95% CI) | 1.10 [‐4.22, 6.42] |
| 5 Tardive dyskinesia: 3b. Average endpoint score on modified Simpson TDRS (low score = better) | Other data | No numeric data | ||
| 5.1 deanol ‐ more than 6 weeks | Other data | No numeric data | ||
| 6 Tardive dyskinesia: 4a. Deterioration (as assessed by rater) | 8 | 147 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.11 [0.55, 2.24] |
| 6.1 deanol ‐ less than 6 weeks | 2 | 30 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.67 [0.48, 5.76] |
| 6.2 deanol ‐ more than 6 weeks | 2 | 11 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.67 [0.20, 2.18] |
| 6.3 donepezil ‐ less than 6 weeks | 1 | 10 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.67 [0.06, 7.85] |
| 6.4 lecithin ‐ less than 6 weeks | 2 | 36 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.0 [0.16, 6.31] |
| 6.5 meclofenoxate ‐ more than 6 weeks | 1 | 60 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.87 [0.18, 19.55] |
| 7 Tardive dyskinesia: 4b. Deterioration (as assessed by self report) | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 7.1 lecithin ‐ less than 6 weeks | 1 | 30 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.0 [0.13, 68.26] |
| 8 Global outcome: Death for any reason | 11 | 278 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 8.1 deanol ‐ less than 6 weeks | 4 | 83 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 8.2 deanol ‐ more than 6 weeks | 2 | 11 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 8.3 galantamine ‐ more than 6 weeks | 1 | 38 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 8.4 lecithin ‐ less than 6 weeks | 3 | 86 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 8.5 meclofenoxate ‐ more than 6 weeks | 1 | 60 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 9 Global outcome: Intervention not useful as assessed by Global Usefulness Rating (GUR) | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 9.1 meclofenoxate ‐ more than 6 weeks | 1 | 60 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.89 [0.59, 1.32] |
| 10 Global state: Average endpoint score on CGI (low score = better) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 10.1 lecithin ‐ less than 6 weeks | 1 | 31 | Mean Difference (IV, Fixed, 95% CI) | ‐0.43 [‐1.36, 0.50] |
| 11 Mental state: Deterioration | 5 | 77 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.5 [0.10, 2.61] |
| 11.1 deanol ‐ less than 6 weeks | 1 | 10 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.33 [0.02, 6.65] |
| 11.2 deanol ‐ more than 6 weeks | 2 | 11 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.2 [0.08, 18.75] |
| 11.3 lecithin ‐ less than 6 weeks | 2 | 56 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.33 [0.01, 7.81] |
| 12 Adverse effects: Any | 4 | 106 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.56 [0.15, 2.14] |
| 12.1 donepezil ‐ less than 6 weeks ‐ any adverse events | 1 | 10 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 12.2 lecithin ‐ more than 6 weeks ‐ any other adverse effects, undesirable body odour, sedation | 2 | 36 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 12.3 meclofenoxate ‐ more than 6 weeks ‐ any adverse events | 1 | 60 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.56 [0.15, 2.14] |
| 13 Adverse effects: Various specific | 8 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 13.1 deanol ‐ less than 6 weeks ‐ gastric adverse effects | 5 | 61 | Risk Ratio (M‐H, Fixed, 95% CI) | 9.0 [0.55, 147.95] |
| 13.2 deanol ‐ less than 6 weeks ‐ sedation, periferal cholinergic effects, undesirable body odour | 6 | 94 | Risk Ratio (M‐H, Fixed, 95% CI) | 6.83 [0.99, 47.25] |
| 13.3 lecithin ‐ less than 6 weeks ‐ GI adverse effects | 2 | 36 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 14 Leaving the study early | 12 | 288 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.09 [0.56, 2.10] |
| 14.1 deanol ‐ less than 6 weeks | 4 | 83 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.33 [0.02, 6.65] |
| 14.2 deanol ‐ more than 6 weeks | 2 | 11 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.2 [0.08, 18.75] |
| 14.3 donepezil ‐ less than 6 weeks | 1 | 10 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 14.4 galantamine ‐ more than 6 weeks | 1 | 38 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.0 [0.96, 9.39] |
| 14.5 lecithin ‐ less than 6 weeks | 3 | 86 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.5 [0.17, 1.45] |
| 14.6 meclofenoxate ‐ more than 6 weeks | 1 | 60 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
Comparison 2. CHOLINERGIC DRUGS versus OTHER CHOLINERGIC DRUGS.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Tardive dyskinesia: 2a. Not any improvement (as assessed by rater) ‐ less than 6 weeks | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 1.1 deanol 2g vs deanol 1g | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 2 Global outcome: Death for any reason ‐ less than 6 weeks | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 2.1 deanol 2g vs deanol 1g | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3 Leaving the study early ‐ less than 6 weeks | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 3.1 deanol 2g vs deanol 1g | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Beckham 1981.
| Methods | Allocation: randomised, no details.
Blindness: double, described and adequate.
Duration: 11 days.
Design: parallel.
Raters: 1 blinded rater, frequency of dyskinesia count rated from videotapes presented in random order. Setting: mostly inpatients, some outpatients, USA. |
|
| Participants | Diagnosis: schizophrenia (21), affective disorder (3), OBS (7), neurosis (2). History: TD present and stable > 6 months, antipsychotic dose stable > 4 months, mean duration psychiatric ill 17 years (range 1‐45), CPE dose (mg/day) mean 420 mg (SD 430) N = 50. Sex: all male. Age: mean 55 years, range 23‐77. | |
| Interventions | 1. Lecithin: dose 60 g/day containing phosphatidylcholine 33 g/day. N = 25. 2. Placebo. N = 25. Effort made to keep antipsychotic medication stable during study, 7 received anticholinergic (‐parkinsonian) medication. | |
| Outcomes | TD symptoms: CGI.
Leaving the study early. Unable to use ‐ Frequency of dyskinetic movement count (frequency of one selected movement/minute in 4 body areas counted visually from videotapes) (validation unsure, no SD). Adverse effects (reporting unspecific). Patient's subjective assessments (not reported). |
|
| Notes | ITT analysis not performed for continuous outcomes (CGI), results reported only for N = 31 who completed study (lecithin group 15, control group 16).
Sample attrition well reported.
Author contacted 2002, awaiting further information. Sponsorship source: Supported in part by a grant by the Veterans Administration. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "Patients were randomly assigned", further details not reported. |
| Allocation concealment (selection bias) | Unclear risk | Allocation concealment not reported. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | "double blind". "Only a member of the hospital pharmacy staff was aware of each patient's group assignment during the study. The investigator, patients, ward nurses, and physicians were all blind to patient status." "The control substance was a mixture of crushed graham cracker and corn oil which, when mixed with milk, resembled the lecithin mixture in taste, appearance, and viscosity. The mixtures were further disguised and made more palatable by the addition of artificial sweetener and vanilla extract." |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | "double blind". "Only a member of the hospital pharmacy staff was aware of each patient's group assignment during the study. The investigator, patients, ward nurses, and physicians were all blind to patient status." "Treatment effect was assessed by blind evaluation of randomly sequenced videotapes wade during standard examinations before, during, and after treatment." "the sole rater was blind to patient treatment assignment". |
| Incomplete outcome data (attrition bias) All outcomes | High risk | High drop‐out rate: 24%. 38/50 participants completed the trial (reasons reported per intervention group). Moreover, only 31/50 (62%) were included in the analysis (reasons reported). |
| Selective reporting (reporting bias) | Low risk | Dissertation. All outcomes seem to have been reported. |
| Other bias | Unclear risk | The groups seem to have had differences in their baseline dental status. |
Caroff 2007.
| Methods | Allocation: randomised, no details.
Blindness: double‐blind, no details.
Duration: 30 weeks (2 weeks baseline, 12 weeks followed by 4 weeks wash‐out then crossed over to another 12 weeks).
Design: cross‐over.
Raters: no details. Setting: Patients treated in the Department of Veteran Affairs Medical Center, USA. |
|
| Participants | Diagnosis: TD (research criteria), long‐duration schizophrenia (DSM‐IV criteria). History: Clinical diagnosis of TD lasting at least 3 months; treatment with antipsychotic drugs at least for 3 months. N = 38 Sex: all male. Age: mean 56.4 (SD 9.9) years. |
|
| Interventions | 1. Galantamine: dose 4 mg twice daily for 4 weeks followed by 8 mg twice daily for 4 weeks, and 12 mg twice daily for an additional 4 weeks (followed by 4 weeks washout ad 12 weeks placebo). N = 19.
2. Placebo: 12 weeks placebo (followed by 4 weeks "washout" and 12 weeks galantamine). N = 19. Antipsychotics dose stable at least one month prior to the start of the study for oral medications and within 2 months for depot medications. Patients remained on a stable dose of antipsychotics throughout the study. Two patients were not receiving antipsychotics during the study. Any anticholinergic drugs or vitamin supplements were discontinued 2 weeks prior to randomisation. |
|
| Outcomes | TD symptoms: total AIMS Leaving the study early Unable to use ‐ no report from first phase before crossing over separately: Simpson‐Angus Scale, BAS, BPRS, MMSE. |
|
| Notes | Sponsorship source: Supported by a grant from Ortho‐McNeil Neurologics, Inc. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "randomized controlled trial," further details not reported. |
| Allocation concealment (selection bias) | Unclear risk | Allocation concealment not reported. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | "double‐blind," details not reported. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | "double blind", details not reported. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | "Overall, 10 (31.3%) of 32 patients receiving galantamine dropped out, and 6 (23.1%) of 26 patients receiving placebo dropped out. Twelve patients dropped out during phase 1 (galantamine, N=9; placebo, N=3), and 4 dropped out during phase 2 (galantamine, N = 1; placebo, N=3)." |
| Selective reporting (reporting bias) | High risk | Although the protocol specified that SAS (secondary outcome) and BAS should have been reported at the end of three months (phase I), data not reported per phase. Also data for BPRS not reported per phase. |
| Other bias | Unclear risk | Insufficient information to make a judgement. |
de Montigny 1979.
| Methods | Allocation: randomised, no details.
Blindness: double, no details.
Duration: 3 weeks.
Design: parallel.
Raters: ESRS rated independently by 2 psychiatrists. Setting: from long‐term wards, Canada. |
|
| Participants | Diagnosis: chronic schizophrenia. TD: significant (CGI Scale TD). History: TD moderate to severe, maintenance antipsychotic treatment > 6 years, CPE dose range 0 mg to 1850 mg/day, duration of TD not reported. N = 20. Sex: 10 M, 10 F. Age: Median 61 years, range 34‐73 years. | |
| Interventions | 1. Deanol: dose increased from 600 mg to 1500 mg/day during first week, constant thereafter. N = 10, for three weeks. 2. Placebo. N = 10. Antipsychotic dose stable during study, no other psychotropics permitted. | |
| Outcomes | Adverse effects.
Leaving the study early. Death Unable to use ‐ TD symptom scores: ESRS (no SD). Mental state scores: BPRS (no SD). |
|
| Notes | Sponsorship source: Sponsorship source not reported Analysis of ESRS scores in publication did not detect significant treatment effect. No difference between treatments regarding parkinsonism. There was significant increase in mean schizophrenic subscore of BPRS in deanol‐treated group. Authors contacted ‐ no reply. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "Patients were randomly assigned", further details not reported. |
| Allocation concealment (selection bias) | Unclear risk | Allocation concealment not reported. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | "double‐blind" Details not reported. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | "The ESRS was completed independently by two psychiatrists during the same interview and a final rating was made by consensus." "double‐blind", further details of blinding not reported. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | "All subjects completed the 3‐week trial" |
| Selective reporting (reporting bias) | High risk | TD symptoms (ESRS) and Mental State (BPRS) reported as means only. |
| Other bias | Low risk | The study seems to have been free of other sources of bias. |
Gelenberg 1990.
| Methods | Allocation: randomised, procedure conducted independently by trial statistician, stratified by maintenance antipsychotic drug therapy.
Blindness: double, adequate. 1 blinded rater assessed TD and psychopathology. 1 open rater assessed side‐effects and distributed medication.
Duration: 18‐20 weeks (4 weeks baseline, 8 weeks followed by 2‐4 weeks washout and then crossed to another 8 weeks).
Design: cross‐over. Setting: patients recruited from mental health centres and private physicians, USA. |
|
| Participants | Diagnosis: schizophrenia (9), bipolar (6), major depression (3), generalised anxiety disorder (1), brief reactive psychosis (1), no psychiatric diagnosis (1). TD diagnosed by psychiatrist and neurologist using criteria. History: TD present 6 months ‐ 17 years (median 1.5 years). N = 21. Sex: 10 M, 11 F. Age: median 47 years, range 19‐70 years. | |
| Interventions | 1. Lecithin: containing PC 20 g/day. N = 5 (completers).*
2. Placebo. N = 9 (completers).* Antipsychotics stable during trial. No anticholinergics permitted. Patients took the following concomitant psychoactive medications during the trial: antipsychotic agents alone (N = 7), antipsychotic drugs plus lithium (N = 3), antipsychotic drugs plus trazodone (N = 1), antipsychotic drugs plus an antianxiety agent (N = 1), antianxiety drugs alone (N = 1), antianxiety drugs plus lithium (N = 3), and lithium alone (N = 1). |
|
| Outcomes | TD symptoms: AIMS. Unable to use ‐ Global impression: CGI (not reported). Movement disorders: TAKE (reported only final summary scores from both segments, after cross‐over). Mental state: BPRS, HAM‐D (reported only final summary scores from both segments, after cross‐over). Adverse effects (reported only final summary scores from both segments, after cross‐over). Leaving study early (reported only final summary scores from both segments, after cross‐over). |
|
| Notes | * No information given on how many were originally allocated to each group. 14 of 21 completed the trial. Sponsorship source: Funded by National Institute of Mental Health grant, the Arbour Research Foundation, and the Center for Brain Sciences. ITT analysis not performed for AIMS scores (results reported only for completers). Physiology (lab‐tests, ECG, serum choline) monitored during trial. No clinically important changes in lab variables or vital signs during study. Serum choline levels doubled during lecithin treatment. Authors contacted, awaiting further information. Details of allocation procedure from authors. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "random‐order," "patients were stratified by whether they were on maintenance antipsychotic drug therapy." Details of sequence generation not reported. |
| Allocation concealment (selection bias) | Low risk | Allocation concealment not reported, but procedure confirmed as adequate from study authors. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | "double‐blind". "Several lecithin preparations were used during the study. We started with frappes prepared with chunks of 55% PC. That preparation was succeeded by chunks, chicken soup, and granola bars that contained 80% to 100% PC. Placebo included corn oil in frappes, ground corn flakes, and matching chicken noodle soup and granola bars." Unclear if the lecithin and placebo preparations were identical (color, taste, smell...)."The 14 completers were asked to fill out a questionnaire in which they specified (l) which of the two medications they thought was most helpful, (2) what effects (if any) they noted on their mood, and (3) whether they could guess which of the two medications was lecithin and which was placebo. Seven of the 14 patients felt that one treatment was definitely more helpful than the other; of those, 6 indicated that lecithin was the more helpful treatment." |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | "We used two clinical raters, one blind rater who assessed TD and psychopathology and one open rater who rated side effects and distributed medication." "Both the blind rater and the patient completed Clinical Global Impressions and Improvement ratings at each visit and the blind rater assessed extrapyramidal effects with the Target Abnormal Kinetic Effects (TAKE) scale." |
| Incomplete outcome data (attrition bias) All outcomes | High risk | "Fourteen patients‐7 men and 7 women‐completed at least 3 visits on the second leg of the trial. Data from these 14 completers were used in the efficacy analyses." Number completed the first period and number completed the trial not reported. 14/21 participants were entered to the analyses (approximately 33% drop out). |
| Selective reporting (reporting bias) | High risk | Clinical Global Impressions and Improvement, Target Abnormal Kinetic Effects (TAKE) scale, Mental State (BPRS and HAM‐D), adverse effects, and leaving the study early not fully reported. |
| Other bias | Low risk | The study seems to be free from other sources of bias. |
George 1981.
| Methods | Allocation: randomised, stratified by severity of TD.
Blindness: double.
Duration: 4 weeks.
Design: parallel.
Raters: Videotapes presented in random order and rated independently by 2 raters. Setting: chronic psychiatric hospital residents, Australia. |
|
| Participants | Diagnosis: chronic psychiatric hospital residents suffering from oral TD; having been treated with antipsychotics. History: No information about medication status and dose prior to study, or duration of TD. N = 33. Sex: 8 M, 25 F. Age: range 49 ‐ 89 years, mean ˜ 70 years. | |
| Interventions | 1. Deanol: dose 2000 mg/day for four weeks. N = 11.
2. Deanol: dose 1000 mg/day for four weeks. N = 11.
3. Placebo four weeks. N = 11. Seven participants on antipsychotics during trial, CPE range 50 mg to 800 mg/day. Other concomitant medication not reported. |
|
| Outcomes | TD symptoms.
Adverse effects.
Leaving study early. Unable to use ‐ TD symptom scores: local scale (not validated). |
|
| Notes | Sponsorship source: The drug used in this trial was supplied by Riker Laboratories Pty. Ltd. who in addition, provided a grant for expenses involved in this project. In the review, the two deanol groups are analysed as one group. Authors contacted ‐ no reply. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "randomly assigned", further details not reported. |
| Allocation concealment (selection bias) | Unclear risk | Allocation concealment not reported. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | "double‐blind", further details not reported. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | "The baseline rating of filming 1 and the ratings of filming 2, 3 and 4 were carried out by randomizing. All film segments and showing them unidentified to the raters on the 30th day of the study." |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | "all patients completed the trial." |
| Selective reporting (reporting bias) | High risk | TD symptoms reported only as means, Adverse events not fully reported. |
| Other bias | Unclear risk | "One subject in Group A showed 'substantial improvement', however, on preliminary and baseline rating that patient was one of the less severely afflicted. In Group B one patient also showed 'substantial improvement' and this patient was receiving thioridazine 200mg three times a day in addition to deanol." Possible confounding variables. |
Jackson 1978.
| Methods | Allocation: randomised, no details.
Blindness: double.
Duration: 32 weeks (withdrawal of antipsychotics/single antipsychotic in constant dose followed by 4 weeks baseline, 12 weeks then crossed over to another 12 weeks).
Design: cross‐over. Setting: long‐term inpatients, USA. Raters: videotapes presented in random temporal sequence and rated independently by 4 psychiatrists using AIMS. |
|
| Participants | Diagnosis: schizophrenia + TD (Global AIMS rating of moderate to severe). History: mean duration ill ˜22 years (range 18‐30), high dose antipsychotic drugs over extended periods of time. N = 6. Sex: all female. Age: mean 48 years, range 34‐59. | |
| Interventions | 1. Deanol: dose gradually increased to 1500 mg/day over 4 weeks. N = 4. 2. Placebo. N = 2. Maintained on single, stable antipsychotic dose during study. No other psychotropics or anticholinergics permitted. | |
| Outcomes | TD symptoms: AIMS.
Mental state.
Adverse effects.
Leaving study early. Unable to use ‐ Mental state scores: MIBS (not reported). Parkinsonism scores: SAS scale (not reported). |
|
| Notes | No participants developed clinical parkinsonism.
People leaving the study early may not have been reported. Sponsorship source: Sponsorship source not reported |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "subjects were randomly assigned", further details not reported. |
| Allocation concealment (selection bias) | Unclear risk | Allocation concealment not reported. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | "double‐blind", details not reported. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | "A 20‐minute videotape of the patient sitting alone and then of an examination following the schema for the AIMS, by the same psychiatrist known to the patient. At the end of the study, the three videotapes for each patient were presented in random temporal sequence and rated "blind" by 4 psychiatrists using the AlMS." "Additional AIMS ratings were made by the same psychiatrist every 4 weeks on the ward without the disturbance of the videotape equipment. A weekly Global AIMS and Missouri In‐Patient Behavior Seale (MlBS) was per formed by a ward nurse" |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Insufficient information to judge. |
| Selective reporting (reporting bias) | High risk | AIMS scores reported as means only. Data for Simpson Angus Scale not reported: "there was no significant change in the Simpson and Angus ratings". Data for MIBS not reported: "There was no significant or sustained change in the Missouri In‐Patient Behavior Scale ratings" |
| Other bias | High risk | "During the 32 weeks of the study, interrater variability, day‐to‐day changes in patient condition, and non‐drug related trends across time reduced the power of the single crossover design to the point where it would be unlikely to detect any but the most clearcut changes in a single patient." |
Jackson 1979.
| Methods | Allocation: randomised, no details.
Blindness: double, described and adequate.
Duration: 2 weeks (preceded by 2‐4 weeks pre‐entry).
Design: cross‐over. Setting: long‐term inpatients, USA. Raters: videotapes presented in random temporal sequence and rated blind and independently by 2 psychiatrists using AIMS. |
|
| Participants | Diagnosis: long‐term schizophrenia + TD (moderate or severe on AIMS global rating). History: antipsychotics continuously > 4 years (range 4‐23), duration of TD not reported. N = 6. Sex: 1 M, 5 F. Age: mean 57 years, range 49‐60. | |
| Interventions | 1. Lecithin: dose 50 g/day containing PC 35 g/day. N = 3. 2. Placebo. N = 3. Antipsychotics stable during study. No other psychotropics or anticholinergics permitted. | |
| Outcomes | TD symptoms: AIMS.
Mental state.
Adverse effects.
Leaving study early. Unable to use ‐ Mental state scores: BPRS, MIBS (not reported). |
|
| Notes | Sponsorship source: Sponsorship source not reported One person withdrawn early due to nausea and vomiting on a lecithin/water/orange flavour mix. Protocol changed to lecithin/ice cream/chocolate mix ‐ well tolerated! Physiology (blood pressure, serum choline) monitored. Serum choline increased substantially during lecithin. Physiological assessment showed no evidence of adverse effects. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "randomly assigned", further details not reported. |
| Allocation concealment (selection bias) | Unclear risk | Allocation concealment not reported |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | "double‐blind"; "Each dose of lecithin or placebo was prepared in a coded bottle independent of the patient clinical staff, and raters". "No attempt was made to systematically and objectively rate body odour, but no significant change, and particularly no "fishy odour," was noted by the subjects, ward staff, or raters." |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | "Each dose of lecithin or placebo was prepared in a coded bottle independent of the patient clinical staff, and raters". "At the end of the study the 12 videotapes for each patient were presented in random temporal sequence and rated blind and independently by 2 psychiatrists using the AIMS...two raters' total AIMS scores, rated blind and independently from videotapes.." |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Figure 1 reports data from all 6 participants. ITT is not mentioned. One participant was withdrawn from the study; reason reported. |
| Selective reporting (reporting bias) | High risk | BPRS and MIBS data not reported. |
| Other bias | Unclear risk | Insufficeint information to make a judgement. 1/6 participants was antipsychotic‐free throughout the study. |
Jahanian 2014.
| Methods | Allocation: "randomly assigned" no details reported.
Blindness: "double blind" no details reported.
Design: not reported.
Duration: "eight weeks". Setting: "Razi Psychiatric Center, Iran". |
|
| Participants | Diagnosis: Patients with schizophrenia and TD based on DSM‐IV‐TR diagnosed by psychiatrist. N = 40. Age: range 18‐65 years Sex: not reported |
|
| Interventions | 1. Rivastigmine: dose: 1.5 mg twice daily. N = 20 2. Placebo: no details reported. N = 20. |
|
| Outcomes | TD symptoms: AIMS | |
| Notes | Sponsorship source: "no financial support". | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "Randomly". No details reported. |
| Allocation concealment (selection bias) | Unclear risk | Not reported. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | "Double blind". No details reported. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | "Double blind". No details reported. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Not reported. |
| Selective reporting (reporting bias) | Low risk | Outcomes have been reported based on the registered protocol IRCT2012092910964N1. |
| Other bias | Unclear risk | Insufficient information to make a judgement |
Kocher 1980.
| Methods | Allocation: randomised, no details.
Blindness: double, not described.
Duration: 4 weeks.
Design: cross‐over.
Raters: two independent raters. Setting: long‐term inpatients. |
|
| Participants | Diagnosis: schizophrenia (17), senile dementia (3) + TD (diagnosed by 2 physicians). History: antipsychotic medication >5 yrs. N=20. Sex: 10 M, 10 F. Age: average 67 years, range 42‐82. | |
| Interventions | 1. Deanol: dose gradually increased to 1500 mg/day. N =1 0. 2. Placebo. N = 10. Antipsychotic dose stable during trial, antiparkinsonian (‐cholinergic) medication used by some. | |
| Outcomes | TD symptoms.
Adverse effects.
Leaving study early. Unable to use ‐ TD symptom scores: local scale (not validated). |
|
| Notes | Deanol well tolerated. Authors contacted ‐ no reply. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "randomised", no details. |
| Allocation concealment (selection bias) | Unclear risk | No allocation concealment details. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | "double blind" not described. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Two independent raters assessed outcomes. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No dropouts in study. |
| Selective reporting (reporting bias) | Unclear risk | No study protocol and the results are not proper reported. |
| Other bias | Unclear risk | No further information provided. |
Lucius 1976.
| Methods | Allocation: matched pairs were randomised. Allocation procedure conducted independently by hospital pharmacist and not reported to trialists.
Blindness: double, unclear.
Duration: 5 weeks, preceded by pre‐entry period 1 week.
Design: cross‐over. Setting: long‐term inpatients. Raters: two independent raters under standardised conditions. |
|
| Participants | Diagnosis: schizophrenia (8), bipolar (1), cerebral sclerosis (1) + TD (diagnosed by 3 physicians using criteria). History: mean duration antipsychotic drugs ˜12 years (range 2‐19), mean CPE dose ˜177 mg/day (100 mg to 225 mg). N = 20 (please see notes). Sex: 2 M, 8 F. Age: mean 62 years (28‐75). | |
| Interventions | 1. Deanol: dose gradually increased to 1500 mg/day. N = 5. 2. Placebo. N = 5. Antiparkinsonians ceased 8 days before trial. | |
| Outcomes | TD symptoms.
Mental state.
Adverse effects.
Leaving study early. Unable to use ‐ TD symptom scores: local scale (not validated). |
|
| Notes | Original study N = 20. Due to information about toxic effects of clozapine in July 1975, antipsychotic medication abruptly changed. In dissertation, detailed individual patient data supplied. Data extracted for 10 participants whose antipsychotic medication was stable during study. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "randomised" No further details. |
| Allocation concealment (selection bias) | Low risk | "pharmacy‐controlled allocation, identical sequentially number drug containers". |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Identical capsules planned, but apparently differences in form and taste. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | "two independent raters under standardised conditions". |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Dropouts with reasons reported, but unclear in which treatment phase of cross‐over study. |
| Selective reporting (reporting bias) | Unclear risk | No protocol available and the study outcomes are unclear if all were reported. |
| Other bias | Unclear risk | Unclear the cross‐over phases. |
Ogunmefun 2009.
| Methods | Allocation: "randomized" Blinding: "double blind" Design: cross‐over* Duration: Trial I*: 12 weeks (4 weeks, 4 weeks washout, then crossed over to another 4 weeks); Trial II: 16 weeks (6 weeks, 4 weeks washout, then crossed over to another 6 weeks) Setting: not reported. |
|
| Participants | Diagnosis: TD diagnosis according to Schooler and Kane research diagnostic criteria History: average duration that participants experienced TD was 6.4 years (range, 2‐10 years). N (Trial I) = 7; N (Trial II) = 5* Age: mean 61.4 years Sex: 7 M, 3 F. |
|
| Interventions | Trial I: 5 mg** donepezil daily (N = 4) vs placebo (N = 3). Trial II: 10 mg donepezil daily (N = 3) vs placebo (N = 2). Permitted to stay on current antipsychotic medication, but not allowed to take anticholinergic medication during the study. |
|
| Outcomes | TD symptoms: improved/deteriorated, AIMS scale scores Adverse event Leaving the study early Unable to use ‐ SAS, BPRS, MMSE (data not fully reported) |
|
| Notes | *Two individuals participated in both studies, in which case, their data from the earlier 5 mg study were used. **Because there was no significant effect of donepezil 5 mg daily on dyskinetic movements the same trial design was continued but with increased daily dose to 10 mg. We have analysed the two doses together. Study author kindly replied to our request for outcome‐ and 'Risk of bias' data. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "Random digit generation: even‐‐donepezil; odd—placebo" (personal communication). |
| Allocation concealment (selection bias) | Unclear risk | No details reported. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | "double‐blind", no further details reported. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | One to two raters blindly scored subjects for each outcome scale. "Allocation was concealed by not telling rater about allocation. Therefore, the AIMS rater rated movements viewed on videotapes not knowing whether subject was taking donepezil or placebo and not knowing the time order of the videotape sessions." (personal communication) |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | 2/10 participants discontinued, not reported reasons or from which group. |
| Selective reporting (reporting bias) | High risk | MMSE and BPRS not reported. |
| Other bias | High risk | |
Price 1982.
| Methods | Allocation: randomised, no details.
Blindness: double, described and adequate.
Duration: 9‐11 days.
Design: parallel.
Raters: one blinded rater. Setting: inpatients, USA. |
|
| Participants | Diagnosis: schizophrenia (69%), OBS (29%), bipolar (2%) + TD (diagnosed by criteria), thorough evaluation to rule out differential diagnostic categories. History: mean duration of antipsychotic treatment 17 (SD 8.5) years (range 2‐26 years). TD for at least 3 months. N = 45. Sex: all male. Age: mean 56 years, range 26‐77 years. | |
| Interventions | 1. Lecithin: dose 60 g/day containing PC dose of 33 g/day. N = 15. 2. Placebo. N = 15. 3. No‐treatment control group. N = 15. Antipsychotics stable, anticholinergics used by 7 participants. | |
| Outcomes | TD symptoms.
Adverse effects.
Leaving study early. Unable to use ‐ TD symptom scores: Simpson TDRS, SRTDRS (reported in ANCOVA tables, unable to extract data). |
|
| Notes | Sponsorship source: Sponsorship source not reported. Review uses data only from lecithin and placebo groups for whom blinding adequate and reporting consistent. (N = 15 + 15 = 30). Author contacted to confirm lack of additional data. 60% of participants overlapped with Beckham 1981 study. Extensive neuropsychological and motor tests performed. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "randomly selected". Details not reported |
| Allocation concealment (selection bias) | Unclear risk | Allocation concealment not reported. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | "double‐blind". "Although the subjects assigned to one of the treatment groups were informed that they would receive either the lecithin treatment or a placebo control treatment, neither the patients nor the researcher knew to which group any individual had been assigned." "The placebo substance resembled the lecithin mixture in taste, .appearance, and thickness" The no treatment group's participants could not have been blinded |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Rater and self‐report were used as outcome measures. Research personnel and participants seem to have been blinded to the assignment. Self‐report ratings were not recorded for the "no treatment" group. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | All participants completed the trial. |
| Selective reporting (reporting bias) | High risk | Dissertation. All outcomes seem to have been reported. However, adverse effects expected to be reported in such trials have not been reported. Data for TD scores are not extractable. |
| Other bias | Low risk | "A one‐way analysis of variance was performed on subject variables to determine if there were initial differences among the groups. These analyses show no significant differences for age... , duration of antipsychotic treatment..., or initial symptom severity. A chi‐square analysis of diagnostic categories demonstrates no significant difference among the groups...". The study seems to have been free of other sources of bias. |
Tarsy 1977.
| Methods | Allocation: randomised, by table of random numbers, concealment unclear.
Blindness: double.
Duration: 16 weeks (8 weeks then crossed over to another 8 weeks).
Design: cross‐over. Setting: outpatients and inpatients, USA. Raters: one rater. |
|
| Participants | Diagnosis: psychiatric disorder. History: mean duration of TD ˜13 months, none chronically institutionalised, phenothiazines discontinued in 4 people 1‐11 months before trial. N = 5. Sex: all male. Age: mean 54.8 years. | |
| Interventions | 1. Deanol: dose 1000 mg/day for 4 weeks, then 2000 mg/day for next 4 weeks. N = 4.
2. Placebo. N = 1. Concomitant medication not reported. |
|
| Outcomes | TD symptoms: modified Simpson TDRS. Mental state. Adverse effects. Leaving study early. | |
| Notes | Sponsorship source: Supported by Veterans Administration research funds. Osvaldo N. Re, MD, and Riker Laboratories provided assistance. No parkinsonian adverse effects or mood changes observed. One person died suddenly at home due to acute aspiration in second cross‐over period (which is not included in review analysis), by which time he had completed 8 weeks of deanol treatment and 4 weeks of placebo treatment ‐ e.g. 4 weeks after cessation of deanol. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "Order of treatment was determined by a table of random numbers". |
| Allocation concealment (selection bias) | Unclear risk | Allocation concealment not reported. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | "double blind" no details reported. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | "double blind" no details reported. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | All participants completed the study. |
| Selective reporting (reporting bias) | Unclear risk | Outcomes have not been clearly defined to make a judgement. |
| Other bias | Unclear risk | Insufficient information to make a judgement. |
Yagi 1990.
| Methods | Allocation: randomised, blocks of 4.
Blindness: double, described and adequate.
Duration: 8 weeks preceded by 2 weeks pre‐entry period.
Design: parallel, multicentre study.
Raters: not reported for primary outcome, additional videotapes rated independently by blinded raters. Setting: 97% long‐term inpatients, Japan. |
|
| Participants | Diagnosis: schizophrenia (90%), other (10%). History: TD > 3 months, stable during 2 weeks pre‐entry period, mean duration of TD for 97% > 1 year, for 80% > 3 years, mean duration of antipsychotic drugs 63% > 5 years. N = 60. Sex: 27 M, 33 F. Age: range 30‐79 years. | |
| Interventions | 1. Meclofenoxate hydrochloride (MF): dose 900 mg/day. N = 31. 2. Placebo. N = 29. Antipsychotics stable, antidepressants, minor tranquillisers, antiparkinsonian drugs were used but doses stable. | |
| Outcomes | TD symptoms: AIMS, FGIR.
Global improvement: GUR.
Adverse effects.
Leaving study early. Unable to use ‐ Mental state scores: BPRS (hypochondriasis item scored only, not all participants assessed). |
|
| Notes | For blood test no differences between MF and placebo groups. According to Overall Safety Rating MF caused no severe adverse effects, as did not placebo. Assistance with translation provided by Prof Toshiaki Furukawa, Nagoya, Japan. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "randomized" No further information |
| Allocation concealment (selection bias) | Unclear risk | randomised in blocks of 4 |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | "double blind" not described |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | "double blind", details on blinding not reported for primary outcome |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | All subjects completed the trial |
| Selective reporting (reporting bias) | Unclear risk | Outcomes are reported but not with all the necessary information |
| Other bias | Unclear risk | Insufficient information to make a judgement. |
Scales: AIMS = Abnormal Involuntary Movement Scale BAS = Barnes Akathisia Scale BPRS = Brief Psychiatric Rating Scale CGI = Clinical Global Impressions DSM IV = Diagnostic and Statistical Manual, 4th edition ESRS = Extrapyramidal Symptom Rating Scale HAM‐D = Hamilton Rating Scale for Depression MIBS = Missouri In‐Patient Behavior Scale MMSE = Mini‐Mental State Examination SAS = Simpson Angus Scale SRTDRS = Self‐Report Tardive Dyskinesia Rating Scale STDRS = Simpson (Rockland) Tardive Dyskinesia Rating Scale TAKE = Target Abnormal Kinetic Effects
Other abbreviations: ANCOVA = Analysis of covariance CPE = Chlorpromazine equivalent ECG = Electrocardiogram GI = gastrointestinal ITT = intention‐to‐treat OBS = Organic Brain Syndrome PC = Phosphatidylcholine SD = standard deviation TD = Tardive dyskinesia
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Anderson 1982 | Allocation: publication does not specify if trial was randomised; authors contacted to confirm lack of additional data. |
| Bartels 1981 | Allocation: not randomised. |
| Branchey 1979 | Allocation: no mention of randomisation; authors contacted twice, no reply. |
| Caroff 2001 | Allocation: not randomised. |
| Casey 1975 | Allocation: not randomised, case study. |
| Casey 1977 | Allocation: not randomised, ABAB design. |
| Casey 1979 | Allocation: not randomised, clinical trial. |
| Chien 1978 | Allocation: randomised. Participants: people with TD. Intervention: sodium valproate versus oxypertine versus deanol. Outcomes: unable to extract data from first cross‐over phase (TD improvement, AIMS, Leaving the study early); unable to identify up‐to‐date study author contact details. |
| Crane 1975 | Allocation: not randomised, case series. |
| Curran 1975 | Allocation: not randomised, case study. |
| Davis 1975 | Allocation: not randomised, case study. |
| Davis 1976 | Allocation: not randomised, cohort study, AB(A). |
| Davis 1977 | Allocation: not randomised, AB design. |
| Davis 1978 | Allocation: not randomised, cohort study, AB. |
| De Silva 1975 | Allocation: not randomised, case reports. |
| Domino 1985 | Allocation: randomised, cross‐over. Participants: people with TD (not all had mental illness). Intervention: phosphatidylcholine (lecithin) versus placebo. Outcomes: AIMS, Physician´s Global Impression of Patient´s Mental Illness, Nurse´s Global Impression of Patient´s Mental Illness, ESS and mouth movements frequency count, plasma and RBC choline concentration; unable to extract results from the first segment before cross‐over; author contacted to confirm lack of additional data. |
| Escobar 1975 | Allocation: not randomised, case studies. |
| Fann 1974 | Allocation: not randomised, clinical trial. |
| Fann 1975 | Allocation: not randomised, cohort study. |
| Fann 1976 | Allocation: not randomised, case series. |
| Gelenberg 1979 | Allocation: not randomised, cohort study. |
| Gelenberg 1989 | Allocation: randomised. Participants: persistent TD (research criteria). Schizophrenia, schizoaffective disorder; bipolar disorder; major depression with psychotic features; attention deficit disorder and atypical psychosis. Interventions: CDP‐Choline versus placebo. Outcomes: no outcome data has been provided for the first period before cross‐over; author contacted ‐ no additional information received. |
| Granacher 1975 | Allocation: not randomised, case series. |
| Growdon 1977 | Allocation: not randomised. |
| Hanus 1993 | Allocation: not randomised, open clinical study. |
| Ingram 1983 | Allocation: not randomised, open clinical study. |
| Izumi 1986 | Allocation: not randomised, open‐study. |
| Joe 1985 | Allocation: randomised. Participants: people with chronic schizophrenia diagnosed by DSM III who had taken antipsychotic drugs for at least 3 months, abnormal involuntary body movement in at least one part of body (face, lip and perioral, jaw, tongue, upper extremity, lower extremity, trunk (neck, shoulder, hips)) rated at least 2 point, who has no other kind of neurological disease which may cause the abnormal involuntary movement. Interventions: Lecithin versus placebo. Outcomes: no outcome data provided for first period before cross‐over; author contacted ‐ no additional information received. |
| Jus 1978 | Allocation: randomised, cross‐over. Participants: people with TD. Interventions: deanol versus lithium carbonate versus placebo. Outcomes: AIMS, TD symptom rating scale, CGI, BPRS, NOSIE, vital signs, lab values; impossible to extract data from segment before cross‐over; authors contacted to confirm lack of additional data. |
| Klawans 1974 | Allocation: not randomised, case series. |
| Kumar 1976 | Allocation: not randomised, case study. |
| Laterre 1975 | Allocation: not randomised, case study. |
| Lieberman 1988 | Allocation: randomised. Participants: people with TD. Interventions: physostigmine vs bromocriptine vs benztropine vs haloperidol. Outcomes: no outcome data provided for first period before cross‐over; study author contacted ‐ no additional information received. |
| Lonowski 1979 | Allocation: not randomised, controlled clinical trial. |
| Marsalek 1994 | Allocation: not randomised, open‐trial. |
| Marsalek 1997 | Allocation: randomised. Participants: people with TD (17 schizophrenia, 5 schizoaffective disorder and 1 atypical psychosis). Interventions: 7‐methoxytacrine (7‐MEOTA) vs placebo. Outcomes: therapeutic efficacy and adverse events ‐ no usable data from this brief report; unable to identify up‐to‐date contact details of authors. |
| Mehta 1976 | Allocation: not randomised, case reports. |
| Moore 1980 | Allocation: randomised. Participants: people with TD. Interventions: methscopolamine i.m.+ physostigmine i.v. versus saline i.m. + benztropine i.v. all received deanol thereafter. |
| Nasrallah 1984 | Allocation: not randomised, cohort study, ABA. |
| Nasrallah 1986 | Allocation: randomised. Participants: schizophrenia, paranoid disorder, and schizoaffective disorder + persistent TD Interventions: AMPT vs L‐DOPA vs choline chloride vs valproic acid vs hydroxytryptophan. Outcomes: no outcome data provided for first period before cross‐over; author contacted ‐ no additional information received. |
| Noring 1984 | Allocation: not randomised, controlled single‐dose trial. |
| Penovich 1978 | Allocation: randomised, cross‐over. Participants: people with TD. Interventions: deanol versus placebo. Outcomes: locally developed TD severity scale; impossible to extract results from before cross‐over; author contacted to confirm lack of additional data. |
| Perez Cruet 1981 | Allocation: randomised, cross‐over. Participants: chronic psychiatric disorders; severe persistent TD of more than six months. Interventions: lecithin versus placebo. Outcomes: no outcome data reported for first treatment phase before cross‐over; authors contacted but no new information received. |
| Ray 1982 | Allocation: not randomised, case series. |
| Rektor 1988 | Allocation: not randomised. |
| Simpson 1977 | Allocation: randomised. Participants: antipsychotic medication for some participants abruptly stopped 4 weeks before start of trial. |
| Tamminga 1977 | Allocation: not randomised, ABA design. |
| Volavka 1986 | Allocation: not randomised. |
| Zapletalek 1989 | Allocation: not randomised, open‐study. |
Abbreviations: AIMS = Abnormal Involuntary Movement Scale BPRS = Brief Psychiatric Rating Scale CGI = Clinical Global Impressions DSM IV = Diagnostic and Statistical Manual, 4th edition ESS = Emergent Symptom Scale (adverse effects) i.m. = intramuscular i.v. = intravenous NOSIE = Nurses´ Observation Scale for Inpatient Evaluation RBC = Red blood cell TD = Tardive dyskinesia
Differences between protocol and review
The protocol as published with this review has evolved over time. The revisions of protocol are in line with the development of RevMan and in keeping with Cochrane guidance. We think the revisions have greatly improved and enhanced this review. We do not think, however, that it has materially affected our conduct of the review or interpretation of the results.
There was a substantial update to the protocol in the 2017 review update. The biggest changes to affect the review were to:
broaden the inclusion criteria, and adding the comparison 'Cholinergic medication versus any other intervention for the treatment of tardive dyskinesia';
change the title from 'Cholinergic medication for neuroleptic‐induced tardive dyskinesia' to 'Cholinergic medication for antipsychotic‐induced tardive dyskinesia';
update the list of outcomes following consultation with consumers; and
add a 'Summary of findings' table.
Previous methods are reproduced in Appendix 1.
Contributions of authors
Irina Tammenmaa‐ Aho ‐ searching for trials, evaluating trials, data extraction, analysis, writing of final report (2002).
Rosie Asher ‐ study screening, study selection, data extraction (2017).
Karla Soares‐Weiser ‐ protocol development, author of first version of review (2002 ‐ 2017).
Hanna Bergman ‐ study selection, data extraction and assimilation, summary of findings, report writing (2017 update).
Sources of support
Internal sources
Queensland Health, Australia.
CAPES ‐ Ministry of Education, Brazil.
-
Clinic of Psychiatry, Helsinki University Central Hospital (HUCS), Finland.
At the time of writing the review, employed Irina Tammenmaa as a resident physician.
-
Enhance Reviews Ltd., UK.
Logistics support for Hanna Bergman.
External sources
Universidade Federal de Sao Paulo, Brazil.
FinOHTA, STAKES, Finland.
-
NIHR HTA Project Grant, reference number: 14/27/02, UK.
Salary support for Hanna Bergman. Support for patient involvement. Support for traceable data database.
Declarations of interest
Irina Tammenmaa‐Aho ‐ None.
Karla Soares‐Weiser ‐ is the Deputy Editor‐in‐Chief for Cochrane and Cochrane Innovations. When the NHIR HTA programme grant was awarded that included to update this review, Karla was the Managing Director of Enhance Reviews Ltd.
Hanna Bergman ‐ worked for Enhance Reviews Ltd. during preparation of this review and was paid for her contribution to this review. Enhance Reviews Ltd. is a private company that performs systematic reviews of literature. HB works for Cochrane Response, an evidence consultancy that takes commissions from healthcare guideline developers and policy makers.
Rosie Asher ‐ worked for Enhance Reviews Ltd. during preparation of this review and was paid for her contribution to this review.
New search for studies and content updated (no change to conclusions)
References
References to studies included in this review
Beckham 1981 {published data only}
- Beckham BJ. Lecithin therapy for tardive dyskinesia [dissertation]. Denton, Texas: North Texas State University, 1981. [Google Scholar]
Caroff 2007 {unpublished data only}
- Caroff SN, Waljer P, Campbell C, Lorry A, Petro C, Lynch K, et al. Treatment of tardive dyskinesia with galantamine: a randomized controlled crossover trial. Journal of Clinical Psychiatry 2007;68(3):410‐5. [DOI] [PubMed] [Google Scholar]
- NCT00164242. Treatment of tardive dyskinesia with galantamine. www.ClinicalTrials.gov 2005.
de Montigny 1979 {published data only}
- Montigny C, Chouinard G, Annable L. Ineffectiveness of deanol in tardive dyskinesia: a placebo controlled study. Psychopharmacology 1979;65:219‐23. [DOI] [PubMed] [Google Scholar]
Gelenberg 1990 {published and unpublished data}
- Gelenberg AJ, Dorer DJ, Wojcik JD, Falk WE, Brotman AW, Leahy L. A crossover study of lecithin treatment of tardive dyskinesia. Journal of Clinical Psychiatry 1990;51(4):149‐53. [PubMed] [Google Scholar]
George 1981 {published data only}
- George J, Pridmore S, Aldous D. Double blind controlled trial of deanol in tardive dyskinesia. Australian and New Zealand Journal of Psychiatry 1981;15:68‐71. [DOI] [PubMed] [Google Scholar]
Jackson 1978 {published data only}
- Jackson IV. Cholinergic enhancement in tardive dyskinesia. Current Therapeutic Research 1978;24(6):725‐33. [Google Scholar]
Jackson 1979 {published data only}
- Jackson IV, Davis LG, Cohen RK, Nuttall EA. Lecithin administration in tardive dyskinesia: clinical and biomedical correlates. Biological Psychiatry 1981;16(1):85‐90. [PubMed] [Google Scholar]
- Jackson IV, Nuttall EA, Ibe IO, Perez‐Cruet J. Treatment of tardive dyskinesia with lecithin. American Journal of Psychiatry 1979;136(11):1458‐60. [DOI] [PubMed] [Google Scholar]
Jahanian 2014 {published data only}
- Jahanian AA, Rezaei O, Fadai F, Yaraghchi A. The effectiveness of rivastigmine in reducing tardive dyskinesia symptoms in patients with schizophrenia. Iranian Journal of Psychiatry and Clinical Psychology 2014;20:29‐34. [Google Scholar]
Kocher 1980 {published data only}
- Kocher R, Hobi V, Linder M, Studer K. Therapy with dimethylaminoethanol (Deanol) in late dyskinesias induced by neuroleptics [Zur Therapie mit Dimethylaminoäthanol (Deanol) bei neuroleptikainduzierten Spätdyskinesien]. Schweizer Archiv fur Neurologie, Neurochirurgie und Psychiatrie 1980;126(1):103‐9. [PubMed] [Google Scholar]
Lucius 1976 {published data only}
- Bockenheimer S, Lucius G. Deanol in tardive dyskinesia: a double‐blind study (author's transl) [Zur Therapie mit Dimethylaminoäthanol (Deanol) bei neuroleptikainduzierten extrapyramidalen Hyperkinesen]. Archiv fur Psychiatrie und Nervenkrankheiten 1976;222(1):69‐75. [DOI] [PubMed] [Google Scholar]
- Lucius G. Uber die therapeutische Wirksamkeit von Dimethylaminoaethanol bei neuroleptikainduzierten Späthyperkinesen [dissertation]. Freiburg im Breisgau, Deutschland: Der Albert‐Ludwigs‐Universität Freiburg im Breisgau, 1978. [Google Scholar]
Ogunmefun 2009 {published data only}
- Ogunmefun A, Hasnain M, Alam A, Osuala T, Regenold WT. Effect of donepezil on tardive dyskinesia. Journal of Clinical Psychopharmacology 2009;29(1):102‐4. [DOI] [PubMed] [Google Scholar]
Price 1982 {published and unpublished data}
- Price LA. Lecithin treatment for tardive dyskinesia: a clinical evaluation [dissertation]. Denton, Texas: North Texas State University, 1982. [Google Scholar]
Tarsy 1977 {published data only}
- Tarsy D, Bralower M. Deanol acetamidobenzoate treatment in choreiform movement disorders. Archives of Neurology 1977;34:756‐8. [DOI] [PubMed] [Google Scholar]
Yagi 1990 {published data only}
- Ojima Y, Tsubaki M, Yagi G, Kamishima K, Miura S. Experimental design and analysis for determination of improvement rating by video imaging ‐ A double‐blind placebo‐controlled study for meclofenoxate hydrochloride (Lucidril) in tardive dyskinesia. Rinsho Hyoka (Clinical Evaluation) 1991;19(2):267‐76. [Google Scholar]
- Yagi G, Kamishima K, Miura S. Meclofenoxate hydrochloride (Lucidril) in tardive dyskinesia ‐ A double‐blind placebo‐controlled study. Rinsho Hyoka (Clinical Evaluation) 1990;18(3):455‐79. [Google Scholar]
- Yagi G, Kamizima K, Miura S. Meclofenoxate (lucidril) in tardive dyskinesia – a double‐blind placebo‐controlled study. Proceedings of the 17th Collegium Internationale Neuro‐Psychopharmacologicum Congress; 1990 Sep 10‐14; Kyoto, Japan:303.
References to studies excluded from this review
Anderson 1982 {published data only (unpublished sought but not used)}
- Anderson BG, Reker D, Ristich M, Friedman E, Banay‐Schwartz M, Volavka J. Lecithin treatment of tardive dyskinesia ‐ a progress report. Psychopharmacology Bulletin 1982;18(1):87‐8. [Google Scholar]
- Yackulic CF, Anderson BG, Reker D, Webb E, Volavka J. The safety of lecithin diet supplementation in schizophrenic patients. Biological Psychiatry 1982;17(12):1445‐8. [PubMed] [Google Scholar]
Bartels 1981 {published data only}
- Bartels M, Mezger G, Schmalzing G, Schonle PW. Long‐term treatment of tardive dyskinesia with lecithin. Proceedings of the Symposium der Arbeitsgemeinschaft für Neuropsychopharmakologie und Pharmakopsychiatrie. Nuernberg, Germany, 1981.
Branchey 1979 {published data only}
- Branchey MH, Branchey LB, Bark NM, Richardson MA. Lecithin in the treatment of tardive dyskinesia. Communications in Psychopharmacology 1979;3:303‐7. [PubMed] [Google Scholar]
Caroff 2001 {published data only}
- Caroff SN, Campbell EC, Havey J, Sullivan KA, Mann SC, Gallop R. Treatment of tardive dyskinesia with donepezil: A pilot study. Journal of Clinical Psychiatry 2001;62(10):772‐5. [DOI] [PubMed] [Google Scholar]
- Caroff SN, Campbell EC, Havey JC, Sullivan KA, Katz IR, Mann SC. Treatment of tardive dyskinesia with donepezil [letter]. Journal of Clinical Psychiatry 2001;62(2):128‐9. [DOI] [PubMed] [Google Scholar]
Casey 1975 {published data only}
- Casey DE, Denney D. Deanol in the treatment of tardive dyskinesia. American Journal of Psychiatry 1975;132(8):864‐7. [DOI] [PubMed] [Google Scholar]
Casey 1977 {published data only}
- Casey DE, Denney D. Pharmacological characterization of tardive dyskinesia. Psychopharmacology 1977;54:1‐8. [DOI] [PubMed] [Google Scholar]
Casey 1979 {published data only}
- Casey DE. Mood alterations during deanol therapy. Psychopharmacology 1979;62:187‐91. [DOI] [PubMed] [Google Scholar]
Chien 1978 {published data only}
- Chien CP, Jung K, Ross‐Townsend A. Efficacies of agents related to GABA, dopamine and acetylcholine in the treatment of tardive dyskinesia. Psychopharmacology Bulletin 1978;14(2):20‐2. [PubMed] [Google Scholar]
Crane 1975 {published data only}
- Crane GE. Deanol for tardive dyskinesia. New England Journal of Medicine 1975;292:926. [DOI] [PubMed] [Google Scholar]
Curran 1975 {published data only}
- Curran DJ, Nagaswami S, Mohan KJ. Treatment of phenothiazine induced bulbar persistent dyskinesia with deanol acetamidobenzoate. Diseases of the Nervous System 1975;36:71‐3. [PubMed] [Google Scholar]
Davis 1975 {published data only}
- Davis KL, Berger PA, Hollister LE. Choline for tardive dyskinesia. New England Journal of Medicine 1975;293:152. [DOI] [PubMed] [Google Scholar]
Davis 1976 {published data only}
- Davis KL, Hollister LE, Barchas JD, Berger PA. Choline in tardive dyskinesia and Huntington's disease. Life Sciences 1976;19(10):1507‐15. [DOI] [PubMed] [Google Scholar]
Davis 1977 {published data only}
- Davis KL, Berger PA, Hollister LE. Deanol in tardive dyskinesia. American Journal of Psychiatry 1977;134(7):807. [DOI] [PubMed] [Google Scholar]
Davis 1978 {published data only}
- Davis KL, Berger PA. Pharmacological investigations of the cholinergic imbalance hypotheses of movement disorders and psychosis. Biological Psychiatry 1978;13(1):23‐49. [PubMed] [Google Scholar]
De Silva 1975 {published data only}
- Silva L, Huang CY. Deanol in tardive dyskinesia. British Medical Journal 1975;3(5981):466. [DOI] [PMC free article] [PubMed] [Google Scholar]
Domino 1985 {published data only (unpublished sought but not used)}
- Domino EF, May WW, Demetriou S, Mathews B, Tait S, Kovacic B. Lack of clinically significant improvement of patients with tardive dyskinesia following phosphatidylcholine therapy. Biological Psychiatry 1985;20:1189‐96. [DOI] [PubMed] [Google Scholar]
Escobar 1975 {published data only}
- Escobar JI, Kemp KF. Dimethylaminoethanol for tardive dyskinesia. New England Journal of Medicine 1975;292:317‐8. [DOI] [PubMed] [Google Scholar]
Fann 1974 {published data only}
- Fann WE, Lake CR, Gerber CJ, McKenzie GM. Cholinergic suppression of tardive dyskinesia. Psychopharmacologia (Berlin) 1974;37:101‐7. [DOI] [PubMed] [Google Scholar]
Fann 1975 {published data only}
- Fann WE, Sullivan JL, Miller RD, McKenzie GM. Deanol in tardive dyskinesia: a preliminary report. Psychopharmacologia (Berlin) 1975;42:135‐7. [DOI] [PubMed] [Google Scholar]
Fann 1976 {published data only}
- Fann WE, Stafford JR, Thornby JI, Richman BW. Chronic deanol administration in tardive dyskinesia. Clinical Pharmacology and Therapeutics 1976;19:106. [Google Scholar]
Gelenberg 1979 {published data only}
- Gelenberg AJ, Doller‐Wojcik JC, Growdon JH. Choline and lecithin in the treatment of tardive dyskinesia: preliminary results from a pilot study. American Journal of Psychiatry 1979;136(6):772‐6. [DOI] [PubMed] [Google Scholar]
Gelenberg 1989 {published and unpublished data}
- Gelenberg AJ, Wojcik J, Falk WE, Bellinghausen B, Joseph AB. CDP‐Choline for the treatment of tardive dyskinesia: A small negative series. Comprehensive Psychiatry 1989;30(1):1‐4. [DOI] [PubMed] [Google Scholar]
Granacher 1975 {published data only}
- Granacher RP, Baldessarini RJ, Cole JO. Deanol for tardive dyskinesia. New England Journal of Medicine 1975;292:926‐7. [DOI] [PubMed] [Google Scholar]
Growdon 1977 {published data only (unpublished sought but not used)}
- Growdon JH. Effects of choline on tardive dyskinesia and other movement disorders. Psychopharmacology Bulletin 1978;14(4):55‐6. [PubMed] [Google Scholar]
- Growdon JH, Hirsch MJ, Wurtman RJ, Wiener W. Oral choline administration to patients with tardive dyskinesia. New England Journal of Medicine 1977;297(10):524‐7. [DOI] [PubMed] [Google Scholar]
- Wurtman RJ, Growdon JH. Dietary enhancement of CNS neurotransmitters. Hospital Practice 1978;13:71‐7. [DOI] [PubMed] [Google Scholar]
Hanus 1993 {published data only}
- Hanus H, Tuma I, Fusek J, Patocka J. Treatment of tardive dyskinesias with 7‐metoxytacrine ‐ II [Lecba tardivnich dyskinez 7‐metoxytakrinem ‐ II]. Sbornik vedeckych praci Lekarske fakulty Karlovy univerzity v Hradci Kralove. Supplementum. 1993;36(1‐2):47‐53. [PubMed] [Google Scholar]
Ingram 1983 {published data only}
- Ingram NAW, Newgreen DB. The use of tacrine for tardive dyskinesia. American Journal of Psychiatry 1983;140(12):1629‐31. [DOI] [PubMed] [Google Scholar]
Izumi 1986 {published data only}
- Izumi K, Tominaga H, Koja T, Nomoto M, Shimizu T, Sonoda H, et al. Meclofenoxate therapy in tardive dyskinesia: A preliminary report. Biological Psychiatry 1986;21:151‐60. [DOI] [PubMed] [Google Scholar]
Joe 1985 {published data only}
- Joe SH, Suh KY, Lee BY. Effect of lecithin on tardive dyskinesia. Korea University Medical Journal 1985;22(3):197‐206. [Google Scholar]
Jus 1978 {published data only (unpublished sought but not used)}
- Jus A, Villeneuve A, Gautier J, Jus K, Villeneuve C, Pires P, et al. Deanol, lithium and placebo in the treatment of tardive dyskinesia: A double‐blind crossover study. Neuropsychobiology 1978;4:140‐9. [DOI] [PubMed] [Google Scholar]
Klawans 1974 {published data only}
- Klawans HL, Rubovits R. Effect of cholinergic and anticholinergic agents on tardive dyskinesia. Journal of Neurology, Neurosurgery, and Psychiatry 1974;27:941‐7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Kumar 1976 {published data only}
- Kumar BB. Treatment of tardive dyskinesia with deanol. American Journal of Psychiatry 1976;133(8):978. [Google Scholar]
Laterre 1975 {published data only}
- Laterre EC, Fortemps E. Deanol in spontaneous and induced dyskinesias. Lancet 1975;1(7919):1301. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Lieberman 1988 {published data only}
- Lieberman J, Pollack S, Lesser M, Kane J. Pharmacologic characterization of tardive dyskinesia. Journal of Clinical Psychopharmacology 1988;8(4):254‐60. [PubMed] [Google Scholar]
Lonowski 1979 {published data only}
- Lonowski DJ, Sterling FE, King HA. Electromyographic assessment of dimethylaminoethanol (deanol) in treatment of tardive dyskinesia. Psychological Reports 1979;45:415‐9. [DOI] [PubMed] [Google Scholar]
Marsalek 1994 {published data only}
- Marsalek M, Filip V, Praskova H, Karen P. An open trial with 7‐methoxytacrine in tardive dyskinesia. European Neuropsychopharmacology 1994;4(3, Special issue):369. [Google Scholar]
Marsalek 1997 {published data only}
- Marsalek M, Filip V, Petrovsky M, Klar I, Filipova M, Klaschka J. 7‐MEOTA in the treatment of tardive dyskinesia. Double‐blind placebo controlled study. Homeostasis ‐PRAHA‐ 39th, Psychopharmacological meeting, Jesenik, Spa; Czech Republi 1997;38(1):7. [Google Scholar]
Mehta 1976 {published data only}
- Mehta D, Mehta S, Mathew P. Failure of deanol in treating tardive dyskinesia. American Journal of Psychiatry 1976;133(12):1467. [DOI] [PubMed] [Google Scholar]
Moore 1980 {published data only}
- Moore DC, Bowers MB. Identification of a subgroup of tardive dyskinesia patients by pharmacologic probes. American Journal of Psychiatry 1980;137(10):1202‐5. [DOI] [PubMed] [Google Scholar]
Nasrallah 1984 {published data only}
- Nasrallah HA, Dunner FJ, Smith RE, McCalley‐Whitters M, Sherman AD. Variable clinical response to choline in tardive dyskinesia. Psychological Medicine 1984;14:697‐700. [DOI] [PubMed] [Google Scholar]
Nasrallah 1986 {published data only}
- Nasrallah HA, Dunner FJ, McCalley‐Whitters M, Smith RE. Pharmacologic probes of neurotransmitter systems in tardive dyskinesia: Implications for clinical management. Journal of Clinical Psychiatry 1986;47(2):56‐9. [PubMed] [Google Scholar]
Noring 1984 {published data only}
- Noring U, Juul Povlsen U, Casey DE, Gerlach J. Effect of a cholinomimetic drug (RS 86) in tardive dyskinesia and drug‐related parkinsonism. Psychopharmacology 1984;84:569‐71. [DOI] [PubMed] [Google Scholar]
Penovich 1978 {published data only (unpublished sought but not used)}
- Penovich P, Morgan JP, Kerzner B, Karch F, Goldblatt D. Double‐blind evaluation of deanol in tardive dyskinesia. JAMA 1978;239(19):1997‐8. [PubMed] [Google Scholar]
Perez Cruet 1981 {published data only}
- Perez‐Cruet J, Menendez I, Alvarez‐Ghersi J, Falcon JR, Valderrabano O, Castro‐Urrutia EC, et al. Double‐blind study of lecithin in the treatment of persistent tardive dyskinesia. Boletin Asociacion Medica Puerto Rico 1981;73(11):531‐7. [PubMed] [Google Scholar]
Ray 1982 {published data only}
- Ray R, Ramakrishnan N, Rao BSS. Oral choline in tardive dyskinesia. Indian Journal of Medical Research 1982;76:628‐31. [PubMed] [Google Scholar]
Rektor 1988 {published data only}
- Rektor J. Cholinergic system in the pathophysiology of tardive dyskinesias [Cholinergni system v patofyziologii tardivnich dyskinezi]. Ceskoslovenska Psychiatrie 1988;84(5):289‐96. [PubMed] [Google Scholar]
Simpson 1977 {published data only}
- Simpson GM, Voitashevsky A, Young MA, Lee JH. Deanol in the treatment of tardive dyskinesia. Psychopharmacology 1977;52:257‐61. [DOI] [PubMed] [Google Scholar]
Tamminga 1977 {published data only}
- Tamminga CA, Smith RC, Ericksen SE, Chang S, Davis JM. Cholinergic influences in tardive dyskinesia. American Journal of Psychiatry 1977;134(7):769‐74. [DOI] [PubMed] [Google Scholar]
Volavka 1986 {published data only}
- Volavka J, O'Donnell J, Muragali R, Anderson BG, Gaztanaga P, Boggiano W, et al. Lithium and lecithin in tardive dyskinesia: an update. Psychiatry Research 1986;19:101‐4. [DOI] [PubMed] [Google Scholar]
Zapletalek 1989 {published data only}
- Zapletalek M, Hanus H, Fusek J, Hrdina V. First experience with the application of 7‐methoxytacrine to psychiatric patients. Activitas Nervosa Superior 1989;31(4):305‐6. [PubMed] [Google Scholar]
Additional references
Alabed 2011
- Alabed S, Latifeh Y, Mohammad HA, Rifai A. Gamma‐aminobutyric acid agonists for neuroleptic‐induced tardive dyskinesia. Cochrane Database of Systematic Reviews 2011, Issue 4. [DOI: 10.1002/14651858.CD000203.pub3] [DOI] [PubMed] [Google Scholar]
Alphs 1983
- Alphs LD, Davis JM. Cholinergic treatments for tardive dyskinesia. Modern Problems in Pharmacopsychiatry 1983;21:168‐86. [DOI] [PubMed] [Google Scholar]
Altman 1996
- Altman DG, Bland JM. Detecting skewness from summary information. BMJ 1996;313(7066):1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
Andreassen 2000
- Andreassen OA, Jorgensen HA. Neurotoxicity associated with neuroleptic‐induced oral dyskinesias in rats. Implications for tardive dyskinesia?. Progress in Neurobiology 2000;61(5):525‐41. [DOI] [PubMed] [Google Scholar]
Andreassen 2001
- Andreassen OA, Meshul CK, Moore C, Jorgensen HA. Oral dyskinesias and morphological changes in rat striatum during long‐term haloperidol administration. Psychopharmacology (Berlin) 2001;157(1):11‐9. [DOI] [PubMed] [Google Scholar]
APA 1992
- American Psychiatric Association. Tardive dyskinesia: a task force report of the American Psychiatric Association. Washington DC: American Psychiatric Association, 1992. [Google Scholar]
Armitage 1991
- Armitage P. Should we cross off the crossover?. Journal of Clinical Pharmacology 1991;32:1‐2. [DOI] [PMC free article] [PubMed] [Google Scholar]
Ascher‐Svanum 2008
- Ascher‐Svanum H, Zhu B, Faries D, Peng X, Kinon BJ, Tohen M. Tardive dyskinesia and the 3‐year course of schizophrenia: results from a large, prospective, naturalistic study. Journal of Clinical Psychiatry 2008;69:1580‐8. [DOI] [PubMed] [Google Scholar]
Ballesteros 2000
- Ballesteros J, Gonzalez‐Pinto A, Bulbena A. Tardive dyskinesia associated with higher mortality in psychiatric patients: results of a meta‐analysis of seven independent studies. Journal of Clinical Psychopharmacology 2000;20(2):188‐94. [DOI] [PubMed] [Google Scholar]
Barnes 1993
- Barnes TRE, Edwards JG. The side‐effects of antipsychotic drugs. I. CNS and neuromuscular effects. In: Barnes TRE editor(s). Antipsychotic Drugs and their Side‐effects. London: Harcourt Brace & Company, 1993. [Google Scholar]
Bergen 1989
- Bergen JA, Eyland EA, Campbell JA. The course of tardive dyskinesia in patients on long‐term neuroleptics. British Journal of Psychiatry 1989;154:523‐8. [DOI] [PubMed] [Google Scholar]
Bergman 2017
- Bergman H, Walker DM, Nikolakopoulou A, Soares‐Weiser K, Adams CE. Systematic review of interventions for treating or preventing antipsychotic‐induced tardive dyskinesia. Health Technol Assess 2017 Aug;21(43):1‐218. [DOI] [PMC free article] [PubMed] [Google Scholar]
Bhoopathi 2006
- Bhoopathi PS, Soares‐Weiser K. Benzodiazepines for neuroleptic‐induced tardive dyskinesia. Cochrane Database of Systematic Reviews 2006, Issue 3. [DOI: 10.1002/14651858.CD000205.pub2] [DOI] [PubMed] [Google Scholar]
Birks 2006a
- Birks JS. Cholinesterase inhibitors for Alzheimer's disease. Cochrane Database of Systematic Reviews 2006, Issue 1. [DOI: 10.1002/14651858.CD005593] [DOI] [PMC free article] [PubMed] [Google Scholar]
Birks 2006b
- Birks J, Harvey RJ. Donepezil for dementia due to Alzheimer's disease. Cochrane Database of Systematic Reviews 2006, Issue 1. [DOI: 10.1002/14651858.CD001190.pub2] [DOI] [PubMed] [Google Scholar]
Birks 2015
- Birks JS, Chong LY, Grimley Evans J. Rivastigmine for Alzheimer's disease. Cochrane Database of Systematic Reviews 2015, Issue 9. [DOI: 10.1002/14651858.CD001191.pub4] [DOI] [PMC free article] [PubMed] [Google Scholar]
Bland 1997
- Bland JM. Statistics notes. Trials randomised in clusters. BMJ 1997;315:600. [DOI] [PMC free article] [PubMed] [Google Scholar]
Boissel 1999
- Boissel JP, Cucherat M, Li W, Chatellier G, Gueyffier F, Buyse M, et al. The problem of therapeutic efficacy indices. 3. Comparison of the indices and their use [Apercu sur la problematique des indices d'efficacite therapeutique, 3: comparaison des indices et utilisation. Groupe d'Etude des Indices D'efficacite]. Therapie 1999;54(4):405‐11. [PUBMED: 10667106] [PubMed] [Google Scholar]
Cadet 1989
- Cadet JL, Lohr JB. Possible involvement of free radical in neuroleptic‐induced movement disorders. Annals of the New York Academy of Sciences 1989;570:176‐85. [DOI] [PubMed] [Google Scholar]
Casey 1995
- Casey DE. Tardive dyskinesia: pathophysiology. In: Bloom FE, Kupfer DJ editor(s). Psychopharmacology. The Fourth Generation of Progress. New York: Raven Press, 1995. [Google Scholar]
Casey 1999
- Casey DE. Tardive dyskinesia and atypical antipsychotic drugs. Schizophrenia Research 1999;35:S31‐S36. [DOI] [PubMed] [Google Scholar]
Cavallaro 1993
- Cavallaro R, Regazzetti MG, Mundo E, Brancato V, Smeraldi E. Tardive dyskinesia outcomes: clinical and pharmacologic correlates of remission and persistence. Neuropsychopharmacology 1993;8(3):233‐9. [DOI] [PubMed] [Google Scholar]
Chong 2009
- Chong SA, Tay JA, Subramaniam M, Pek E, Machin D. Mortality rates among patients with schizophrenia and tardive dyskinesia. Journal of Clinical Psychopharmacology 2009;29:5‐8. [DOI] [PubMed] [Google Scholar]
Chouinard 2008
- Chouinard G, Chouinard VA. Atypical antipsychotics: CATIE study, drug‐induced movement disorder and resulting iatrogenic psychiatric‐like symptoms, supersensitivity rebound psychosis and withdrawal discontinuation syndromes. Psychotherapy and Psychosomatics 2008;77(2):69‐77. [DOI] [PubMed] [Google Scholar]
Clarke 2001
- Clarke M, Oxman AD, editors. Cochrane Reviewers´ Handbook 4.1.4 [updated October 2001]. Cochrane Database of Systematic Reviews. Oxford, England: Update Software, 2001, issue 4. [MEDLINE: ]
Cloud 2014
- Cloud LJ, Zutshi D, Factor SA. Tardive dyskinesia: therapeutic options for an increasingly common disorder. Neurotherapeutics 2014;11(1):166‐76. [DOI] [PMC free article] [PubMed] [Google Scholar]
Correll 2004
- Correll CU, Leucht S, Kane JM. Lower risk for tardive dyskinesia associated with second‐generation antipsychotics: a systematicreview of 1‐year studies. American Journal of Psychiatry 2004;161(3):414‐25. [DOI] [PubMed] [Google Scholar]
Deeks 2000
- Deeks J. Issues in the selection for meta‐analyses of binary data. Proceedings of the 8th International Cochrane Colloquium; 2000 Oct 25‐28; Cape Town. Cape Town: The Cochrane Collaboration, 2000.
Divine 1992
- Divine GW, Brown JT, Frazier LM. The unit of analysis error in studies about physicians' patient care behavior. Journal of General Internal Medicine 1992;7(6):623‐9. [DOI] [PubMed] [Google Scholar]
Donner 2002
- Donner A, Klar N. Issues in the meta‐analysis of cluster randomized trials. Statistics in Medicine 2002;21:2971‐80. [DOI] [PubMed] [Google Scholar]
Egger 1997
- Egger M, Davey Smith G, Schneider M, Minder C. Bias in meta‐analysis detected by a simple, graphical test. BMJ 1997;315:629‐34. [DOI] [PMC free article] [PubMed] [Google Scholar]
El‐Sayeh 2006
- El‐Sayeh HG, Lyra da Silva JP, Rathbone J, Soares‐Weiser K. Non‐neuroleptic catecholaminergic drugs for neuroleptic‐induced tardive dyskinesia. Cochrane Database of Systematic Reviews 2006, Issue 1. [DOI: 10.1002/14651858.CD000458.pub2] [DOI] [PubMed] [Google Scholar]
Elbourne 2002
- Elbourne D, Altman DG, Higgins JPT, Curtina F, Worthingtond HV, Vaile A. Meta‐analyses involving cross‐over trials: methodological issues. International Journal of Epidemiology 2002;31(1):140‐9. [DOI] [PubMed] [Google Scholar]
Essali 2011
- Essali A, Deirawan H, Soares‐Weiser K, Adams CE. Calcium channel blockers for neuroleptic‐induced tardive dyskinesia. Cochrane Database of Systematic Reviews 2011, Issue 11. [DOI: 10.1002/14651858.CD000206.pub3] [DOI] [PubMed] [Google Scholar]
Fernandez 2001
- Fernandez HH, Krupp B, Friedman JH. The course of tardive dyskinesia and parkinsonism in psychiatric inpatients: 14‐year follow‐up. Neurology 2001;56:805‐7. [DOI] [PubMed] [Google Scholar]
Fleiss 1984
- Fleiss JL. The crossover study. The Design and Analysis of Clinical Experiments. Chichester: John Wiley & Sons, 1984. [Google Scholar]
Furukawa 2006
- Furukawa TA, Barbui C, Cipriani A, Brambilla P, Watanabe N. Imputing missing standard deviations in meta‐analyses can provide accurate results. Journal of Clinical Epidemiology 2006;59(7):7‐10. [DOI] [PubMed] [Google Scholar]
Gerlach 1988
- Gerlach J, Casey DE. Tardive dyskinesia. Acta Psychiatrica Scandinavica 1988;77:369‐78. [DOI] [PubMed] [Google Scholar]
Glazer 1990
- Glazer WM, Morgenstern H, Schooler N, Berkman CS, Moore DC. Predictors of improvement in tardive dyskinesia following discontinuation of neuroleptic medication. British Journal of Psychiatry 1990;157:585‐92. [DOI] [PubMed] [Google Scholar]
Glazer 2000
- Glazer WM. Review of incidence studies of tardive dyskinesia associated with typical antipsychotics. Journal of Clinical Psychiatry 2000;61(suppl 4):15‐20. [PubMed] [Google Scholar]
Grimm 2001
- Grimm JW, Chapman MA, Zahm DS, See RE. Decreased choline acetyltransferase immunoreactivity in discrete striatal subregions following chronic haloperidol in rats. Synapse 2001;39(1):51‐7. [DOI] [PubMed] [Google Scholar]
Gulliford 1999
- Gulliford MC. Components of variance and intraclass correlations for the design of community‐based surveys and intervention studies: data from the Health Survey for England 1994. American Journal of Epidemiology 1999;149:876‐83. [DOI] [PubMed] [Google Scholar]
Guy 1976a
- Guy W. Abnormal Involuntary Movement Scale (AIMS). In: Guy W editor(s). ECDEU Assessment Manual for Psychopharmacology. US Dept Health, Education and Welfare publication (ADM) 76‐338. Rockville, MD: National Institute of Mental Health, 1976:534‐7. [Google Scholar]
Guy 1976b
- Guy W. Clinical Global Impressions (CGI). In: Guy W editor(s). ECDEU Assessment Manual for Psychopharmacology. US Dept Health, Education and Welfare publication (ADM) 76‐338. Rockville, MD: National Institute of Mental Health, 1976:217‐22. [Google Scholar]
Higgins 2003
- Higgins JP, Thompson SG, Deeks JJ, Altman DG. Measuring inconsistency in meta‐analyses. BMJ 2003;327:557‐60. [DOI] [PMC free article] [PubMed] [Google Scholar]
Higgins 2011
- Higgins JPT, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.0.2 [updated September 2011]. The Cochrane Collaboration, 2011. Available from www.cochrane‐handbook.org..
Jadad 1996
- Jadad A, Moore A, Carroll D, Jenkinson C, Reynolds DJM, Gavanagh DJ, et al. Assessing the quality of reports of randomized clinical trials: Is blinding necessary?. Controlled Clinical Trials 1996;17:1‐12. [DOI] [PubMed] [Google Scholar]
Jeste 2000
- Jeste DV. Tardive dyskinesia in older patients. Journal of Clinical Psychiatry 2000;61(suppl 4):27‐32. [PubMed] [Google Scholar]
Kane 1982
- Kane JM, Smith JM. Tardive dyskinesia: prevalence and risk factors, 1959 to 1979. Archives of General Psychiatry 1982;39:473‐81. [DOI] [PubMed] [Google Scholar]
Kay 1986
- Kay SR, Opler LA, Fiszbein A. Positive and Negative Syndrome Scale (PANSS) Manual. North Tonawanda, NY: Multi‐Health Systems, 1986. [Google Scholar]
Leon 2006
- Leon AC, Mallinckrodt CH, Chuang‐Stein C, Archibald DG, Archer GE, Chartier K. Attrition in randomized controlled clinical trials: methodological issues in psychopharmacology. Biological Psychiatry 2006;59(11):1001‐5. [PUBMED: 16905632] [DOI] [PubMed] [Google Scholar]
Leucht 2005
- Leucht S, Kane JM, Kissling W, Hamann J, Etschel E, Engel RR. What does the PANSS mean?. Schizophrenia Research 2005;79(2‐3):231‐8. [PUBMED: 15982856] [DOI] [PubMed] [Google Scholar]
Leucht 2005a
- Leucht S, Kane JM, Kissling W, Hamann J, Etschel E, Engel R. Clinical implications of brief psychiatric rating scale scores. British Journal of Psychiatry 2005;187:366‐71. [PUBMED: 16199797] [DOI] [PubMed] [Google Scholar]
Leucht 2009
- Leucht S, Kissling W, Davis JM. Second‐generation antipsychotics for schizophrenia: can we resolve the conflict?. Psychological Medicine 2009;39(10):1591‐602. [DOI] [PubMed] [Google Scholar]
Li 2015
- Li Y, Hai S, Zhou Y, Dong BR. Cholinesterase inhibitors for rarer dementias associated with neurological conditions. Cochrane Database of Systematic Reviews 2015, Issue 3. [DOI: 10.1002/14651858.CD009444] [DOI] [PMC free article] [PubMed] [Google Scholar]
Lieberman 1996
- Lieberman JA, Fleishhacker W. Introduction. British Journal of Psychiatry 1996;168(Supplement 29):7‐8. [Google Scholar]
Loy 2006
- Loy C, Schneider L. Galantamine for Alzheimer's disease and mild cognitive impairment. Cochrane Database of Systematic Reviews 2006, Issue 1. [DOI: 10.1002/14651858.CD001747.pub3] [DOI] [PMC free article] [PubMed] [Google Scholar]
Maher 2012
- Maher AR, Theodore G. Summary of the comparative effectiveness review on off‐label use of atypical antipsychotics. Journal of Managed Care Pharmacy 2012;18(5 Suppl B):S1‐20. [DOI] [PMC free article] [PubMed] [Google Scholar]
Maidment 2006
- Maidment I, Fox C, Boustani M. Cholinesterase inhibitors for Parkinson's disease dementia. Cochrane Database of Systematic Reviews 2006, Issue 1. [DOI: 10.1002/14651858.CD004747.pub2] [DOI] [PubMed] [Google Scholar]
Marshall 2000
- Marshall M, Lockwood A, Bradley C, Adams C, Joy C, Fenton M. Unpublished rating scales: a major source of bias in randomized controlled trials of treatments for schizophrenia. British Journal of Psychiatry 2000;176:249‐52. [DOI] [PubMed] [Google Scholar]
Martins 2011
- Martins ES, Rosso A, Coutinho E, Adams C, Huf G. Prevalence of tardive dyskinesia and all‐cause mortality amongst patients in a large psychiatirc institute in Rio de Janeiro. Revista de Psiquiatria Clínica 2011;38:44. [Google Scholar]
Miller 2007
- Miller DD, Eudicone JM, Pikalov A, Kim E. Comparative assessment of the incidence and severity of tardive dyskinesia in patients receiving aripiprazole or haloperidol for the treatment of schizophrenia: a post hoc analysis. Journal of Clinical Psychiatry 2007;68(12):1901‐6. [DOI] [PubMed] [Google Scholar]
Miller 2008
- Miller DD, Caroff SN, Davis SM, Rosenheck RA, McEvoy JP, Saltz BL, et al. Extrapyramidal side‐effects of antipsychotics in a randomised trial. Brtish Journal of Psychiatry 2008;193(4):279‐88. [DOI] [PMC free article] [PubMed] [Google Scholar]
Moher 2001
- Moher D, Schulz KF, Altman D. The CONSORT statement: revised recommendations for improving the quality of reports of parallel‐group randomized trials. JAMA 2001;285:1987‐91. [DOI] [PubMed] [Google Scholar]
NICE 2014
- NICE. Psychosis and schizophrenia in adults: treatment and management. NICE clinical guideline 178 (guidance.nice.org.uk/cg178) 2014.
Overall 1962
- Overall JE, Gorham DR. The brief psychiatric rating scale. Psychological Reports 1962;10:799‐812. [Google Scholar]
Pocock 1983
- Pocock SJ. Crossover trials. Clinical trials. A Practical Approach. Chichester: John Wiley & Sons, 1983. [Google Scholar]
Rolinski 2012
- Rolinski M, Fox C, Maidment I, McShane R. Cholinesterase inhibitors for dementia with Lewy bodies, Parkinson's disease dementia and cognitive impairment in Parkinson's disease. Cochrane Database of Systematic Reviews 2012, Issue 3. [DOI: 10.1002/14651858.CD006504.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Rosenheck 2007
- Rosenheck RA. Evaluating the cost‐effectiveness of reduced tardive dyskinesia with second‐generation antipsychotics. British Journal of Psychiatry : the journal of mental science 2007;191:238‐45. [DOI] [PubMed] [Google Scholar]
Schooler 1993
- Schooler NR, Keith SJ. Clinical research for the treatment of schizophrenia. Psychopharmacology Bulletin 1993;29:431‐46. [PubMed] [Google Scholar]
Schulz 1995
- Schulz KF, Chalmers I, Hayes RJ, Altman DG. Empirical evidence of bias: dimensions of methodological quality associated with estimates of treatment effects in controlled trials. JAMA 1995;273:408‐12. [DOI] [PubMed] [Google Scholar]
Schünemann 2008
- Schünemann HJ, Oxman AD, Vist GE, Higgins JPT, Deeks JJ, Glasziou P, et al. Chapter 12: Interpreting results and drawing conclusions. In: Higgins JPT, Green S editor(s). Cochrane Handbook for Systematic Reviews of Interventions. The Cochrane Collaboration, 2008:359‐83. [Google Scholar]
Simpson 1979
- Simpson GM, Lee JH, Zoubok B, Gardos G. A rating scale for tardive dyskinesia. Psychopharmacology 1979;64:171‐9. [DOI] [PubMed] [Google Scholar]
Smith 1980
- Smith JM, Balessarini RJ. Changes in prevalence, severity and recovery in tardive dyskinesia with age. Archives of General Psychiatry 1980;37:1368‐73. [DOI] [PubMed] [Google Scholar]
Soares‐Weiser 1997
- Soares‐Weiser K, Mobsy C, Holliday E. Anticholinergic medication for neuroleptic‐induced tardive dyskinesia. Cochrane Database of Systematic Reviews 1997, Issue 2. [DOI: 10.1002/14651858.CD000204] [DOI] [PubMed] [Google Scholar]
Soares‐Weiser 2003
- Soares‐Weiser K, Joy C. Miscellaneous treatments for neuroleptic‐induced tardive dyskinesia. Cochrane Database of Systematic Reviews 2003, Issue 2. [DOI: 10.1002/14651858.CD000208] [DOI] [PubMed] [Google Scholar]
Soares‐Weiser 2006
- Soares‐Weiser K, Rathbone J. Neuroleptic reduction and/or cessation and neuroleptics as specific treatments for tardive dyskinesia. Cochrane Database of Systematic Reviews 2006, Issue 1. [DOI: 10.1002/14651858.CD000459.pub2] [DOI] [PubMed] [Google Scholar]
Soares‐Weiser 2011
- Soares‐Weiser K, Maayan N, McGrath J. Vitamin E for neuroleptic‐induced tardive dyskinesia. Cochrane Database of Systematic Reviews 2011, Issue 2. [DOI: 10.1002/14651858.CD000209.pub2] [DOI] [PubMed] [Google Scholar]
Tammenmaa 2004
- Tammenmaa IA, Sailas E, McGrath JJ, Soares‐Weiser K, Wahlbeck K. Systematic review of cholinergic drugs for neuroleptic‐induced tardive dyskinesia: a meta‐analysis of randomized controlled trials. Progress in Neuropsychopharmacology & Biological Psychiatry 2004 Nov;28(7):1099‐107. [DOI] [PubMed] [Google Scholar]
Tarsy 2011
- Tarsy D, Lungu C, Baldessarini RJ. Epidemiology of tardive dyskinesia before and during the era of modern antipsychotic drugs. Handbook of Clinical Neurology 2011;100:601‐16. [DOI] [PubMed] [Google Scholar]
Taylor 2009
- Taylor D, Paton C, Kapur S. The Maudsley Prescribing Guidelines (10th Edition). London: Informa Healthcare, 2009. [Google Scholar]
Ukoumunne 1999
- Ukoumunne OC, Gulliford MC, Chinn S, Sterne JAC, Burney PGJ. Methods for evaluating area‐wide and organistation‐based intervention in health and health care: a systematic review. Health Technology Assessment 1999;3(5):1‐75. [PubMed] [Google Scholar]
Woods 2010
- Woods SW, Morgenstern H, Saksa JR, Walsh BC, Sullivan MC, Money R, et al. Incidence of tardive dyskinesia with atypical versus conventional antipsychotic medications: a prospective cohort study. Journal of Clinical Psychiatry 2010;71(4):463‐74. [DOI] [PMC free article] [PubMed] [Google Scholar]
Wurtman 1978
- Wurtman RJ, Growdon JH. Dietary enhancement of CNS neurotransmitters. Hospital Practice 1978 Mar;13(3):71. [DOI] [PubMed] [Google Scholar]
Xia 2009
- Xia J, Adams CE, Bhagat N, Bhagat V, Bhoopathi P, El‐Sayeh H, et al. Loss to outcomes stakeholder survey: the LOSS study. Psychiatric Bulletin 2009;33(7):254‐7. [Google Scholar]
References to other published versions of this review
McGrath 1997
- McGrath JJ, Soares‐Weiser K. Cholinergic medication for neuroleptic‐induced tardive dyskinesia. Cochrane Database of Systematic Reviews 1997, Issue 2. [DOI: 10.1002/14651858.CD000207] [DOI] [PubMed] [Google Scholar]
Soares‐Weiser 1999
- Soares‐Weiser K, McGrath JJ. The treatment of tardive dyskinesia ‐ a systematic review and meta‐analysis. Schizophrenia Research 1999;39(1):1‐16. [DOI] [PubMed] [Google Scholar]
Tammenmaa 2002
- Tammenmaa I, McGrath J, Sailas E, Soares‐Weiser K. Cholinergic medication for neuroleptic‐induced tardive dyskinesia. Cochrane Database of Systematic Reviews 2002, Issue 3. [DOI: 10.1002/14651858.CD000207; MEDLINE: ] [DOI] [PubMed] [Google Scholar]