

Abstract
PURPOSE
To compare the risk of neoplastic progression by germline mutation status versus family history without a known germline mutation (familial risk) among individuals with an increased risk for pancreatic cancer who are undergoing surveillance.
METHODS
Of 464 high-risk individuals in the Cancer of the Pancreas Screening program at Johns Hopkins Hospital who were undergoing pancreatic surveillance, 119 had a known deleterious germline mutation in a pancreatic cancer susceptibility gene; 345 met family history criteria for pancreatic surveillance but were not known to harbor a germline mutation. We used next-generation sequencing to identify previously unrecognized germline mutations among these 345 individuals. We compared the development of pancreatic cancer, high-grade dysplasia, or clinically worrisome features, adjusting for competing mortality, among all germline mutation carriers with the risk of progression in a cohort without a known germline mutation.
RESULTS
Fifteen (4.3%) of 345 individuals classified as having familial risk had a previously unrecognized pancreatic cancer susceptibility gene mutation (nine that involved ATM, two BRCA2, one BRCA1, one PALB2, one TP53, and one CPA1). The cumulative incidence of pancreatic cancer, high-grade dysplasia, or worrisome features on pancreatic imaging was significantly higher in the germline mutation risk group (n = 134) than in the familial risk group (n = 330 [for pancreatic cancer, hazard ratio, 2.85; 95% CI, 1.0 to 8.18; P = .05]).
CONCLUSION
The cumulative incidence of pancreatic cancer is significantly higher among individuals with an identifiable deleterious germline mutation in a pancreatic cancer susceptibility gene than it is among individuals with a strong family history but no identified mutation. Gene testing of individuals who meet criteria for pancreatic surveillance on the basis of their family history may better define those most at risk for neoplastic progression.
INTRODUCTION
Pancreatic ductal adenocarcinoma is the third most common cause of cancer death in the United States, with a 5-year survival of only approximately 8%.1 Most patients with pancreatic cancer are diagnosed at an advanced stage, and patients diagnosed with advanced disease have a 5-year survival rate of less than 5% and a median survival of less than 12 months.2 Early detection of pancreatic cancer and its precursors may be the most effective way of reducing mortality as a result of the disease.3,4 The International Cancer of the Pancreas Screening (CAPS) Consortium5 recommends selective screening for high-risk individuals (HRIs) with an estimated 5% or higher lifetime risk of developing pancreatic cancer. Some patients are candidates for pancreatic surveillance because they carry a deleterious germline mutation in a known familial pancreatic cancer susceptibility gene (including BRCA2, ATM, BRCA1, PALB2, CDKN2A, MLH1, MSH2, MSH6, STK11, PRSS1, and TP53).6-14 Two additional genes that encode for pancreatic enzymes, CPA1 (a known pancreatitis susceptibility gene) and CPB1, also have been implicated as pancreatic cancer susceptibility genes.15 Of these genes, the lifetime risk of developing pancreatic cancer is highest among individuals who carry germline mutations in PRSS1,13 STK11,10 and CDKN2A.16 Despite our improved understanding of the genetic basis for the aggregation of pancreatic cancer in families, inherited gene mutations explain only a small portion (less than 20%) of the familial clustering of pancreatic cancer.17
Most individuals in the CAPS program undergo pancreatic screening on the basis of their strong family history of pancreatic cancer. The estimated risk of developing pancreatic cancer among individuals with a family history increases with the number of affected first-degree relatives (FDRs).18 On the basis of these risks, pancreatic surveillance is recommended for those with at least one FDR and one second-degree relative (SDR) with pancreatic cancer.5
In many programs, surveillance commences at age 50 years for these germline mutation carriers (excluding STK11, CDKN2A, and PRSS1) and at age 55 years for those whose risk is solely on the basis of their family cancer history.18 Knowledge of an individual’s gene mutation status may help to better estimate pancreatic cancer risk. For example, a European study of 411 HRIs detected pancreatic cancer in 13 (7.3%) of 178 CDKN2A mutation carriers compared with three (1.4%) of 214 individuals at risk on the sole basis of their family history.3 This study did not routinely gene test patients in the family history group, so some may have been misclassified.
We and others have reported that deleterious germline mutations in pancreatic cancer susceptibility genes are commonly (5% to 10%) identified in patients with pancreatic cancer.14,19-21 Patients with a family history of pancreatic cancer are approximately twice as likely to have such a germline mutation as those without a family history, although most germline mutation carriers who develop pancreatic cancer do not report a family history of pancreatic cancer and do not have family histories that suggest an inherited cancer syndrome.14,19 Some patients who undergo pancreatic surveillance because of their family history may carry a germline mutation in a pancreatic cancer susceptibility gene. The purpose of the current study was to determine whether individuals with a deleterious germline mutation are at greater risk of progression than individuals with a strong family history of pancreatic cancer but no known germline mutation.
METHODS
Study Design and Patients
This study included 464 HRIs who were prospectively enrolled in the CAPS studies4,22-24 (ClinicalTrials.gov identifiers: NCT00438906, NCT00714701, and NCT02000089) between 1998 and 2017 (Data Supplement) who provided samples for germline DNA analysis. The current CAPS5 study enrolls individuals who meet recommended guidelines for pancreatic surveillance.5 These guidelines recommend surveillance for individuals who meet familial risk criteria (ie, FDRs with pancreatic cancer) from familial pancreatic cancer kindred (ie, two FDRs with pancreatic cancer), in other words, someone who has at least one FDR and one SDR with pancreatic cancer who are at least 55 years old or 10 years younger than the youngest with pancreatic cancer in the family. The enrollment criteria for CAPS4 (2009 to 2013) was similar to CAPS5 (CAPS5 enrollment includes ATM mutation carriers). Earlier CAPS studies required a stronger family history of pancreatic cancer among familial risk individuals for enrollment (having either two FDRs or one FDR and two SDRs with pancreatic cancer), and the age eligibility was younger (40 years for CAPS1 [1998 to 2001] and 50 years for CAPS2 [2001 to 2004] and CAPS3 [2006 to 2009]). For individuals with a germline mutation in a familial pancreatic cancer susceptibility gene, pancreatic surveillance is recommended for those who are at least 30 years old who meet clinical criteria for Peutz-Jeghers syndrome and those with a germline mutation in BRCA2, PALB2, CDKN2A, ATM, BRCA1, MLH1, MSH2, and MSH6 and who are at least 50 years old or 10 years younger than the youngest with pancreatic cancer in the family. Patients with CDKN2A mutations are recommended for pancreatic screening (typically starting at age 40 years) irrespective of family history, whereas for those with BRCA2, ATM, PALB2, and mismatch repair gene mutations, it is recommended if there is a family history of pancreatic cancer in a close blood relative (FDR or SDR). Pancreatic surveillance also is recommended for individuals with hereditary pancreatitis, who are at least 50 years old, or who are 20 years since their first attack of pancreatitis.
A detail description of follow-up and clinical management of HRIs has been published previously.4 Pancreatic surveillance is up to date for most patients in the CAPS program. If pancreatic surveillance is not up to date, follow-up to determine status (alive or not) is determined from the medical record, CAPS study team communications, or updates provided by participation in the National Familial Pancreas Tumor Registry.25 For patients who continued surveillance beyond their baseline visit, follow-up was up to date for 94%; status within the past 2 years was known for 96% (all but 20 patients). The characteristics of these 20 patients were similar to the overall study population (mean ± standard deviation age, 59 ± 10 years, 13 familial, seven germline, seven male). Patients not able to continue surveillance in CAPS are offered referral to a more local screening program. Two patients enrolled in CAPS were diagnosed with pancreatic cancer at their baseline evaluation; both had symptoms attributable to their pancreatic cancer, were not considered to have screen-detected cancer, and were not included in the incidence analysis. The final diagnoses were made by surgical pathology or cytology. All pathologic diagnoses were made by an expert pathologist (R.H.H.) using the WHO classification system,26 including patients who underwent pancreatic resection at another institution. If the pathologic specimen had multiple pancreatic lesions, the highest pathologic grade present was used for end point analysis. Worrisome features found by pancreatic imaging (either main pancreatic duct dilation of 5 to 9 mm, pancreatic cyst size greater than or equal to 3 cm, cyst growth rate greater than or equal to 5 mm diameter/2 years, thickened enhanced cyst walls, nonenhanced mural nodules, abrupt change in main pancreatic duct caliber with distal pancreatic atrophy, or lymphadenopathy) were defined according to revisions of international consensus Fukuoka guidelines for the management of intraductal papillary mucinous neoplasm of the pancreas.27 This study was approved by the Johns Hopkins institutional review board, and written informed consent was provided from all enrolled patients.
DNA Extraction
Genomic DNA was extracted from either peripheral blood mononuclear cells or frozen normal tissue from pancreatic resection specimens (duodenum, spleen, or pancreas) as previously described.19
Next-Generation Sequencing
Known pancreatic cancer susceptibility genes (BRCA2, ATM, PALB2, BRCA1, CDKN2A, MLH1, MSH2, PRSS1, STK11, TP53, CPA1, and CPB1), the cancer susceptibility genes MSH6 and BUB1B (Appendix Table A1, online only), and two candidate genes CEL and CTRB215 were sequenced using an AmpliSeq (Thermo Fisher Scientific, Waltham, MA) custom panel. Next-generation sequencing was performed with 540 chips (Ion S5 System; Thermo Fisher Scientific) or with P1v3 chips (Ion Proton; Thermo Fisher Scientific), according to manufacturer’s protocols as previously described.28,29 Eighteen DNA samples yielded very low read coverage and were excluded as uninformative.
All candidate potentially deleterious variants were validated by Sanger sequencing, (primers listed in Appendix Table A2, online only), which was performed at the Johns Hopkins Genetic Resources Core Facility. Variant prevalence information was obtained from public databases, including ExAC Browser by the Exome Aggregation Consortium30 and Exome Variant Server by the National Heart, Lung, and Blood Institute Exome Sequencing Project.31 ClinVar was used to classify the variants and rare nonsynonymous variants. Variants of uncertain significance (VUSs) in CPA1 and CPB1 were evaluated functionally as previously described15 (Data Supplement).
Statistical Analysis
The characteristics of CAPS participants at baseline were summarized and compared between those with and without germline mutations. Differences between groups were evaluated using Fisher’s exact test for categorical variables and t tests for continuous measures. Time to pancreatic cancer diagnosis; pancreatic cancer or high-grade dysplasia diagnosis; detection of worrisome features; and a composite outcome of first occurrence of pancreatic cancer, high-grade dysplasia, or worrisome features were calculated as time from CAPS enrollment to diagnosis/detection date. Those without the outcome were censored at the date of last follow-up. Patients with worrisome features or pancreatic cancer at baseline were excluded from the respective time-to-event analyses. Cumulative incidence of time-to-event outcomes were estimated using Fine and Gray’s method, which accounted for death as a competing event.32 Differences in time-to-event outcomes between those with and without a germline mutation were estimated using proportional subdistribution hazards models that adjusted for age at baseline CAPS enrollment and sex. Competing mortality was calculated using the date of last contact or date and cause of death. The cumulative probabilities of a diagnosis of pancreatic cancer, pancreatic cancer or high-grade dysplasia, and detection of worrisome features on pancreatic imaging at ages 60, 65, 70, 75, and 80 years were calculated using age of diagnosis or age at last follow-up. All participants were included in these calculations. The t test (one-sided) was used to determine whether BiP levels were elevated in variant-transfected cells versus wild-type–transfected cells. Statistical analysis and graphic presentations were performed using R version 3.4.2 (www.R-project.org) and JMP 13 software (SAS Institute, Cary, NC). P < .05 were considered statistically significant.
RESULTS
Identification of Previously Unrecognized Germline Mutations in Pancreatic Cancer Susceptibility Genes
Of 345 HRIs under surveillance initially on the basis of their family history of pancreatic cancer, 15 (4.3%) were found to have a deleterious germline mutation in a known pancreatic cancer susceptibility gene by sequencing analysis (Table 1). These 15 patients included two with the Ashkenazi Jewish founder BRCA2 mutation (C.5946delT), one with the Ashkenazi Jewish founder BRCA1 mutation (C.68_69delAG), two with PALB2 mutations, nine with an ATM mutation, and one with a TP53 mutation. Sixty-six VUSs also were identified (Appendix Table A3, online only). Only definitively deleterious germline changes were considered significant in this study.
TABLE 1.
Identification of Previously Unrecognized Germline Mutations in Pancreatic Cancer Susceptibility Genes

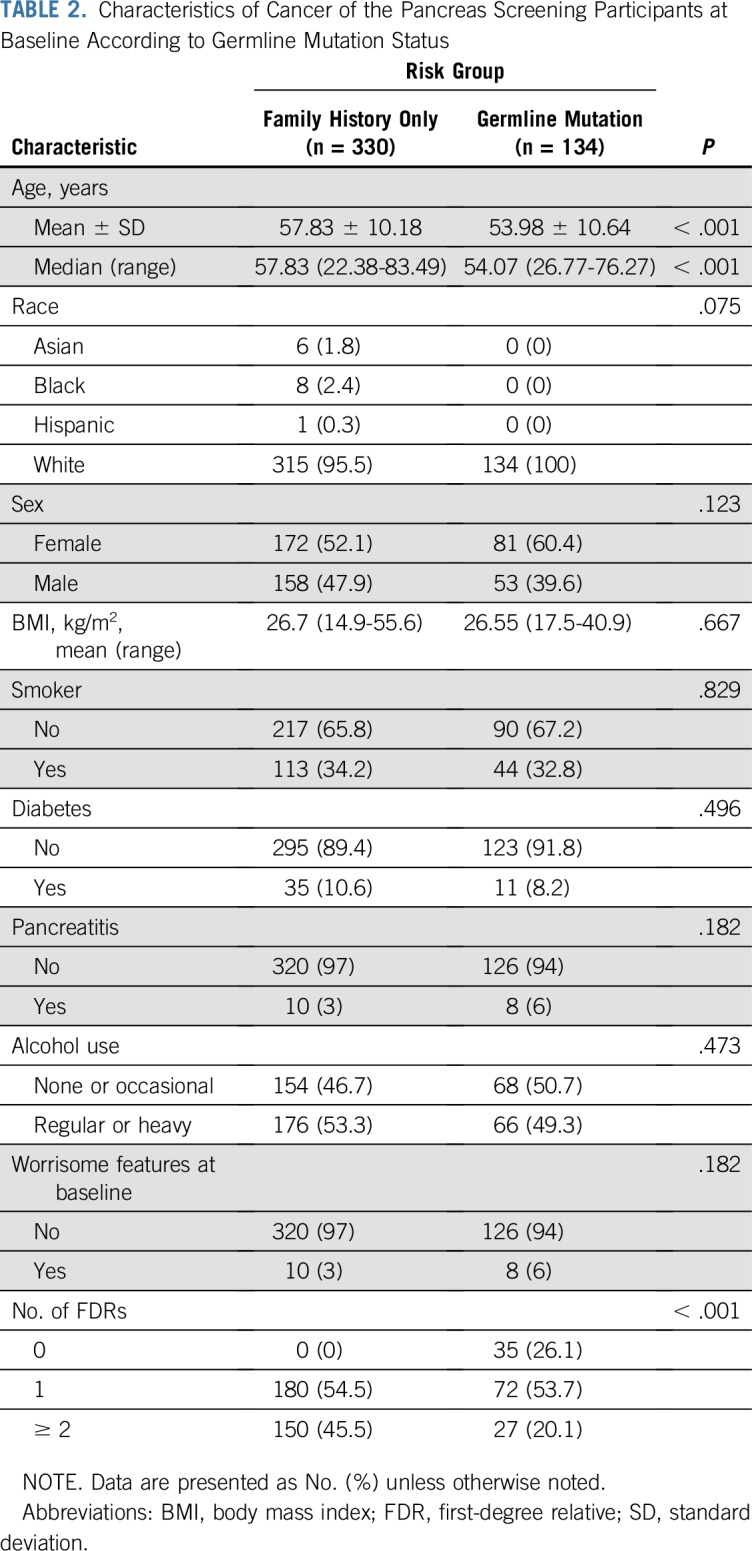
After gene testing the cohort, individuals were reclassified into those with a known deleterious germline mutation and those without (Data Supplement). Overall, 330 individuals were classified as having risk on the sole basis of their family history (familial risk group), and 134 were classified as having a deleterious germline mutation (Appendix Table A4, online only). Characteristics of both groups are listed in Table 2, and more detailed information about each mutation carrier is listed in Appendix Table A5 (online only). At the time of their baseline screening, individuals with a germline mutation were significantly younger than those in the familial risk group (mean ± standard deviation, 54.0 ± 10.6 v 57.9 ± 10.2 years of age, respectively; P < .001). The median follow-up period of pancreas surveillance in the germline mutation group and the familial risk group was similar (2.4 v 3.2 years, respectively; P = .21). The familial risk group was more likely than the germline mutation group to have multiple FDRs with pancreatic cancer (46% v 20% with two or more FDRs; P < .001). There were other no significant differences in the characteristics of the two groups.
TABLE 2.
Characteristics of Cancer of the Pancreas Screening Participants at Baseline According to Germline Mutation Status
Neoplastic Progression in the Familial Risk and Germline Mutation Groups
Estimates of time to a pancreatic cancer diagnosis, time to a diagnosis of pancreatic cancer or high-grade dysplasia, and time to the detection of worrisome features on imaging stratified by germline mutation status are listed in Table 3. The cumulative incidence of pancreatic cancer in the germline mutation group was higher than in the familial risk group, adjusted for age and sex and accounting for death as a competing event (hazard ratio [HR], 2.85; 95% CI, 1.0 to 8.18; P = .05; Fig 1). The likelihood of developing pancreatic cancer or high-grade dysplasia was also significantly higher in the germline mutation group than in the familial risk group (HR, 2.81; 95% CI, 1.17 to 6.76; P = .02; Data Supplement). Similar results also were found when the presence of clinically worrisome features in the pancreas was included as an end point (HR, 3.13; 95% CI, 1.71 to 5.6; P < .001; Data Supplement). Within the germline mutation group, BRCA2 mutation carriers were significantly more likely than familial risk individuals to have neoplastic progression (to worrisome features, high-grade dysplasia, or pancreatic [HR, 2.44; P = .05]), as were BRCA1 mutation carriers (HR, 7.49; P < .001) but not PALB2 or ATM mutation carriers (Appendix Table A6, online only), although with the small number of carriers of these latter mutations, the power to detect any differences was limited. Estimates of the cumulative incidence of being diagnosed with pancreatic cancer, high-grade dysplasia, and worrisome features by ages 60, 65, 70, 75, and 80 years are listed in Table 4.
TABLE 3.
Estimates of Time to PDAC, PDAC or HGD, and Detection of Worrisome Features According to Germline Mutation Status

FIG 1.

Estimates of time to pancreatic cancer diagnosis according to germline mutation status. CAPS, Cancer of the Pancreas Screening; HR, hazard ratio; PDAC, pancreatic ductal adenocarcinoma.
TABLE 4.
Estimates of Cumulative Incidence of PDAC, PDAC or HGD, and Detection of Worrisome Features by Age, According to Germline Mutation Status

Characteristics of HRIs who developed pancreatic cancer or high-grade dysplasia are listed in Table 5. Four of six individuals in the germline mutation group who developed pancreatic cancer during surveillance had unresectable disease at diagnosis, and in three of these individuals, the germline mutation was identified after their pancreatic cancer diagnosis. One individual of particular interest was a 60-year-old female with two FDRs with pancreatic cancer who had a missense variant (c.640G>T; p.D214Y) in CPA1 that was classified as deleterious after functional analysis. When expressed in HEK293 cells, this variant produced a protein with impaired secretion that induced endoplasmic reticulum stress15 (Appendix Table A7, online only; Data Supplement). This patient did not have a clinical history of pancreatitis, but her endoscopic ultrasound showed many features of chronic pancreatitis (seven of nine endoscopic ultrasound features). She also had other features suggestive of pancreatitis, including a 4.7-mm main pancreatic duct dilation in the neck of her pancreas and a 10-mm cyst in the tail (Data Supplement). Four other patients had a VUS in CPA1 or CPB1 (one in CPA1 and three in CPB1). These variants were tested as previously described15 for their effect on secretion of their protein product and endoplasmic reticulum stress); all four variants produced normal function and so were classified as benign (Appendix Table A7).
TABLE 5.
Characteristics of High-Risk Individuals With PDAC or HGD

DISCUSSION
We find that the risk of neoplastic progression within the pancreas is higher among individuals with a known deleterious germline mutation than in individuals with a strong family history alone. Furthermore, the five patients diagnosed with pancreatic cancer or a precursor lesion with high-grade dysplasia before the age of 55 years were all germline mutation carriers. These latter results support the recommendations of the International CAPS Consortium consensus that pancreatic surveillance of individuals who carry a deleterious germline mutation should start at an earlier age than those under surveillance for their pancreatic cancer family history.5 The earlier age of enrollment for germline mutation carriers than for familial risk individuals would not be expected to create a bias in the determination of outcome between the two groups because recommendations for ongoing follow-up were the same for both groups, each individual’s surveillance intervals are determined by the nature of any abnormalities detected during surveillance.
Our results indicate that germline mutation status can be used to help to predict the risk of neoplastic progression and raise the question of whether patients eligible for pancreatic surveillance on the basis of current clinical guidelines should all undergo germline gene testing. National Comprehensive Cancer Network guidelines for gene testing patients with newly diagnosed pancreatic cancer have been revised recently on the basis of recent studies that found the prevalence of germline mutations in patients with pancreatic cancer to be significant (5% or higher) and that family history of pancreatic or other cancers is not a reliable indicator of which individuals harbor these mutations.33 Furthermore, relatives of patients with pancreatic cancer can be identified as having a germline mutation after cascade testing of close blood relatives of these germline mutation carriers, and these individuals in turn may benefit from early detection screening. Our results give some guidance to health care providers who are considering offering panel gene testing to their patients with a family history of pancreatic cancer who meet family history criteria for pancreatic surveillance.
Although our results indicate that germline mutation carriers have a higher risk of neoplastic progression than those with familial risk alone, the risk of neoplastic progression was still significant among those with familial risk alone (approximately 16% of individuals in the familial risk group had progressed to pancreatic cancer by age 80 years; Table 4). Thus, we would recommend that individuals who meet family history criteria who do not have an identifiable germline mutation after gene testing still undergo pancreatic surveillance.
The study has some limitations. First, although overall pancreatic cancer risk may be higher among germline mutation carriers than among those with familial risk, this risk is not similarly elevated for all germline mutation carriers; some genes confer a higher risk when mutated than others, and some mutations are more deleterious than others, and variants in other genes can modify disease risk. Although pancreatic cancer risk associated with germline mutation has been estimated for several genes, larger cohorts of mutation carriers are needed to better determine this risk. Second, our study population may not be representative of all populations who meet eligibility criteria for pancreatic screening. As gene testing becomes more widespread, more individuals are expected to be identified as having deleterious germline variants associated with pancreatic cancer risk. Pancreatic cancer risk may not necessarily be as high in the wider population of patients who harbor relevant germline alterations without a significant family history of pancreatic cancer.
In conclusion, among a cohort of individuals undergoing pancreatic surveillance, the cumulative incidence of pancreatic cancer and high-grade dysplasia is significantly higher in individuals who carry a deleterious germline mutation than in individuals with a strong family history but without an identifiable germline mutation. The findings provide better risk stratification and improved clinical decision making with regard to recommendations for pancreatic and other cancer surveillance.
ACKNOWLEDGMENT
We thank all Cancer of the Pancreas Screening study participants.
Appendix
TABLE A1.
Target Regions of the 16 Genes

TABLE A2.
Oligonucleotides Used for Polymerase Chain Reaction Amplification for Sanger Sequencing

TABLE A3.
VUSs

TABLE A4.
Known Germline Mutations in Pancreatic Cancer Susceptibility Genes

TABLE A5.
Category of High-Risk Individuals for Pancreatic Ductal Adenocarcinoma
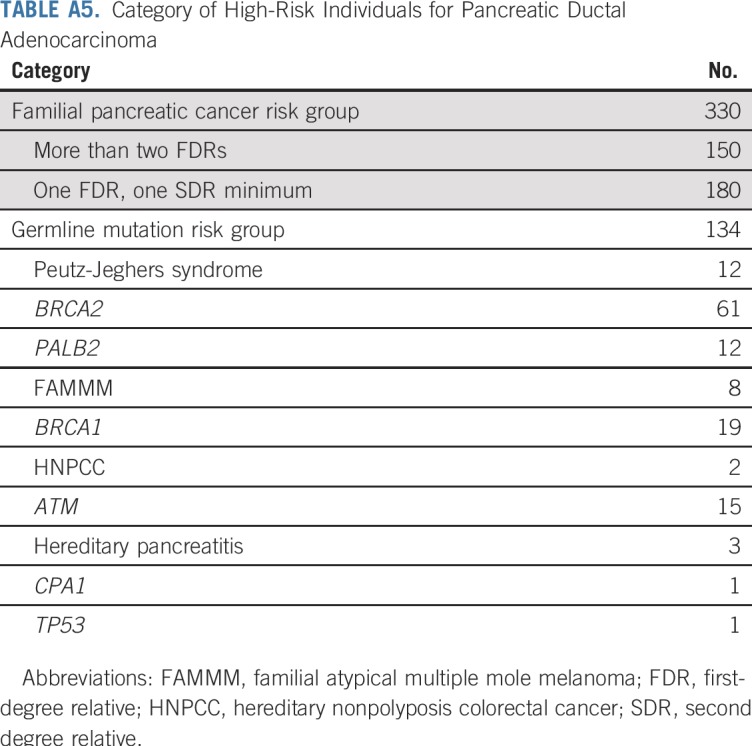
TABLE A6.
Estimates of Time to PDAC, PDAC or HGD, or Detection of Worrisome Features According to Germline Mutation Status

TABLE A7.
CPA1 and CPB1 Variants

Footnotes
Supported by National Institutes of Health Grant No. U01210170, R01CA176828, and CA62924; Susan Wojcicki and Dennis Troper; the Pancreatic Cancer Action Network; Stand Up to Cancer; the V Foundation for Cancer Research; and the Rolfe Pancreatic Cancer Foundation. M.G. is the Sol Goldman Professor of Pancreatic Cancer Research.
AUTHOR CONTRIBUTIONS
Conception and design: Toshiya Abe, Marcia I. Canto, Michael Goggins
Financial support: Alison P. Klein, Michael Goggins
Administrative support: Michael Goggins
Provision of study material or patients: Alison P. Klein, Michael Goggins
Collection and assembly of data: Toshiya Abe, Koji Tamura, Madeline Ford, Patrick McCormick, Miguel Chuidian, Jose Alejandro Almario, Michael Borges, Anne Marie Lennon, Eun Ji Shin, Alison P. Klein, Marcia I. Canto, Michael Goggins
Data analysis and interpretation: Toshiya Abe, Amanda L. Blackford, Koji Tamura, Michael Borges, Eun Ji Shin, Alison P. Klein, Ralph H. Hruban, Michael Goggins
Manuscript writing: All authors
Final approval of manuscript: All authors
Accountable for all aspects of the work: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
Deleterious Germline Mutations Are a Risk Factor for Neoplastic Progression Among High-Risk Individuals Undergoing Pancreatic Surveillance
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or ascopubs.org/jco/site/ifc.
Miguel Chuidian
Research Funding: C2 Therapeutics (I), PENTAX Medical (I)
Travel, Accommodations, Expenses: EndoGastric Solutions (I)
Eun Ji Shin
Consulting or Advisory Role: Boston Scientific, Medtronic, C2 Therapeutics
Alison P. Klein
Consulting or Advisory Role: OptumInsight
Ralph H. Hruban
Leadership: miDIAGNOSTICS
Research Funding: Applied Materials
Marcia I. Canto
Research Funding: C2 Therapeutics, Cosmo Pharmaceuticals
Patents, Royalties, Other Intellectual Property: Royalties from UpToDate online
Michael Goggins
Patents, Royalties, Other Intellectual Property: Royalty related to licensing as a codiscoverer of PALB2 as a pancreatic cancer susceptibility gene to Myriad Genetics
No other potential conflicts of interest were reported.
REFERENCES
- 1.Rahib L, Smith BD, Aizenberg R, et al. Projecting cancer incidence and deaths to 2030: The unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res. 2014;74:2913–2921. doi: 10.1158/0008-5472.CAN-14-0155. [DOI] [PubMed] [Google Scholar]
- 2.Vincent A, Herman J, Schulick R, et al. Pancreatic cancer. Lancet. 2011;378:607–620. doi: 10.1016/S0140-6736(10)62307-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vasen H, Ibrahim I, Ponce CG, et al. Benefit of surveillance for pancreatic cancer in high-risk individuals: Outcome of long-term prospective follow-up studies from three European expert centers. J Clin Oncol. 2016;34:2010–2019. doi: 10.1200/JCO.2015.64.0730. [DOI] [PubMed] [Google Scholar]
- 4. doi: 10.1053/j.gastro.2018.05.035. Canto MI, Almario JA, Schulick RD, et al: Risk of neoplastic progression in high-risk individuals undergoing long-term surveillance for pancreatic cancer. Gastroenterology 155:740-751, 2018 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. doi: 10.1136/gutjnl-2012-303108. Canto MI, Harinck F, Hruban RH, et al: International Cancer of the Pancreas Screening (CAP) Consortium summit on the management of patients with increased risk for familial pancreatic cancer. Gut 62:339-347, 2013 [Erratum: Gut 63:1978, 2014] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Goggins M, Schutte M, Lu J, et al. Germline BRCA2 gene mutations in patients with apparently sporadic pancreatic carcinomas. Cancer Res. 1996;56:5360–5364. [PubMed] [Google Scholar]
- 7.Roberts NJ, Jiao Y, Yu J, et al. ATM mutations in patients with hereditary pancreatic cancer. Cancer Discov. 2012;2:41–46. doi: 10.1158/2159-8290.CD-11-0194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kastrinos F, Mukherjee B, Tayob N, et al. Risk of pancreatic cancer in families with Lynch syndrome. JAMA. 2009;302:1790–1795. doi: 10.1001/jama.2009.1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jones S, Hruban RH, Kamiyama M, et al. Exomic sequencing identifies PALB2 as a pancreatic cancer susceptibility gene. Science. 2009;324:217. doi: 10.1126/science.1171202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Giardiello FM, Brensinger JD, Tersmette AC, et al. Very high risk of cancer in familial Peutz-Jeghers syndrome. Gastroenterology. 2000;119:1447–1453. doi: 10.1053/gast.2000.20228. [DOI] [PubMed] [Google Scholar]
- 11.Lowenfels AB, Maisonneuve P, DiMagno EP, et al. Hereditary pancreatitis and the risk of pancreatic cancer. J Natl Cancer Inst. 1997;89:442–446. doi: 10.1093/jnci/89.6.442. [DOI] [PubMed] [Google Scholar]
- 12. doi: 10.1038/gim.2014.153. Zhen DB, Rabe KG, Gallinger S, et al: BRCA1, BRCA2, PALB2, and CDKN2A mutations in familial pancreatic cancer: A PACGENE study. Genet Med 17:569-577, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rebours V, Boutron-Ruault MC, Schnee M, et al. Risk of pancreatic adenocarcinoma in patients with hereditary pancreatitis: A national exhaustive series. Am J Gastroenterol. 2008;103:111–119. doi: 10.1111/j.1572-0241.2007.01597.x. [DOI] [PubMed] [Google Scholar]
- 14.Hu C, Hart SN, Polley EC, et al. Association between inherited germline mutations in cancer predisposition genes and risk of pancreatic cancer. JAMA. 2018;319:2401–2409. doi: 10.1001/jama.2018.6228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Tamura K, Yu J, Hata T, et al: Mutations in the pancreatic secretory enzymes CPA1 and CPB1 are associated with pancreatic cancer. Proc Natl Acad Sci U S A 115:4767-4772, 2018. [DOI] [PMC free article] [PubMed]
- 16.Vasen HF, Gruis NA, Frants RR, et al. Risk of developing pancreatic cancer in families with familial atypical multiple mole melanoma associated with a specific 19 deletion of p16 (p16-Leiden) Int J Cancer. 2000;87:809–811. [PubMed] [Google Scholar]
- 17.Roberts NJ, Klein AP. Genome-wide sequencing to identify the cause of hereditary cancer syndromes: With examples from familial pancreatic cancer. Cancer Lett. 2013;340:227–233. doi: 10.1016/j.canlet.2012.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Klein AP, Brune KA, Petersen GM, et al. Prospective risk of pancreatic cancer in familial pancreatic cancer kindreds. Cancer Res. 2004;64:2634–2638. doi: 10.1158/0008-5472.can-03-3823. [DOI] [PubMed] [Google Scholar]
- 19.Shindo K, Yu J, Suenaga M, et al. Deleterious germline mutations in patients with apparently sporadic pancreatic adenocarcinoma. J Clin Oncol. 2017;35:3382–3390. doi: 10.1200/JCO.2017.72.3502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lowery MA, Wong W, Jordan EJ, et al. Prospective evaluation of germline alterations in patients with exocrine pancreatic neoplasms. J Natl Cancer Inst. 2018;110:1067–1074. doi: 10.1093/jnci/djy024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brand R, Borazanci E, Speare V, et al. Prospective study of germline genetic testing in incident cases of pancreatic adenocarcinoma. Cancer. 2018;124:3520–3527. doi: 10.1002/cncr.31628. [DOI] [PubMed] [Google Scholar]
- 22.Canto MI, Goggins M, Yeo CJ, et al. Screening for pancreatic neoplasia in high-risk individuals: An EUS-based approach. Clin Gastroenterol Hepatol. 2004;2:606–621. doi: 10.1016/s1542-3565(04)00244-7. [DOI] [PubMed] [Google Scholar]
- 23. doi: 10.1016/j.cgh.2006.02.005. Canto MI, Goggins M, Hruban RH, et al: Screening for early pancreatic neoplasia in high-risk individuals: A prospective controlled study. Clin Gastroenterol Hepatol 4:766-781, 2006; quiz 665 . [DOI] [PubMed] [Google Scholar]
- 24. Canto MI, Hruban RH, Fishman EK, et al: Frequent detection of pancreatic lesions in asymptomatic high-risk individuals. Gastroenterology 142:796-804, 2012; quiz e14-e5 . [DOI] [PMC free article] [PubMed]
- 25. Johns Hopkins Medicine: NFPTR. http://pathology.jhu.edu/pancreas/nfptr/index.php.
- 26. Bosman F, Carneiro F, Hruban R, et al: WHO Classification of Tumours of the Digestive System, Volume 3 (ed 4). Geneva, Switzerland, WHO, 2010. [Google Scholar]
- 27.Tanaka M, Fernández-del Castillo C, Adsay V, et al. International consensus guidelines 2012 for the management of IPMN and MCN of the pancreas. Pancreatology. 2012;12:183–197. doi: 10.1016/j.pan.2012.04.004. [DOI] [PubMed] [Google Scholar]
- 28.Suenaga M, Yu J, Shindo K, et al. Pancreatic juice mutation concentrations can help predict the grade of dysplasia in patients undergoing pancreatic surveillance. Clin Cancer Res. 2018;24:2963–2974. doi: 10.1158/1078-0432.CCR-17-2463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yu J, Sadakari Y, Shindo K, et al. Digital next-generation sequencing identifies low-abundance mutations in pancreatic juice samples collected from the duodenum of patients with pancreatic cancer and intraductal papillary mucinous neoplasms. Gut. 2017;66:1677–1687. doi: 10.1136/gutjnl-2015-311166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Exome Aggregation Consortium: ExAC Brower. http://exac.broadinstitute.org.
- 31. University of Washington: NHLBI Exome Sequencing Project: Exome Variant Server. http://evs.gs.washington.edu/EVS.
- 32.Fine J, Gray RJ. A proportional hazards model for the subdistribution of a competing risk. J Am Stat Assoc. 1999;94:496–509. [Google Scholar]
- 33.National Comprehensive Cancer Network . NCCN Clinical Practice Guidelines in Oncology. Genetic/Familial High-risk Assessment: Breast and Ovarian version I.2019. Plymouth Meeting, PA: National Comprehensive Cancer Network; 2018. [DOI] [PubMed] [Google Scholar]