

Abstract
We have reported that the major histocompatibility molecule HLA-DQ2 (DQA1*05:01/DQB1*02:01) is relatively resistant to HLA-DM (DM), a peptide exchange catalyst for MHC class II. Here, we analyzed the role of DQ2/DM interaction in the generation of DQ2-restricted gliadin epitopes, relevant to celiac disease, or DQ2-restricted viral epitopes, relevant to host defense. We used paired human antigen presenting cells (APC), differing in DM expression (DMnull vs DMhigh) or differing by expression of wild type DQ2 versus a DM-susceptible, DQ2 point mutant DQ2α+53G. The APC pairs were compared for their ability to stimulate human CD4+ T cell clones. Despite higher DQ2 levels, DMhigh APC attenuated T cell responses compared to DMnull APC after intracellular generation of 4 tested gliadin epitopes. DMhigh APC expressing the DQ2α+53G mutant further suppressed these gliadin-mediated responses. The gliadin epitopes were found to have moderate affinity for DQ2, and even lower affinity for the DQ2 mutant, consistent with DM suppression of their presentation. In contrast, DMhigh APC significantly promoted the presentation of DQ2-restricted epitopes derived intracellularly from inactivated herpes simplex virus type 2 (HSV-2), influenza hemagglutinin and human papillomavirus (HPV) E7 protein. When extracellular peptide epitopes were used as antigen, the DQ2 surface levels and peptide affinity were the major regulators of T cell responses. The differential effect of DM on stimulation of the 2 groups of T cell clones implies differences in DQ2 presentation pathways associated with non-pathogen and pathogen-derived antigens in vivo.
Introduction
Presentation of peptides to CD4+ T cells by major histocompatibility complex class II (MHCII) proteins (HLA-DR, -DQ and -DP in humans) is a key initiating step in immune responses. Allelic variations in MHCII proteins influence antigen presentation of both self and foreign proteins, with consequences for autoimmunity/tolerance and host defense (1). DQ2 (DQA1*05:01/ DQB1*02:01; also called DQ2.5) is an allele of particular interest in this regard. This allele confers genetic risk to several autoimmune diseases including type 1 diabetes and celiac disease (CD) (2, 3). CD is a disorder caused by immunue intolerance to ingested cereal gluten proteins of wheat (consisting of gliadin and glutenin subcomponents), barley and rye. In susceptible individuals, DQ2 proteins preferentially bind and present to CD4+ T cells (4, 5) certain proline-rich gluten peptides that have been posttranslationally modified (i.e. deamidation of glutamine) by the enzyme transglutaminase 2 (TG2) (6–9). While DQ2 is associated with vigorous anti-gluten T cell responses, DQ2 also is associated with poor responses to several vaccines (10–12) and with failure to control hepatitis virus C (13) and hepatitis virus B (14).
Peptide presentation by MHCII, including DQ2, is influenced by interaction with antigen presentation co-factors, invariant chain (Ii) and HLA-DM (DM). Ii, a class II chaperone, directs nascent MHCII/Ii oligomers from the endoplasmic reticulum (ER) to MHCII containing compartments (MIIC), where it is processed to CLIP, a nested set of class-II-associated invariant chain peptides that bind the groove (15). For most class II allelic proteins, the displacement of CLIP in MIIC is catalyzed by DM that transiently associates with MHCII, protects peptide-receptive MHCII from degradation by peptide exchange when peptides are available, and edits bound peptides in favor of high affinity ones for cell surface presentation (16). Work from our laboratories (17, 18), confirmed by others (19), showed that DQ2 has reduced interaction with HLA-DM, compared to most other alleles.
DM can enhance or suppress the presentation of specific MHCII-peptide complexes. In general, MHCII-peptide complexes with lower intrinsic stability are DM-susceptibile (20–22), but not all high-stability complexes are DM-resistant (23). Immunodominant epitopes preferentially survive DM editing within endosomal compartments and trigger CD4+ T cell activation (16). In contrast, other potential epitopes are removed from MHCII through DM catalysis and fail to elicit T cell responses (24), except when they get presented through pathways that avoid interaction with DM (25). We hypothesized that the DM-resistant feature of DQ2 likely contributes to the escape of gliadin peptides from extensive DM editing. In addition, DQ2 has the special ability to stably bind proline-rich gliadin peptides (5) that utilize TG2-deamidated residues as DQ2-binding anchors (26); together these unique features of DQ2 may allow gliadin presentation to disease-driving CD4+ T cells. In vitro, DQ2 resistance to DM can be overcome by increased DM concentrations or prolonged exposure to DM (17). We produced transfectants expressing high levels of DM and showed that intracellular DM/DQ2 interaction occurs in these cells (18). In addition, DQ2 has a natural deletion in the region involved in interaction with HLA-DM (5); we found that insertion of arginine or glycine at α53 (DQ2+53R, DQ2+53G) confers DM sensitivity to the mutant DQ2 proteins, providing another tool to test the effects of increased DM/DQ2 interaction on antigen presentation (18).
Here, we analyze DM effects on DQ2 presentation of 4 gliadin epitopes to T cell clones isolated from CD patients. We also test the response of DQ2-restricted CD4+ T cells specific for viral peptides from 3 different viruses. Strikingly, we found different consequences of DM activity for these 2 groups of T cell clones, with suppression of gliadin presentation and enhancement of viral peptide presentation. These results imply key differences in DQ2 antigen presentation pathways operating in CD compared to host defense against viral infection.
Materials and Methods
Cell lines:
The T x B hybrid APC cell lines, T2 (MHCII−/DM−) and T2DM (MHCII−/DM+) stably expressing WT DQ2 (T2.DQ2 and T2.DQ2.DM) or mutant DQ2 (T2.DQ2α+53G.DM) have been established previously (18). They were cultured in complete Iscove’s Modified Dulbecco’s Media (IMDM, Thermo Fisher Scientific, Waltham, MA) supplemented with 10% (v/v) fetal bovine serum (FBS), 2 mM L-glutamine and maintained at 37°C in a humidified atmosphere of 5% CO2. DQ2-restricted T cell clones specific for different gliadin epitopes were isolated, as described (27). These include: TCC819.392 (specific for α1a, see below for a decription of different gliadin epitopes), TCC820.250 (specific for α2), TCC820.270 (specific for γ1) and TCC820.59 (specific for γ4d); the various epitopes are described in Antigens section below. 1A.B.25, a DQ2-restricted T cell clone recognizing amino acids 431–440 (EVDMTPADAL) of VP16 (gene UL48) of herpes simplex virus type 2 (HSV-2), isolated for a previous study was used (28). DQ2-restricted influenza-specific T cell clones (Clone 5) that recognize amino acids 97–113 of hemagglutinin (HA) protein from A/New Caledonia/20/99 (H1N1) were isolated as described (29). DQ2-restricted human papillomavirus type 16 (HPV16) specific T cell clone (Clone 60) recognizing E735–50 was from Dr. van der Burg (30).
Antigens:
High-molecular-weight fraction of gluten was prepared using a previously described method (18). Briefly, gluten proteins were digested with pepsin and trypsin/chymotrypsin. The pepsin/trypsin-treated gluten (PT-gluten) was further deamidated with TG2 in the presence of CaCl2. HSV-2 was UV-inactivated and prepared as described previously (28). H1 Hemagglutinin (HA) protein with C-terminal histidine tag from influenza A/New Caledonia/20/1999 (H1N1) was obtained from BEI Resources (NR-48873). Recombinant HPV16 protein E7 was purchased from CUSABIO (Wuhan, China). An α-gliadin fragment (α1/α2: LQLQPFPQPELPYPQPELPY), containing the overlapping T cell epitopes, α1a (underscored) and α2 (bolded) (31, 32) was synthesized and provided by Dr. Xi Jin in the laboratory of Dr. Chaitan Khosla, Department of Chemistry, Stanford University, Palo Alto, CA. Synthetic peptides containing the gliadin T cell epitopes (underscored) (27, 33), γ1 (P1213: pyroEPEQPQQSFPEQERP), synthesized by GLBiochem (Shanghai, China) and γ4d (P1936: PFPQPEQPFCEQPQR), synthesized at the University of Oslo, Norway. Glutamine (Q) at the N terminus of peptide P1213 is unstable and tends to spontaneously convert to pyroglutamic acid (pyroE); therefore P1213 was synthesized with pyroE at the N terminus. The following peptides were synthesized by Genscript (Piscataway, NJ): HA97–113 (YPGYFADYEELREQLSS), biotinylated MHCIα49–63 (APWIEQEGPEYWDQE) (18), biotinylated Ii81–104 (CLIP1: LPKPPKPVSKMRMATPLLMQALPM), biotinylated Ii92–107 (CLIP2: RMATPLLMQALPMGAL) (17), biotinylated P1269 (QLQPFPQPELPY) containing α1a, biotinylated PS1200 (PQPELPYPQPQS) containing α2 (34), biotinylated P1213 (pyroEPEQPQQSFPEQERP) containing γ1 (31), biotinylated P1936 (PFPQPEQPFCEQPQR) containing γ4d (35). HPV16 E722–56 (LYCYEQLNDSSEEEDEIDGPAGQAEPDRAHYNIVT) was provided by Dr. van der Burg (30).
Pulse/chase and immunoprecipitation:
Pulse/chase analysis was performed as previously described (36). Briefly, 6×107 T2.DQ2 or T2.DQ2.DM cells were washed with complete Cys/Met-free RPMI medium (containing 10% FBS and 2 mM L-glutamine), and then re-suspended in complete Cys/Met-free RPMI medium for starving at 37°C for at least 1 h. After starving, cells were pulsed with 150 µCi/ml Express [35S] labeling mix (PerkinElmer, Waltham, MA) for 3 h. One half of the pulsed cells were washed and pelleted without chasing (t=0 time point), and the other half of pulsed cells were chased overnight in complete RPMI medium and then washed before pelleting (t=24 h time point). Cell pellets were lysed with lysis buffer (50 mM Tris-HCl, 150 mM NaCl, 1% IGEPAL CA630 from Sigma, St. Louis, MO; PMSF and complete protease inhibitors from Thermo Fisher Scientific, Waltham, MA). DQ2 proteins in clear cell lysates were then immunoprecipitated using protein G beads (GE Healthcare, Chicago, IL) that were pre-coupled with anti-DQ monoclonal antibody (mAb), SPV-L3. After incubation with cell lysate overnight at 4°C, protein G beads were pelleted, washed 5 times with cold PBS, and boiled in reducing sample buffer containing 62.5 mM Tris-HCl (pH 6.8), 1% SDS, 3% glycerol, 0.007% bromophenol blue, and 1% 2-ME. DQ2 proteins eluted from the beads were separated by 12% SDS-PAGE gel electrophoresis, and separated bands representing target proteins were visualized by exposing dried gels to radiography film (Kodak, Rochester, NY).
Peptide loading capture ELISA assay:
Soluble DM, DQ2-CLIP1 and DQ2α+53G-CLIP1 were generated as previously described (37). For the time course of spontaneous peptide loading, 50 nM of thrombin cleaved sDQ2-CLIP1 or sDQ2α+53G-CLIP1 were incubated with 50 µM biotinylated peptides (CLIP1, CLIP2, MHCIα, P1269, PS1200, P1213, or P1936) in the reaction buffer (100 mM acetate buffer pH 4.6, 150 mM NaCl, 1% NaCl, 1% BSA, 0.5% NP40, 0.1% NaN3) supplemented with 1x EDTA-free protease inhibitor cocktail (Thermo Fisher Scientific). The peptide loading reaction took place at 37°C for the indicated time. At each time point, two volumes of neutralization buffer (100 mM Tris-Cl pH 8.0, 150 mM NaCl, 1% NaCl, 1% BSA, 0.5% NP40, 0.1% NaN3) were added to terminate the reaction. 100 µl of the neutralized mixture was then transferred to a 96-well plate pre-coated with DQ-specific mAb SPV-L3 and incubated at RT for 1 h. After 5 washes with wash buffer (0.05% Tween 20 in PBS), 100 µl PBS containing 1% BSA and 1:1000 diluted europium-labeled streptavidin (PerkinElmer, Waltham, MA) were added to each well and left at RT for 1 h. After another 5 washes, 100 µl of enhancement solution (PerkinElmer, Waltham, MA) were then applied, and the time-resolved fluorescence signal associated with europium in each well was measured using a plate reader (Tecan, Männedorf, Switzerland). In order to examine DM effects, 50 nM of thrombin cleaved sDQ2-CLIP1 or sDQ2α+53G-CLIP1 were incubated with 10 µM biotinylated peptides (CLIP2 or MHCIα), in the presence of 50, 200, or 800 nM of soluble DM at 37°C for 1 h, and then captured by SPV-L3 for quantification of biotinylated peptides as detailed above.
T cell proliferation assays:
Frozen T cell clones were thawed, washed and rested in IMDM with 10% FBS, 2mM L-glutamine, 2% human serum, and 1% penicillin/streptomycin at 37°C in a humidified atmosphere of 5% CO2 for at least 1 h. APC including T2.DQ2, T2.DQ2.DM, T2.DQ2α+53G.DM, 9.5.3, and 9.5.3.DM were irradiated (12,000 rads) or fixed with 1% paraformaldehyde for 5 minutes on ice followed by two PBS washes. After irradiation/fixation, APC (50,000 cells) were incubated with antigen and T cell clones (50,000 cells). [3H]-thymidine (1 µCi per well; PerkinElmer, Waltham, MA) was added after 48 h of incubation. Cells were harvested after another 16–18 h, using the Tomtec Harvester (Hamden, CT), and thymidine incorporation (cpm) was measured by Wallac 1450 Microbeta Trilux Liquid Scintillation and Luminescence Counter (PerkinElmer, Waltham, MA).
For CFSE staining, thawed T cell clones were rested in RPMI 1640 with 10% human serum, 1% penicillin/streptomycin for 3 days after thawing. Cells were washed with PBS, and then labelled with 1 µM CFSE (carboxyfluorescein diacetate succinimidyl ester) in 5% FBS containing PBS for 5 min at 37°C. Labelling reaction was quenched by washes with ice cold 20% FBS in PBS twice. CFSE-labelled T cells were resuspended in RPMI 1640 with 5% human serum, 1% penicillin/streptomycin and cocultured with irradiated T2.DQ2 and T2.DQ2.DM at 1:1 ratio (APC: T cell). The co-cultures were stimulated with indicated peptides at 10 µg/ml or with antigen and incubated for 10 days. Cells were washed with flow buffer (2% BSA in PBS) and stained with PI before analysis on LSR II cytometer at Stanford Shared FACS facility. Data were analyzed by FlowJo version 10 for Windows (FlowJo, LLC).
Flow cytometry:
Surface DQ2 was directly stained by PE-conjugated anti-DQ mAb (Ia3, Leinco Technologies, St. Louis, MO) or indirectly stained using a primary antibody SPV-L3 (mouse anti-DQ) or 2.12.E11 (mouse anti-DQ2) followed by a secondary Ab, PE-conjugated goat anti-mouse IgG (Thermo Fisher Scientific, Waltham, MA). CLIP1 peptide (Ii amino acids 81–103, LPKPPKPVSKMRMATPLLMQALP) that is associated with DQ2 was stained by FITC-conjugated anti-CLIP mAb (CerCLIP; BD Biosciences, San Jose, CA). Directly labeled mAbs were used in case of co-staining of multiple surface proteins. DM was detected using PE-conjugated anti-DM mAb (MaP.DM1, BD Biosciences, San Jose, CA) either on the surface of untreated cells or in cells that were fixed and permeabilized using the Cytofix/Cytoperm kit (BD Pharmingen, San Jose, CA). Fluorescently labeled cells were analyzed using a FACScan flow cytometer (BD Biosciences, San Jose, CA), and data were analyzed using the FlowJo software (FlowJo, LLC).
Statistical analyses:
Data were analyzed for statistical significance using Prism software (version 4.0; GraphPad Software, San Diego, CA) and are expressed as mean±standard deviation. Statistical significance and p values are indicated on the figures where appropriate. p values <0.05 were considered statistically significant.
Results
Increased DM overcomes poor interaction with the resistant DQ2 allele
We previously constructed DMnull and DMhigh APC cell lines expressing DQ2 as the only MHCII allele, without or with DM: T2.DQ2 and T2.DQ2.DM, respectively (18). The amount of DM available for mediating interaction with DQ2 in T2.DQ2.DM cells is higher than in, for example, activated B cells, as modelled by B lymphoblastoid cell lines. This is not only because the transfected cells were selected to overexpress DM, but also because other MHCII, which could compete with DQ2 for interaction with DM, are not expressed in these cells. In physiologic APC, co-dominantly expressed MHCII molecules with higher affinity for DM compromise DQ2 access to DM. The effects of increased DM/DQ2 ratios and consequent increased DM/DQ2 interaction in the T2.DQ2.DM transfectant include DM chaperoning of DQ2, which increases surface DQ2 levels [(18), Figure 1A, supplementary Figure 1A, B], and DM catalysis of CLIP removal from DQ2, which reduces CLIP-associated DQ2 during biosynthesis. To observe the release of DQ2-associated CLIP, we immunoprecipitated DQ2 from T2.DQ2.DM and T2.DQ2 cells in a pulse-chase analysis (Figure 1B). Freshly synthesized, metabolically labelled DQ2/CLIP complexes were observed within 1 day in T2.DQ2, but are undetectable in T2.DQ2.DM. In a related result, we also found that DQ2/CLIP complexes are nearly absent at the cell surface of T2.DQ2.DM [Figure 1C, supplementary Figure 1C, D]. Together, these results indicate that increased DM abundance can overcome its poor reactivity to DQ2.
Figure 1. Effect of DM in DQ2-CLIP1 association.
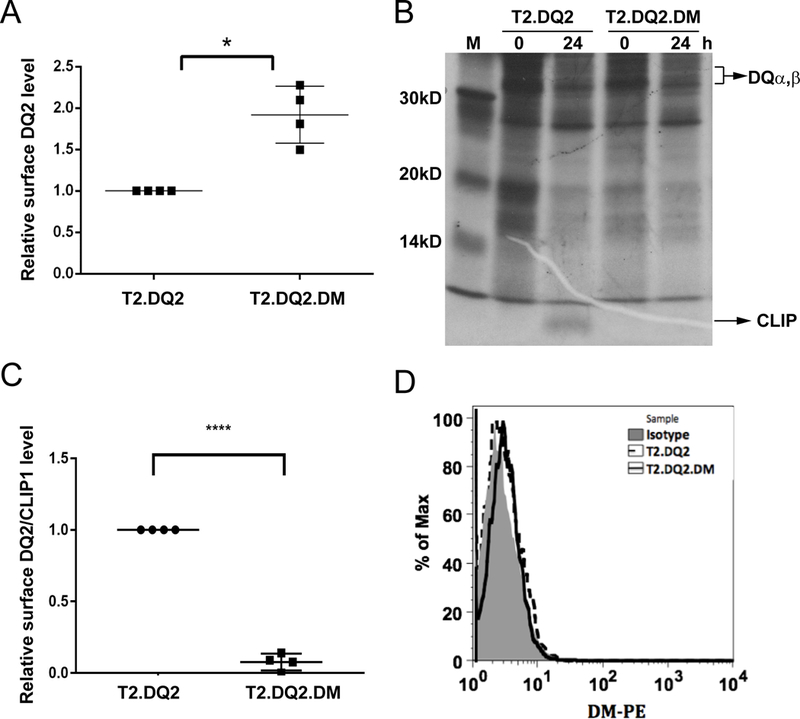
(A) Surface DQ2 levels were measured by Ia3.DQ-PE staining of T2.DQ2 and T2.DQ2.DM cells followed by flow cytometric analysis; geometric mean fluorescence intensity (MFI) of DQ2 in T2.DQ2.DM was compared to T2.DQ2. * P< 0.05. (B) Time course of CLIP association with metabolically-labeled DQ2, immunoprecipitated from T2.DQ2 and T2.DQ2.DM cells. Cell lysates of metabolically-labeled cells (3× 106 cell equivalents/lane) of the indicated cell lines were immunoprecipitated with anti-DQ mAb, SPVL3, at indicated times and analyzed by SDS-PAGE. Representative images from one of two independent experiments are shown. (C and D) Cells were stained for surface DQ2-associated CLIP1 (C) and surface DM (D) levels with CerCLIP-FITC and Map.DM1-PE, respectively, before flow cytometry. **** P< 0.0001. (D) Isotype control antibody served as controls. Representative results (histograms) from one of three experiments with similar results are shown.
Abundant DM attenuates presentation of PT-gluten by DQ2+ cells
DM is a lysosomal resident and is typically undetectable on the surface of APCs at physiological conditions, due to the presence of a lysosomal sorting motif at DMβ cytoplasmic tail (38). To test whether the overexpression of DM in the transfected T2 cells led to presence of DM at the cell surface, we used anti-DM mAb to stain the surface of T2.DQ2.DM. Flow cytometric analysis showed negligible levels of staining, compared with control lines that were DM-deficient (Figure 1D). This implied that CLIP removal occurred intracellularly, likely in MIIC, rather than at the cell surface.
Given the intracellular activity of DM in T2.DQ2.DM, these cells provided a tool to analyze the effect of DM on DQ2-restricted presentation to T cells of various antigens that require intracellular processing. To test gliadin epitope presentation, we used PT-gluten (pepsin/trypsin treated-gluten, see methods), which is a mixture of relatively large (molecular weight >10 kDa), proteolytic fragments of gluten protein. PT-gluten was also treated with transglutaminase-2, which converts selected glutamines to glutamic acid, mimicking the naturally occurring deamidation of gluten in the small intestine. As responding CD4+ T cells, we used a panel of 4 gliadin-specific T cell clones. These clones were isolated from the small intestine of CD patients, and each is specific for one of the following epitopes: α1a, α2, γ1 and γ4d (short for DQ2.5-glia-α1a, DQ2.5-glia-α2, DQ2.5-glia-γ1 and DQ2.5-glia-γ4d, respectively, as named elsewhere (35). Also, please see Supplementary Table 1. These gliadin epitopes are implicated in CD pathogenesis, as they are recognized by intestinal T cells of the majority of adults with CD (39).
Previous work by our group (Sollid group) has shown that TG2-treated PT-gluten requires further processing to generate robust presentation of the relevant T cell epitopes for clones with the specificities we used here. This was demonstrated by the ability of irradiated cells to significantly out-perform fixed cells as APC (31, 40). Of note, however, PT-gluten antigen preparations also include a low level of fragments that are stimulatory without further processing, presumably binding to surface DQ2 of fixed cells (31). We confirmed the need for processing for our PT-gluten preparation and also the low level of T cell stimulation by fixed cells, presumably from the availability of some smaller fragments in the preparation (Supplementary Figure 2).
We next tested the influence of DM on the presentation of intracellularly processed gliadin, using irradiated DQ2+ T2 transfectants. The gliadin-specific human T cell clones were co-cultured with irradiated T2.DQ2 or T2.DQ2.DM cells in the presence of deamidated PT-gluten, and the T cell proliferation was quantified. Both irradiated T2.DQ2 and T2.DQ2.DM cells were able to process PT-gluten and present the corresponding gliadin epitopes to activate the T cell clones in a dose-dependent manner. However, T cell proliferation for all 4 clones was diminished significantly in the presence of DM (Figure 2), despite the higher level of DQ2 in T2.DQ2.DM cells (Figure 1A).
Figure 2. Reduction of PT-gluten presentation by DQ2+ cells in the presence of DM.
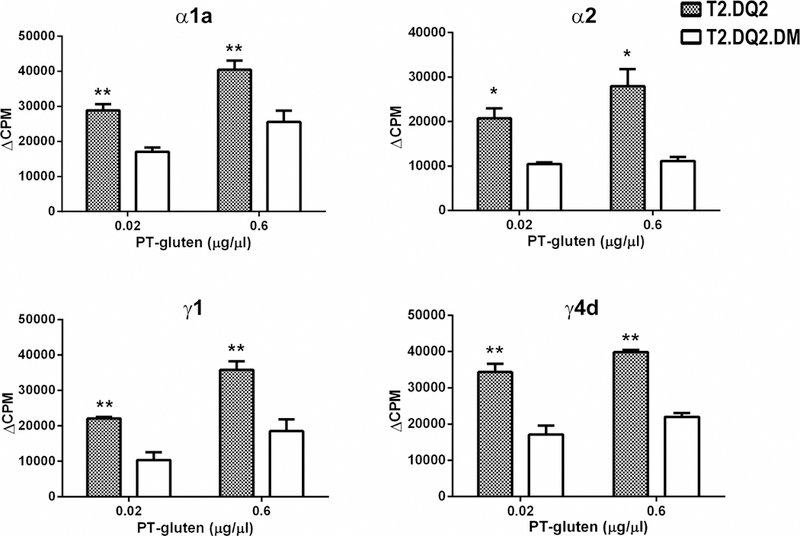
Stimulation of each of the four DQ2-restricted, α1a, α2, γ1, or γ4d specific-T cells was suppressed by the presence of DM. T2.DQ2 or T2.DQ2.DM cells were irradiated and then incubated with 0.02 or 0.6 μg/μl of PT-gluten and gliadin specific-T cell clones, as indicated. Cells were cultured for 48 hours and then [3H]-thymidine was added for another 16–18 hours and [3H]-thymidine incorporation was measured. Background CPM from the no antigen condition are subtracted, and ΔCPM are shown. Each condition was done in triplicate and all experiments were repeated at least twice with similar results. Mean and standard deviations (SD) from one representative experiment is shown. * P< 0.05, ** P< 0.01. Background cpm from the no antigen condition were subtracted from the values with antigen.
DM suppression of gliadin presentation is APC type-independent
To eliminate the possibility that TxB hybrid T2 cells provided an environment that uniquely allowed DM action on resistant MHCII alleles, we repeated the DQ2-restricted presentation of PT-gluten using another pair of DQ2+ APC lines: a DMnull EBV-transformed B cell line 9.5.3 (DR3+/DQ2+/DP4+/DM−) and its repaired DMhigh-transfectant 9.5.3.DMhigh (41). Similar to the differences observed using DQ2+ T2 cells as APC, the expression of DM increased the surface level of DQ2 in 9.5.3.DMhigh compared to that of 9.5.3 (Figure 3A); and 9.5.3.DMhigh substantially lacked cell surface CLIP1/MHCII complexes (Figure 3B), despite the presence of other MHCII alleles (DR3, DP4). Also, similar to the DM localization in T2.DQ2.DM, very little if any DM was detected on the surface of 9.5.3.DMhigh (Figure 3C). We then compared the capacity of irradiated 9.5.3.DMhigh and 9.5.3 to present PT-gluten and to stimulate the proliferation of gliadin-specific T cell clones. Consistent with our previous findings, significant attenuation of T cell proliferation was observed when DMhigh APC were used (Figure 3D), despite the higher levels of DQ2 in 9.5.3.DMhigh cells. Taken together, these findings argue that presentation of gliadin epitopes α1a, α2, γ1 and γ4d, after intracellular processing from gluten-derived products by gut APC, would be limited by functional interaction with DM. Consequently, the poor interaction of DQ2 with DM may contribute to CD pathogenesis.
Figure 3. DM decreases PT-gluten presentation by DQ2 independent of APC cells.
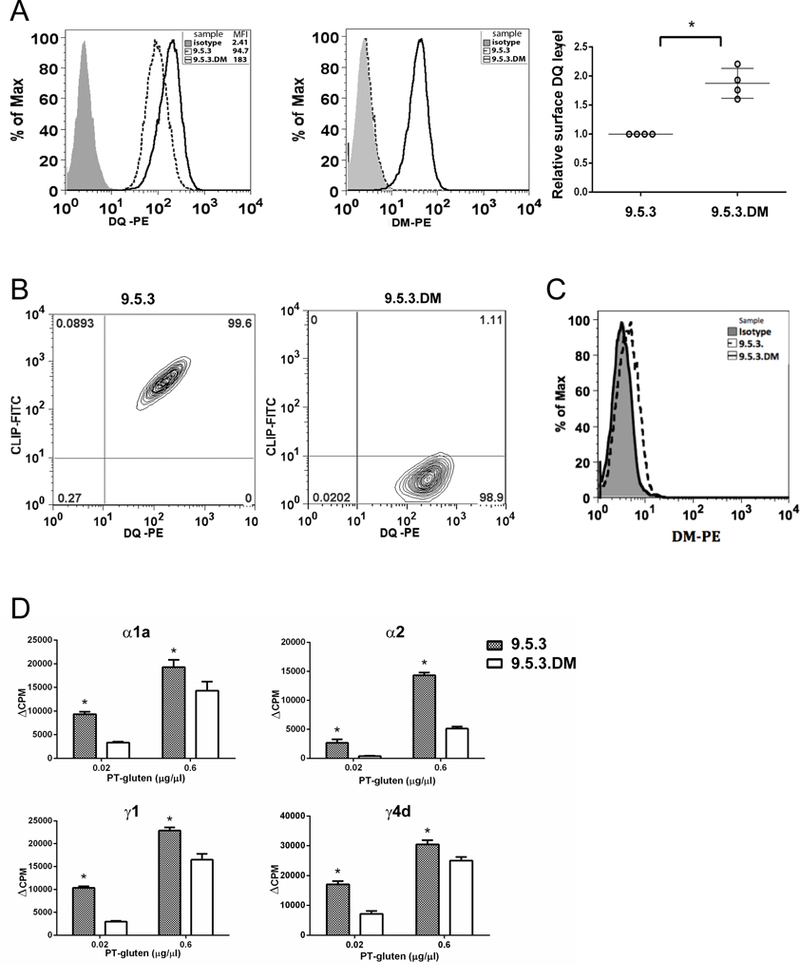
(A) Expression of WT DQ2 and DM in 9.5.3 and 9.5.3.DM cells. Surface DQ2 and intracellular DM were stained respectively by mouse anti-human DQ (Ia3-PE) and mouse anti-human DM (MaP.DM1-PE). Staining was repeated several times, and MFI of DQ2 was normalized to the level on 9.5.3, which was considered as 1.0. *P<0.05. (B) Surface expression of class II-bound CLIP1 in 9.5.3 (left) and 9.5.3.DM (right) cells. CLIP1 was stained by mouse anti-human CLIP1 (CerCLIP.1-FITC) followed by flow cytometric analysis. (C) Surface DM levels were measured by Map.DM1-PE staining of 9.5.3.DM cells and flow cytometric analysis; 9.5.3 cells and isotype control antibody served as controls. Representative results (histograms) from one of three experiments with similar results are shown. (D) Suppression of gliadin-specific T cells (TCC819.392 for α1a, TCC820.250 for α2, TCC820.270 for γ1 and TCC820.59 for γ4d, respectively) by DM was also observed with 9.5.3.DM as APC. The experiment was performed as described in Figure 2, except that 9.5.3 or 9.5.3.DM were used as antigen presenting cells. Each condition was done in triplicate and all experiments were repeated at least twice with similar results (means ± SD). * P< 0.05. Background cpm from the no antigen condition were subtracted from the values with antigen.
A point mutant DQ2 with increased DM interaction suppresses gliadin presentation
As another approach to evaluate the suppressive effect of DM/DQ2 interaction on gliadin peptide presentation, we compared gluten presentation by wild type DQ2 with presentation by the DQ2α+53G mutant with increased DM susceptibility (18). T cell proliferation of all four gliadin epitope-specific clones was significantly diminished when the T2 transfectant expressing DQ2α+53G was used as the APC (Figure 4A). T2.DQ2.DMhigh and T2.DQ2α+53G.DMhigh express comparable levels of intracellular DM (18), ruling out an effect caused by DM abundance in the mutant. In addition, the decrease in T cell proliferation stimulated by T2.DQ2α+53G.DMhigh did not result from lower DQ expression, as T2.DQ2α+53G.DMhigh cells express significantly higher levels of surface DQ compared to T2.DQ2.DMhigh cells (Figure 4B). Notably, in the absence of DM, T2.DQ2α+53G expressed less surface DQ than T2.DQ2 (18), therefore, the increased DQ expression on the surface of T2.DQ2α+53G.DM vs T2.DQ2.DM likely reflects another restored DM action; the enhanced DM/DQ2α+53G interaction allows DM to stabilize and rescue these MHCII molecules in MIIC (42, 43).
Figure 4. DQ2α+53G mutant with increased DM affinity reduces PT-gluten presentatio0n by DQ2.
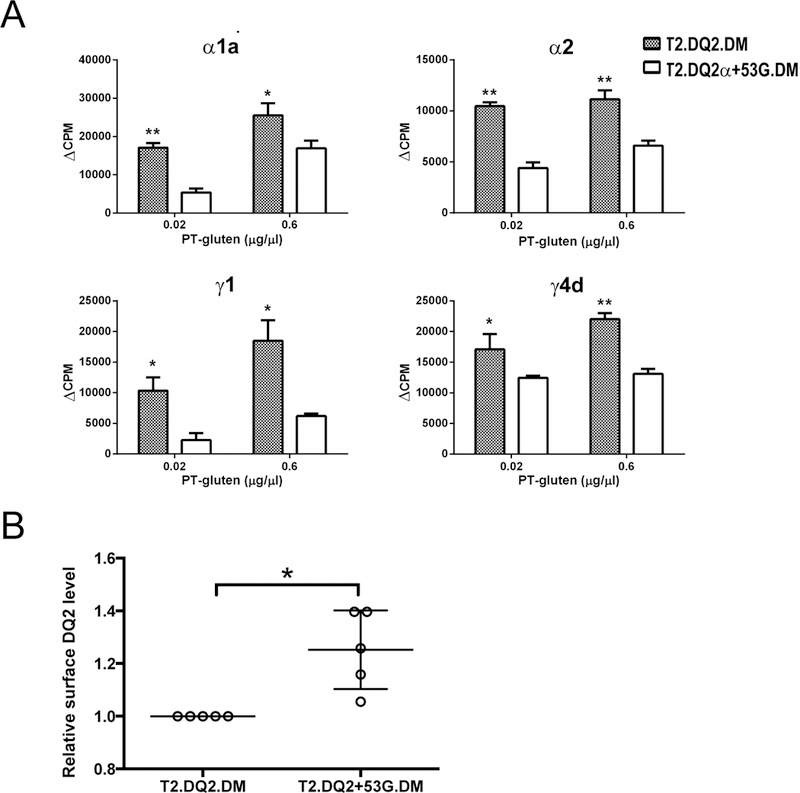
(A) T cell proliferation was decreased when gliadin was presented by the mutated DQ2 molecule, DQ2α+53G. T2.DM cells expressing WT DQ2 or DQ2α+53G mutant were irradiated and then incubated with transglutaminase-treated PT-gluten. Gliadin-specific (α1a, α2, γ1, γ4d) T cell clone (TCC819.392, TCC820.250, TCC820.270, and TCC820.59, respectively) proliferation was assessed by measuring [3H]-thymidine incorporation, as described for Figure 2. (B) Expression of DQ2 in T2.DQ2.DM and T2.DQ2α+53G.DM cells. T2.DQ2.DM and T2.DQ2α+53G.DM cells were surface-stained with mouse anti-human DQ (Ia3; DQ-PE) or isotype control and analyzed by flow cytometry. Staining was repeated several times, and median MFI of DQ2 was normalized to the level in T2.DQ2.DM, which was considered as 1.0. * P< 0.05.
Poor DQ binding of gliadin peptides coordinates with DM suppression
In addition to the increased DM/DQ interaction, the potential impaired binding of DQ2 mutant to gliadin peptides may also contribute to the DM-mediated suppression of T cell proliferation in response to gliadin peptides presented by T2.DQ2α+53G.DMhigh APCs. To test this hypothesis, we ranked 4 gliadin peptides and 3 reference peptides based on their relative binding to soluble, recombinant DQ2α+53G as compared to wild type DQ2, using a peptide loading assay. In this assay, biotinylated peptides in excess will competitively replace CLIP1 peptide, engineered to be pre-loaded in the peptide-binding groove, but exchangeable after cleavage of the covalent linker. Reference peptides include a DQ2-binding peptide derived from the MHCIα protein (18), CLIP1 (Ii81–104), CLIP2 (Ii92–107). The relative binding of peptides followed the order: CLIP2 > MHCIα ≈ CLIP1 ≈ P1269 (α1a) ≈ P1936 (γ4d) > PS1200 (α2) ≈ P1213 (γ1) for wild type DQ2; and CLIP2 > MHCIα > CLIP1 > P1936 (γ4d) > P1269 (α1a) ≈ PS1200 (α2) ≈ P1213 (γ1) for DQ2α+53G (Figure 5A). Unlike wild type DQ2, which is capable of binding to all four gliadin peptides, DQ2α+53G barely bound gliadin peptides, except for a poor binder, P1936 containing the γ4d epitope. These findings confirmed that reduced binding of gliadin epitopes to mutant DQ2 coordinated with increased DM/DQ interaction for DM-mediated suppression of gliadin presentation by T2.DQ2α+53G.DMhigh.
Figure 5. Differential peptide binding to WT DQ2 and DQ2α+53G and the influence of DM.
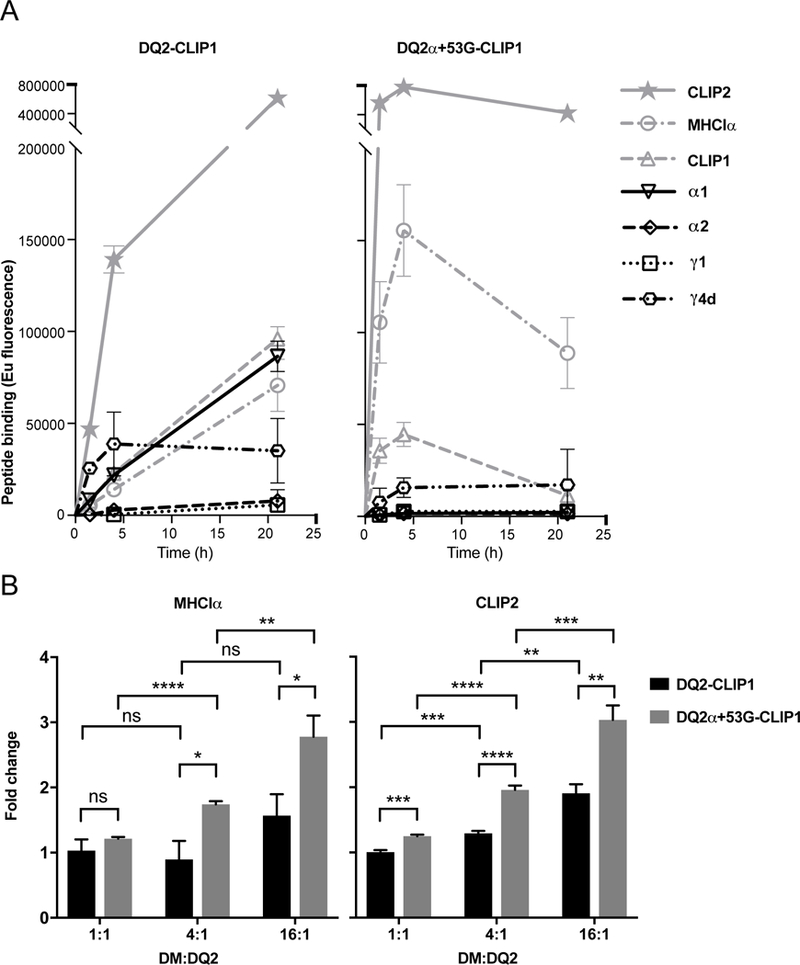
(A) Relative binding affinity of gliadin peptides. Thrombin cleaved soluble DQ2-CLIP1 or DQ2α+53G-CLIP1 was incubated with the indicated biotinylated peptides (1000x more concentrated than DQ-CLIP1 proteins) at 37°C. At the indicated time points, displacement of CLIP1 by the biotinylated peptide was measured by capture ELISA (see Methods) and represented as time-resolved europium fluorescence. (B) Effect of DM on displacement of weak binders. Thrombin cleaved soluble DQ2-CLIP1 or DQ2α+53G-CLIP1 was incubated with biotinylated MHCIα or CLIP2 peptides (200x more concentrated than DQ2-CLIP1 proteins) in the presence of soluble DM at the indicated ratio to DQ2 proteins at 37°C for 1 h. Displacement of CLIP1 by MHCIα or CLIP2 was measured as in (A) and represented as fold change over the corresponding no DM conditions. Data are represented as mean ± SEM. n=3. * P< 0.05, ** P< 0.01, *** P< 0.001, **** P< 0.0001, ns: no significance.
An interesting finding of the in vitro study is that although wild type DQ2 bound gliadin peptides, none of these epitopes associated with DQ2 better than CLIP1(≈MHCIα<CLIP2). The intermediate binding capability (reflecting lower relative affinities compared to MHCIα) of tested gliadin peptides for DQ2 is the likely basis of reduced presentation of these peptides by DMhigh APC and the associated attenuation of T cell proliferation. We next used the in vitro binding assay to test whether a moderate affinity peptide like CLIP1 bound to DQ2 can be efficiently edited by DM and replaced by higher affinity competitor peptides (e.g., MHCIα or CLIP2) when DM is abundant, regardless of DM susceptibility of DQ2 proteins. The catalytic effect of DM on peptide loading is best observed at early time points (37); therefore, we performed binding experiments with DM at 1 h and measured the fold change in binding to binding in the absence of DM (Figure 5B). When present at lower ratios to DQ2 (1:1 to 4:1), DM had little effect on peptide loading to wild type DQ2, in contrast to DQ2α+53G, that is DM susceptible. However, at DM:DQ2 ratio of 16:1, DM enhanced peptide loading, especially of CLIP2, onto wild type DQ2, pre-loaded with CLIP1 (Figure 5B). These results agree with our findings using DMhigh APCs and support the idea that with sufficient DM editing, the presentation of gliadin peptides would be suppressed, as their relative binding capacities to DQ2 (≤ CLIP1) are not as strong as one would expect for a DM-resistant epitope (see discussion). Notably, the substantially increased DM:DQ2 ratio needed to drive interaction of soluble wild type DQ2 and DM molecules is higher than that required for interaction of membrane bound molecules (44). In our T cell assays, the DM:DQ2 ratio in DMhigh APC is sufficient to replace intermediate affinity gliadin peptides with higher affinity peptides available in the late endosomal peptide loading compartments, although this would not happen at constitutive DM:DQ2 ratios in professional APC.
Presentation of exogenous gliadin peptides
To distinguish effects of gliadin peptide binding to DQ2 and DM/DQ2 interaction and to confirm that both effects underlie the DM-mediated suppression observed in DMhigh APCs, we also tested presentation of exogenously-loaded gliadin peptides, which bypass intracellular processing and DM editing, Unlike results with PT-gluten, which depends on intracellular processing and peptide loading, the presentation of all four gliadin peptides by T2.DQ2.DMhigh increased T cell stimulation to varying degrees (Figure 6A). This is consistent with peptide loading occurring at the cell surface or in early endosomes. At these sites, DM action is typically low, due to lower steady-state DM levels and higher pH than in lysosomes, and DQ2 expression levels primarily contribute to the amount of gliadin peptide that is loaded and presented to T cells. Exogenous peptide presentation also allowed us to evaluate the contribution of DQ2-peptide binding affinity to gliadin presentation. We compared T2.DQ2α+53G.DM to T2.DQ2.DM for the capacity to present the four gliadin peptides to T cell clones. In this comparison, the decreased T cell stimulation by the mutant APC (Figure 6B) likely results from reduced peptide loading or less stable binding DQ2α+53G. An efficiently DM-edited peptide repertoire, consisting of mostly stably bound peptides in association with surface DQ2α+53G, also could contribute to poor peptide exchange for exogenous gliadin peptides. Together with analyses on the presentation of PT-gluten, these findings implicate increased DM abundance, the restoration of DM interaction with DQ2, and reduced peptide binding as sources of suppressed stimulation of DQ2-restricted, gliadin-specific T cells from CD patients.
Figure 6. The effect of DM on the presentation of gliadin peptides.
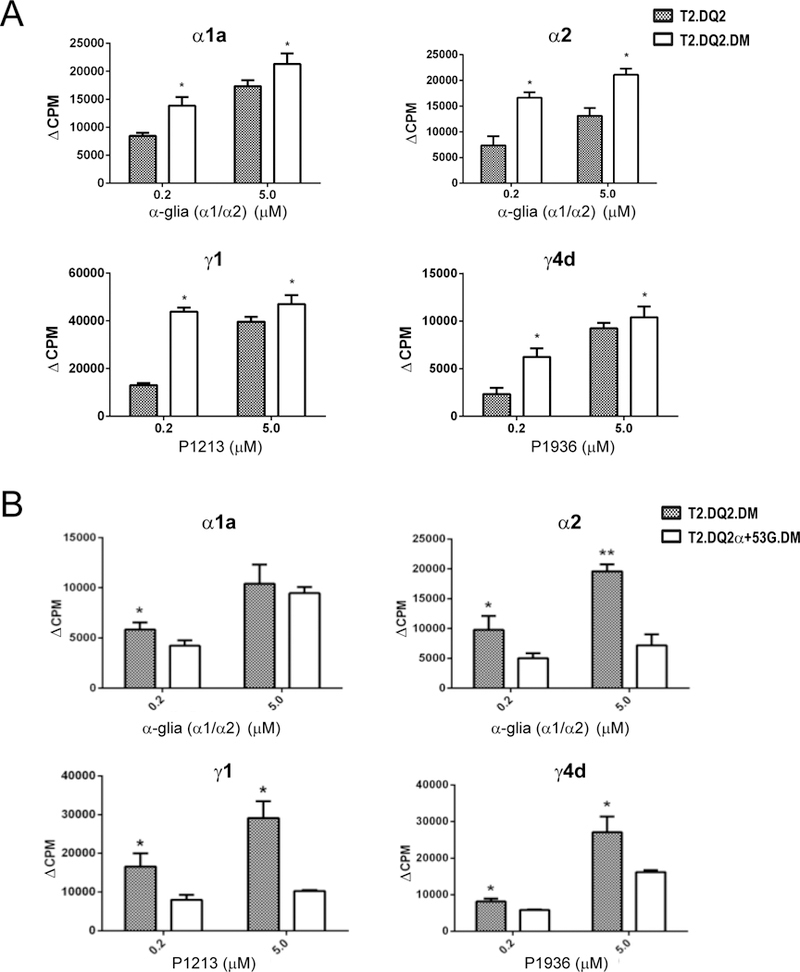
T2.DQ2 (A), T2.DQ2.DM (A and B) and T2.DQ2α+53G.DM (B) cells were irradiated and then incubated overnight with indicated T cell clones and corresponding gliadin peptides, including α-glia (α1/α2) containing α1a and α2, P1213 containing γ1, P1936 containing γ4d. T cell proliferation was then assessed by measuring [3H]-thymidine incorporation, as described for Figure 2. Data shown are representative of at least two independent experiments (means ± SD). * P< 0.05.
Differential presentation of gluten and viral antigens
The association of DQ2 with suboptimal responses to some viruses (10, 13, 14, 45) raised the possibility that its reduced interaction with DM might also lead to presentation of moderate affinity (DM-sensitive) viral peptides, whose unstable binding to DQ2 would reduce the surface half-life of the DQ2/peptide complex and compromise CD4+ T cell responses (20, 46). We therefore assessed the effect of DM on the activation of DQ2-restricted T cell clones specific for epitopes from viral proteins. 1A.B.25 is a T cell clone specific for a DQ2-restricted VP16 epitope (aa 431–440) derived from HSV-2 proteins. Its DQ2 restriction was validated by selective inhibition using anti-DQ, but not anti-DR or anti-DP mAb, presentation by allogeneic APC matched only at DQ2, and strong peptide binding to both purified DQ2 proteins and DQ2-expressing cell lines (38, 47). In contrast to PT-gluten presentation to gliadin-specific T cell clones, the presentation of UV-inactivated HSV-2 by irradiated T2.DQ2.DMhigh significantly increased the proliferation of 1A.B.25 (>5 fold) compared to the stimulation using T2.DQ2 (Figure 7A). This dramatic increase was unlikely due only to the modest (~2x) DM-dependent increase in surface DQ2 expression. Rather this result suggested improved presentation of the high affinity VP16-derived peptide (48) by DMhigh APC. We next tested a DQ2-restricted T cell clone specific for hemagglutinin (HA97–113). Using native HA from influenza A/New Caledonia/20/1999 (H1N1) as antigen, we observed increased T cell proliferation with T2.DQ2.DMhigh compared to T2.DQ2 cells as APC, again with the difference not likely to be fully explained by different levels of surface DQ2. The comparable levels of T cell proliferation in response to peptide (HA97–113) presentation despite higher DQ2 on the DMhigh APC is consistent with reduced peptide loading in the presence of an edited DQ2-associated peptide repertoire in these cells (Figure 7B). This result also argues that DMnull APC are comparable to DMhigh APC when the intracellular processing and presentation pathway is by-passed. Last, we tested a DQ2-restricted CD4 T cell clone specific for an epitope within the E7 protein of human papillomavirus (HPV) type 16 (E722–56). Similar to the other viral peptide specific T cells, proliferation was enhanced using DMhigh APC when antigen was provided as intact protein, particularly at lower doses of antigen, where DM editing has a greater effect on the level of MHCII/peptide complex. T cell proliferation was not significantly different using extracellular peptide-pulsing of DMnull and DMhigh APC lines (Figure 7B and 7C, Supplementary Figure 3A and 3B). Thus, DM enhanced presentation of three different, DQ2-restricted viral epitopes after intracellular processing and peptide loading.
Figure 7. DM facilitates the presentation of DQ2-restricted viral antigens.
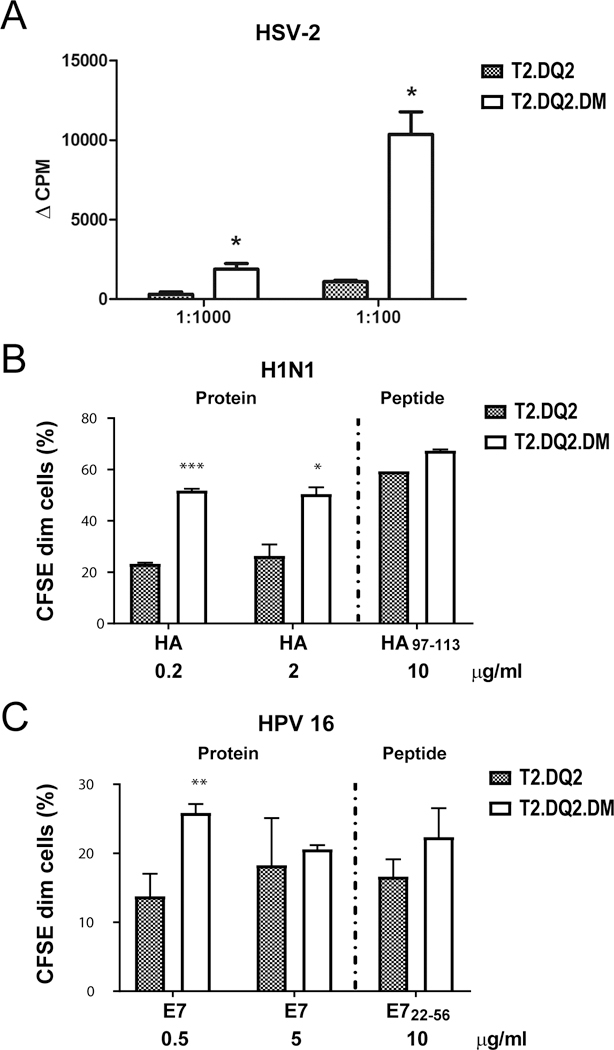
(A) T2.DQ2 or T2.DQ2.DM cells were irradiated and then incubated with inactivated HSV-2 virus and CD4 T-cell clone 1A.B.25, the DQ2-restricted T cell clone specific for VP16431–440 of HSV-2 for 48 hours. Proliferation was measured by [3H]-thymidine incorporation, as described for Figure 2. (B and C) H1N1- and HPV- specific T cell clones were labelled with CFSE before co-culture with T2.DQ2 or T2.DQ2.DM and incubation with (B) recombinant HA proteins from H1N1 A/New Caledonia/20/1999 or peptide HA97–113 and (C) recombinant E7 protein from HPV16 or E722–56, respectively. After 10 days, cells were harvested and analyzed by flow cytometry for the proliferation. Data shown are representative of at least two independent experiments (means ± SD). * P< 0.05, ** P< 0.01, *** P< 0.001.
Discussion
In this paper, we report differential proliferation of DQ2-restricted, antigen-specific CD4+ T cells in response to antigen presented by paired DMhigh/DMnull APC. Each pair is comprised of isogenic lines differing only in the expression of DM. Also, the lines are polyclonal, so that an unusual clonal phenotype arising from a retroviral insertion site is unlikely to influence the assay results. The use of these model APC provides a robust approach to isolate the effect of DM on presentation of DQ2-restricted epitopes.
Using a panel of DQ2-restricted T cell clones specific for different non-self epitopes, we observe a striking pattern: gliadin epitopes associated with developing autoimmunity in CD are DM-sensitive (suppressing their presentation), and viral epitopes related to host defense are DM-resistant (promoting their presentation). Results broadly similar to ours, though not specifically related to presentation by DQ2, show DM antagonism for HLA-DR*04:01-restricted epitopes from glutamate decarboxylase (GAD273–285), a type 1 diabetes autoantigen (49), and type II collagen (CII261–273), a rheumatoid arthritis autoantigen (50), whereas numerous pathogen-derived peptides are DM-resistant (51, 52). Interestingly, in an in vitro antigen presentation assay system, distinct paths for peptide processing and selection were observed for pathogen-derived proteins compared to autoantigens (23).
Expression of DM at higher than physiological levels in cells allowed us to determine that, after intracellular generation, the DQ2/gliadin complexes we studied are DM senstive. Some disease-associated gliadin epitopes, for example, the γ1 epitope, are generated by intracellular processing from larger gliadin fragments produced by digestive enzymes (53). These observations raise the question as to how such gliadin peptides get presented, as they must, because T cells specific for these complexes can be isolated. Unlike our DMhigh APC lines, professional APC (dendritic cells, macrophages and B cells), express substoichiometric DM levels relative to MHCII molecules (54), and the interaction time for DM and class II molecules in endosomal compartments is thought to be limited (55). In addition, in professional APC, access to DM is further compromised for DQ2 by competition with other alleles, including HLA-DR3, which is linked in the DR3/DQ2 haplotype (56), and interacts efficiently with DM (17). Indeed, in B cells expressing DR3, DQ2 and DM, DQ2 is primarily associated with CLIP peptides even at the cell surface, whereas DR3 is effectively edited and carries heterogeneous (non-CLIP) peptide cargo (17). Constitutive DM levels in APC in the small intestine are likely insufficient for effective intracellular editing of DQ2-gliadin complexes, providing a path for presentation of these DM-senstive complexes to T cells.
We also addressed the impact of DM/DQ2 interaction on DM-sensitive epitopes using a mutant, DQ2α+53G, which is a better DM substrate than wild type DQ2. Unlike most MHCII, DQ2 (DQA1*0501/ DQB1*02:01) has a deletion in the alpha chain alpha helix that leads to loss of a hydrogen bond with the peptide backbone that normally stabilizes the complex in the region of the P1 pocket (57). Gliadin-derived epitopes place a proline residue in the P1 pocket of DQ2 and stabilize binding with alternative hydrogen bonds elsewhere in the binding groove (5, 58). However, crystal structures show that the DQ2α53 deletion affects a region predicted to interact with DM (5, 59), and thus likely contributes to diminished DM susceptibility of DQ2. DQ2α+53G rescues DM susceptibility, but as shown here also reduces gliadin peptide binding. Thus, for wild type DQ2, the combination of impaired DM interaction and sufficient gliadin peptide binding lead to efficient presentation of DM-sensitive gliadin peptides.
Our analysis using APC to directly present epitope peptides also showed that DM-sensitive gliadin peptides may be presented to T cells after loading by secondary pathways that avoid the DM/DQ2 interaction. Some gliadin peptides generated in the digestive tract can activate specific CD4+ T cells without further proteolytic processing in intracellular, late endosomal, MHCII compartments (31). Thus, one scenario is binding of such peptides at the plasma membrane or in early endosomes. In these locations, DM editing of peptide/MHC complexes is inefficient, due to low steady state DM levels and pH (6.5–7.4), which is unfavorable for DM action (37). In addition, reduced efficiency of DM editing of the DQ2-bound peptide repertoire during biosynthesis yields surface DQ2/peptide complexes that are particularly susceptible to exchange and presentation of extracellular peptides (60).
Dramatically, we demonstrated that, in contrast to gluten-derived gliadin peptides, three viral antigens triggered significantly increased proliferation of epitope-specific T cell clones in response to DMhigh APC presentation of the corresponding epitopes. Another DQ2-restricted T cell clone specific for EBV gp350 peptide was also found (by one of us) to be stimulated 8-fold more effectively by DMhigh compared to DMnull APC after incubation with native protein antigen (52). In vivo, the presentation of these DQ2-restricted, high affinity epitopes could occur even at low DM levels through competition with CLIP½ and/or endosomal peptides. More likely, perhaps, is that during viral infection DM activity is increased in activated APC (61, 62). In the specific case of immune activation by viruses, this can arise by direct infection of APC, such as DC infection by influenza (63) or B cell infection by EBV (64). Notably, EBV gp42, which is essential for viral entry and binds MHCII, preferentially binds to DQ2 (65). In addition, and especially in the case of viruses like HSV, which may not infect professional APC, detection of viral antigen by professional APC or the microenvironment/cytokine milieu of infected cells may be routes to APC activation and increased DM levels (66–70).
In summary, we propose that, in celiac disease, insufficient DM accessibility by DQ2, impaired DM/DQ2 interaction, moderately increased DQ2/peptide affinity, and bypassing of DM peptide editing all contribute to the uniquely selective DQ2 presentation of DM-sensitive gliadin epitopes. In contrast, presentation of DM-resistant epitopes that form more stable complexes with DQ2 likely relies less on the above mechanisms, as DM editing positively affects presentation of these epitopes. Our findings suggest that the elevation of DM expression in peripheral APC (particularly during infection) may benefit self tolerance by attenuating presentation of DM-sensitive epitopes, while boosting presentation of DM-resistant pathogen-derived epitopes and aiding in host defense.
Supplementary Material
Key Points.
High DM overcomes poor DM/DQ2 interaction to inhibit DQ2 presentation of gliadin.
Relative low strength of gliadin peptide-DQ2 binding contributes when DM is high.
In contrast, abundant DM promotes DQ2 presentation of DM-resistant viral antigen.
Acknowledgments
We thank Dr. Robert Belshe at the Center for Vaccine Development, Saint Louis University, for providing inactivated A/New Caledonia/20/99 (H1N1) influenza. We also thank Dr. Xi Jin from the laboratory of Dr. Chaitan Khosla, Department of Chemistry, Stanford University, for synthesizing gliadin peptides. Also, we thank the Vaccine and Infectious Diseases Division, Fred Hutchinson Cancer Research Center, Seattle, WA 98109 for providing inactivated HSV-2 antigen and T cell clone.
1. This work is supported by funding from the NIH, 5R21DK079163–02 and the Daylight Foundation (to E.D.M.), Stanford Deans Postdoctoral Fellowship (to T.H.), Immunology Program Training Grant, 5T32AI07290–24 (to T.H.), F32 (to T.H.), Stiftelsen Kristian Gerhard Jebsen (project SKGJ-MED-017) (S.W.Q. and L.M.S.) , the Research Council of Norway (project 179573/V40 through the Centre of Excellence funding scheme and project 233885 to S.W.Q. and L.M.S.), the NovoNordisk Foundation (to W.J.), NIH-P01A030071 (to D.M.K.) and R01 AI094019 (to D.M.K.).
3. Abbreviations used in this paper:
- DQ2
HLA-DQ2 (DQA1*05:01, DQB1*02:01)
- DM
HLA-DM
- DR
HLA-DR
- DP
HLA-DP
- MHCII
major histocompatibility complex class II
- MIIC
MHCII containing compartment
- CII
type II collagen
- APC
antigen presenting cells
- Ii
invariant chain
- PT-gluten
pepsin- and trypsin/chymotrypsin-digested gluten
- CLIP
class-II-associated invariant chain peptides
- TG2
transglutaminase 2
- WT
wild type
- CD
celiac disease
- HSV-2
herpes simplex virus, type 2
- H1N1
Hemagglutinin Type 1 and Neuraminidase Type 1
- EBV
Epstein-Barr virus
- HA
hemagglutinin
- VP
viral protein
- gp
glycoprotein
- DC
dendritic cells
Footnotes
Footnotes:
References:
- 1.Matzaraki V, Kumar V, Wijmenga C, and Zhernakova A. 2017. The MHC locus and genetic susceptibility to autoimmune and infectious diseases. Genome Biol 18: 76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nepom GT, and Erlich H. 1991. MHC class-II molecules and autoimmunity. Annu Rev Immunol 9: 493–525. [DOI] [PubMed] [Google Scholar]
- 3.Thorsby E 1997. Invited anniversary review: HLA associated diseases. Hum Immunol 53: 1–11. [DOI] [PubMed] [Google Scholar]
- 4.Mujica-Mota RE, Roberts M, Abel G, Elliott M, Lyratzopoulos G, Roland M, and Campbell J. 2015. Common patterns of morbidity and multi-morbidity and their impact on health-related quality of life: evidence from a national survey. Qual Life Res 24: 909–918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kim CY, Quarsten H, Bergseng E, Khosla C, and Sollid LM. 2004. Structural basis for HLA-DQ2-mediated presentation of gluten epitopes in celiac disease. Proc Natl Acad Sci U S A 101: 4175–4179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Molberg O, McAdam SN, Korner R, Quarsten H, Kristiansen C, Madsen L, Fugger L, Scott H, Noren O, Roepstorff P, Lundin KE, Sjostrom H, and Sollid LM. 1998. Tissue transglutaminase selectively modifies gliadin peptides that are recognized by gut-derived T cells in celiac disease. Nat Med 4: 713–717. [DOI] [PubMed] [Google Scholar]
- 7.van de Wal Y, Kooy Y, van Veelen P, Pena S, Mearin L, Papadopoulos G, and Koning F. 1998. Selective deamidation by tissue transglutaminase strongly enhances gliadin-specific T cell reactivity. J Immunol 161: 1585–1588. [PubMed] [Google Scholar]
- 8.Dendrou CA, Petersen J, Rossjohn J, and Fugger L. 2018. HLA variation and disease. Nat Rev Immunol 18: 325–339. [DOI] [PubMed] [Google Scholar]
- 9.Molberg O, Kett K, Scott H, Thorsby E, Sollid LM, and Lundin KE. 1997. Gliadin specific, HLA DQ2-restricted T cells are commonly found in small intestinal biopsies from coeliac disease patients, but not from controls. Scand J Immunol 46: 103–109. [DOI] [PubMed] [Google Scholar]
- 10.Amirzargar AA, Mohseni N, Shokrgozar MA, Arjang Z, Ahmadi N, Yousefi Behzadi M, Amanzadeh A, and Shokri F. 2008. HLA-DRB1, DQA1 and DQB1 alleles and haplotypes frequencies in Iranian healthy adult responders and non-responders to recombinant hepatitis B vaccine. Iran J Immunol 5: 92–99. [PubMed] [Google Scholar]
- 11.Alper CA, Kruskall MS, Marcus-Bagley D, Craven DE, Katz AJ, Brink SJ, Dienstag JL, Awdeh Z, and Yunis EJ. 1989. Genetic prediction of nonresponse to hepatitis B vaccine. N Engl J Med 321: 708–712. [DOI] [PubMed] [Google Scholar]
- 12.Martinetti M, De Silvestri A, Belloni C, Pasi A, Tinelli C, Pistorio A, Salvaneschi L, Rondini G, Avanzini MA, and Cuccia M. 2000. Humoral response to recombinant hepatitis B virus vaccine at birth: role of HLA and beyond. Clin Immunol 97: 234–240. [DOI] [PubMed] [Google Scholar]
- 13.Audet M, Piardi T, Cag M, Navarro F, Ornis S, Cinqualbre J, Wolf P, and Panaro F. 2011. Hepatitis C recurrence after liver transplantation: has the human leukocyte antigen mismatching at individual loci a role? J Gastroenterol Hepatol 26: 1772–1778. [DOI] [PubMed] [Google Scholar]
- 14.Albayrak A, Ertek M, Tasyaran MA, and Pirim I. 2011. Role of HLA allele polymorphism in chronic hepatitis B virus infection and HBV vaccine sensitivity in patients from eastern Turkey. Biochem Genet 49: 258–269. [DOI] [PubMed] [Google Scholar]
- 15.Blum JS, Wearsch PA, and Cresswell P. 2013. Pathways of antigen processing. Annu Rev Immunol 31: 443–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mellins ED, and Stern LJ. 2014. HLA-DM and HLA-DO, key regulators of MHC-II processing and presentation. Curr Opin Immunol 26: 115–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fallang LE, Roh S, Holm A, Bergseng E, Yoon T, Fleckenstein B, Bandyopadhyay A, Mellins ED, and Sollid LM. 2008. Complexes of two cohorts of CLIP peptides and HLA-DQ2 of the autoimmune DR3-DQ2 haplotype are poor substrates for HLA-DM. J Immunol 181: 5451–5461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hou T, Macmillan H, Chen Z, Keech CL, Jin X, Sidney J, Strohman M, Yoon T, and Mellins ED. 2011. An Insertion Mutant in DQA1*0501 Restores Susceptibility to HLA-DM: Implications for Disease Associations. J Immunol 187: 2442–2452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhou Z, Reyes-Vargas E, Escobar H, Rudd B, Rockwood AL, Delgado JC, He X, and Jensen PE. 2016. Type 1 diabetes associated HLA-DQ2 and DQ8 molecules are relatively resistant to HLA-DM mediated release of invariant chain-derived CLIP peptides. Eur J Immunol 46: 834–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lazarski CA, Chaves FA, Jenks SA, Wu S, Richards KA, Weaver JM, and Sant AJ. 2005. The kinetic stability of MHC class II:peptide complexes is a key parameter that dictates immunodominance. Immunity 23: 29–40. [DOI] [PubMed] [Google Scholar]
- 21.Hall FC, Rabinowitz JD, Busch R, Visconti KC, Belmares M, Patil NS, Cope AP, Patel S, McConnell HM, Mellins ED, and Sonderstrup G. 2002. Relationship between kinetic stability and immunogenicity of HLA-DR4/peptide complexes. Eur J Immunol 32: 662–670. [DOI] [PubMed] [Google Scholar]
- 22.Lazarski CA, Chaves FA, and Sant AJ. 2006. The impact of DM on MHC class II-restricted antigen presentation can be altered by manipulation of MHC-peptide kinetic stability. J Exp Med 203: 1319–1328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kim A, Hartman IZ, Poore B, Boronina T, Cole RN, Song N, Ciudad MT, Caspi RR, Jaraquemada D, and Sadegh-Nasseri S. 2014. Divergent paths for the selection of immunodominant epitopes from distinct antigenic sources. Nat Commun 5: 5369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nanda NK, and Sant AJ. 2000. DM determines the cryptic and immunodominant fate of T cell epitopes. J Exp Med 192: 781–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rinderknecht CH, Lu N, Crespo O, Truong P, Hou T, Wang N, Rajasekaran N, and Mellins ED. 2010. I-Ag7 is subject to post-translational chaperoning by CLIP. Int Immunol 22: 705–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sollid LM, and Jabri B. 2011. Celiac disease and transglutaminase 2: a model for posttranslational modification of antigens and HLA association in the pathogenesis of autoimmune disorders. Curr Opin Immunol 23: 732–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Qiao SW, Bergseng E, Molberg O, Jung G, Fleckenstein B, and Sollid LM. 2005. Refining the rules of gliadin T cell epitope binding to the disease-associated DQ2 molecule in celiac disease: importance of proline spacing and glutamine deamidation. J Immunol 175: 254–261. [DOI] [PubMed] [Google Scholar]
- 28.Koelle DM, Corey L, Burke RL, Eisenberg RJ, Cohen GH, Pichyangkura R, and Triezenberg SJ. 1994. Antigenic specificities of human CD4+ T-cell clones recovered from recurrent genital herpes simplex virus type 2 lesions. J Virol 68: 2803–2810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gebe JA, Novak EJ, Kwok WW, Farr AG, Nepom GT, and Buckner JH. 2001. T cell selection and differential activation on structurally related HLA-DR4 ligands. J Immunol 167: 3250–3256. [DOI] [PubMed] [Google Scholar]
- 30.van der Burg SH, Ressing ME, Kwappenberg KM, de Jong A, Straathof K, de Jong J, Geluk A, van Meijgaarden KE, Franken KL, Ottenhoff TH, Fleuren GJ, Kenter G, Melief CJ, and Offringa R. 2001. Natural T-helper immunity against human papillomavirus type 16 (HPV16) E7-derived peptide epitopes in patients with HPV16-positive cervical lesions: identification of 3 human leukocyte antigen class II-restricted epitopes. Int J Cancer 91: 612–618. [DOI] [PubMed] [Google Scholar]
- 31.Qiao SW, Bergseng E, Molberg O, Xia J, Fleckenstein B, Khosla C, and Sollid LM. 2004. Antigen presentation to celiac lesion-derived T cells of a 33-mer gliadin peptide naturally formed by gastrointestinal digestion. J Immunol 173: 1757–1762. [DOI] [PubMed] [Google Scholar]
- 32.Xia J, Siegel M, Bergseng E, Sollid LM, and Khosla C. 2006. Inhibition of HLA-DQ2-mediated antigen presentation by analogues of a high affinity 33-residue peptide from alpha2-gliadin. J Am Chem Soc 128: 1859–1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sollid LM, Qiao SW, Anderson RP, Gianfrani C, and Koning F. 2012. Nomenclature and listing of celiac disease relevant gluten T-cell epitopes restricted by HLA-DQ molecules. Immunogenetics 64: 455–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Anderson RP, Degano P, Godkin AJ, Jewell DP, and Hill AV. 2000. In vivo antigen challenge in celiac disease identifies a single transglutaminase-modified peptide as the dominant A-gliadin T-cell epitope. Nat Med 6: 337–342. [DOI] [PubMed] [Google Scholar]
- 35.Raki M, Dahal-Koirala S, Yu H, Korponay-Szabo IR, Gyimesi J, Castillejo G, Jahnsen J, Qiao SW, and Sollid LM. 2017. Similar Responses of Intestinal T Cells From Untreated Children and Adults With Celiac Disease to Deamidated Gluten Epitopes. Gastroenterology 153: 787–798 e784. [DOI] [PubMed] [Google Scholar]
- 36.Hou T, Rinderknecht CH, Hadjinicolaou AV, Busch R, and Mellins E. 2013. Pulse-chase analysis for studies of MHC class II biosynthesis, maturation, and peptide loading. Methods Mol Biol 960: 411–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jiang W, Strohman MJ, Somasundaram S, Ayyangar S, Hou T, Wang N, and Mellins ED. 2015. pH-susceptibility of HLA-DO tunes DO/DM ratios to regulate HLA-DM catalytic activity. Sci Rep 5: 17333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Potter PK, Copier J, Sacks SH, Calafat J, Janssen H, Neefjes JJ, and Kelly AP. 1999. Accurate intracellular localization of HLA-DM requires correct spacing of a cytoplasmic YTPL targeting motif relative to the transmembrane domain. Eur J Immunol 29: 3936–3944. [DOI] [PubMed] [Google Scholar]
- 39.Arentz-Hansen H, Korner R, Molberg O, Quarsten H, Vader W, Kooy YM, Lundin KE, Koning F, Roepstorff P, Sollid LM, and McAdam SN. 2000. The intestinal T cell response to alpha-gliadin in adult celiac disease is focused on a single deamidated glutamine targeted by tissue transglutaminase. J Exp Med 191: 603–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Quarsten H, Molberg O, Fugger L, McAdam SN, and Sollid LM. 1999. HLA binding and T cell recognition of a tissue transglutaminase-modified gliadin epitope. Eur J Immunol 29: 2506–2514. [DOI] [PubMed] [Google Scholar]
- 41.Morris P, Shaman J, Attaya M, Amaya M, Goodman S, Bergman C, Monaco JJ, and Mellins E. 1994. An essential role for HLA-DM in antigen presentation by class II major histocompatibility molecules. Nature 368: 551–554. [DOI] [PubMed] [Google Scholar]
- 42.Busch R, Rinderknecht CH, Roh S, Lee AW, Harding JJ, Burster T, Hornell TM, and Mellins ED. 2005. Achieving stability through editing and chaperoning: regulation of MHC class II peptide binding and expression. Immunol Rev 207: 242–260. [DOI] [PubMed] [Google Scholar]
- 43.Busch R, De Riva A, Hadjinicolaou AV, Jiang W, Hou T, and Mellins ED. 2012. On the perils of poor editing: regulation of peptide loading by HLA-DQ and H2-A molecules associated with celiac disease and type 1 diabetes. Expert Rev Mol Med 14: e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Weber DA, Dao CT, Jun J, Wigal JL, and Jensen PE. 2001. Transmembrane domain-mediated colocalization of HLA-DM and HLA-DR is required for optimal HLA-DM catalytic activity. J Immunol 167: 5167–5174. [DOI] [PubMed] [Google Scholar]
- 45.Svensson M, Ramelius A, Nilsson AL, Delli AJ, Elding Larsson H, Carlsson A, Forsander G, Ivarsson SA, Ludvigsson J, Kockum I, Marcus C, Samuelsson U, Ortqvist E, Lernmark A, and g. Better Diabetes Diagnosis study. 2014. Antibodies to influenza virus A/H1N1 hemagglutinin in Type 1 diabetes children diagnosed before, during and after the SWEDISH A(H1N1)pdm09 vaccination campaign 2009–2010. Scand J Immunol 79: 137–148. [DOI] [PubMed] [Google Scholar]
- 46.Fallang LE, Bergseng E, Hotta K, Berg-Larsen A, Kim CY, and Sollid LM. 2009. Differences in the risk of celiac disease associated with HLA-DQ2.5 or HLA-DQ2.2 are related to sustained gluten antigen presentation. Nat Immunol 10: 1096–1101. [DOI] [PubMed] [Google Scholar]
- 47.Rinderknecht CH, Roh S, Pashine A, Belmares MP, Patil NS, Lu N, Truong P, Hou T, Macaubas C, Yoon T, Wang N, Busch R, and Mellins ED. 2010. DM influences the abundance of major histocompatibility complex class II alleles with low affinity for class II-associated invariant chain peptides via multiple mechanisms. Immunology 131: 18–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Koelle DM, Johnson ML, Ekstrom AN, Byers P, and Kwok WW. 1997. Preferential presentation of herpes simplex virus T-cell antigen by HLA DQA1*0501/DQB1*0201 in comparison to HLA DQA1*0201/DQB1*0201. Hum Immunol 53: 195–205. [DOI] [PubMed] [Google Scholar]
- 49.Lich JD, Jayne JA, Zhou D, Elliott JF, and Blum JS. 2003. Editing of an immunodominant epitope of glutamate decarboxylase by HLA-DM. J Immunol 171: 853–859. [DOI] [PubMed] [Google Scholar]
- 50.Amria S, Hajiaghamohseni LM, Harbeson C, Zhao D, Goldstein O, Blum JS, and Haque A. 2008. HLA-DM negatively regulates HLA-DR4-restricted collagen pathogenic peptide presentation and T cell recognition. Eur J Immunol 38: 1961–1970. [DOI] [PubMed] [Google Scholar]
- 51.Nanda NK, and Bikoff EK. 2005. DM peptide-editing function leads to immunodominance in CD4 T cell responses in vivo. J Immunol 175: 6473–6480. [DOI] [PubMed] [Google Scholar]
- 52.Mellins E, Smith L, Arp B, Cotner T, Celis E, and Pious D. 1990. Defective processing and presentation of exogenous antigens in mutants with normal HLA class II genes. Nature 343: 71–74. [DOI] [PubMed] [Google Scholar]
- 53.Shan L, Molberg O, Parrot I, Hausch F, Filiz F, Gray GM, Sollid LM, and Khosla C. 2002. Structural basis for gluten intolerance in celiac sprue. Science 297: 2275–2279. [DOI] [PubMed] [Google Scholar]
- 54.Schafer PH, Green JM, Malapati S, Gu L, and Pierce SK. 1996. HLA-DM is present in one-fifth the amount of HLA-DR in the class II peptide-loading compartment where it associates with leupeptin-induced peptide (LIP)-HLA-DR complexes. J Immunol 157: 5487–5495. [PubMed] [Google Scholar]
- 55.Guerra CB, Busch R, Doebele RC, Liu W, Sawada T, Kwok WW, Chang MD, and Mellins ED. 1998. Novel glycosylation of HLA-DRalpha disrupts antigen presentation without altering endosomal localization. J Immunol 160: 4289–4297. [PubMed] [Google Scholar]
- 56.Price P, Witt C, Allcock R, Sayer D, Garlepp M, Kok CC, French M, Mallal S, and Christiansen F. 1999. The genetic basis for the association of the 8.1 ancestral haplotype (A1, B8, DR3) with multiple immunopathological diseases. Immunol Rev 167: 257–274. [DOI] [PubMed] [Google Scholar]
- 57.Ghosh P, Amaya M, Mellins E, and Wiley DC. 1995. The structure of an intermediate in class II MHC maturation: CLIP bound to HLA-DR3. Nature 378: 457–462. [DOI] [PubMed] [Google Scholar]
- 58.Bergseng E, Xia J, Kim CY, Khosla C, and Sollid LM. 2005. Main chain hydrogen bond interactions in the binding of proline-rich gluten peptides to the celiac disease-associated HLA-DQ2 molecule. J Biol Chem 280: 21791–21796. [DOI] [PubMed] [Google Scholar]
- 59.Pos W, Sethi DK, Call MJ, Schulze MS, Anders AK, Pyrdol J, and Wucherpfennig KW. 2012. Crystal structure of the HLA-DM-HLA-DR1 complex defines mechanisms for rapid peptide selection. Cell 151: 1557–1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhou Z, and Jensen PE. 2013. Structural Characteristics of HLA-DQ that May Impact DM Editing and Susceptibility to Type-1 Diabetes. Front Immunol 4: 262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Adler LN, Jiang W, Bhamidipati K, Millican M, Macaubas C, Hung SC, and Mellins ED. 2017. The Other Function: Class II-Restricted Antigen Presentation by B Cells. Front Immunol 8: 319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Vander Lugt B, Khan AA, Hackney JA, Agrawal S, Lesch J, Zhou M, Lee WP, Park S, Xu M, DeVoss J, Spooner CJ, Chalouni C, Delamarre L, Mellman I, and Singh H. 2014. Transcriptional programming of dendritic cells for enhanced MHC class II antigen presentation. Nat Immunol 15: 161–167. [DOI] [PubMed] [Google Scholar]
- 63.Hargadon KM, Zhou H, Albrecht RA, Dodd HA, Garcia-Sastre A, and Braciale TJ. 2011. Major histocompatibility complex class II expression and hemagglutinin subtype influence the infectivity of type A influenza virus for respiratory dendritic cells. J Virol 85: 11955–11963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Taylor GS, Long HM, Brooks JM, Rickinson AB, and Hislop AD. 2015. The immunology of Epstein-Barr virus-induced disease. Annu Rev Immunol 33: 787–821. [DOI] [PubMed] [Google Scholar]
- 65.Li Q, Bu W, Gabriel E, Aguilar F, Hoshino Y, Miyadera H, Hess C, Hornung RL, Roy A, and Cohen JI. 2017. HLA-DQ beta1 alleles associated with Epstein-Barr virus (EBV) infectivity and EBV gp42 binding to cells. JCI Insight 2: e85687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Baharom F, Thomas S, Bieder A, Hellmer M, Volz J, Sandgren KJ, McInerney GM, Karlsson Hedestam GB, Mellman I, and Smed-Sorensen A. 2015. Protection of human myeloid dendritic cell subsets against influenza A virus infection is differentially regulated upon TLR stimulation. J Immunol 194: 4422–4430. [DOI] [PubMed] [Google Scholar]
- 67.Munz C 2014. Dendritic cells during Epstein Barr virus infection. Front Microbiol 5: 308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zhao X, Deak E, Soderberg K, Linehan M, Spezzano D, Zhu J, Knipe DM, and Iwasaki A. 2003. Vaginal submucosal dendritic cells, but not Langerhans cells, induce protective Th1 responses to herpes simplex virus-2. J Exp Med 197: 153–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Min YH, Lee ST, Choi KM, Hahn JS, and Ko YW. 2000. Surface expression of HLA-DM on dendritic cells derived from CD34-positive bone marrow haematopoietic stem cells. Br J Haematol 110: 385–393. [DOI] [PubMed] [Google Scholar]
- 70.Pezeshki AM, Cote MH, Azar GA, Routy JP, Boulassel MR, and Thibodeau J. 2011. Forced expression of HLA-DM at the surface of dendritic cells increases loading of synthetic peptides on MHC class II molecules and modulates T cell responses. J Immunol 187: 74–81. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.