

Abstract
Electron-deficient chemicals (electrophiles) react with compounds that have one or more unshared valence electron pairs (nucleophiles). The resulting covalent reactions between electrophiles and nucleophiles (e.g., Michael addition, SN2 reactions) are important, not only to Organic Chemistry, but also to the fields of Molecular Biology and Toxicology. Specifically, covalent bond formation is the operational basis of many critically important cellular processes; e.g., enzyme function, neurotransmitter release, and membrane-vesicle fusion. Given this context it is understandable that these reactions are also relevant to Toxicology, since a significant number of xenobiotic chemicals are toxic electrophiles that can react with endogenous nucleophilic residues. Therefore, the purpose of this Review is to discuss electrophile-nucleophile chemistry as it pertains to cell injury and resulting organ toxicity. Our discussion will involve an introduction to the Hard and Soft, Acids and Bases (HSAB) theory of Pearson. The HSAB concept provides a framework for calculation of quantum chemical parameters that classify the electrophile and nucleophile covalent components according to their respective electronic nature (softness/hardness) and reactivity (electrophilicity/nucleophilicity). The calculated quantum indices in conjunction with corroborative in vivo, in chemico (cell free) and in vitro research can offer an illuminating approach to mechanistic discovery. Accordingly, we will provide examples that demonstrate how this approach has been used to discern mechanisms and sites of electrophile action.
Keywords: nucleophiles; environmental pollution; toxicity; α,β-unsaturated aldehydes; type-2 alkenes; electrophilic toxicants
1.0. INTRODUCTION
Electrophiles are electron-deficient chemicals that appear to be involved in toxicity through formation of covalent adducts via electron-rich biological nucleophiles. Human populations are exposed to a complex mixture of electrophilic toxicants derived from environmental, industrial, pharmaceutical and agricultural sources (e.g., 1, 4-naphthoquinone, acrolein, methyl mercury and chlorpyrophos oxon). This diverse exposure could represent potentially serious human health risk that is possibly complicated by synergistic or additive interactions among constituent electrophiles (e.g., Abraham et al., 2011; Bhatnagar, 2006; Bucham, 2016; Andrews and Clary, 1986; Bisesi, 1994; Dejamett et al., 2014; Faroon et al., 2008). Despite the potential risk of electrophile exposure, the mechanistic details of target selection and resulting cytotoxicity are not sufficiently understood. To address this information gap we used parameters derived from the hard and soft, acids and bases (HSAB) theory of Pearson to determine electronic disposition (softness, hardness) and reactivity of the electrophilic component (electrophilic index). In addition, we calculated the respective nucleophilic indices, which provide a measure of the propensity for an electrophile to react with a given nucleophile. Thus, in accordance with HSAB principles, we showed that electrophilic toxicants preferentially formed covalent adducts with nucleophiles of similar softness/hardness (see LoPachin et al, 2012). The toxicological relevance of these quantum mechanical parameters was established in corroborative in chemico and in vivo studies (e.g., see LoPachin et al., 2007a, b). Electrophilic toxicants can therefore be divided into groups according to their respective soft or hard demeanor and corresponding nucleophilic targets. Although electrophilic reactivity is the important determinant of toxic potency, the accuracy of this parameter is dependent upon intervening physicochemical variables that limit target accessibility; e.g., steric hindrance, solubility. Also to be considered in this Review, we will discuss the growing realization that toxic electrophiles do not target specific types of proteins or organelle. Instead, they cause toxicity by disabling protein constituents of an electrophile-receptive proteome. We propose that application of HSAB principles represents a rational basis for determining mechanisms of electrophile toxicity and for predicting the toxicity associated with new or unknown chemicals (see also; Anders, 2017; Schultz et al, 2006; Schwobel et al., 2011; Zhang et al., 2016). Understanding electrophile-based mechanisms of toxicity is also important for development of environmental remediation/avoidance strategies and for devising pharmacotherapeutic approaches to certain diseases. We will begin with a brief discussion of human exposure patterns to electrophilic chemicals that have multiple environmental and/or endogenous sources.
2.0. POSSIBLE SOURCES OF HUMAN EXPOSURE
Human populations are exposed to electrophilic chemicals derived from both anthropogenic (e.g., automobile exhaust, industrial pollution and drug-based toxicity) and natural sources (e.g., wood combustion, certain cheeses, cooking) and there is growing evidence that such exposures can have significant toxic consequences (Faroon et al., 2008; Stevens et al., 2008; Adams et al., 2008; Woodruff et al., 2007; Kumagai and Abiko, 2017; LoPachin and Gavin, 2014, 2012; Leikauf, 2002; Feron et al., 1991; Abraham et al., 2011 Andrews and Clary, 1986 ). For example, electrophiles such as methyl mercury (MeHg), formaldehyde, acrolein and methyl vinyl ketone (MVK) are pervasive contaminants of the ambient environment (air, water, soil; e.g., see Bisesi, 1994; O’Brien et al., 2005; Kehrer and Biswal; 2000). Many chemicals used in the manufacturing and agricultural industries are electrophiles (e.g., n-propylbromide, vinyl chloride) or electrophile-producing protoxicants (e.g., n-hexane, chlorpyrifos) and therefore incidental exposure to these compounds represents a potential source of toxicity (Burcham, 2016; Samet and Wages, 2018; Kumagai and Abiko, 2017; LoPachin and DeCaprio, 2005; LoPachin and Gavin, 2015). Mainstream cigarette smoke, as well as second- and third-hand smoke, contain electrophiles from broad chemical classes; e.g., acrolein, acrylonitrile and cadmium (Bahl et al., 2016; Fujioka and Shibamoto, 2006; Werley et al., 2008). These constituents appear to be directly involved in the toxic consequences of smoking (Bahl et al., 2016; Llewellyn et al., 2009; van der Toorn et al., 2013). The therapeutic benefits of certain clinically important drugs (e.g., acetaminophen, cyclophosphamide, atorvastatin) are limited by biotransformation of the parent compounds to reactive electrophilic metabolites that subsequently produce toxicity; e.g., acetaminophen→N-acetyl-p-benzoquinone imine, cyclophosphamide →acrolein (Kalgutkar and Dalvie, 2015; Stachulski et al., 2012; Gurtoo et al., 1981). Cisplatin and other platinum (Pt)-based antineoplastic drugs (e.g., carboplatin, oxaliplatin) are highly effective and widely used in the treatment of solid tumors mainly of the testis, ovary, cervix, neck and bladder. Cisplatin chemotherapy is, however, frequently (40% - 50%) associated with a painful dose-dependent chemotherapy-induced peripheral neuropathy (CIPN; Seretny et al., 2014; Staff et al., 2017). This adverse outcome is likely mediated by the soft electrophilic character of platinum, which causes irreversible damage to sensory neurons in the dorsal root ganglion (Qing et al., 1996; Wang et al., 1996).
Growing evidence indicates that acrolein, 4-hydroxy-2-nonenal (HNE) and other unsaturated aldehyde electrophiles mediate the oxidative stress-induced pathogenic processes that appear to underlie many disease and tissue injury states; e.g., Alzheimer’s disease, atherosclerosis, diabetes and spinal cord injury (reviewed in Csala et al., 2015; LoPachin et al., 2008a, 2009a; Moghe et al., 2015; O’Brien et al., 2005; Shi et al., 2011). In this regard, research also suggests that inflammatory responses which often accompany pathogenic processes are mediated by endogenous electrophiles; e.g., acrolein, crotonaldehyde, acetaldehyde (Noerager et al., 2015; van der Toorn et al., 2013; Yin et al., 2015). Additional research indicates that environmentally-derived electrophilic aldehydes (e.g., acrolein, crotonaldehyde) with similar mechanisms of toxicity might act additively or synergistically with their endogenous counterparts to accelerate the onset and development of certain diseases (e.g., see Conklin et al., 2010; Wang et al., 2008; Dejarnett et al., 2015; Luo et al., 2007). Thus, humans are exposed to electrophilic toxicants through contact with diverse sources; e.g., atmospheric, personal, pharmaceutical and occupational environments. The possibility that environmental and endogenous electrophiles might interact to augment toxicity is a significant concern (see ahead).
3.0. TOXICOLOGICAL CONSEQUENCES OF COVALENT REACTIONS.
The formation of drug-receptor complexes in pharmacology involves short distance forces such as hydrogen-bonding, hydrophobic or van der Waals interactions. Reversible drug-receptor occupancy alters activity of the respective signal-transduction pathway, which subsequently initiates a change in cell physiology. In contrast, many toxic chemicals and/or their active metabolites exhibit electron deficient centers and are therefore classified as electrophiles (electron seeking). These chemicals form covalent bonds (e.g., 1,4-Michael addition) with electron rich nucleophilic sites; e.g., the sulfhydryl thiolate state of cysteine, ε-amino group of lysine or the N2 nitrogen of deoxyguanosine. Exposure of biological systems to electrophiles can cause cytotoxicity since the formation of covalently bonded adducts with specific nucleophilic sites can irreversibly disable the functions of enzymes, DNA, cytoskeletal proteins and other biological macromolecules (Fig. 1). Thus, for example, abundant evidence now indicates that α,β-unsaturated carbonyl derivatives of the type-2 alkene chemical class (e.g., acrolein, acrylamide, 4-hydroxy-2-nonenal) cause toxicity by forming Michael adducts with anionic sulfhydryl thiolate sites in the active zones of many cysteine-regulated enzymes (e.g., see Doorn and Petersen, 2002, 2003; Eliuk et al., 2007; Fritz et al., 2011, 2013; LoPachin et al., 2009a; Martyniuk et al., 2011; Seiner et al., 2007).
Figure 1.

This figure is a schematic representation of the major mechanistic aspects of a soft electrophile-initiated toxic cascade. Soft electrophiles preferentially form Michael adducts with soft nucleophilic thiolate residues. This promotes initial cellular GSH depletion and inactivation of proteins that are constituents of specific soft electrophile responsive proteomes. The resulting mitochondrial injury initiates cellular oxidative stress via generation of superoxide anions (O2−.) and hydrogen peroxide (H2O2). Through metal-catalyzed Fenton reactions, these free radicals generate highly reactive hydroxyl radicals (OH−.) that can cause direct macromolecular damage. In addition, the hydroxyl and superoxide radicals can initiate peroxidation of membrane polyunsaturated fatty acids to yield α,β-unsaturated aldehydes (e.g., acrolein, 4-hydroxy-2-nonenal). As soft electrophiles, these endogenous aldehyde toxicants contribute to the cellular electrophile burden and can thereby augment cytotoxicity. Electrophile toxicity can be muted by activation of antioxidant cellular stress responses (e.g., sirtuin 1 deacetylase enzyme, Keap1/Nrf2 pathway). Hard-hard covalent interactions cause cytotoxicity via a common mechanism; i.e., disruption of discrete hard electrophile-sensitive cellular proteomes. Our recent research has shown that soft electrophile-mediated cascades (e.g., acrolein exposure; NAPQI intoxication) can be prevented by multifunctional enolate-forming compounds (e.g., 2’, 4’ 6’-trihydroxyacetophenone) that act as soft nucleophilic surrogate targets (LoPachin et al., 2016). Thus, since many pathogenic processes are mediated by soft electrophiles, soft nucleophilic scavengers could be highly effective cytoprotectants.
Covalent electrophile-nucleophile reactions are not arbitrary and are instead relatively selective as predicted by the HSAB theory of Pearson (1990). According to HSAB principles, electrophilic and nucleophilic species are classified as either “soft” or “hard” based on polarizability or the ease with which corresponding electron density can be delocalized to form a covalent bond. Remote electrons that are less influenced by the nucleus or those that occupy a larger volume (cloud) are more readily displaced into new bonding patterns. For example, the type-2 alkenes are designated as soft electrophiles because the delocalized π electrons are mobile. The corresponding electron cloud is stretched over four nuclear centers (C=C-C=0) based on the orbital interactions of the electron-withdrawing carbonyl group and the alkene moiety. As a consequence, the extended electron cloud is easily distorted and is by definition soft (polarizble). In contrast, hard electrophilic toxicants (e.g., 2,5-hexanedione, chlorpyrifos, and vinyl chloride) have highly localized non-extended charge densities at specific electron deficient centers. These chemicals are therefore characterized by low electron polarizability.
Nucleophiles are also designated as either soft or hard based on the polarizability of corresponding frontier shell electrons. Elements with large atomic radii such as sulfur have outer-shell electrons that are relatively far from the nucleus and are consequently highly polarizable. Thiol ionization (i.e., SH→S−) and the consequential expansion of the anionic cloud yield the relatively soft (easily polarizable) sulfhydryl thiolate nucleophile. In contrast, nitrogen and oxygen nucleophiles have relatively small atomic radii and, accordingly, their electron clouds are less susceptible to distortion. Such atoms are therefore harder nucleophiles; e.g., the N2 nitrogen of deoxyguanosine.
4.0. QUANTITATIVE HSAB DESCRIPTORS OF COVALENT REACTIONS.
Covalent bond formation between reacting chemicals involves the electronic properties of the respective outermost orbitals. Consequently, the most important orbitals are the highest energy orbital that contains electrons (HOMO = Highest Occupied Molecular Orbital) and the lowest energy orbital that is vacant (LUMO = Lowest Unoccupied Molecular Orbital). The formation of a covalent adduct can be described as the overlap of the respective frontier orbitals and the transfer of electron density from the donating HOMO of the nucleophile to the recipient LUMO of the electrophile. The respective energies of the frontier molecular orbitals (ELUMO and EHOMO) are known and can be used to calculate corresponding hardness (η =[ELUMO - EHOMO]/2] and softness (σ = 1/η). Within this context, softness is an index of the relative ease with which electron density is transferred from the nucleophile to the electrophile during covalent bond formation. This parameter is related to the rate of the adduct-forming reaction. Finally, values of σ and η can be combined with other HSAB descriptors to estimate the propensity of an electrophile to undergo an adduct reaction. Specifically, the electrophilic index (ω) is a comprehensive measure of electrophilicity that combines softness and chemical potential (μ): ω = ½ σμ2. The latter parameter (μ = [ELUMO + EHOMO] / 2) represents the ability of an electrophilic or nucleophilic species to undergo chemical change. Calculations of electrophilicity can provide quantitative information about the transition state energies involved in toxicant-protein covalent bond formation. Thus, values for ω correspond to the rate constant (k) of these adduct reactions and, as a consequence, are directly related to toxicant potency (see LoPachin et al., 2007a,b; 2009a). With respect to the nucleophile, the corresponding molecular orbital energies can also be used to calculate nucleophilic softness and chemical potential. In addition, the likelihood that a given nucleophile (A) will form an adduct with a given electrophile (B) can be predicted by calculating a nucleophilicity index (ω−); ω− = ηA (μA - μB)2/2(ηA + ηB)2. This parameter considers the hardness (η) and chemical potential (μ) of both the electrophilic and nucleophilic reactants. The electrophilic (ω) and nucleophilic (ω−) indices have been demonstrated to be reliable descriptors for a variety of electrophile-nucleophile interactions (LoPachin and Gavin 2012; LoPachin et al., 2008; 2012).
5.0. HSAB PRINCIPLES: PROVIDING MECHANISTIC INSIGHT INTO ELECTROPHILE TOXICITY
The HSAB model stipulates that toxic electrophiles will react preferentially with nucleophilic biological targets of comparable softness or hardness. Thus, for example, the conjugated α,β-unsaturated carbonyl structure of acrylamide (ACR), methyl vinyl ketone (MVK) and other type-2 alkenes is a soft electrophile that forms Michael-type adducts via second-order addition reactions with soft nucleophilic side chains of peptide amino acids (Table 1). Results from detailed studies suggest that unsaturated alkenes react faster with cysteine sulfhydryl groups than with respective primary and secondary nitrogen nucleophiles on lysine ε-amino groups and imidazole side chains of histidine. These kinetic differences indicate that cysteine residues are the preferred sites of type-2 alkene adduct formation; e.g., see Cai et al., 2009; Doorn and Petersen, 2003; LoPachin et al., 2007a,b; Martyniuk et al., 2011. However, it is important to recognize that not all cysteine sulfhydryl groups are functionally relevant and, therefore, it cannot be assumed that covalent adduction of these residues has toxicological significance (LoPachin and Barber, 2006). Substantial evidence, nonetheless, indicates that type-2 alkenes and other electrophiles; e.g., the ortho-quinone metabolite of dopamine; the acetaminophen metabolite, N-acetyl-p-benzoquinone imine (NAPQI) cause cytotoxicity via a common molecular mechanism involving the formation of irreversible adducts at specific cysteine residues (e.g., Cys280 of sirtuin 3; Cys152 of glyceraldehyde 3-phosphate dehydrogenase (GAPDH); Cys374 of actin; Dalle-Donne et al., 2007; Fritz et al., 2011; Leeming et al., 2015; Martyniuk et al., 2011). At physiological conditions (pH = 7.4), cysteine sulfhydryl groups exist mostly in the weakly nucleophilic thiol (0) state (Table 2) and are therefore not kinetically favorable targets for soft type-2 alkene electrophiles. Basic research complemented by calculations of HSAB parameters have demonstrated that amino acid nitrogen groups (histidine, lysine) are hard, relatively weak nucleophiles and therefore are unfavorable targets for soft electrophiles (Barber and LoPachin, 2004; Barber et al., 2007; LoPachin et al., 2007a,b; LoPachin et al., 2009). Ionization of cysteine sulfhydryl groups, in contrast, yields the anionic thiolate (−1). As reflected in the corresponding higher ω− values, this is a soft, highly nucleophilic state that reacts correspondingly faster with soft type-2 alkene electrophiles (Table 2). It is noteworthy that as a second-order reaction, the relative rates (k2) of these soft-soft covalent adduct reactions will also vary as a function of the inherent reactivity (electrophilicity) of the electrophile component.
Table 1.
Calculated quantum mechanical parameters for selected electrophilic toxicants (parent compound or metabolite).a
| Compound | Structure | Softness (σ, 10−3 eV−1 |
Hardness (η, eV) |
Electrophilicity (ω, eV) |
|---|---|---|---|---|
| APAP | 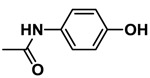 |
217.86 | 4.59 | 0.92 |
| NAPQI (reactive metabolite of APAP) |  |
523.56 | 1.91 | 7.08 |
| n-Hexane (industial solvent) | 184.67 | 5.42 | 0.71 | |
| 2,5-Hexanedione (metabolite of n-hexane) | 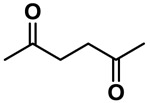 |
314.96 | 3.18 | 2.01 |
| Valproate (pharmaceutical) |  |
404.04 | 4.48 | 0.009 |
| Acrolein (industrial and endogeneous unsaturated aldehyde derivative | 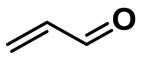 |
371.75 | 2.69 | 3.57 |
| Acrylamide (food contaminant) |  |
327.33 | 3.06 | 2.52 |
| Cisplatin (platinum-based antipeoplastic) |  |
400 | 2.50 | 3.43 |
Ground state equilibrium geometries were calculated for each structure with DF B3LYP-6-31G* in water from 6-31G* initial geometries. Values obtained were used to calculate σ and ω (see text).
Table 2.
Calculated quantum mechanical parameters for selected nucleophilic targets.a
| Compound | Structure of R-group |
Softness (σ, eV−1) |
Hardness (η, eV) |
Nucleophilicity w/ Acrolein (ω−, eV) |
Nucleophilicity w/ Chlorpyrifos (ω−, eV) |
|---|---|---|---|---|---|
| Cysteine | 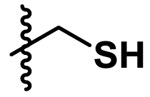 |
1.724 | 0.58 | 0.0502 | 0.0421 |
| Cysteine anion |  |
0.601 | 1.67 | 0.6388 | 0.6097 |
| Serine |  |
0.289 | 3.46 | 0.0610 | 0.0485 |
| Serine anion |  |
0.373 | 2.68 | 0.9790 | 0.9414 |
| Lysine cation |  |
0.259 | 3.86 | 0.0632 | 0.0505 |
| Lysine | 0.296 | 3.38 | 0.0830 | 0.0684 | |
| Histidine cation |  |
0.321 | 3.115 | 0.0092 | 0.0045 |
| Histidine |  |
0.298 | 3.36 | 0.1146 | 0.0976 |
Ground state equilibrium geometries were calculated for each structure with DF B3LYP-6-31G* in water from 6-31G* initial geometries. Values obtained were used to calculate σ and ω (see text).
With respect to hard-hard interactions, the neurotoxic n-hexane metabolite, 2,5-hexanedione (2,5-HD), is a hard electrophile (Table 2) that preferentially forms 2,5-dimethyl-pyrrole adducts with hard nucleophilic nitrogen atoms of the ε-amino groups on lysine residues of neurofilaments and other cytoskeletal proteins. These aberrant proteins populated the characteristic giant axonal swellings in distal peripheral nerves of intoxicated humans and laboratory animals. The swellings were presumed to be responsible for the γ-diketone axonopathy associated with subchronic occupational exposure to n-hexane (DeCaprio et al., 1997; Graham et al., 1991). Accordingly, previous studies (reviewed in LoPachin and DeCaprio, 2004, 2005) demonstrated the presence of abundant high molecular weight neurofilament derivatives in nervous tissue preparations from 2,5-HD-intoxicated animals. However, more recent research has shown that these abnormal proteins were common to nervous tissue samples from both control and 2,5-HD intoxicated animals. As a result, the corresponding pathognomonic relevance is uncertain. Subsequent studies (Zhang et al., 2010) indicated that 2,5-HD selectively impaired the binding of microtubule-associated proteins (MAPs) to microtubules through adduction of lysine residues that mediate these protein-protein interactions. The critical role of MAPs in cytoskeletal structure and function suggests that these lysine residues are toxicologically relevant targets for 2,5-HD. Hard-hard reactions also play a critical role in the antineoplastic mechanism of platinum (Pt)-based chemotherapy. Specifically, Pt has hard electrophilic attributes that can form adducts with hard nucleophilic residues on DNA (e.g., guanine N7). Pt binding of DNA disrupts transcription which leads to cancer cell death (Wang et al., 1996).
6.0. THE CYSTEINE-CENTERED CATALYTIC TRIAD: A MOLECULAR TARGET FOR SOFT ELECTROPHILE TOXICITY
Our discussion has thus far indicated that the soft sulfhydryl thiolate state is the preferential target for the type-2 alkenes and other soft electrophiles. However, at the intracellular pH range (7.0-7.4), sulfhydryl groups exist largely in the non-reactive thiol (0) state. What therefore is the molecular condition that creates an available thiolate protein target? Highly nucleophilic sulfhydryl thiolate groups are found in cysteine-centered catalytic triads and other microenvironments that lower side-chain pKa values (discussed in LoPachin and Gavin, 2012; LoPachin et al., 2009; LoPachin and Barber, 2006). The aforementioned selective binding of soft electrophiles to specific cysteines suggests that these residues exist in a pKa lowering microenvironment. Research has revealed that these specialized amino acid configurations are located in the enzyme active zones where the nucleophilicity of the thiolate is involved in protein function. For example, GAPDH catalyzes the conversion of glyceraldehyde 3-phosphate (G3P) to d-glycerate 1,3-bisphosphate. The initial step in this process is dependent upon Cys152 which mediates nucleophilic attack on the G3P carbonyl to yield a hemithioacetal intermediate. Normally, this reaction is regulated by reversible binding of redox modulators, e.g., nitric oxide (NO), hydrogen peroxide (H2O2), at thiolate acceptors of enzyme catalytic triads. In this regard, Cys152 of GAPDH is a thiolate-based nitric oxide (NO) acceptor (Mohr et al., 1994). More broadly, NO signaling regulates synaptic strength by modulating the activity of proteins involved in the synaptic vesicle cycle; e.g., NEM-sensitive factor (neurotransmitter release), the dopamine-transporter (re-uptake) and the vesicular monoamine transporter (vesicular monoamine transporter; Kiss 2000; Rudkouskaya et al., 2010). Our research suggested that these and other synaptic proteins were inactivated by ACR and that this leads to a disruption of neurotransmission at CNS synapses (Barber et al., 2004, 2007; LoPachin et al., 2004, 2007a,b, 2009). Since the ionization states of cysteine residues in catalytic triads play a direct role in enzyme function, irreversible acceptor adduction by ACR and other soft electrophiles will block redox signaling and directly inhibit enzyme function. Depending on the proteome affected (see ahead), this can disable broad cytophysiological processes thereby leading to cell damage and toxicity. It is important to note that electrophiles discriminate targets based on the favorability of the corresponding covalent reaction. This favorability is determined by the kinetics of the reaction which includes the relative reactivities (ω; ω−) of the electrophile and nucleophile components.
7.0. THE CELLULAR ELECTROPHILE-RESPONSIVE PROTEOME
The research discussed thus far indicates that the toxicology of soft electrophiles involves formation of irreversible adducts with soft nucleophilic thiolate groups located in active zones of many proteins. There is now evidence that electrophile-induced toxicity is mediated by the inactivation of multiple protein types by a given electrophile (e.g., see Barber et al., 2007; Barber and LoPachin, 2004). The size of the affected proteome is determined by the relative electrophilicity (ω) of the toxicant. Accordingly, a highly reactive electrophile (large ω) will form adducts with a board range of nucleophilic targets that vary with respect to nucleophilicity (ω−). In contrast, toxicants with lower electrophilicity (ω) will react preferentially with those target sites exhibiting higher nucleophilicity. The restricted availability of reactive targets reduces the size of the responsive proteome (see Martyniuk et al., 2011). The sensitive proteins are collectively known as an electrophile-responsive proteome (Ceaser et al., 2004; Higdon et al., 2012). Also contributing to the proteome size are additional physicochemical attributes such as active site accessibility, pKa and turnover of the constituent proteins. In essence, the proteome vulnerability serves to amplify the toxicological consequences of individual protein dysfunction.
Our research (Barber et al., 2007; Barber and LoPachin, 2004) identified a presynaptic ACR-sensitive proteome that included adduct formation with cysteine-regulated proteins mediating: 1) synaptic vesicle cycling; 2) mitochondrial/glycolytic energy production and 3) protein degradation. When considered in toto, the distribution of prote6in adducts clearly incorporated the respective proteomes for synaptic vesicles and presynaptic active zone (Burre and Volknandt, 2007; Morciano et al., 2009). Accordingly, our functional studies indicated that unsaturated carbonyl derivatives (e.g., ACR, acrolein and HNE) had broad inhibitory effects on presynaptic vesicle cycling (Barber et al., 2004; LoPachin et al., 2004, 2006, 2007a,b, 2009) that corresponded to the onset and development of neurological deficits. That soft electrophiles react with soft nucleophilic thiolate sites on proteins within a given proteome appears to be a general mechanistic pattern. For example, the severe liver damage associated with acetaminophen (Tylenol™) overdose is mediated by the highly reactive metabolite, N-acetyl-p-benzoquinone imine (NAPQI). This soft electrophile causes hepatocyte injury by depleting glutathione (GSH) and by forming Michael adducts with soft nucleophilic thiolate sites on liver cell proteins. Research suggests that the NAPQI-sensitive proteome incorporates specific cysteine-directed proteins from a number of subcellular organelles, pathways and regions; e.g., cytoplasm, mitochondria (Dietze et al., 1997; Hoffmann et al., 1985; Leeming et al., 2015; Reid et al., 2005). Therefore, putative mechanisms of toxicity should incorporate the concept of electrophile (soft or hard)-induced damage to responsive cellular proteomes.
Alternatively, the proteome can be narrowly populated with respect to nucleophile target diversity. For example, the organophosphate insecticide, chlorpyrifos (Cpf), is metabolized to chlorpyrifos oxon (Cpo), a highly reactive hard electrophile. Acetyl cholinesterase (AChE) activity is selectively inhibited by Cpo adduction of serine oxyanion, a hard nucleophile located at the terminus of the AChE gorge. Because this enzyme is responsible for acetylcholine (ACh) metabolism, the irreversible inhibition of this enzyme causes cholinergic neurotoxicity through an increase in synaptic acetylcholine. Cpo can selectively gain access to the nucleophilic terminus because it exhibits many of the spatial and electronic characteristics of ACh and accordingly, interacts with AChE as an ACh analogue (Ripoll et al, 1993).
8.0. PHYSICOCHEMICAL AND CELLULAR FEATURES THAT INFLUENCE SOFT ELECTROPHILE TOXICITY
Although the relative electrophilicity (ω) of a chemical is an important determinant of corresponding toxicity, it cannot be assumed that this chemical will carry the risk of toxicity since other physicochemical, toxicokinetic and cellular features can influence the covalent reaction with a nucleophile. Thus, for example, in previous studies of toxic unsaturated carbonyl derivatives (LoPachin et al., 2008a,b), we found that the corresponding electrophilicity (ω) and softness (σ) values for 4-hydroxy-2-nonenal (HNE) exceeded those for acrolein or methyl vinyl ketone (MVK; Table 1). This suggested that HNE was a more significant toxicant than either acrolein or MVK. However, when the respective toxic potencies (IC50 of synaptosomal sulfur depletion) and adduct rate constants (k2) were determined as indices of actual toxicity, the HNE values were significantly lower than predicated by the aforementioned HSAB calculations (Table 1; LoPachin et al., 2009a,b; LoPachin and Gavin, 2014). Whereas this inconsistency might suggest that ω and σ cannot accurately predict chemical reactivity and therefore toxicity, the extended alkane tail of HNE could impede access to the active sites of many enzymes. In this regard, the reduced toxicity might be caused by unfavorable reaction kinetics that could arise from steric hindrance.
Also influencing the onset and development of toxicity is the induction of glutathione transferase (GST), a Phase II enzyme that catalyzes GSH-electrophile conjugation. Other physicochemical attributes such as solubility and acid-base equilibrium can also influence the correspondence between experimentally derived electrophile behavior and that expected based on HSAB calculations (LoPachin et al., 2012; LoPachin and Gavin, 2014). These types of disagreements are expected since the HSAB algorithms do not incorporate these physicochemical and toxicokinetic properties. Nonetheless, results can be correctly interpreted since these mitigating physicochemical traits can be recognized by their structural characteristics; e.g., compare the structural differences that characterize acrolein (non-hindered), citral (hindered) and crotonaldehyde (partially hindered; LoPachin and Gavin, 2014).
In addition to the aforementioned physicochemical and toxicokinetic features, certain cell-based characteristics can also shape electrophile toxicity. In this regard, ACR, methyl acrylate (MA), ethyl methacrylate (EMA) are unlikely toxicants given their low electrophilic reactivity and consequential slow adduct formation (Table 2). Nonetheless, we showed that ACR intoxication was associated with nerve terminal dysfunction and eventual degeneration in rat brain and spinal cord (LoPachin et al., 2003; LoPachin and Gavin, 2012). Animals Intoxicated over a very broad ACR exposure-range (1.0-50 mg/kg/d) expressed hindlimb skeletal muscle weakness, decreased grip strength, gait incoordination and weight loss (LoPachin et al., 2002). Early studies revealed that neurotoxicity was a selective effect of ACR since other organ toxicity (e.g., liver, kidney) were not identified (LoPachin et al., 2003).
The presynaptic focus revealed in our studies did not appear to be due to ACR targeting of nerve terminals, rather it involved the influence of cell-specific features that predispose nerve terminals to soft electrophile toxicity. Thus, ACR is a water-soluble unsaturated alkene with a large volume of distribution that includes the CNS (Barber et al., 2001). Presynaptic neurotransmission is a complex process that is highly vulnerable to electrophile attack since it involves the coordinated function of multiple NO/cysteine-regulated proteins (LoPachin et al., 2003; 2008; LoPachin and Barber, 2006; LoPachin and Gavin, 2012, 2014; 2015). The nerve terminal is therefore a target rich environment for soft electrophiles. This nerve region is also anatomically separated from the cell body and is therefore devoid of transcriptional or translational capabilities. As a consequence, this distal region is limited with respect to mounting transcription-based reparative or protective reactions; e.g., the Nrf2-Keap1 antioxidant response (Zhang et al., 2011). In the absence of synthetic processes, the nerve terminal proteome must be maintained by perikaryal protein synthesis and subsequent anterograde transport. The turnover rates of many presynaptic proteins are slow (Calakos and Scheller, 1996; Katyare and Shallom, 1988), presumably as an attempt to limit cell body stress through reduced material expenditure and increased efficiency. However, research indicates that because of slow turnover rates proteins inactivated through cysteine adduct formation are replaced slowly leading to a gradual deficit of normal functioning proteins. In this regard, proteomic analyses (Barber and LoPachin, 2004; Barber et al., 2007) have shown that presynaptic cysteine adducts build-up in correspondence with the cumulative development of ACR neurological symptoms.
9.0. SUMMARY
Toxic electrophilic chemicals are an endemic component of the human biosphere. Specifically, humans are exposed to complex mixtures of soft and hard electrophiles that are derived from a variety of anthropogenic and natural sources. It is likely that this diverse exposure carries significant health risks and possibly augments development of disease processes that are mediated by endogenous electrophiles. Yet, despite this potential, the mechanism(s) of electrophile toxicity has not been fully elucidated. Our understanding of the electrophile-sensitive proteome also requires further refinement. In particular, it is important to know the physicochemical features of the electrophile/nucleophile reactants that shape the size and corresponding toxicological consequences of the inhibited proteome. In this regard, the responsive proteome might include electrophile sensitive proteins of mitochondrial origin, it should be realized, however, that this might not necessarily constitute specific targeting of this organelle. Also not well defined are the sequence of cytotoxic events that lead to cell injury and the intervening role of endogenous cytoprotective pathways (e.g., sirtuin 1 deacetylase enzyme, Keap1/Nrf2 pathway) in tempering this electrophile toxicity (see related discussions in Fritz and Petersen, 2013; Grimsrud et al., 2008; LoPachin and DeCaprio, 2005). Furthermore, it is of significant toxicological relevance to determine whether electrophiles can interact additively or synergistically. Thus, electrophiles from different sources might interact leading to acceleration of the disease processes or environmental toxicity. Defining mechanisms of electrophile toxicity is critical toward deciphering the relationship between electronic structure (soft, hard) and electrophile reactivity (electrophilicity). Such information represents a rational basis for predicting toxicity associated with new or unknown chemicals. A mechanistic understanding is also important for development of environmental remediation/avoidance strategies and for devising pharmacotherapeutic approaches to certain disease processes that involve environmental and/or endogenous electrophiles (e.g., see LoPachin et al., 2016).
Achieving an adequate level of mechanistic understanding however appears daunting when the diversity of electrophilic chemicals is presumed to reflect a diversity of corresponding molecular mechanisms. As presented in this Perspective, we have used calculated HSAB parameters (σ, η, ω and ω−) and knowledge of intervening physicochemical features to untangle the search for a rational electrophile mechanism (Fig. 1). Thus, we have realized that ACR, HNE, methylacrylate (MA) and acrolein are members of the same chemical family; i.e., α,β-unsaturated carbonyl derivatives. HSAB-based calculations of softness (σ) and hardness (σ) indicate that these chemicals are soft electrophiles of variable electrophilicity (ω; Table 1). According to HSAB principles, soft electrophiles preferentially form covalent adducts with the highly nucleophilic soft thiolate state of cysteine. In support of this theory, we and others have shown that ACR, HNE, MA and acrolein form Michael adducts with the thiolate state of cysteine-centered catalytic triads. Research has demonstrated that irreversible covalent reactions are responsible for protein inhibition and subsequent toxicity. We propose that our application of HSAB principles represents a rational approach toward deciphering molecular mechanisms of toxic electrophiles.
10.0.
FUNDING
The research discussed in this Perspective was supported by NIH grants from the National Institutes of Environmental Health Sciences RO1 ES03830-30; RO1 ESO7912-11.
ABBREVIATIONS
- HSAB
hard and soft, acids and bases
- HIV
human immunodeficiency virus
- DNA
deoxyribonucleic acid
- RNA
ribonucleic acid
- FMO
frontier molecular orbital
- LUMO
lowest unoccupied molecular orbital
- HOMO
highest occupied molecular orbital
- Cys
cysteine
- EA
ethyl acrylate
- MMA
methyl methacrylate
- ELUMO
LUMO energy
- PTP1B
protein tyrosine phosphate 1B
- LD50
lethal oral dose for 50% of the population
- HNE
4-hydroxy-2-nonenal
- ONE
4-oxy-2-nonenal
- NAC
N-acetylcysteine
- NSF
N-ethylmaleimide sensitive factor
- SNAP-25
Synaptosomal-associated protein of 25 kDa
- AGEs
advanced glycation end products
- CMC
S-(carbonxymethyl)cysteine
- SIRT3
mitochondrial sirtuin3
- GSTP1-1
glutathione S-transferase P1-1
- GAPDH
glyceraldehydes 3-phosphate dehydrogenase
- NO
nitric oxide
- H2O2
hydrogen peroxide
- Nrf2/Keap1
nuclear factor erythroid 2-related factor 2/kelch-like erythroid cell-derived protein with CNS homology-associated protein 1
- eV
electron volt
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
11.0. REFERENCES
- 1.Anders MW (2017) Diacetyl and related flavorant α-diketones: biotransformation, cellular interaction and respiratory-tract toxicity. Toxicology 388: 21–29. [DOI] [PubMed] [Google Scholar]
- 2.Abraham K, Andres S, Palavenskas R, Berg K and Appel KE (2011) Toxicology and risk assessment of acrolein in food. Mol. Nutr. Food Res 55, 1277–1290. [DOI] [PubMed] [Google Scholar]
- 3.Adams TB, Gavin CL, Taylor SV, Waddell WJ, Cohen SV, Feron VJ, Goodman, Rietjens IMCM, Marnett LJ, Portoghese PS and Smith RL (2008) The FEMA GRAS assessment of α,β-unsaturated aldehydes and related substances used as flavor ingredients. Food Chem. Toxicol 46, 2935–2967. [DOI] [PubMed] [Google Scholar]
- 4.Andrews LS and Clary JJ (1986) Review of the toxicity of multifunctional acrylates. J. Toxicol. Environ. Health 19, 149–164. [DOI] [PubMed] [Google Scholar]
- 5.Bahl V, Weng N, Schick SF, Sleiman M, Whitehead J, Ibarra A and Talbot P. (2016) Cytotoxicity of third-hand smoke and identification of acrolein as a volatile thirdhand smoke chemical that inhibits cell proliferaction. Toxicol Sci 150; 234–246. [DOI] [PubMed] [Google Scholar]
- 6.Barber DS and LoPachin RM (2004) Proteomic analysis of acrylamide-protein adduct formation in rat brian synaptosomes. Toxicol Appl Pharmacology 201, 120–136. [DOI] [PubMed] [Google Scholar]
- 7.Barber DS, Stevens S and LoPachin RM (2007) Proteomic analysis of rat striatal synaptosomes during acrylamide intoxication at a low dose-rate. Toxicol Sci 100:156–167 [DOI] [PubMed] [Google Scholar]
- 8.Bhatnagar A (2006) Environmental cardiology: studying mechanistic links between pollution and heart disease. Circ. Res 99, 692–705. [DOI] [PubMed] [Google Scholar]
- 9.Bisesi MS (1994) Esters. 3. Esters of alkenylcarboxylic acids and monoalcohols In Patty’s Industrial Hygiene and Toxicology, 4th ed. (Clayton GD, Clayton FE, Eds.). Vol. 11, pp. 2999–3007. John Wiley and Sons, New York. [Google Scholar]
- 10.Burcham BC (2016) Acrolein and human disease: untangling the knotty exposure scenarios accompanying several diverse disorders. Chem Res Toxiol 30, 145–161. [DOI] [PubMed] [Google Scholar]
- 11.Burre J and Volknandt W (2007) The synaptic vesicle proteome. J Neurochem 101, 1448–1462. [DOI] [PubMed] [Google Scholar]
- 12.Cai J, Bhatnagar A and Pierce WM (2009) Protein modification by acrolein: formation and stability of cysteine adducts. Chem Res Toxicol 22, 708–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ceaser EK, Moellering DR, Ramachandran SSA, Landar A, Venkartraman ALCrawford J, Patel R, Dickinson DA, Ulasova E,Ji S and Darley-Usmar VM (2004) Mechanisms of signal transduction mediated by oxidized lipids: the role of the electrophile-responsive proteome. Biochem Soc Trans 32, 151–155. [DOI] [PubMed] [Google Scholar]
- 14.DeCaprio AP, Kinney EA, Fowke JH (1997) Regioselective binding of 2,5-hexanedione to high-molecular-weight rat neurofilament proteins in vitro. Tox Appl Pharmacol 145, 211–217. [DOI] [PubMed] [Google Scholar]
- 15.Dietze EC, Schafer A, Omischinski JG and Nelson SD (1997) Inactivation of glyceraldehyde-3-phophate dehydrogenase by a reactive metabolite of acetaminophen and mass spectral characterization of an arylated active site peptide. Chem Res Toxicol 10, 1097–1103. [DOI] [PubMed] [Google Scholar]
- 16.Doorn JA, and Petersen DR (2002) Covalent modification of amino acid nucleophiles by the lipid peroxidation products 4-hydroxynonenal and 4-oxo-2-nonenal. Chem. Res. Toxicol 15, 1445–1450. [DOI] [PubMed] [Google Scholar]
- 17.Doorn JA and Petersen DR (2003) Covalent adduction of nucleophilic amino acids by 4-hydroxynonenal and 4-oxononenal. Chem-Biol. Interact 143–144, 93–100. [DOI] [PubMed] [Google Scholar]
- 18.Calakos N, Scheller RJ (1996) Synaptic vesicle biogenesis, docking and fusion: a molecular description. Physiol Rev 76, 1–29. [DOI] [PubMed] [Google Scholar]
- 19.Conklin DJ, Barski OA, Lesgards J-F, Juvan P, Rezen T, Rozman D, Prough RA, Vladykovskaya E, Liu S, Srivastava S and Bhatnagar A (2010) Acrolein consumption induces systemic dyslipidemia and lipoprotein modification. Toxicol. Appl. Pharmacol 24, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Csala M, Kardon T, Legeza B, Lizak B, Mandl J, Margittai E, Puskas F, Szaraz P, Szelenyi P, Banhegy G. (2015) On the role of 4-hydroxynonenal in health and disease. Biochimica [DOI] [PubMed] [Google Scholar]
- 21.Dalle-Donne I, Vistoli G, Gamberoni L, Giustarini D, Colmbo R, Facino RM, Rossi R, Milzani A and Aldini G (2007) Actin Cys374 as a nucleophilic target of α,β-unsaturated aldehydes. Free Rad. Biol. Med 42, 583–598. [DOI] [PubMed] [Google Scholar]
- 22.Dejamett N, Conklin DJ, Riggs W, Myers JA et al. (2014) Acrolein exposure is associated with increased cardiovascular disease risk. J Am Heart Assoc 10.1161/JAHA.114.000934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Eliuk SM, Renfrow MB, Shonsey EM, Barnes S and Kim H (2007) Active site modifications of the brain isoform of creatine kinase by 4-hydroxy-2-nonenal correlate with reduced enzyme activity: mapping of modified sites by Fourier transform-ion cyclotron resonance mass spectrometry. Chem Res Toxicol 20: 1260–1268. [DOI] [PubMed] [Google Scholar]
- 24.Faroon O, Roney N and Taylor J (2008) Acrolein environmental levels and potential for human exposure. Toxicol. Indust. Health 24, 543–564. [DOI] [PubMed] [Google Scholar]
- 25.Feron VJ, Til HP and de Vrijer F (1991) Aldehydes: occurrence, carcinogenic potential, mechanism of action and risk assessment. Mut. Res 259, 363–385. [DOI] [PubMed] [Google Scholar]
- 26.Fritz KS and Petersen DR (2013) An overview of the chemistry and biology of reactive aldehydes. Free Rad. Biol. Med 59, 85–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fritz KS, Galligan JJ, Smathers RL, Roede JR, Shearn CT, Peigan P and Petersen DR (2011) 4-Hydroxynonenal inhibits SIRT3 via thiol-specific modification. Chem. Res. Toxicol 24, 651–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fujioka K and Shibamoto T (2006) Determination of toxic carbonyl compounds in cigarette smoke. Environ. Toxicol 21, 47–54. [DOI] [PubMed] [Google Scholar]
- 29.Graham DG, Clair G, Amarnath MB and Anthony DC (1991) Molecular mechanisms of γ-diketone neuropathy. Adv Exp Med Biol 283, 427–431. [DOI] [PubMed] [Google Scholar]
- 30.Grimsrud PA, Xie H, Griffin TJ and Bernlohr DA (2008) Oxidative stress and covalent modification of protein with bioactive aldehydes. J Biol Chem 283, 21837–21841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gurtoo HL, Hipkens JH and Sharma SD (1981) Role of glutathione in the metabolism-dependent toxicity and chemotherapy of cyclophosphamide. Cancer Res 41, 3584–3591. [PubMed] [Google Scholar]
- 32.Higdon AN, Landar A, Barnes S and Darley-Usmar VM (2012) The electrophile responsive proteome: Integrating proteomics and lipidomics with cellular function. Antiox. Redox Signal. 17, 1580–1589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hoffmann KJ, Streeter J, Axworthy DV and Baillie TA (1985) Identification of the major covalent adduct formed in vitro and in vivo between acetaminophen and mouse liver proteins. Mol Pharmacol 27, 566–573. [PubMed] [Google Scholar]
- 34.Kalgutkar AS and Dalvie D (2015) Predicting toxicities of reactive metabolite-positive drug candidates. Annu Rev Pharmacol Toxicol 55: 35–54. [DOI] [PubMed] [Google Scholar]
- 35.Katyare SS, Shallom JM (1988) Altered cerebral protein turnover in rats following prolonged in vio treatment with nicotine. J Neurochem 50, 1356–1363. [DOI] [PubMed] [Google Scholar]
- 36.Kehrer JP and Biswal SS (2000) The molecular effects of acrolein. Toxicol. Sci 57, 6–15. [DOI] [PubMed] [Google Scholar]
- 37.Kiss JP (2000) Role of nitric oxide in the regulation of monoaminergic neurotransmission. Brain Res Bull 52, 459–466. [DOI] [PubMed] [Google Scholar]
- 38.Kumagai Y and Abiko Y (2017) Environmental electrophiles: protein adducts, modulation of redox signaling and interaction with persulfides/plysulfides. Chem Res Toxicol 30, 203–219. [DOI] [PubMed] [Google Scholar]
- 39.Leikauf GD 2002. Hazardous air pollutants and asthma. Environ. Health Persp 110 (suppl.4), 505–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Leeming MG, Gamon LK, Wille U, Donald WA and O’Hair RAJ (2015) What are the potential sites of protein arylation by N-acetyl-p-benzoquinone imiine (NAPQI)? Chem Res Toxicol 22, 2224–2233. [DOI] [PubMed] [Google Scholar]
- 41.Llewellyn DJ, Lang IA, Langa KM, Naughton F and Matthews FE (2009) Exposure to second hand smoke and cognitive impairment in non-smokers: national cross sectional study with cotinine measurement. BMJ 338:b462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.LoPachin RM, Balaban CD and Ross JF (2003) Acrylamide axonopathy revisited. Toxicol Appl Pharmacol 188, 135–153.. [DOI] [PubMed] [Google Scholar]
- 43.LoPachin RM, Ross JF, Reid ML, Das S, Mansukhani S and Lehing EJ (2002) Neurotoxicological evaluation of toxic axonopathies in rats: acrylamide and 2,5-hexanedione. NeuroToxicology 23, 95–110. [DOI] [PubMed] [Google Scholar]
- 44.LoPachin RM, Schwarcz AL, Mansukhani S and Das S (2004) In vivo and in vitro effects of acrylamide on synaptosomal neurotransmitter uptake and release. NeuroToxicology. 25, 349–363. [DOI] [PubMed] [Google Scholar]
- 45.LoPachin RM and De Caprio AP (2004) γ-Diketone neuropathy: axon atrophy and the role of cytoskeletal protein adduction. Tox Appl Pharmacol 199: 20–34. [DOI] [PubMed] [Google Scholar]
- 46.LoPachin RM and De Caprio AP (2005) Protein adduct formation as a molecular mechanism in neurotoxicity. Tox Sci 86, 214–225. [DOI] [PubMed] [Google Scholar]
- 47.LoPachin RM and Barber DS. (2006) Synaptic cysteine sulfhydryl groups as targets of electrophilic neurotoxicants. Tox. Sci 94, 240–255. [DOI] [PubMed] [Google Scholar]
- 48.LoPachin RM, Barber DS, Geohagen BC, Gavin T, He D and Das S (2007a) Structure-toxicity analysis of type-2 alkenes: in vitro neurotoxicity. Tox. Sci 95, 136–146. [DOI] [PubMed] [Google Scholar]
- 49.LoPachin RM, Gavin T, Geohagen BC and Das S (2007b) Neurotoxic mechanisms of electrophilic type-2 alkenes: soft-soft interactions described by quantum mechanical parameters. Tox. Sci 98, 561–570. [DOI] [PubMed] [Google Scholar]
- 50.LoPachin RM, Barber DS and Gavin T (2008a) Molecular mechanisms of the conjugated α,β-unsaturated carbonyl derivatives: relevance to neurotoxicity and neurodegenerative diseases. Tox. Sci 104, 235–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.LoPachin RM, Gavin T and Barber DS (2008b) Type-2 alkenes mediate synaptotoxicity in neurodegenerative diseases. NeuroToxicology 29, 871–882. [DOI] [PubMed] [Google Scholar]
- 52.LoPachin RM, Gavin T, Petersen DR and Barber DS (2009a) Molecular mechanisms of 4-hydroxy-2-nonenal and acrolein toxicity: nucleophilic targets and adduct formation. Chem. Res. Toxicol 22, 1499–1508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.LoPachin RM, Gavin T and Geohagen BC (2009b) Synaptosomal toxicity and nucleophilic targets of 4-hydroxy-2-nonenal. Tox. Sci 107, 171–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.LoPachin RM and Gavin T (2012) Molecular mechanism of acrylamide neurotoxicity: lessons learned from organic chemistry. Environ. Health Persp 120, 1650–1657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.LoPachin RM, Gavin T, DeCaprio A and Barber DS (2012) Application of the hard and soft, acids and bases (HSAB) theory to toxicant-target interactions. Chem. Res. Toxicol 25, 239–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.LoPachin RM and Gavin T (2014) Molecular mechanisms of aldehyde toxicity: a chemical perspective. Chem Res Toxicol 27: 1081–1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.LoPachin RM and Gavin T (2015) Reactions of electrophiles with nucleophilic thiolate sites: relevance to pathophysiological mechanisms and remediation. Free Rad Res 50: 195–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.LoPachin RM, Geohagen B, Nordstrom LU and Gain T (2016) Enolate-forming compounds as a novel approach to cytoprotection. Chem Res Toxicol 29: 2096–2107. [DOI] [PubMed] [Google Scholar]
- 59.Luo J, Hill BG, Gu Y, Cai J, Srivastava S and Bhatnager A (2007). Mechanisms of acrolein-induced myocardial dysfunction: implications of environmental and endogenous aldehyde exposure. Am J Physiol Heart Cir, Physiol 293:H3673–H3684. [DOI] [PubMed] [Google Scholar]
- 60.Moghe A, Ghare S, Lamoreau B, Mohammad M, Barve S, McClain C and Joshi-Barve S (2015) Molecular mechanisms of acrolein toxicity: relevance to human disease. Toxicol Sci 143: 242–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mohr S, Stamler JS and Brune B (1994) Mechanism of covalent modification of glyceraldehyde-3-phosphate dehydrogenase at its active site thiol by nitric oxide, peroxynitrite and related nitrosating agents FEBS Lett 348, 223–227. [DOI] [PubMed] [Google Scholar]
- 62.Morciano M, Beckhaus T, Karas M, Zimmermann H and Volkandt W (2009) J Neurochem 108, 662–675. [DOI] [PubMed] [Google Scholar]
- 63.Martyniuk CJ, Fang B, Koomen JM, Gavin T, LoPachin RM and Barber DS (2011) Molecular mechanisms of α,β-unsaturated carbonyl toxicity: cysteine-adduct formation correlates with loss of enzyme function. Chem. Res. Toxicol 24, 2302–2311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Noerager BD, Xu X. Davis VA. Jones CW. Okafor S, Whitehead A, Blalock JE, Jackson PL (2015) A potential role for acrolein in neurtrophil-mediated chronic inflammation. Inflammation 10.1007/s10753-015-213-2. [DOI] [PubMed] [Google Scholar]
- 65.O’Brien PJ, Diraki AG and Shangari N (2005) Aldehyde sources, metabolism, molecular toxicity mechanisms, and possible effects on human health. Crit. Rev. Toxicol 35, 609–662. [DOI] [PubMed] [Google Scholar]
- 66.O’Toole TE, Conklin DJ and Bhatnagar A (2008) Environmental risk factors for heart disease. Res. Environ. Health 23, 167–202. [DOI] [PubMed] [Google Scholar]
- 67.Pearson RG (1990). Hard and soft acids and bases – the evolution of a chemical concept. Coord. Chem. Rev 100, 403–425. [Google Scholar]
- 68.Qing WG, Powell KL and MacLeod MC (1996) Kinetics of the reaction of a potential chemopreventive agent, 2,6-dithiopurine and its major metabolite, 2,6-dithiouric acid, with multiple classes of electrophilic toxicants. Chem Res Toxicol.9: 1298–1304. [DOI] [PubMed] [Google Scholar]
- 69.Reid AV, Kurten RC, McCullough SS, Brock RW, Hinson JA (2005) Mechanisms of acetaminophen-induced hepatotoxicity: role of oxidative stress and mitochondrial permeability transition in freshly isolated mouse hepatocytes. J Pharmacol Exp Ther 312: 509–516. [DOI] [PubMed] [Google Scholar]
- 70.Ripoll DR, Faerman CH, Axelsen PH and Silman I. and Sussman JL (1993) An electrostatic mechanism for substrate guidance down the aromatic gorge of acetylcholinesterase. Proc Natl Acad Sci 90, 5128–5132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Rudkouskaya A, Sim V, Shah AA, Feustel PJ, Jourd’heuil D, Mongin AA (2010) Long-lasting inhibition of presynaptic metabolism and neurotransmitter release by protein S-nitrosylation. Free Radic Biol Med 49, 757–769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Samet JM and Wages PA (2018) Oxidative stress from environmental exposures. Curr Op Toxicol 7, 60–66. [PMC free article] [PubMed] [Google Scholar]
- 73.Schultz TW, Carlson RE, Cronin MTD, Hermens JLM, Johnson R, O’Brien PJ, Roberts DW, Siraki A, Wallace KB and Veith GD (2006) A conceptual framework for predicting the toxicity of reactive chemicals: modeling soft electrophilicity. SAR QSAR Eviron. Res 17, 413–428. [DOI] [PubMed] [Google Scholar]
- 74.Schwobel JAH, Koleva YK, Enoch SJ, Bajot F, Hewitt M, Madden JC, Roberts DW, Schultz TW and Cronin MTD (2011) Measurement and estimation of electrophilic reactivity for predictive toxicology. Chem. Rev. 111, 2562–2596. [DOI] [PubMed] [Google Scholar]
- 75.Seiner DR, LaButti JN and Gates KS (2007) Kinetics and mechanism of proteintyrosine phosphatase B inactivation by acrolein. Chem Res Toxicol 20: 1315–1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Seretny M, Currie GL, Sena ES, Ramnarine S, Grant R, MacLeod MR, Colvin LA and Gallon M. (2014) Incidence, prevalence and predictors of chemotherapy-induced peripheral neuropathy: a systematic review and meta-analysis. Pain 155: 2461–2470. [DOI] [PubMed] [Google Scholar]
- 77.Shi Y, Sun W, McBride JJ, Cheng JX and Shi R (2011) Acrolein induces myelin damage in mammalian spinal cord. J Neurochem 117: 554–564. [DOI] [PubMed] [Google Scholar]
- 78.Stachulski AV, Baillie TA, Park BK, Obach RS, Dalvie DK, Williams DP, Srivastava A, Regan SL, Antoine DJ, Goldring EP, Chia AJL, Kitteringham NR, Randle LE, Callan H, Castrejon JLFarrell J, Naisbitt DJ and Lennard MS (2012) The generation, detection and effects of reactive drug metabolites. Med Res Rev 11:1–96. [DOI] [PubMed] [Google Scholar]
- 79.Staff NP, Grisold A, Grisold W and Windebank AJ (2017) Chemotherapy-induced peripheral neuropathy: a current review. Ann Neurol 81: 772–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Stevens JF and Maier CS (2008) Acrolein: sources, metabolism and biomolecular interactions relevant to human health and disease. Mol. Nutr. Food Res 52, 7–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Van der Toorn M, Slebos D-J, de Bruin HG, Gras R, Rezayat D, Lorge L, Sndra K and von Oosterhout (2013) Resp Res 14:45–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Werley MS, Freelin SA, Wrenn SE, Gerstenberg B, Roemer E, Schramke H, van Miert E, Vanscheeuwijck P, Weber S and Coggins CRE (2008) Smoke chemistry, in vitro and in vivo toxicology evaluations of the electrically heated cigarette smoking system series K. Reg. Toxicol. Pharmacol 52, 122–139. [DOI] [PubMed] [Google Scholar]
- 83.Wang K, Lu J and Li R (1996) The events that occur when cisplatin encounters cells. Coorrd Chem Rev 151: 53–88. [Google Scholar]
- 84.Wang G-W, Guo Y, Vondriska TM, Zhang J, Zhang S, Tsai LL, Zong NC, Bolli R, Bhatnagar A and Prabhu SD (2008) Acrolein consumption exacerbates myocardial ischemic injury and blocks nitric oxide-induced PKCε signaling and cardioprotection. J. Mol. Cellular Cardiol 44, 1016–1022. [DOI] [PubMed] [Google Scholar]
- 85.Woodruff TJ, Wells EM, Holt EW, Burgin DE and Axelrad DA (2007) Estimating risk from ambient concentrations of acrolein across the United States. Environ. Health Perspect 115, 410–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Yin G, Wang Y, Cen X-m, Yang M, Liang Y and Xie QB (2015) Lipid peroxidation-mediated inflammation promotes cell apoptosis through activation of NF-κB pathway in rheumatoid arthritis synovial cells. Med Inflamm dx/doi/org/10.1155/2015/460310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Zhang L, Gavin T, De Caprio AP and LoPachin RM, (2010) γ-Diketone axonopathy: analyses of cytoskeletal motors and highways in CNS myelinated axons. Tox Sci 86, 214–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Zhang L, Gavin T, Barber DS and LoPachin RM (2011) Role of the Nrf2-ARE pathway in acrylamide neurotoxicity. Toxicol Lett 205, 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Zhang J, Wang C, Ji L and Liu W. (2016) Modeling of toxicity-relevant electrophilic reactivity for guanine with epoxides: estimating the hard and soft acids and bases (HSAB) parameter as a predictor. Chem Res Toxicol 29: 841–850 [DOI] [PubMed] [Google Scholar]