

Enhancers at critical craniofacial development genes are established by p63 and enriched for SNPs associated with CL/P.
Abstract
The transcription factor p63 is a key mediator of epidermal development. Point mutations in p63 in patients lead to developmental defects, including orofacial clefting. To date, knowledge on how pivotal the role of p63 is in human craniofacial development is limited. Using an inducible transdifferentiation model, combined with epigenomic sequencing and multicohort meta-analysis of genome-wide association studies data, we show that p63 establishes enhancers at craniofacial development genes to modulate their transcription. Disease-specific substitution mutation in the DNA binding domain or sterile alpha motif protein interaction domain of p63, respectively, eliminates or reduces establishment of these enhancers. We show that enhancers established by p63 are highly enriched for single-nucleotide polymorphisms associated with nonsyndromic cleft lip ± cleft palate (CL/P). These orthogonal approaches indicate a strong molecular link between p63 enhancer function and CL/P, illuminating molecular mechanisms underlying this developmental defect and revealing vital regulatory elements and new candidate causative genes.
INTRODUCTION
The transcription factor p63, a member of the p53 family of proteins, is a key mediator of epithelial cell processes, including establishment and maintenance of epithelial identity (1). Deletion of Trp63 (encoding the p63 protein) in mice leads to developmental and morphological defects in the squamous epithelia and epidermis, leading to abnormal craniofacial development, truncated limbs, and loss of salivary glands, hair follicles, and teeth (2, 3). In humans, germline missense mutations in p63 cause ectodermal dysplastic syndromes, which are characterized by orofacial clefting and limb malformations (4–6). These mutations are largely clustered in either the DNA binding domain (DBD) or the protein-protein interaction sterile alpha motif (SAM) domain; amino acid substitutions show clear differences in the physical location and severity of phenotypes (7). p63 can bind directly to chromatin to change the chromatin landscape during keratinocyte differentiation (8–10). However, the role of p63 in de novo epithelial lineage commitment and how p63 amino acid mutations lead to defects in this process remain unknown.
Mutations in p63 in humans are rare; however, orofacial clefting [with its distinct subtypes cleft lip ± cleft palate (CL/P) and cleft palate only (CPO)] is a key feature of mutant p63 syndromes and is one of the most common congenital defects worldwide, with an approximate prevalence of 1:700 births (11). The etiology of orofacial clefting is complex, with involvement of genetic and environmental factors. To date, more than 400 different human syndromes have been reported showing orofacial clefting as part of their phenotypic spectrum. However, more than 70% of CL/P cases and more than 50% of CPO cases are nonsyndromic, presenting as isolated phenotypes with the absence of other affected organs and systems, and hence, their etiology remains largely unclear (11–14). The commonality and complexity of the constellation of orofacial clefting syndromes suggest involvement of p63-linked regulatory dysfunction.
Murine models have been crucial in understanding lip and palate development. Nonetheless, mutations and knockout (KO) of proteins involved in human CL/P in mice rarely show clefting of the lip, with clefting primarily observed in the secondary palate (15, 16). Genome-wide association studies (GWAS) and other genetic approaches in patients with nonsyndromic CL/P (nsCL/P) have identified 40 risk loci that show robust associations of single-nucleotide polymorphisms (SNPs) with nsCL/P (14, 17–20). A provocative finding is that most of the SNPs associated with nsCL/P are not found within protein-coding exons but rather at intronic and intergenic regions, and for the vast majority of these SNPs, the molecular mechanism underlying nsCL/P pathogenesis remains unknown. A single case in a family with CL/P has been reported in which a substitution mutation in p63 disrupts its binding to an enhancer and affects transcription of IRF6 (21). Whether CL/P-associated SNPs function in a similar way is unknown, and most broadly, it remains to be elucidated how general is enhancer disruption in developmental diseases.
In this study, we investigated the role of p63 in human craniofacial development and in the pathogenesis of orofacial clefting. We used an inducible transdifferentiation model that reveals p63 interaction with chromatin when expressed de novo and p63 regulation of gene expression. Our strategy encompassed a combination of epigenomic assays, including assay for transposase-accessible chromatin sequencing (ATAC-seq), chromatin immunoprecipitation sequencing (ChIP-seq) of p63, and the active enhancer histone modification H3K27ac, along with analysis of nsCL/P GWAS data. Our results reveal p63 establishment of enhancers, displaying increased chromatin accessibility and de novo H3K27 acetylation at critical craniofacial genes, and demonstrate that substitution mutations in p63 abrogate this function. We validate known genes and identify novel candidate genes and the enhancers that regulate them via p63, which might play a role in nsCL/P pathogenesis. Last, we show that enhancers established by p63 are highly enriched for SNPs associated with nsCL/P, providing novel mechanistic insights into regulation of pathogenesis by CL/P-associated SNPs.
RESULTS
p63 remodels chromatin to establish enhancers, up-regulating epithelial and inflammation genes
To determine whether p63 has the ability to remodel the chromatin landscape to establish epithelial enhancers, we set up a doxycycline-inducible system to express the ∆NP63α isoform (hereafter referred to as p63) in human BJ dermal fibroblasts [CRL-2522, American Type Culture Collection (ATCC)]. After 48 hours, p63 reaches an expression level comparable to endogenous levels observed in undifferentiated normal human epidermal keratinocytes, and expression levels are maintained without increase at 72 hours (fig. S1A). This p63 isoform was selected because it is the most abundant isoform in epithelial cells, is not expressed in fibroblasts, and has been demonstrated to play a fundamental role as a regulator of epithelial fate and keratinocyte differentiation (22–24).
Expression of p63 was induced for 72 hours, followed by p63 chromatin precipitation (ChIP-seq to assess p63 binding genome-wide), ATAC-seq (to assess open chromatin), and H3K27 acetylation (H3K27ac) ChIP-seq (Fig. 1A). We observed an increase in ATAC-seq chromatin accessibility and H3K27 acetylation (H3K27ac) at p63-bound sites in fibroblasts expressing p63 compared to control fibroblasts with empty vector (Fig. 1B and fig. S1B). Forty thousand sites were bound by p63, of which 40% were previously closed chromatin sites becoming open only after ectopic expression of p63 (17,156 of 27,441 peaks; Fig. 1C and fig. S1B), while a smaller proportion was unchanged or closed after p63 expression (14,991 open before and after and 10,302 closed). Further, 40% of these p63-bound sites were flanked by H3K27ac, with approximately 60% of these sites showing de novo H3K27ac (12,071 of 20,190; Fig. 1D and fig. S1B). Partitioning of p63-bound sites by distance to transcription start sites (TSSs) revealed that most p63 sites are located at intergenic and intronic regions at a median distance of 27 kb (Fig. 1, E and F), a distribution that is characteristic of enhancers. Overlaying published micrococcal nuclease datasets in human fibroblasts (25) with our p63 ChIP sites revealed that 55% of sites bound by p63 when ectopically expressed are nucleosome-occupied in fibroblasts, and 75% of these become open and devoid of nucleosomes upon ectopic expression of p63 (fig. S1C), expanding on previous work that showed that p63 has the ability to bind to closed chromatin and remodel the enhancer landscape during keratinocyte differentiation (8). Overall, these data reveal that de novo binding of p63 to chromatin leads to global changes in the enhancer landscape, specifically to the establishment of enhancers characterized by de novo chromatin accessibility and by H3K27ac flanking p63-bound sites.
Fig. 1. p63 remodels chromatin to establish enhancers and up-regulate epithelial and inflammation genes.
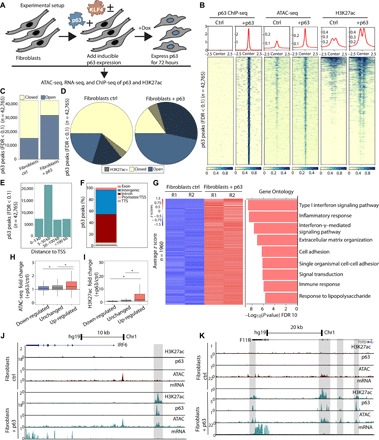
(A) Experimental setup showing inducible expression of p63 and downstream epigenomic analyses. Dox, doxycycline. (B) Heatmap of p63 ChIP-seq, ATAC-seq, and H3K27ac ChIP-seq (±2.5 kb from peak center) in control fibroblasts expressing an empty vector (ctrl) and in fibroblasts expressing p63 for 72 hours. (C) Graph of called p63 peaks partitioned into open and closed chromatin according to ATAC-seq results in fibroblasts ctrl and fibroblasts + p63. (D) Pie chart depicting differences in chromatin landscape at p63 called peaks in ctrl and after ectopic expression of p63 showing clear increase in chromatin accessibility (blue, open) and H3K27ac (patterned lines). (E) Distance to nearest TSS for all p63 peaks. (F) Partitioning of p63 peaks into different genomic features. (G) Heatmap of z-scored RNA sequencing (RNA-seq) results for 1960 genes up-regulated (fold change > 1.5; FDR < 0.05) upon ectopic expression of p63 and top Gene Ontology (GO) categories showing enrichment of epithelial and inflammation categories among up-regulated genes. (H and I) Boxplots showing increased chromatin accessibility and H3K27ac at p63/KLF4 sites ±250 bp near up-regulated genes (*P < 10 × 10−10). (J) UCSC genome browser tracks showing transcriptional activation of IRF6 (interferon regulatory factor 6) and de novo H3K27ac and ATAC-seq signal at an enhancer upstream of IRF6 in fibroblasts with ectopic expression of p63 (gray highlighted box). Chr1, chromosome 1. (K) UCSC genome browser tracks showing transcriptional activation of F11R and de novo H3K27ac and ATAC-seq signal within introns of F11R in fibroblasts with ectopic expression of p63 (gray highlighted boxes).
We examined the transcriptional consequence of these changes in the chromatin landscape, via RNA sequencing (RNA-seq) after ectopic expression of p63 for 72 hours in fibroblasts. A total of 1960 genes were up-regulated upon ectopic expression of p63 [fold change > 1.5; false discovery rate (FDR) < 0.05; Fig. 1G], validating known p63 targets (IRF6, LCE1C, IVL, ITGB4, and SHH) (26, 27) and identifying more than 1000 genes previously not known to be regulated by p63 (full list in table S1). Gene Ontology (GO) analysis of all up-regulated genes showed enrichment of cell-cell adhesion categories characteristic of epithelial cells and important for epithelial lineage specification (Fig. 1G). Other categories were interferon, inflammation, and immune pathways (Fig. 1G), consistent with previous reports showing that p63 modulates epidermal development and inflammation programs and plays a crucial role in T cell development through establishment of the thymic epithelium (28–31). In addition, 1479 genes were down-regulated upon expression of p63 (fold change > 1.5; FDR < 0.05; fig. S1D), consistent with previous reports of p63 playing a repressive role at specific genes (COL6A2, CITED2, and CDH13) (27, 28) and uncovering more than 1000 new genes down-regulated by p63.
We compared changes in the chromatin landscape to transcription, finding that chromatin accessibility and H3K27ac increased at enhancers and promoters of genes that became up-regulated upon expression of p63 in fibroblasts (Fig. 1, H and I). We found the largest fold change in chromatin accessibility in genes that became up-regulated (Fig. 1H and fig. S1E). H3K27ac decreased in genes that were down-regulated and increased markedly in genes that were up-regulated (Fig. 1I and fig. S1F). To validate these findings, we examined the previously known p63-regulated gene IRF6 (21, 32, 33): The p63-dependent IRF6 enhancer showed de novo chromatin accessibility and H3K27ac, and establishment of this enhancer was accompanied by de novo activation of IRF6 expression in fibroblasts (Fig. 1J). We also observed enhancer establishment and transcriptional activation of genes encoding the F11 receptor (F11R) and integrin subunit a7 (ITGA7) (Fig. 1K and fig. S1G). Thus, our data provide the first evidence that p63 can establish enhancers, revealing known and new gene targets of p63 linked to epithelial cells and inflammation and uncovering enhancers that might regulate their expression.
p63 and Kruppel-like factor 4 coestablish keratinocyte-specific enhancers to convert fibroblasts into keratinocyte-like cells
These results demonstrate that p63 can establish chromatin structures required for expression of key epithelial specification genes. However, previous findings show that conversion of fibroblasts into keratinocyte-like epithelial cells requires coexpression of p63 and Kruppel-like factor 4 (KLF4), and these keratinocyte-like epithelial cells share substantial morphology, functional characteristics, and gene expression patterns with basal epithelial keratinocytes (34). To understand the molecular mechanisms and chromatin changes that underlie this crucial conversion, we extended our system to temporal coexpression of p63 and KLF4 in fibroblasts driven by the same doxycycline-induced promoter (see Fig. 1A and fig. S2A). Keratin 14 (KRT14), a key biomarker of basal keratinocytes, was up-regulated only when p63 and KLF4 were coexpressed and was not up-regulated with expression of either p63 or KLF4 (Fig. 2, A and C, and fig. S2B). Our results thus confirm the requirement for both factors to achieve conversion of fibroblasts into keratinocyte-like cells.
Fig. 2. p63 and KLF4 coestablish keratinocyte-specific enhancers to convert fibroblasts into keratinocyte-like cells.
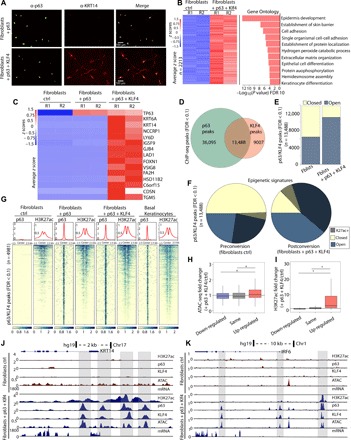
(A) Immunofluorescence of p63 and KRT14 in fibroblasts + p63 and fibroblasts + p63 + KLF4 showing that KRT14 is only up-regulated when both p63 and KLF4 are expressed. (B) Heatmap of z-scored RNA-seq results for 2213 genes up-regulated (fold change > 1.5; FDR < 0.05) upon ectopic expression of p63 + KLF4 showing high enrichment of epidermal and skin-related GO categories. (C) Comparison of transcriptional regulation of the top 15 genes up-regulated in fibroblasts + p63 + KLF4 showing that both factors are required for up-regulation of keratinocyte-specific genes. (D) Venn diagram showing that p63 and KLF4 are cobound in 13,488 loci in the genome. (E) Graph of p63/KLF4 peaks partitioned into open and closed chromatin according to peaks called from ATAC-seq showing increased chromatin accessibility after 72 hours at cobound sites. Fblsts, fibroblasts. (F) Pie charts showing stark increase in chromatin accessibility and H3K27ac at p63/KLF4 peaks. (G) Heatmap showing H3K27ac enrichment flanking p63/KLF4 peaks (±2.5 kb from peak center) shared in p63 ChIP-seq of basal keratinocytes, comparing fibroblasts, fibroblasts + p63, and fibroblasts + p63 + KLF4, and reanalyzed basal keratinocyte data from (8). (H and I) Boxplots showing increased chromatin accessibility and H3K27ac at p63/KLF4 sites near up-regulated genes (*P < 10 × 10−10). (J and K) UCSC genome browser tracks showing de novo H3K27ac and ATAC-seq signal at enhancer and promoter regions close to keratinocyte genes KRT14 and IRF6, as well as transcriptional activation (gray highlighted box).
Broad GO analysis of the 2213 genes up-regulated upon conversion showed high enrichment of gene classes encompassing epidermal development, cell adhesion, and other epithelial specific programs (Fig. 2B and table S2). Many genes critical for keratinocytes, including FOXN1, DSC2, and DSG1, were newly expressed (Fig. 2C and table S2), and others became up-regulated over 1000-fold, including KRT14, KRT6a, GJB4, and LAD1 (Fig. 2C). As with ectopic expression of p63 alone, approximately 1000 genes were down-regulated with ectopic expression of p63 + KLF4 (fold change > 1.5; FDR < 0.05; fig. S2C), including previously known repressed targets (CITED2, SATB2, TGFB1I1, and SHOX2) (27). Overall, the fold-decreased enrichment and GO category specificity of down-regulated genes were much lower compared to those of the up-regulated genes (fig. S2C). Many key genes required for epidermal development and epithelial lineage specification were only up-regulated in the combined system, including JAG2, GRHL3, and SFN (fig. S2D and table S2), and importantly, their dysregulation has been implicated in developmental malformations including skeletal dysplasias and orofacial clefting (29, 35, 36). In addition, the top 15 up-regulated genes (via fold change p63 + KLF4/ctrl) by the combined system include keratinocyte-specific genes (e.g., KRT6a and KRT14) that are key mediators of cell-cell adhesion programs (e.g., CDSN, LAD1, and GJB4), as well as keratinocyte differentiation (e.g., TGM5 and FA2H), all of which were not up-regulated when p63 was expressed without KLF4 (Fig. 2C).
To determine whether changes in transcription were a result of upstream enhancer establishment by p63 and KLF4, we induced expression of both proteins for 72 hours, followed by ATAC-seq and ChIP-seq of p63, KLF4, and H3K27ac. Our data revealed that p63 bound to over 40,000 sites in the genome and KLF4 bound to approximately 25,000 sites (Fig. 2D and fig. S2, E and F). Because both proteins are needed for transdifferentiation, we hypothesized that cobound sites might play a crucial role in establishing key keratinocyte-specific enhancers. Our ChIP-seq experiments revealed that 13,488 sites were cobound by p63 and KLF4 (Fig. 2D), primarily at intronic, intergenic, and promoter regions (fig. S2, G and H). Sites cobound by p63 and KLF4 showed a clear increase in chromatin accessibility at 80% of the sites; 30% of the sites became newly open after ectopic expression of p63 and KLF4 (Fig. 2E and fig. S2I) and 20% of shared sites became newly decorated by H3K27ac (Fig. 2F and fig. S2I). Direct comparison of specific shared p63/KLF4 bound peaks in basal keratinocytes (8) to fibroblasts, to fibroblasts + p63, and to fibroblasts + p63 + KLF4 revealed highest similarity between H3K27ac marked enhancers in keratinocytes and those in fibroblasts + p63 + KLF4 (Fig. 2G and fig. S2J). These results show that p63 and KLF4 function together to establish chromatin structures concordant with those in basal keratinocytes.
Comparison of chromatin changes to gene expression revealed decreased chromatin accessibility at down-regulated genes, as well as at genes that remained unchanged (fig. 2J). Chromatin accessibility was increased only at promoters and enhancers of genes that were up-regulated upon conversion (Fig. 2H and fig. S2K). Furthermore, H3K27ac decreased in genes that were down-regulated and profoundly increased in up-regulated genes, reaching a more than 20-fold increase in converting cells compared to controls (Fig. 2I and fig. S2L). At the local level, a genome browser view of the KRT14 locus exemplifies these marked changes in chromatin landscape and transcription (Fig. 2J). In fibroblasts + p63 + KLF4, establishment of enhancers occurred at intronic and intergenic regions around KRT14. The establishment of these enhancers was accompanied by a stark increase in KRT14 expression (Fig. 2J). As mentioned above, IRF6 is a key gene for CL/P, and KLF4 was also cobound with p63 to the IRF6 enhancer with accompanying chromatin changes (Fig. 2K).
Overall, our results show that p63 and KLF4 convert fibroblasts into keratinocyte-like cells via coestablishment of keratinocyte-specific enhancers. Specifically, p63 alone was able to establish epithelial enhancers to up-regulate 32% of genes up-regulated by p63 + KLF4 (710 of 2214 up-regulated genes; Fig. 1F and table S1; e.g., IRF6, F11R, and ITGA7). However, both p63 and KLF4 were required to establish keratinocyte-specific enhancers and up-regulate their respective genes, such as KRT14, KRT6A, and JAG2. Because fibroblasts do not express p63, this transdifferentiation model constitutes a tractable system to investigate mechanisms underlying wild-type (WT) and mutant p63 function in keratinocyte enhancer specification and to uncover key disease-specific processes. Because p63 represents a key transcription factor driving epithelial specification and because p63 mutations are implicated in CL/P and other orofacial developmental anomalies, we focused on further characterization of p63.
Disease-specific p63 mutations found in patients with craniofacial malformations show defects in enhancer establishment
Heterozygous point mutations in the DBD and SAM domains of p63 have been shown to lead to limb and craniofacial malformations in patients (4–6). While mutations within the DBD are likely to be deleterious because of loss of DNA binding (37–39), mutations within the SAM domain are likely to lead to dysregulation of protein-protein interactions needed for epithelial lineage specification (40–42).
We selected two point mutations for this study: p63R304W (Fig. 3A, orange), a point mutation in the DBD occurring in patients with ectrodactyly ectodermal dysplasia cleft lip/palate syndrome (EEC) (7), and p63I537T (Fig. 3A, blue), a point mutation in the SAM domain found in patients with ankyloblepharon-ectodermal dysplasia and cleft lip/palate syndrome (AEC) (39). Since characterized patients carry heterozygous p63 mutations, our homozygous model captures a severe scenario, with mutation of all p63 molecules within a tetramer; this approach allows investigation of substitution mutations in a simplified context.
Fig. 3. Disease-specific p63 mutations found in patients with craniofacial malformations show defects in enhancer establishment.
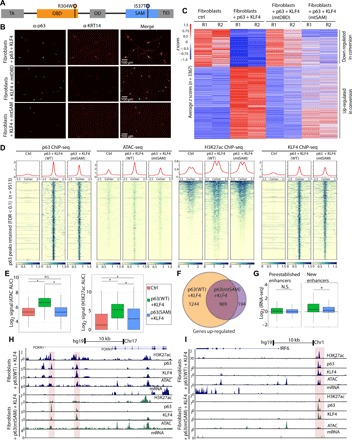
(A) Linear schematic of p63, showing domain structure and selected patient-derived mutations. TA, transactivation; OD, oligomerization domain; TID, terminal inhibitory domain. (B) Immunofluorescence of p63 and KRT14 in fibroblasts + fibroblasts + KLF4 and +WT p63, mtDBD, or mtSAM showing that KRT14 is not up-regulated when the DBD is mutated and induction of KRT14 is lower in mtSAM. (C) Heatmap of z-scored 3367 differentially expressed genes (fold change > 1.5; FDR < 0.05) between fibroblasts ctrl and fibroblasts + p63 + KLF4 showing that mtDBD has almost no change in transcriptional profile and mtSAM shows a transcriptional profile in between fibroblasts ctrl and fibroblast + WT p63 + KLF4. (D) Heatmap showing a stark reduction in chromatin accessibility (ATAC-seq signal) in mtSAM compared to WT p63, decreased H3K27ac, and retained p63 and KLF4 binding. (E) Boxplots showing no increase in chromatin accessibility for mtSAM, as well as reduced enrichment of H3K27ac flanking p63/KLF4 peaks. AUC, area under the curve; N.S., not significant. (F) Venn diagram showing that more than 1000 genes are no longer up-regulated in mtSAM + KLF4 and about 200 genes are up-regulated de novo by this mutant. (G) Boxplots showing that the RNA-seq signal of genes close to p63/KLF4 retained peaks separated into peaks at preestablished enhancers in fibroblasts and newly established enhancers after ectopic expression of p63/KLF4. (H and I) UCSC genome browser tracks showing defects (red boxes) in establishing open chromatin and inducing gene expression at FOXN1 and IRF6.
We introduced p63R304W (mtDBD) or p63I537T (mtSAM) into our inducible conversion system with coexpressed KLF4 for 72 hours (Fig. 1A). Cellular immunofluorescence for KRT14 revealed defects in conversion for both mutants (Fig. 3B). As expected, the deleterious effects of mtDBD were most severe, showing no induction of KRT14 expression after 72 hours, whereas mtSAM showed reduced KRT14 expression compared to WT p63 (Fig. 3B). RNA-seq revealed that either substitution mutation poorly induced the top 15 genes up-regulated by WT p63 + KLF4, including epithelial-specific genes KRT6A, GJB4, LAD1, and CDSN (fig. S3A and table S3). Further, among the 3367 differentially expressed genes in WT p63 + KLF4, nearly all failed to be induced in the mtDBD + KLF4 compared to control fibroblasts (Fig. 3C). Hence, loss of p63 binding to DNA leads to complete loss of epithelial enhancer establishment in fibroblasts and nearly abrogates differential gene expression [16 of 3367 genes became up-regulated (table S3)]. In contrast, mtSAM showed an intermediate expression pattern between WT + KLF4 and control fibroblasts, indicating specific defects in enhancer establishment and downstream epithelial gene induction (Fig. 3C). Given this latter interesting specificity in defects, we focused on the SAM mutant p63I537T to dissect which deficiencies underlie the disease-specific phenotypes observed in AEC.
We investigated the effect of the mtSAM on p63 binding genome-wide. Among the 13,488 p63/KLF4 DNA binding sites in WT p63 + KLF4 conversion (Fig. 2D), most (71%; 9513) of the p63 binding peaks were retained in mtSAM + KLF4 and only 29% (3975) of p63 bound peaks were lost (fig. S3B). The partial loss of mtSAM binding to DNA may be due to increased protein aggregation caused by SAM domain mutations within p63 (43). However, despite largely maintaining binding to DNA (Fig. 3D, left), there was no increase in chromatin accessibility (Fig. 3D, ATAC-seq) at retained mtSAM/KLF4 sites [Fig. 3, D and E (left)]. Furthermore, while most H3K27ac peaks were retained (Fig. 3D), overall, the amplitude of H3K27ac peaks increased only modestly compared to WT + KLF4 (Fig. 3E, right). Together with the observation that KLF4 binding is retained in most of these peaks (Fig. 3D), these results suggest that the SAM domain of p63 is crucial for chromatin accessibility and establishment of H3K27ac in epithelial enhancer specification.
We examined how these chromatin changes affect downstream gene regulation of the closest genes to p63/KLF4-bound sites. RNA-seq analysis revealed that 50% of genes up-regulated by p63/KLF4 were no longer up-regulated in mtSAM (~1200 genes; Fig. 3F), including key genes involved in craniofacial development, such as GRHL3, JAG2, LAMB3, and IRF6 (fig. S2C and table S3). We assessed whether genes that were able to maintain activation in mtSAM/KLF4 had preexisting enhancers, which might explain their continued expression. Genes that maintained expression with mtSAM/KLF4 were near preestablished and H3K27ac predecorated enhancers (Fig. 3G, left). In contrast, genes near newly established enhancers failed to be up-regulated in mtSAM/KLF4 (Fig. 3G, right). In addition, ~200 genes were aberrantly up-regulated in mtSAM/KLF4, including genes involved in craniofacial development in nonepithelial cell types, e.g., BMP2, VAX2, and GAD1 (Fig. 3F and table S3).
At the local level, FOXN1 (Fig. 3H), IRF6 (Fig. 3I), and LAMB3 (fig. S3D) showed defects in enhancer establishment despite retained binding of mtSAM and KLF4. In particular, chromatin remained largely closed with mtSAM (in red highlighted box) and reduced H3K27ac flanking these sites, and these defects in establishing normal enhancer chromatin landscape lead to profound failure in up-regulating associated genes FOXN1, IRF6, and LAMB3 (Fig. 3, H and I, and fig. 3, A and C).
Together, our chromatin and transcription data show a marked difference in the roles of the DBD and SAM domains of p63. The DBD is essential for enhancer establishment due to loss of function in DNA binding. While the SAM mutant is able to bind to DNA, there are profound defects in both chromatin opening and establishment of H3K27ac, correlating with defective up-regulation of proximal epithelial genes. Thus, because the SAM domain mutant I537T is found prominently in AEC syndrome, our results identify a potential explicit molecular mechanism, in which mtSAM drives pathogenesis through aberrant enhancer establishment (despite maintaining DNA binding), leading to low levels of specific critical craniofacial genes, such as IRF6, JAG2, and GRHL3.
p63 regulates expression of critical genes associated with cleft lip/palate
While EEC and AEC are uncommon syndromes (5, 6), orofacial clefting is a key feature of these pathologies, and clefting is a common developmental defect throughout the world (11). Mouse genetic models (including KOs of Trp63 and Irf6) have been crucial in understanding lip and palate development; however, they do not fully recapitulate defects observed in patients (15, 16). To date, 16 genes have been strongly implicated in nsCL/P via related phenotypes in mouse KO and knockdown (KD) studies (table S4A) (44). Of the 16 reported genes, 5 were up-regulated in our transdifferentiation model (ESRP1, IRF6, FOLR1, FGFR8, and WNT9B; see Fig. 2K, fig. S2D, and tables S2 and S4A), and ESRP1, IRF6, and FOLR1 also showed robust dysregulation in mtSAM (fig. S4A). Of these genes, only IRF6 was previously reported to be regulated by p63 (21, 27, 28), providing novel mechanistic insights into clefting pathogenesis via p63 mtSAM from our genome-wide study.
Genetic Association Database studies (45) of genes up-regulated in our transdifferentiation model identified cleft lip and/or cleft palate as the most highly enriched disease-associated category (Fig. 4A), with 76 associated genes identified, including genes known to cause nsCL/P (e.g., WNT9B, JAG2, and CDH1; full list in table S4B) (29, 35, 46, 47). Many of these 76 genes have been identified as causal for orofacial clefting through human syndromes and family studies, while others are in proximity of SNPs involved in CL/P but are not known to be causally or functionally involved with orofacial clefting.
Fig. 4. p63 regulates expression of critical genes associated with orofacial clefting.
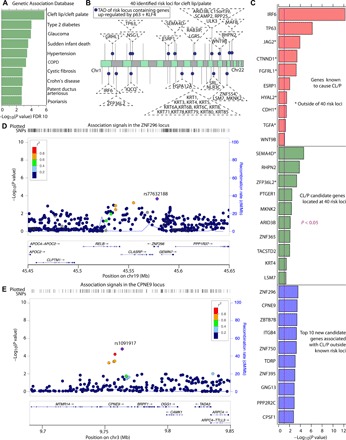
(A) Genetic association database results showing that cleft lip/cleft palate is the most enriched disease category among the 2213 genes up-regulated (fold change > 1.5; FDR < 0.05) by p63 and KLF4. (B) Forty identified risk loci for cleft lip/palate indicated by black lines at the respective chromosomes with dark blue circles at topologically associated domains (TADs) containing at least one gene up-regulated by both p63 and KLF4 (respective genes in dashed triangles). (C) List showing association values of top 10 genes calculated by gene-based analysis with MAGMA as implemented in FUMA (80) in the following categories: red, genes known to cause CL/P with asterisk marking genes outside of the known 40 risk loci; green, CL/P candidate genes located at 40 risk loci with significant (P < 0.05) association to CL/P; blue, new candidate genes associated with CL/P outside of known risk loci. (D and E) Regional association plots of ZNF296 and CPNE9, showing nsCL/P-associated SNPs with nominal significant gene-based P value (<0.05) within the genes and up to 200 kb away from TSSs.
To date, GWAS and other approaches have identified 40 human risk loci for nsCL/P with conclusive evidence (table S4C) (14, 17–20). These 40 loci are estimated to explain about 25% of the heritability reported for nsCL/P. However, the causative genes and mechanisms at most of these loci are unknown. While previous studies have shown the importance of neural crest cells in CL/P development (14), different tissue systems are expected to contribute to this complex disease. In particular, keratinocytes from patients with CL/P have recently been reported to display differentiation defects (48–51). On the basis of the key observation that p63 + KLF4 establish enhancers to regulate keratinocyte-specific genes, including critical craniofacial genes (see fig. S2D and table S2), we investigated the genes located within the topologically associated domain (TAD) (52, 53) overlapping each of the 40 risk loci. The reason why gene association within TADs is examined is that gene regulatory enhancer-promoter contacts occur primarily within TADs (52–54). We found that within the associated TAD of 17 of these risk loci, at least 1 gene, and a total of 37 genes were up-regulated in our transdifferentiation model (Fig. 4B and table S4C). We conclude that crucial regulatory interactions occur within TADs encompassing genes induced by p63/KLF4.
To analyze whether any of the 37 genes (Fig. 4B) located at the risk loci and/or their enhancer regions might contribute to nsCL/P, we used SNPs and their P values from a recent GWAS on nsCL/P (14) to compute gene-based association P values using MAGMA (55) (table S4D). We first validated our approach by examining genes up-regulated by p63 + KLF4 within and outside of the 40 risk loci that have been shown to cause nsCL/P (21, 47, 56). We identified significant P values for known causal genes within the 40 risk loci, such as in the 1q32 locus (IRF6), 17q21 (WNT9B), and 8q22.1 (ESRP1) loci (Fig. 4C, red). Our approach also identified significant P values for known causal genes outside of the risk loci, including JAG2, CDH1, and CTNND1 (Fig. 4C, red with asterisk) (29, 46, 57).
Within the 40 risk loci, we identified 10 candidate genes up-regulated by p63 with significant gene-based association P values to nsCL/P (P < 0.05), including SEMA4D, BEST3, ZFP36L2, and KRT4 (Fig. 4C, green). In three of the risk loci, our data point toward a risk gene for nsCL/P that is different from the gene previously suggested: At 2p21, THADA was previously suggested to be the risk gene (58). Our data suggest that it is ZFP36L2 (P < 0.05; Fig. 4C, green with asterisk), a gene shown to play a role in basal keratinocytes (59). At 2p25.1, KLF11 had been suspected to be the nsCL/P risk gene. Our data suggest that it is GRHL1 (P < 0.5), a close family member of GRHL3, a well-established risk gene for cleft palate (60, 61). Further, at risk loci 9q22.2, our data prioritize SEMA4D (P < 10 × 10−5; Fig. 4C, green with asterisk) instead of GADD45G (61) as the probable nsCL/P risk candidate.
Last, to identify novel risk loci and novel causal candidate genes for nsCL/P, we extended our gene-based association P value study to genes up-regulated in our transdifferentiation model outside of the known 40 risk loci. We identified 111 novel candidate genes showing high association to nsCL/P (P < 0.05 computed as above; table S4D). Several identified genes, including ZNF296, CPNE9, ITGB4, and ZNF750 (Fig. 4C, blue), play key roles in epithelial cells but have no known role in CL/P. Regional association plots for ZNF296 and CPNE9 demonstrating the local association structures at these genes in the GWAS data reveal SNPs with nominal statistical significance at each of these genes and also in the surrounding noncoding areas (Fig. 4, D and E). The location of these SNPs suggests that common genetic variants within or near these genes regulated by p63 might alter gene function or expression and, thus, may contribute to nsCL/P, but the genes have escaped detection in genetic studies due to lack of statistical power (such as sample size).
Overall, our transdifferentiation model in human cells provides an orthogonal approach to identify candidate genes in nsCL/P directly regulated by p63. Even at established risk loci, we have used our approach to find key p63-regulated genes, as the most likely CL/P-associated genes (e.g., ESRP1, GRHL1, RHPN2, and ARID3B). Further, we use this approach to identify new nsCL/P-specific candidate genes and the enhancers that regulate them (e.g., ZNF296, CPNE9, ITGB4, and TDRP).
SNPs associated with cleft lip/palate are highly enriched in enhancers established by p63 and KLF4
GWAS of SNPs have identified several genetic variants strongly associated with nsCL/P (14, 17–20). Many of these disease-associated SNPs are located outside of exons, thus suggesting a gene expression regulatory role, such as enhancers and promoters. Since orofacial clefting is a major phenotype in patients with point mutations in p63 and, as we show here, since p63 plays a key role in establishing epithelial enhancers, we explored potential overlap between p63 binding sites and nsCL/P-associated SNPs identified in GWAS. We curated three lists of p63 binding sites: (i) all p63 peaks in fibroblasts + p63, (ii) all p63 peaks in fibroblasts + p63 + KLF4, and (iii) p63/KLF4 shared peaks in fibroblasts + p63 + KLF4 [i.e., a subset of (ii)]. Using the same GWAS dataset as above (14) and the enrichment tool Genomic Regulatory Elements and Gwas Overlap algoRithm (GREGOR) (62), we determined that list (iii)—p63 peaks cobound by KLF4—showed strongest enrichment of nominal significant (P < 0.05) nsCL/P SNPs (Fig. 5A). We focused on H3K27ac flanking these sites as a mark of active enhancers, partitioning H3K27ac peaks into enhancers that are preestablished (already marked by H3K27ac in fibroblasts) or into new enhancers (only become marked by H3K27ac after fibroblast–to–keratinocyte-like conversion) (see Fig. 2, F and G). Enrichment analysis showed significant enrichment (P < 0.05) of nominal significant nsCL/P SNPs only for newly established enhancers (Fig. 5A).
Fig. 5. SNPs associated with nonsyndromic cleft lip with or without cleft palate are highly enriched in enhancers established by p63 and KLF4.
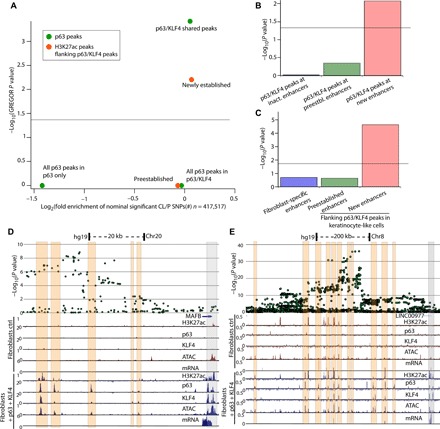
(A) GREGOR analysis showing that p63/KLF4 shared peaks (green) and newly established H3K27ac (orange) flanking these peaks are significantly (P < 0.05) enriched for SNPs associated with nsCL/P. (B) Colocalization of shared p63/KLF4 peaks at inactive (inact.)/preestablished (preestbl.)/new enhancers, with nominal significant nsCL/P-associated GWAS SNPs. The graph shows that only p63/KLF4 peaks flanked by de novo H3K27ac peaks are enriched for nsCL/P-associated SNPs. (C) Colocalization of H3K27ac peaks with nominal significant nsCL/P-associated GWAS SNPs. The graph shows that only new enhancers in converted cells flanking p63/KLF4 peaks are significantly enriched for nsCL/P-associated SNPs. (D and E) Overlay of SNPs and UCSC genome browser tracks highlighting (in orange) that p63/KLF4 peaks strongly colocalize with highly associated nsCL/P SNPs near MAFB and the known 8q24 locus; binding of both proteins at promoters is highlighted in gray.
On the basis of the results from GREGOR, we performed colocalization analysis of nsCL/P SNPs, focusing on the p63/KLF4 shared peaks (14). This analysis further accounts for the strength of association with nsCL/P of each SNP, with highly associated SNPs contributing to a lower overall colocalization P value (see Materials and Methods). We observed significant colocalization (P < 0.05) for p63/KLF4 peaks in which active enhancers are established (newly marked by H3K27ac), whereas shared p63/KLF4 peaks at preestablished enhancers and peaks without H3K27ac did not achieve statistical significance (Fig. 5B).
We next focused on H3K27ac peaks—which are broader than p63/KLF4 peaks—because SNPs close to, but outside of, p63/KLF4 peaks might be in linkage disequilibrium and contribute to risk of CL/P. We performed colocalization analysis of nsCL/P SNPs in H3K27ac peaks in control fibroblasts, in preestablished enhancers, and in new enhancers flanking p63/KLF4 peaks (Fig. 5C). Here, only enhancers newly established by p63/KLF4 showed strong colocalization (P < 10 × 10−4; Fig. 5C), confirming the results obtained in the GREGOR analysis.
Genome browser views overlaid with nsCL/P SNPs allowed visualization of the high colocalization of SNPs at p63/KLF4 peaks and newly established enhancers (ATAC-seq and H3K27ac peaks) close to MAFB (Fig. 5D), at the known 8q24 risk locus (Fig. 5E) (63), at the nsCL/P causing gene IRF6 (fig. S5A), and near RHPN2 (fig. S5B). We activate the genes downstream of these regulatory elements in our transdifferentiation model (see RNA-seq in Fig. 5, D and E, and fig. S5, A and B), indicating that they are up-regulated by the establishment of new enhancers by p63/KLF4.
Overall, GREGOR analysis and colocalization results show that enhancers established by p63/KLF4 are highly enriched for SNPs associated with nsCL/P. These SNPs likely affect downstream expression of epithelial-specific genes and long noncoding RNAs (lncRNAs) required for proper lip and palate development, as observed with IRF6 and mutations in an upstream regulatory element of this gene established by p63 (21). Thus, our unbiased transdifferentiation model in conjunction with GWAS data provides an independent method to identify driver genes. The method also provides novel mechanistic insights into how nsCL/P-related SNPs might be regulating pathogenesis via disruption of p63/KLF4 binding sites crucial for enhancer establishment and downstream gene regulation.
DISCUSSION
Here, we report a transdifferentiation model as a novel approach to determining the role of p63 in craniofacial development. We show that p63 can establish craniofacial enhancers to regulate downstream genes and that this process is disrupted when p63 carries mutations derived from human disease. In particular, our results show a novel mechanism underlying pathogenic p63 mutations, in which failure to open and establish enhancers leads to transcriptional dysregulation. We integrate our epigenomic data with GWAS datasets derived from patients with nsCL/P to identify novel CL/P candidate genes regulated by p63. We also uncover strong enrichment of nsCL/P-associated SNPs at enhancers established by p63, providing mechanistic insight into how these SNPs lead to human pathogenesis.
A key role of transcription factor p63 in mediating epidermal commitment, development, and differentiation has been extensively demonstrated through mouse models and causative point mutations in human disease. In mice, KO of p63 leads to developmental and morphological defects in the squamous epithelia and epidermis, causing abnormal craniofacial development, truncated limbs, and loss of salivary glands, hair follicles, and teeth (2, 3). To date, how p63 engages and modifies chromatin to promote normal development during these early stages is still being elucidated. Here, we report key observations that p63 can establish enhancers and that defects in this process may underlie human craniofacial malformations.
Previous studies of p63’s engagement with chromatin have focused on its role in keratinocyte differentiation (8, 10, 51, 64, 65), while de novo engagement with chromatin has not been extensively addressed due in part to lack of tractable human models. Thus, to examine the first encounter of p63 with chromatin, we expressed p63 ectopically in a temporally controlled manner in human BJ fibroblasts, a cell line that does not express p63. We show that p63 establishes enhancers to up-regulate epithelial and inflammation genes. We next developed an inducible transdifferentiation model based on published reports, showing that p63 coexpressed with KLF4 can convert fibroblasts into keratinocyte-like cells (34). We determined that cobinding of p63 and KLF4 leads to robust establishment of enhancers that correspond remarkably well to bona fide enhancers in basal keratinocytes. Thus, this model system recapitulates the epigenetic conditions of early epithelial development.
Mutations in the p63 DBD in patients with EEC have been previously shown to cause loss of downstream transcriptional activation of known p63 targets (40, 51, 66). Further studies showed that p63 mutations in AEC also disrupted transcription and caused p63 aggregation (40–43). In particular, it has been proposed that aggregation of p63 in AEC leads to decreased binding of p63 to its DNA targets and that this is a key underlying disease mechanism (43). However, it remains unclear how enhancer regulation by p63 is altered by substitution mutations. Our approach shows that one prevalent substitution mutation in the DBD leads to complete loss of p63 binding to chromatin and loss of enhancer establishment. On the other hand, one frequent substitution mutation in the SAM domain largely retains DNA binding but results in defects in chromatin opening, causing dysfunctional enhancer establishment and defective up-regulation of key craniofacial genes. These results suggest that the SAM domain is involved in recruiting chromatin remodelers to displace nucleosomes and establish open chromatin, in line with previous reports suggesting that p63 and the BAF complex cooperate to open chromatin during epidermal differentiation (64). Further experiments are required to identify proteins interacting with the SAM domain of p63 to perform these functions. However, our results point toward a novel function of the p63 SAM domain, as well as a novel molecular mechanism underlying AEC.
While EEC and AEC are relatively rare, one phenotype of both syndromes is cleft lip palate, which also occurs in the absence of p63 mutations in 1:700 births (11). A connection between p63 and CL/P has previously been proposed (32, 67, 68); however, these studies were largely carried out in mice, a model in which KO and KD of p63 do not accurately recapitulate clefting of the lip or palate (2, 3). This could be in part due to extensive rewiring of the p63 response element network between mouse and human, as well as human-specific craniofacial enhancers that were recently reported (69, 70). Further, mouse models of known disease-causing genes such as IRF6, PVRL1 (71), and CDH1 (46) in human also fail to recapitulate clefting of the lip (15, 16, 35, 44), suggesting stark differences in these processes between species and highlighting the need for orthogonal human approaches. In humans, GWAS and other approaches have identified 40 human risk loci for nsCL/P with conclusive evidence. These 40 loci represent about 25% of heritability in nsCL/P cases reported; however, at the majority of these loci, the causative variants and mechanisms are unknown, due in part to a lack of mechanistic data within noncoding regions. Thus, by integrating epigenomic datasets from our transdifferentiation keratinocyte model with GWAS datasets, we gained novel insights into molecular mechanisms underlying nsCL/P.
Overlapping of our results with GWAS datasets from nsCL/P identified key known candidate genes regulated by p63 and newly established enhancers at known risk loci, including IRF6 in the 1q32 locus and WNT9B at 17q21.32. We identified ZFP36L2 and GRHL1 at 2p21 and 2p25.1, respectively, as the lead risk nsCL/P genes despite previous reports focusing on other genes in those loci (58, 61). Our data also prioritized ESRP1 and SEMA4D, known genes in epithelial regulation, as nsCL/P risk candidates at 8q22.1 and 9q22.2 loci in which the risk genes had not been identified. KO studies of ESRP1 in particular showed full penetrance of CL/P in mice (56). Further, by combining GWAS datasets with genes up-regulated in our transdifferentiation model, we identified more than 100 new candidate genes and risk loci for nsCL/P validation studies, including ZNF296, CPNE9, and ITGB4. These genes play important roles in epithelial cells, but their role in nsCL/P had been unknown before our study.
In addition to identifying genes downstream of p63, our analysis also identified the enhancers that regulate them. For instance, FOLR1 and FOLR3 were both up-regulated in our transdifferentiation model following establishment of upstream enhancers by p63 and KLF4. KO of the mouse homolog of FOLR1 gene was shown to lead to orofacial clefting (72); however, mutations in these genes have not been found in humans with CL/P. It is possible that previously uncharacterized mutations at the enhancers of these genes may increase risk of CL/P, which could be mitigated by folic acid supplementation, as is the case in mouse models with KO of folbp1 (72).
Last, using two different colocalization analyses, we found that enhancers established by p63 + KLF4 are enriched for SNPs associated with nsCL/P, providing a novel molecular mechanism for how these SNPs may contribute to disease at a molecular level. For instance, at the nsCL/P risk loci 20q12, MAFB has been proposed to be the main risk gene for nsCL/P (12, 73). However, no functional or mechanistic data have supported this claim. Our integrated data show that p63 and KLF4 cobind and establish enhancers upstream of MAFB, leading to robust de novo expression of MAFB. These enhancers are enriched for nsCL/P-associated SNPs, providing a novel mechanism underlying how these SNPs may affect proper expression of MAFB during lip and palate development. Another example is the known 8q24 risk locus, which shows the strongest effect size across populations of Asian and European origin (63, 74). Previous reports conducted in murine models suggested that this region contains enhancers important for MYC regulation (75); however, deletion of this region resulted in clefting of the lip or palate in less than 7% of murine embryos, despite robust down-regulation of MYC. Our data show that this region contains a number of p63 + KLF4 bound and established enhancers that are enriched for SNPs associated (P < 5.0 × 10−8) with nsCL/P. Further, we show that establishment of enhancers in this region led to up-regulation of an lncRNA, LINC00977, that is not present in mice. In human keratinocytes, it was previously observed that KD of p63 leads to down-regulation of MYC (76). Thus, it is possible that p63 regulates enhancer establishment at 8p24 to up-regulate LINC00977 and modulate MYC expression during lip and palate development. While mechanistic experiments are necessary to confirm this, our data provide novel candidate disease mechanisms underlying nsCL/P. Together, our findings address how p63 binds and remodels chromatin with its first encounter, identifies and prioritizes nsCL/P candidate genes and the enhancers that regulate them, and provides new explanations for how p63 mutations and SNPs associated with nsCL/P underlie disease.
MATERIALS AND METHODS
Cell culture
BJ foreskin fibroblasts (CRL-2522) were obtained from ATCC and were cultured in a 37°C incubator at 3% oxygen in tissue culture medium containing Dulbecco’s modified Eagle’s medium with 10% fetal bovine serum, streptomycin (100 μg/ml), and penicillin (100 U/ml).
Lentivirus and retrovirus infection
Stable cell lines were made by lentivirus and retrovirus infection, as previously described (77). Tetracycline On System was introduced in two steps. First, the reverse tetracycline-controlled transactivator (rtTA) was introduced via lentivirus Lenti CMV rtTA3 Blast (w756-1) (plasmid #26429, Addgene). Lentivirus was transfected with packaging plasmids to human embryonic kidney T293T cells, and viral supernatant was filtered through a 0.45-μm filter, supplemented with polybrene (8 μg/ml), and mixed with trypsinized BJ cells for 24 hours. Cells were selected for 1 week using blasticidin at 4 μg/ml.
We then cloned ∆NP63α-Flag (plasmid #26979, Addgene) and ∆NP63α-Flag cloned with a T2A self-cleaving peptide linker to human KLF4 (plasmid #19777) into a retroviral construct pRetroX-TRE3G-puro construct. We introduced point mutations (I537T and R304W) into p63 via QuikChange II site-directed mutagenesis from Agilent (catalog #200523). Retrovirus constructs were transfected to Phoenix packaging cell line, purified, and used as described above. Infected cells were selected with puromycin for approximately a week.
Transdifferentiation
BJ stable cell lines infected with rtTA and either an empty pRetrox-TRE3G vector, pRetrox-TRE3G-∆NP63α-Flag, or pRetrox-TRE3G-∆NP63α-Flag-T2A-hKLF4 were induced for 72 hours using doxcycycline (1 μg/ml) replenished every 24 hours. A low dose of 1 mM hydroxyurea was added after 24 and 48 hours, as we observed that this improves conversion without affecting control cells. Cells were then collected for Western blot, ATAC-seq, RNA-seq, and ChIP-seq.
Western blot
Cells were lysed in radioimmunoprecipitation assay buffer containing 1% NP-40, 150 mM NaCl, 50 mM tris-Cl (pH 8.0), and 1% SDS supplemented with cOmplete, EDTA-free protease inhibitor (11873580001, Roche). Protein concentration was determined by bicinchoninic acid assay (BCA) protein assay (#23227, Life Technologies), after which equal amounts of proteins were loaded and separated by polyacrylamide gels. Proteins were then transferred to nitrocellulose membranes. Antibodies used in this study were as follows: Flag (M2, F1804, Sigma-Aldrich), hKLF4 (AF3640, R&D Systems), p63 (D2K8X, Cell Signaling Technology), glyceraldehyde-3-phosphate dehydrogenase (10R-G109a, Fitzgerald), and KRT14 (ab7800, Abcam).
Immunofluorescence
Transdifferentiated and control fibroblasts grown on cover glasses were fixed in 4% paraformaldehyde in phosphate-buffered saline (PBS) for 30 min. Cells were then washed twice with PBS and permeabilized with 2.5% Triton X-100 in PBS for 10 min. After two washes, cells were blocked in 10% bovine serum albumin (BSA) in PBS for 1 hour at room temperature. Cells were then incubated at 4°C overnight with primary antibodies in 5% BSA in PBS with 0.1% Tween 20 (PBST). Primary antibodies used were as follows: Flag (M2, F1804, Sigma-Aldrich), hKLF4 (AF3640, R&D Systems), p63 (D2K8X, Cell Signaling Technology), and KRT14 (ab7800, Abcam). The next day, cells were washed 4 × 10 min with PBST, followed by incubation with Alexa Fluor–conjugated secondary antibodies (Life Technologies) in 5% BSA in tris-buffered saline with Tween 20 for 45 min at 37°C. Cells were then washed 3 × 10 min in PBS, incubated with 4′,6-diamidino-2-phenylindole (0.5 μg/ml) in PBS for 5 min, and washed twice with PBS. The slides were then mounted using VECTASHIELD antifade mounting medium (H-1000, Vector Laboratories) and incubated overnight in the dark at room temperature. The slides were then imaged with a Nikon Eclipse 80i fluorescent microscope.
ChIP and RNA preparation
Transdifferentiated and control BJ fibroblasts (population doubling 20 to 30) were cross-linked with formaldehyde (1% final) for 5 min at room temperature or lysed in TRIzol (#15596018, Thermo Fisher Scientific) and snap-frozen for RNA isolation. Cross-linked cells were quenched with glycine (125 mM final) for 5 min, followed by two washes in cold PBS. Nuclei were then isolated from 20 million cells as previously described (78), and chromatin was sheared to 250-bp average size using a Covaris S220. Immunoprecipitations were performed using 500 μg of sheared chromatin lysate and 5 μg of antibodies preconjugated to protein G beads (Invitrogen): H3K27ac (39133, Active Motif), Flag (M2, F1804, Sigma-Aldrich), and hKLF4 (AF3640, R&D Systems). ChIP reactions were incubated for 16 hours at 4°C with rotation and then washed four times in wash buffer [50 mM Hepes-HCl (pH 8), 100 mM NaCl, 1 mM EDTA, 0.5 mM EGTA, 0.1% sodium deoxycholate, and 0.5% N-laurylsarcosine], followed by one wash in ChIP final wash buffer (1× tris-EDTA (TE) Buffer and 50 mM NaCl). Immunoprecipitated DNA was eluted from washed beads, reverse cross-linked overnight, purified, and used to construct libraries. Snap-frozen RNA samples were further extracted using chloroform and QIAGEN RNeasy Mini Kit (#74106) including deoxyribonuclease digestion. Purified RNA was then reverse-transcribed to complementary DNA (cDNA) using the High-Capacity cDNA Reverse Transcription Kit from Applied Biosystems (4368814). cDNA was then quantified and used to construct sequencing libraries.
ChIP-seq/RNA-seq and data analysis
Sequencing libraries for ChIP experiments were prepared using NEBNext Ultra reagents (New England Biolabs). All ChIP samples and input were double-end–sequenced on an Illumina NextSeq 500. Reads were mapped to the reference human genome assembly GRCh37/hg19 using Bowtie2 (maximum fragment size set to 2000 bp). Properly aligned, nonduplicated pairs were retained for peak calling using MACS2 (q value cutoff set to be 0.01). Fragment pileup per million reads for each ChIP library and corresponding input were also generated and subtracted using MACS2 for visualization. Only overlapping peaks shared by biological replicates were used for analysis.
Poly(A) + RNA was isolated using double selection with poly-dT beads, followed by first- and second-strand synthesis. Reads were mapped to the reference human genome assembly GRCh37/hg19 using splice-aware tool STAR (maximum fragment size set to be 2000 bp). Alignments with a mapping quality (MAPQ) score over 10 were counted toward RefSeq (Reference Sequence) genes using featureCounts (default parameters). Fragment pileup per 10 million reads for each RNA library was generated by BedTools (“genomecov -split -bg -scale”) and University of California Santa Cruz (UCSC) tool package (“bedGraphToBigWig”). Pairwise differential expression analysis was implemented using R package DESeq2 with FDR cutoff set to be 0.05 and fold-change cutoff set to be 1.5.
Assay for transposase-accessible chromatin
ATAC-seq experiments were performed as previously described (79) using 100,000 cells and 5 μl of Tn5 transposase (Nextera XT Kit, Illumina) to tagment intact nuclei. Reads were mapped to the reference human genome assembly GRCh37/hg19 using Bowtie2 (maximum fragment size set to be 2000 bp). Properly aligned, nonduplicated pairs were retained, and reads mapped to mitochondria or ENCODE blacklist regions were excluded. For peak calling, we adjusted the read start sites to represent the center of the transposon binding event (all reads aligning to the positive strand were offset by +4 bp, and all reads aligning to the negative strand were offset by −5 bp). Broad peaks were called using MACS2 (q value cutoff set to be 0.01). Fragment pileup per million reads for each ATAC library was also generated using MACS2 for visualization. Similar to the ChIP-seq data, only overlapping peaks shared by biological replicates were used for analysis. To acquire subnucleosomal peaks, only fragments shorter than 120 nt were used.
GO analysis
GO terms of differentially regulated genes were determined using DAVID. We selected the top 10 GO categories based on the lowest FDR.
Intersection
All p63 peak sets were combined by taking the union using BedTools (“bedtools merge”) and categorized into different subsets based on whether they overlap with ATAC-seq or ChIP-seq peaks (overlap by at least 1 bp). Peaks were annotated to genes by the nearest RefSeq TSS using HOMER (default parameters for “annotatePeaks” command). Motif analysis was implemented using HOMER (“findMotifsGenome -size given”).
Statistical analysis
Metaplots and heatmaps centered around any peak set were generated using deepTools (“computeMatrix” and “plotHeatmap”). ChIP-seq or ATAC-seq signal over each peak region was computed using bwtool (unit: per million reads per kilobase) to generate boxplots and bar graphs, and Mann-Whitney test was applied unless otherwise specified.
Analysis of GWAS data for nsCL/P
GWAS data for nsCL/P were retrieved from a previously published study (14), which had included 399 cases, 1318 controls, and 1461 case-parent trios. The SNP data comprised about 8.01 million common variants at a minor allele frequency of >1%. All analyses for the present manuscript were performed using the overall data, i.e., without stratification for ethnicity.
Gene-based P values
While single variants alone might be statistically underpowered to show significant effects, their contribution in sum can be analyzed by aggregating individual variants within a predefined region (for instance, genes). On the basis of the P values obtained from the nsCL/P GWAS, we used MAGMA (v1.06) (55), a principal component–based regression framework that allows for gene-based analysis. We ran the gene-based analysis for the p63 up-regulated genes (n = 250) as implemented in the FUMA online tool (Functional Mapping and Annotation of GWAS) platform (v1.3.2) (80), with default parameters (SNP-wide mean model) and with the 1000 Genome Phase3 as reference panel. In the gene set analyses, curated gene sets and GO terms from MsigDB are tested. For FUMA (≥v1.3.1), 10,655 gene sets (curated gene sets: 4738; GO terms: 5917) from MsigDB v6.1 were used. Bonferroni correction was performed for all the tested gene sets.
Enrichment analyses
To evaluate the enrichment of all nominal significant nsCL/P GWAS SNPs (74) in a priori selected regulatory elements, the GREGOR (v1.4.0) (62) software tool was used. Selection of the regions was performed on the basis of the presence of p63 and/or KLF4 and/or H3K27ac peaks. For analysis of overlay of specific SNPs to specific elements, a colocalization approach was performed as previously suggested (14). Briefly, SNPs were mapped to either “positive regions” (i.e., in p63 + KLF4 ± H3K27ac binding regions) or “negative regions” (all others) and then grouped into nine categories 10−k+1 ≥ P > 10−k for k = 1,…, 8 and 10−8 ≥ P. The frequencies of SNPs located in the positive regions and those outside the annotated regions were counted within each category. The one-sided Cochran-Armitage trend test was applied in R environment with function prop.trend.test of stats package to test for an enrichment of smaller P values within annotated regions based on the resulting 9 × 2 contingency table.
Supplementary Material
Acknowledgments
We thank S. Grant, A. Chessi, E. J. Bhoj, and D. Li from the Children’s Hospital of Philadelphia for valuable conversations about SNPs and discordant phenotypes in twin studies. We also thank P. Sen for critical feedback on this manuscript and B. Capell for important conversations about epithelial cells and p63 function. Funding: This work was funded by NIH grant R01 CA078831 (to S.L.B.), German Research Council grant LU 1944/3-1 (to K.U.L.), German Research Foundation grant FOR 423, and individual grant MA 2546/3-1. E.L.-S. was supported by NIH grant 1 F31 GM123744-01. K.A.A. was supported by NIH grant F32 12461842. M.S. was supported by NIH grant R15GM128049. Author contributions: This study was conceived by S.L.B. and E.L.-S. E.L.-S initiated and led the study with input from K.U.L., M.S., E.M., K.A.A., and S.L.B. M.S. contributed in the design of the initial experiments. E.L.-S. carried out most of the experiments. Z.Z. contributed in experiments. E.L.-S. and Y.L. analyzed high-throughput sequencing data. J.W., M.K., and K.U.L. performed colocalization analysis, SNP statistics, and GREGOR. E.L.-S. and S.L.B. wrote the manuscript. All authors reviewed and commented on the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. The relevant datasets generated during this study have been uploaded to National Center for Biotechnology Information Gene Expression Omnibus and are available under accession numbers GSE126376 (ATAC-seq), GSE126397 (RNA-seq), and GSE126390 (ChIP-seq). Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/5/eaaw0946/DC1
Fig. S1. Ectopic expression of p63 in fibroblasts leads to establishment of enhancers and downstream up-regulation of inflammation and epithelial genes.
Fig. S2. Ectopic coexpression of p63 and KLF4 leads to establishment of keratinocyte-specific enhancers and up-regulation of epithelial genes.
Fig. S3. Mutations in p63 disrupt enhancer establishment and lead to loss of up-regulation of key keratinocyte-specific genes.
Fig. S4. Heatmap of genes regulated by p63 + KLF4 that have been strongly linked to CL/P in murine KD and KO models.
Fig. S5. p63/KLF4 peaks colocalize strongly with highly associated CL/P SNPs near IRF6, RHPN2, and GPATCH1.
Table S1. Genes up-regulated in fibroblasts + p63 compared to control fibroblasts (fold change > 1.5; FDR < 0.05).
Table S2. Genes up-regulated in fibroblasts + p63 + KLF4 compared to control fibroblasts (fold change > 1.5; FDR < 0.05).
Table S3. Mutations in p63 disrupt up-regulation of keratinocyte-specific genes.
Table S4. Causal and candidate genes involved in CL/P identified in our transdifferentiation model.
REFERENCES AND NOTES
- 1.Yang A., Kaghad M., Wang Y., Gillett E., Fleming M. D., Dötsch V., Andrews N. C., Caput D., McKeon F., p63, a p53 homolog at 3q27–29, encodes multiple products with transactivating, death-inducing, and dominant-negative activities. Mol. Cell 2, 305–316 (1998). [DOI] [PubMed] [Google Scholar]
- 2.Yang A., Schweitzer R., Sun D., Kaghad M., Walker N., Bronson R. T., Tabin C., Sharpe A., Caput D., Crum C., McKeon F., p63 is essential for regenerative proliferation in limb, craniofacial and epithelial development. Nature 398, 714–718 (1999). [DOI] [PubMed] [Google Scholar]
- 3.Mills A. A., Zheng B., Wang X. J., Vogel H., Roop D. R., Bradley A., p63 is a p53 homologue required for limb and epidermal morphogenesis. Nature 398, 708–713 (1999). [DOI] [PubMed] [Google Scholar]
- 4.Lopez-Pajares V., Yan K., Zarnegar B. J., Jameson K. L., Khavari P. A., Genetic pathways in disorders of epidermal differentiation. Trends Genet. 29, 31–40 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Celli J., Duijf P., Hamel B. C. J., Bamshad M., Kramer B., Smits A. P. T., Newbury-Ecob R., Hennekam R. C. M., van Buggenhout G., van Haeringen A., Woods C. G., van Essen A. J., de Waal R., Vriend G., Haber D. A., Yang A., McKeon F., Brunner H. G., van Bokhoven H., Heterozygous germline mutations in the p53 homolog p63 are the cause of EEC syndrome. Cell 99, 143–153 (1999). [DOI] [PubMed] [Google Scholar]
- 6.van Bokhoven H., McKeon F., Mutations in the p53 homolog p63: Allele-specific developmental syndromes in humans. Trends Mol. Med. 8, 133–139 (2002). [DOI] [PubMed] [Google Scholar]
- 7.Brunner H. G., Hamel B. C. J., van Bokhoven H., The p63 gene in EEC and other syndromes. J. Med. Genet. 39, 377–381 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kouwenhoven E. N., Oti M., Niehues H., van Heeringen S. J., Schalkwijk J., Stunnenberg H. G., van Bokhoven H., Zhou H., Transcription factor p63 bookmarks and regulates dynamic enhancers during epidermal differentiation. EMBO Rep. 16, 863–878 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rapisarda V., Malashchuk I., Asamaowei I. E., Poterlowicz K., Fessing M. Y., Sharov A. A., Karakesisoglou I., Botchkarev V. A., Mardaryev A., p63 transcription factor regulates nuclear shape and expression of nuclear envelope-associated genes in epidermal keratinocytes. J. Invest. Dermatol. 137, 2157–2167 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lin-Shiao E., Lan Y., Coradin M., Anderson A., Donahue G., Simpson C. L., Sen P., Saffie R., Busino L., Garcia B. A., Berger S. L., Capell B. C., KMT2D regulates p63 target enhancers to coordinate epithelial homeostasis. Genes Dev. 32, 181–193 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mangold E., Ludwig K. U., Birnbaum S., Baluardo C., Ferrian M., Herms S., Reutter H., de Assis N. A., Chawa T. A., Mattheisen M., Steffens M., Barth S., Kluck N., Paul A., Becker J., Lauster C., Schmidt G., Braumann B., Scheer M., Reich R. H., Hemprich A., Pötzsch S., Blaumeiser B., Moebus S., Krawczak M., Schreiber S., Meitinger T., Wichmann H. E., Steegers-Theunissen R. P., Kramer F. J., Cichon S., Propping P., Wienker T. F., Knapp M., Rubini M., Mossey P. A., Hoffmann P., Nöthen M. M., Genome-wide association study identifies two susceptibility loci for nonsyndromic cleft lip with or without cleft palate. Nat. Genet. 42, 24–26 (2010). [DOI] [PubMed] [Google Scholar]
- 12.Beaty T. H., Murray J. C., Marazita M. L., Munger R. G., Ruczinski I., Hetmanski J. B., Liang K. Y., Wu T., Murray T., Fallin M. D., Redett R. A., Raymond G., Schwender H., Jin S. C., Cooper M. E., Dunnwald M., Mansilla M. A., Leslie E., Bullard S., Lidral A. C., Moreno L. M., Menezes R., Vieira A. R., Petrin A., Wilcox A. J., Lie R. T., Jabs E. W., Wu-Chou Y. H., Chen P. K., Wang H., Ye X., Huang S., Yeow V., Chong S. S., Jee S. H., Shi B., Christensen K., Melbye M., Doheny K. F., Pugh E. W., Ling H., Castilla E. E., Czeizel A. E., Ma L., Field L. L., Brody L., Pangilinan F., Mills J. L., Molloy A. M., Kirke P. N., Scott J. M., Arcos-Burgos M., Scott A. F., A genome-wide association study of cleft lip with and without cleft palate identifies risk variants near MAFB and ABCA4. Nat. Genet. 42, 525–529 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jugessur A., Farlie P. G., Kilpatrick N., The genetics of isolated orofacial clefts: From genotypes to subphenotypes. Oral Dis. 15, 437–453 (2009). [DOI] [PubMed] [Google Scholar]
- 14.Ludwig K. U., Böhmer A. C., Bowes J., Nikolic M., Ishorst N., Wyatt N., Hammond N. L., Gölz L., Thieme F., Barth S., Schuenke H., Klamt J., Spielmann M., Aldhorae K., Rojas-Martinez A., Nöthen M. M., Rada-Iglesias A., Dixon M. J., Knapp M., Mangold E., Imputation of orofacial clefting data identifies novel risk loci and sheds light on the genetic background of cleft lip ± cleft palate and cleft palate only. Hum. Mol. Genet. 26, 829–842 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lough K. J., Byrd K. M., Spitzer D. C., Williams S., Closing the gap: Mouse models to study adhesion in secondary palatogenesis. J. Dent. Res. 96, 1210–1220 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yu K., Deng M., Naluai-Cecchini T., Glass I. A., Cox T. C., Differences in oral structure and tissue interactions during mouse vs. human palatogenesis: Implications for the translation of findings from mice. Front. Physiol. 8, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Beaty T. H., Marazita M. L., Leslie E. J., Genetic factors influencing risk to orofacial clefts: Today’s challenges and tomorrow’s opportunities. F1000Res. 5, 2800 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Leslie E. J., Carlson J. C., Shaffer J. R., Butali A., Buxó C. J., Castilla E. E., Christensen K., Deleyiannis F. W. B., Leigh Field L., Hecht J. T., Moreno L., Orioli I. M., Padilla C., Vieira A. R., Wehby G. L., Feingold E., Weinberg S. M., Murray J. C., Beaty T. H., Marazita M. L., Genome-wide meta-analyses of nonsyndromic orofacial clefts identify novel associations between FOXE1 and all orofacial clefts, and TP63 and cleft lip with or without cleft palate. Hum. Genet. 136, 275–286 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mostowska A., Gaczkowska A., Żukowski K., Ludwig K. U., Hozyasz K. K., Wójcicki P., Mangold E., Böhmer A. C., Heilmann-Heimbach S., Knapp M., Zadurska M., Biedziak B., Budner M., Lasota A., Daktera-Micker A., Jagodziński P. P., Common variants in DLG1 locus are associated with non-syndromic cleft lip with or without cleft palate. Clin. Genet. 93, 784–793 (2018). [DOI] [PubMed] [Google Scholar]
- 20.Yu Y., Zuo X., He M., Gao J., Fu Y., Qin C., Meng L., Wang W., Song Y., Cheng Y., Zhou F., Chen G., Zheng X., Wang X., Liang B., Zhu Z., Fu X., Sheng Y., Hao J., Liu Z., Yan H., Mangold E., Ruczinski I., Liu J., Marazita M. L., Ludwig K. U., Beaty T. H., Zhang X., Sun L., Bian Z., Genome-wide analyses of non-syndromic cleft lip with palate identify 14 novel loci and genetic heterogeneity. Nat. Commun. 8, 14364 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fakhouri W. D., Rahimov F., Attanasio C., Kouwenhoven E. N., Ferreira de Lima R. L., Felix T. M., Nitschke L., Huver D., Barrons J., Kousa Y. A., Leslie E., Pennacchio L. A., van Bokhoven H., Visel A., Zhou H., Murray J. C., Schutte B. C., An etiologic regulatory mutation in IRF6 with loss- and gain-of-function effects. Hum. Mol. Genet. 23, 2711–2720 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shalom-Feuerstein R., Lena A. M., Zhou H., De la Forest Divonne S., van Bokhoven H., Candi E., Melino G., Aberdam D., ΔNp63 is an ectodermal gatekeeper of epidermal morphogenesis. Cell Death Differ. 18, 887–896 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Romano R.-A., Smalley K., Magraw C., Serna V. A., Kurita T., Raghavan S., Sinha S., ΔNp63 knockout mice reveal its indispensable role as a master regulator of epithelial development and differentiation. Development 139, 772–782 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Soares E., Zhou H., Master regulatory role of p63 in epidermal development and disease. Cell. Mol. Life Sci. 75, 1179–1190 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Soufi A., Garcia M. F., Jaroszewicz A., Osman N., Pellegrini M., Zaret K. S., Pioneer transcription factors target partial DNA motifs on nucleosomes to initiate reprogramming. Cell 161, 555–568 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Viganò M. A., Lamartine J., Testoni B., Merico D., Alotto D., Castagnoli C., Robert A., Candi E., Melino G., Gidrol X., Mantovani R., New p63 targets in keratinocytes identified by a genome-wide approach. EMBO J. 25, 5105–5116 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pozzi S., Zambelli F., Merico D., Pavesi G., Robert A., Maltère P., Gidrol X., Mantovani R., Vigano M. A., Transcriptional network of p63 in human keratinocytes. PLOS ONE 4, e5008 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Carroll D. K., Carroll J. S., Leong C. O., Cheng F., Brown M., Mills A. A., Brugge J. S., Ellisen L. W., p63 regulates an adhesion programme and cell survival in epithelial cells. Nat. Cell Biol. 8, 551–561 (2006). [DOI] [PubMed] [Google Scholar]
- 29.Jiang R., Lan Y., Chapman H. D., Shawber C., Norton C. R., Serreze D. V., Weinmaster G., Gridley T., Defects in limb, craniofacial, and thymic development in Jagged2 mutant mice. Genes Dev. 12, 1046–1057 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yang X., Lu H., Yan B., Romano R. A., Bian Y., Friedman J., Duggal P., Allen C., Chuang R., Ehsanian R., Si H., Sinha S., van Waes C., Chen Z., ΔNp63 versatilely regulates a broad NF-κB gene program and promotes squamous epithelial proliferation, migration, and inflammation. Cancer Res. 71, 3688–3700 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Candi E., Rufini A., Terrinoni A., Giamboi-Miraglia A., Lena A. M., Mantovani R., Knight R., Melino G., ΔNp63 regulates thymic development through enhanced expression of FgfR2 and Jag2. Proc. Natl. Acad. Sci. U.S.A. 104, 11999–12004 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Thomason H. A., Zhou H., Kouwenhoven E. N., Dotto G. P., Restivo G., Nguyen B. C., Little H., Dixon M. J., van Bokhoven H., Dixon J., Cooperation between the transcription factors p63 and IRF6 is essential to prevent cleft palate in mice. J. Clin. Invest. 120, 1561–1569 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ferretti E., Li B., Zewdu R., Wells V., Hebert J. M., Karner C., Anderson M. J., Williams T., Dixon J., Dixon M. J., Depew M. J., Selleri L., A conserved Pbx-Wnt-p63-Irf6 regulatory module controls face morphogenesis by promoting epithelial apoptosis. Dev. Cell 21, 627–641 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen Y., Mistry D. S., Sen G. L., Highly rapid and efficient conversion of human fibroblasts to keratinocyte-like cells. J. Invest. Dermatol. 134, 335–344 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Suzuki A., Jun G., Abdallah N., Gajera M., Iwata J., Gene datasets associated with mouse cleft palate. Data Brief 18, 655–673 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Peyrard-Janvid M., Leslie E. J., Kousa Y. A., Smith T. L., Dunnwald M., Magnusson M., Lentz B. A., Unneberg P., Fransson I., Koillinen H. K., Rautio J., Pegelow M., Karsten A., Basel-Vanagaite L., Gordon W., Andersen B., Svensson T., Murray J. C., Cornell R. A., Kere J., Schutte B. C., Dominant mutations in GRHL3 cause Van der Woude syndrome and disrupt oral periderm development. Am. J. Hum. Genet. 94, 23–32 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.McGrath J. A., Duijf P. H., Doetsch V., Irvine A. D., de Waal R., Vanmolkot K. R., Wessagowit V., Kelly A., Atherton D. J., Griffiths W. A., Orlow S. J., van Haeringen A., Ausems M. G., Yang A., McKeon F., Bamshad M. A., Brunner H. G., Hamel B. C., van Bokhoven H., Hay–Wells syndrome is caused by heterozygous missense mutations in the SAM domain of p63. Hum. Mol. Genet. 10, 221–229 (2001). [DOI] [PubMed] [Google Scholar]
- 38.Chung J., Grant R. I., Kaplan D. R., Irwin M. S., Special at-rich binding protein-2 (satb2) differentially affects disease-causing p63 mutant proteins. J. Biol. Chem. 286, 40671–40680 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sathyamurthy A., Freund S. M. V., Johnson C. M., Allen M. D., Bycroft M., Structural basis of p63α SAM domain mutants involved in AEC syndrome. FEBS J. 278, 2680–2688 (2011). [DOI] [PubMed] [Google Scholar]
- 40.Rostagno P., Wolchinsky Z., Vigano A. M., Shivtiel S., Zhou H., van Bokhoven H., Ferone G., Missero C., Mantovani R., Aberdam D., Virolle T., Embryonic stem cells as an ectodermal cellular model of human p63-related dysplasia syndromes. Biochem. Biophys. Res. Commun. 395, 131–135 (2010). [DOI] [PubMed] [Google Scholar]
- 41.Huang Y.-P., Kim Y., Li Z., Fomenkov T., Fomenkov A., Ratovitski E. A., AEC-associated p63 mutations lead to alternative splicing/protein stabilization of p63 and modulation of notch signaling. Cell Cycle 4, 1440–1447 (2005). [DOI] [PubMed] [Google Scholar]
- 42.Fomenkov A., Huang Y. P., Topaloglu O., Brechman A., Osada M., Fomenkova T., Yuriditsky E., Trink B., Sidransky D., Ratovitski E., p63α mutations lead to aberrant splicing of keratinocyte growth factor receptor in the hay-wells syndrome. J. Biol. Chem. 278, 23906–23914 (2003). [DOI] [PubMed] [Google Scholar]
- 43.Russo C., Osterburg C., Sirico A., Antonini D., Ambrosio R., Würz J. M., Rinnenthal J., Ferniani M., Kehrloesser S., Schäfer B., Güntert P., Sinha S., Dötsch V., Missero C., Protein aggregation of the p63 transcription factor underlies severe skin fragility in AEC syndrome. Proc. Natl. Acad. Sci. U.S.A. 115, E906–E915 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Juriloff D. M., Harris M. J., Mouse genetic models of cleft lip with or without cleft palate. Birt. Defects Res. A Clin. Mol. Teratol. 82, 63–77 (2008). [DOI] [PubMed] [Google Scholar]
- 45.Becker K. G., Barnes K. C., Bright T. J., Wang S. A., The genetic association database. Nat. Genet. 36, 431–432 (2004). [DOI] [PubMed] [Google Scholar]
- 46.Frebourg T., Oliveira C., Hochain P., Karam R., Manouvrier S., Graziadio C., Vekemans M., Hartmann A., Baert-Desurmont S., Alexandre C., Lejeune Dumoulin S., Marroni C., Martin C., Castedo S., Lovett M., Winston J., Machado J. C., Attié T., Jabs E. W., Cai J., Pellerin P., Triboulet J. P., Scotte M., le Pessot F., Hedouin A., Carneiro F., Blayau M., Seruca R., Cleft lip/palate and CDH1/E-cadherin mutations in families with hereditary diffuse gastric cancer. J. Med. Genet. 43, 138–142 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Juriloff D. M., Harris M. J., Mager D. L., Gagnier L., Epigenetic mechanism causes Wnt9b deficiency and nonsyndromic cleft lip and palate in the A/WySn mouse strain. Birt. Defects Res. A. Clin. Mol. Teratol. 100, 772–788 (2014). [DOI] [PubMed] [Google Scholar]
- 48.Mammadova A., Ackermans M. M., Bloemen M., Oostendorp C., Zhou H., Carels C. E., Von den Hoff J. W., Effects of retinoic acid on proliferation and gene expression of cleft and non-cleft palatal keratinocytes. Eur. J. Orthod. 36, 727–734 (2014). [DOI] [PubMed] [Google Scholar]
- 49.Hixon K., Rhea L., Standley J., Canady F. J., Canady J. W., Dunnwald M., Interferon regulatory factor 6 controls proliferation of keratinocytes from children with Van der Woude syndrome. Cleft Palate Craniofac. J. 54, 281–286 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Degen M., Wiederkehr A., La Scala G. C., Carmann C., Schnyder I., Katsaros C., Keratinocytes isolated from individual cleft lip/palate patients display variations in their differentiation potential in vitro. Front. Physiol. 9, 1703 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Qu J., Tanis S. E. J., Smits J. P. H., Kouwenhoven E. N., Oti M., van den Bogaard E. H., Logie C., Stunnenberg H. G., van Bokhoven H., Mulder K. W., Zhou H., Mutant p63 affects epidermal cell identity through rewiring the enhancer landscape. Cell Rep. 25, 3490–3503.e4 (2018). [DOI] [PubMed] [Google Scholar]
- 52.Way G. P., Youngstrom D. W., Hankenson K. D., Greene C. S., Grant S. F., Implicating candidate genes at GWAS signals by leveraging topologically associating domains. Eur. J. Hum. Genet. 25, 1286–1289 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pervjakova N., Prokopenko I., The TAD-pathway for GWAS signals. Eur. J. Hum. Genet. 25, 1179–1180 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Phillips-Cremins J. E., Sauria M. E. G., Sanyal A., Gerasimova T. I., Lajoie B. R., Bell J. S. K., Ong C. T., Hookway T. A., Guo C., Sun Y., Bland M. J., Wagstaff W., Dalton S., McDevitt T. C., Sen R., Dekker J., Taylor J., Corces V. G., Architectural protein subclasses shape 3D organization of genomes during lineage commitment. Cell 153, 1281–1295 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.de Leeuw C. A., Mooij J. M., Heskes T., Posthuma D., MAGMA: Generalized gene-set analysis of GWAS data. PLOS Comput. Biol. 11, e1004219 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bebee T. W., Park J. W., Sheridan K. I., Warzecha C. C., Cieply B. W., Rohacek A. M., Xing Y., Carstens R. P., The splicing regulators Esrp1 and Esrp2 direct an epithelial splicing program essential for mammalian development. eLife 4, e08954 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cox L. L., Cox T. C., Moreno Uribe L. M., Zhu Y., Richter C. T., Nidey N., Standley J. M., Deng M., Blue E., Chong J. X., Yang Y., Carstens R. P., Anand D., Lachke S. A., Smith J. D., Dorschner M. O., Bedell B., Kirk E., Hing A. V., Venselaar H., Valencia-Ramirez L. C., Bamshad M. J., Glass I. A., Cooper J. A., Haan E., Nickerson D. A., van Bokhoven H., Zhou H., Krahn K. N., Buckley M. F., Murray J. C., Lidral A. C., Roscioli T., Mutations in the epithelial cadherin-p120-catenin complex cause mendelian non-syndromic cleft lip with or without cleft palate. Am. J. Hum. Genet. 102, 1143–1157 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Beaty T. H., Taub M. A., Scott A. F., Murray J. C., Marazita M. L., Schwender H., Parker M. M., Hetmanski J. B., Balakrishnan P., Mansilla M. A., Mangold E., Ludwig K. U., Noethen M. M., Rubini M., Elcioglu N., Ruczinski I., Confirming genes influencing risk to cleft lip with/without cleft palate in a case–parent trio study. Hum. Genet. 132, 771–781 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Prenzler F., Fragasso A., Schmitt A., Munz B., Functional analysis of ZFP36 proteins in keratinocytes. Eur. J. Cell Biol. 95, 277–284 (2016). [DOI] [PubMed] [Google Scholar]
- 60.Cura F., Böhmer A. C., Klamt J., Schünke H., Scapoli L., Martinelli M., Carinci F., Nöthen M. M., Knapp M., Ludwig K. U., Mangold E., Replication analysis of 15 susceptibility loci for nonsyndromic cleft lip with or without cleft palate in an italian population. Birt. Defects Res. A. Clin. Mol. Teratol. 106, 81–87 (2016). [DOI] [PubMed] [Google Scholar]
- 61.Leslie E. J., Taub M. A., Liu H., Steinberg K. M., Koboldt D. C., Zhang Q., Carlson J. C., Hetmanski J. B., Wang H., Larson D. E., Fulton R. S., Kousa Y. A., Fakhouri W. D., Naji A., Ruczinski I., Begum F., Parker M. M., Busch T., Standley J., Rigdon J., Hecht J. T., Scott A. F., Wehby G. L., Christensen K., Czeizel A. E., Deleyiannis F. W. B., Schutte B. C., Wilson R. K., Cornell R. A., Lidral A. C., Weinstock G. M., Beaty T. H., Marazita M. L., Murray J. C., Identification of functional variants for cleft lip with or without cleft palate in or near PAX7, FGFR2, and NOG by targeted sequencing of GWAS loci. Am. J. Hum. Genet. 96, 397–411 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Schmidt E. M., Zhang J., Zhou W., Chen J., Mohlke K. L., Chen Y. E., Willer C. J., GREGOR: Evaluating global enrichment of trait-associated variants in epigenomic features using a systematic, data-driven approach. Bioinformatics 31, 2601–2606 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Grant S. F. A., Wang K., Zhang H., Glaberson W., Annaiah K., Kim C. E., Bradfield J. P., Glessner J. T., Thomas K. A., Garris M., Frackelton E. C., Otieno F. G., Chiavacci R. M., Nah H. D., Kirschner R. E., Hakonarson H., A genome-wide association study identifies a locus for nonsyndromic cleft lip with or without cleft palate on 8q24. J. Pediatr. 155, 909–913 (2009). [DOI] [PubMed] [Google Scholar]
- 64.Bao X., Rubin A. J., Qu K., Zhang J., Giresi P. G., Chang H. Y., Khavari P. A., A novel ATAC-seq approach reveals lineage-specific reinforcement of the open chromatin landscape via cooperation between BAF and p63. Genome Biol. 16, 284 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sethi I., Sinha S., Buck M. J., Role of chromatin and transcriptional co-regulators in mediating p63-genome interactions in keratinocytes. BMC Genomics 15, 1042 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kirschner R. D., Rother K., Müller G. A., Engeland K., The retinal dehydrogenase/reductase retSDR1/DHRS3 gene is activated by p53 and p63 but not by mutants derived from tumors or EEC/ADULT malformation syndromes. Cell Cycle 9, 2177–2188 (2014). [DOI] [PubMed] [Google Scholar]
- 67.McDade S. S., Henry A. E., Pivato G. P., Kozarewa I., Mitsopoulos C., Fenwick K., Assiotis I., Hakas J., Zvelebil M., Orr N., Lord C. J., Patel D., Ashworth A., McCance D. J., Genome-wide analysis of p63 binding sites identifies AP-2 factors as co-regulators of epidermal differentiation. Nucleic Acids Res. 40, 7190–7206 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Richardson R., Mitchell K., Hammond N. L., Mollo M. R., Kouwenhoven E. N., Wyatt N. D., Donaldson I. J., Zeef L., Burgis T., Blance R., van Heeringen S. J., Stunnenberg H. G., Zhou H., Missero C., Romano R. A., Sinha S., Dixon M. J., Dixon J., p63 exerts spatio-temporal control of palatal epithelial cell fate to prevent cleft palate. PLOS Genet. 13, e1006828 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wilderman A., VanOudenhove J., Kron J., Noonan J. P., Cotney J., High-resolution epigenomic atlas of human embryonic craniofacial development. Cell Rep. 23, 1581–1597 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sethi I., Gluck C., Zhou H., Buck M. J., Sinha S., Evolutionary re-wiring of p63 and the epigenomic regulatory landscape in keratinocytes and its potential implications on species-specific gene expression and phenotypes. Nucleic Acids Res. 45, 8208–8224 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Suzuki K., Hu D., Bustos T., Zlotogora J., Richieri-Costa A., Helms J. A., Spritz R. A., Mutations of PVRL1, encoding a cell-cell adhesion molecule/herpesvirus receptor, in cleft lip/palate-ectodermal dysplasia. Nat. Genet. 25, 427–430 (2000). [DOI] [PubMed] [Google Scholar]
- 72.Piedrahita J. A., Oetama B., Bennett G. D., van Waes J., Kamen B. A., Richardson J., Lacey S. W., Anderson R. G. W., Finnell R. H., Mice lacking the folic acid-binding protein Folbp1 are defective in early embryonic development. Nat. Genet. 23, 228–232 (1999). [DOI] [PubMed] [Google Scholar]
- 73.Xiao Y., Taub M. A., Ruczinski I., Begum F., Hetmanski J. B., Schwender H., Leslie E. J., Koboldt D. C., Murray J. C., Marazita M. L., Beaty T. H., Evidence for SNP-SNP interaction identified through targeted sequencing of cleft case-parent trios. Genet. Epidemiol. 41, 244–250 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ludwig K. U., Mangold E., Herms S., Nowak S., Reutter H., Paul A., Becker J., Herberz R., AlChawa T., Nasser E., Böhmer A. C., Mattheisen M., Alblas M. A., Barth S., Kluck N., Lauster C., Braumann B., Reich R. H., Hemprich A., Pötzsch S., Blaumeiser B., Daratsianos N., Kreusch T., Murray J. C., Marazita M. L., Ruczinski I., Scott A. F., Beaty T. H., Kramer F. J., Wienker T. F., Steegers-Theunissen R. P., Rubini M., Mossey P. A., Hoffmann P., Lange C., Cichon S., Propping P., Knapp M., Nöthen M. M., Genome-wide meta-analyses of nonsyndromic cleft lip with or without cleft palate identify six new risk loci. Nat. Genet. 44, 968–971 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Uslu V. V., Petretich M., Ruf S., Langenfeld K., Fonseca N. A., Marioni J. C., Spitz F., Long-range enhancers regulating Myc expression are required for normal facial morphogenesis. Nat. Genet. 46, 753–758 (2014). [DOI] [PubMed] [Google Scholar]
- 76.Wu N., Rollin J., Masse I., Lamartine J., Gidrol X., p63 regulates human keratinocyte proliferation via MYC-regulated gene network and differentiation commitment through cell adhesion-related gene network. J. Biol. Chem. 287, 5627–5638 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Zhu J., Sammons M. A., Donahue G., Dou Z., Vedadi M., Getlik M., Barsyte-Lovejoy D., Al-awar R., Katona B. W., Shilatifard A., Huang J., Hua X., Arrowsmith C. H., Berger S. L., Gain-of-function p53 mutants co-opt chromatin pathways to drive cancer growth. Nature 525, 206–211 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Shah P. P., Donahue G., Otte G. L., Capell B. C., Nelson D. M., Cao K., Aggarwala V., Cruickshanks H. A., Rai T. S., McBryan T., Gregory B. D., Adams P. D., Berger S. L., Lamin B1 depletion in senescent cells triggers large-scale changes in gene expression and the chromatin landscape. Genes Dev. 27, 1787–1799 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Buenrostro J. D., Giresi P. G., Zaba L. C., Chang H. Y., Greenleaf W. J., Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat. Methods 10, 1213–1218 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Watanabe K., Taskesen E., van Bochoven A., Posthuma D., Functional mapping and annotation of genetic associations with FUMA. Nat. Commun. 8, 1826 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/5/eaaw0946/DC1
Fig. S1. Ectopic expression of p63 in fibroblasts leads to establishment of enhancers and downstream up-regulation of inflammation and epithelial genes.
Fig. S2. Ectopic coexpression of p63 and KLF4 leads to establishment of keratinocyte-specific enhancers and up-regulation of epithelial genes.
Fig. S3. Mutations in p63 disrupt enhancer establishment and lead to loss of up-regulation of key keratinocyte-specific genes.
Fig. S4. Heatmap of genes regulated by p63 + KLF4 that have been strongly linked to CL/P in murine KD and KO models.
Fig. S5. p63/KLF4 peaks colocalize strongly with highly associated CL/P SNPs near IRF6, RHPN2, and GPATCH1.
Table S1. Genes up-regulated in fibroblasts + p63 compared to control fibroblasts (fold change > 1.5; FDR < 0.05).
Table S2. Genes up-regulated in fibroblasts + p63 + KLF4 compared to control fibroblasts (fold change > 1.5; FDR < 0.05).
Table S3. Mutations in p63 disrupt up-regulation of keratinocyte-specific genes.
Table S4. Causal and candidate genes involved in CL/P identified in our transdifferentiation model.