

Abstract
The first step in the biosynthetic pathway of vitamin C in plants is the formation, at the level of sugar nucleotide, of l-galactosyl residues, catalyzed by a largely unknown GDP-d-mannose 3",5"-epimerase. By using combined conventional biochemical and mass spectrometry methods, we obtained a highly purified preparation of GDP-d-mannose 3",5"-epimerase from an Arabidopsis thaliana cell suspension. The native enzyme is an 84-kDa dimer, composed of two apparently identical subunits. In-gel tryptic digestion of the enzyme subunit, followed by peptide sequencing and a blast search, led to the identification of the epimerase gene. The closest homolog of the plant epimerase is the BlmG gene product of Streptomyces sp., a putative NDP-d-mannose 5"-epimerase. The plant GDP-d-mannose 3",5"-epimerase is, to our knowledge, a novel member of the extended short-chain dehydrogenase/reductase family. The enzyme was cloned and expressed in Escherichia coli cells.
L-ascorbic acid (l-AA; vitamin C) is present in millimolar concentrations in plants, where it functions as the major antioxidant and as an enzyme cofactor. Consequently, l-AA is involved in many different processes within the plant cell, including hormone and cell-wall biosynthesis, stress resistance, photoprotection, and cell growth (1), and possibly other functions still to be discovered. Humans are unable to synthesize vitamin C because they have an unfunctional gene for l-gulono-1,4-lactone oxidase, which is the last enzyme in the biosynthesis of ascorbate in animals (2). Therefore, plant foods are the major source of this essential micronutrient in the human diet. Knowledge of the biosynthesis of vitamin C will help to understand the complex role of l-AA and to modify its level in plants.
In contrast to the well established pathway in animals (3), the biosynthesis of vitamin C in plants has been a subject of controversy for many years (4). Although l-galactono-1,4-lactone was recognized as a direct precursor of l-AA (5–7), the carbon source for l-galactono-1,4-lactone remained an enigma until recently. The demonstration that d-arabinono-1,4-lactone, the direct precursor of d-erythroascorbic acid (a 5-carbon homolog of l-AA) in yeast, is formed from d-arabinose in a reaction catalyzed by d-arabinose dehydrogenase (8), was undoubtedly of great importance for elucidating the biosynthesis of vitamin C in plants (Fig. 1). d-arabinose and d-arabinono-1,4-lactone are 5-carbon homologs of l-Gal and l-galactono-1,4-lactone, respectively. l-Gal was found in algae and plants, and the corresponding sugar nucleotide, GDP-l-Gal, is known to be formed from GDP-Man as a result of the 3",5"-epimerization reaction (9). According to this line of evidence, Wheeler et al. (10) demonstrated an efficient conversion of exogenous d-[14C]Man and of cold l-Gal into l-AA, and the presence, by analogy with the yeast d-arabinose dehydrogenase, of l-Gal dehydrogenase activity in pea. In the proposed biosynthetic pathway for vitamin C in plants (10), d-mannosyl residues of GDP-d-Man are direct precursors of l-galactosyl residues of GDP-l-Gal; after a release, l-Gal undergoes two sequential oxidation reactions: first to l-galactono-1,4-lactone and then to l-AA (Fig. 1). The involvement of sugar nucleotides is reminiscent of the vitamin C pathway in animals, in which UDP-d-Glc is oxidized at C-6 to UDP-d-GlcUA (3). For both pathways, the mechanism of the sugar release from its nucleotide-bound form (GDP-l-Gal and UDP-d-glucuronate in plants and animals, respectively) is not well understood.
Figure 1.

Proposed de novo pathway for the synthesis of l-AA from d-Man in plants. Enzymes 1, hexokinase; 2, phosphomannomutase; 3, GDP-Man pyrophosphorylase; 4, GDP-Man 3",5"-epimerase; 5, l-Gal dehydrogenase; 6, l-galactono-1,4-lactone dehydrogenase.
The involvement of GDP-Man, the product of the GDP-Man pyrophosphorylase reaction, in the plant l-AA biosynthesis was recently demonstrated (11, 12). However, GDP-Man is used not only for l-AA biosynthesis but also for the biosynthesis of GDP-l-Fuc, cell-wall polysaccharides, and glycoproteins. Therefore, changes in the expression of GDP-Man pyrophosphorylase would probably produce pleiotropic effects that, in turn, could affect indirectly the biosynthesis of vitamin C.
According to the proposed l-AA pathway in plants (Fig. 1), GDP-Man is converted to GDP-β-l-Gal by a GDP-Man 3",5"-epimerase. GDP-l-Gal can be used then for the biosynthesis of vitamin C and of l-Gal-containing glycoconjugates. l-Gal is a relatively rare sugar; it was found as a constituent of structural polysaccharides in some invertebrates (13, 14) and in algae (15). On the contrary, in plants, l-Gal seems to be only a minor structural component, if any (9). It is apparently absent from cell-wall polysaccharides (16) as well as from glycoproteins (17) of wild-type Arabidopsis thaliana, but it could be detected as a substituent of l-Fuc in xyloglucans (18) and in a minor portion of N-glycans (17) of Arabidopsis mur1 mutants, which are known to be deficient in the first enzyme of the GDP-l-Fuc pathway, the GDP-Man 4",6"-dehydratase (19).
Therefore, it can be assumed that, at least in plants, the majority if not all l-galactosyl residues of GDP-l-Gal is channeled into the l-AA pathway. This assumption would imply that the GDP-Man 3",5"-epimerization reaction should be considered as the first step in the vitamin C pathway in plants. The enzyme responsible for the reversible conversion of GDP-d-Man into GDP-l-Gal, the GDP-Man 3",5"-epimerase (Fig. 1, reaction 4), catalyzes a unique double epimerization of the hexosyl residue. Other known 3",5"-epimerase activities, such as those involved in the biosynthesis of GDP-l-Fuc (20) and TDP-l-rhamnose (21), operate on NDP derivatives of 4-keto,6-deoxy-hexoses. The GDP-Man 3",5"-epimerase activity was reported in a snail (13), in the green alga Chlorella pyrenoidosa (22), and in the plants Linum usitatissimum (flax) (22) and pea (10). Only the Chlorella enzyme was studied to a limited extent (23–26), but it has never been purified and the corresponding gene is still unknown.
One of the major difficulties in studying GDP-Man 3",5"-epimerase was the lack of a suitable enzymatic test. Recently, we have developed an easy assay for the epimerase activity (27). Here, we describe the purification of the GDP-Man 3",5"-epimerase from A. thaliana cell suspensions and the identification of the corresponding gene.
Materials and Methods
Reagents.
Guanosine diphospho-d-[U-14C]Man (specific activity 296 mCi/mmol) was purchased from Amersham Pharmacia Biotech (Little Chalfont, Buckinghamshire, UK). All reagents were of analytical grade. Guanosine 5′-diphosphate immobilized on crosslinked 4% beaded agarose and heparin-agarose resin were purchased from Sigma-Aldrich. DEAE-Sepharose Fast Flow, Sephacryl S-200, and Blue Sepharose CL-6B were from Amersham Pharmacia Biotech. Hydroxylapatite Bio-Gel HTP was purchased from Bio-Rad.
Cells.
Arabidopsis thaliana (L.) Heynh. ecotype Columbia cell suspensions were grown as described (27).
Protein Determination.
Protein concentration was determined by the method of Bradford (28), using BSA as standard.
GDP-Man 3",5"-Epimerase Assay and Ascorbic Acid Determination.
The GDP-Man 3",5"-epimerase activity and l-AA were determined by the HPLC method as described (27), with the exception that the concentration of methanol in solvent A was 0.5% and the flow rate was 0.8 ml/min.
Purification of the GDP-Man 3",5"-Epimerase.
Arabidopsis suspension cells (4 days old; 250 g wet weight) were ground with liquid nitrogen to a fine powder in a precooled mortar and extracted with 2 volumes of 0.1 M Tris⋅HCl buffer (pH 7.7) containing 5 mM DTT, 1 mM EDTA, 0.6 M sucrose, 1 mM PMSF, and 1% (wt/vol) polyvinylpolypyrrolidone (buffer A). The 55–70% ammonium sulfate fraction was prepared (27), dissolved in 50 mM Tris⋅HCl buffer (pH 7.7) containing 1 mM EDTA, 0.5 mM DTT, 1 mM PMSF, and 20% glycerol (buffer B), desalted on prepacked NAP-25 columns (Amersham Pharmacia Biotech), and loaded on a DEAE-Sepharose column (1.2 × 18 cm) equilibrated with buffer B for FPLC (Amersham plc). The epimerase activity was eluted with a 200-ml linear gradient from 0 to 200 mM NaCl in buffer B. Active fractions were concentrated to 4 ml by ultrafiltration and applied to a Sephacryl S-200 column (1.6 × 94 cm) equilibrated with buffer B at a flow rate of 0.5 ml/min. The pooled active gel-filtration fractions were applied onto a hydroxylapatite Bio-Gel (1.2 × 5 cm) column equilibrated with 2 mM potassium phosphate buffer (pH 7.2) containing 0.5 mM DTT, 1 mM EDTA, 1 mM PMSF, and 20% glycerol (buffer C). The elution was carried out with 100 ml of a linear gradient from 2 to 500 mM potassium phosphate in buffer C. The active fractions were concentrated and applied on a 3-ml heparin-agarose column equilibrated with buffer B. The nonadsorbed material was applied to a 2-ml column of GDP-agarose equilibrated with buffer B. The obtained flow-through was applied at room temperature to a 3-ml Blue Sepharose CL-6B column equilibrated with buffer B. The column was washed with buffer B and eluted with 1 mM NAD+ in buffer B, followed by 0.5 M NaCl in buffer B. The active fractions from different elution steps (nonretained, 1 mM NAD and 0.5 M NaCl) were pooled separately and concentrated to 30 μl by ultrafiltration. A summary of the purification procedure is presented in Table 1.
Table 1.
Purification of GDP-Man 3",5"-epimerase from an A. thaliana cell suspension
| Purification step | Volume, ml | Total protein, mg | Total activity, units | Specific activity, units/mg of protein | Purification fold | Recovery, % |
|---|---|---|---|---|---|---|
| Crude extract | 538 | 2690 | —* | —* | —* | —* |
| 55–70% Ammonium sulfate fraction | 21 | 474 | 97531 | 206 | 1 | 100 |
| DEAE-Sepharose pooled fractions | 20 | 143 | 57402 | 401 | 1.9 | 59 |
| Sephacryl S-200 pooled fractions | 7 | 11.2 | 4306 | 384 | 1.9 | 4.4 |
| Hydroxylapatite pooled fractions | 6 | 2.7 | 1963 | 727 | 3.5 | 2.0 |
| Heparin-agarose nonadsorbed fraction | 6 | 0.73 | 1050 | 1438 | 7.0 | 1.1 |
| GDP-agarose nonadsorbed fraction | 9 | 0.68 | 827 | 1216 | 6.0 | 0.85 |
| Blue-Sepharose nonadsorbed fraction | 11 | 0.55 | 292 | 531 | 2.6 | 0.30 |
| Blue-Sepharose NAD-eluate | 6 | 0.02 | 272† | 13600 | 66.0 | 0.28 |
| Blue-Sepharose NaCl-eluate | 3 | 0.10 | 173 | 1730 | 8.4 | 0.18 |
Not determined.
Epimerase activity measured in the presence of 1 mM NAD.
PAGE.
Proteins were separated by SDS/PAGE, using 12.5% minigels and the buffer system described by Laemmli (29). Gels were stained with Coomassie brilliant blue R-250.
Peptide Preparation for MS.
Tryptic in-gel digestion of protein bands followed by peptide extraction and concentration were performed according to the procedure of Wilm et al. (30).
Nano-Electrospray Ionization Tandem MS (Nano-ESI MS/MS) of Tryptic Peptides.
Peptide analyses were performed on a Finnigan LCQ quadrupole ion trap mass spectrometer (Finnigan-MAT, San Jose, CA), using disposable capillary needles (Protana, Odense, Denmark) as described (31). Product ion spectra were interpreted manually, and the obtained peptide sequences were submitted to database searching for protein identification by using National Center for Biotechnology Information/blast tools (http://www.ncbi.nlm.nih.gov/blast/).
Matrix-Assisted Laser Desorption Ionization–Time of Flight MS (MALDI-TOF) Peptide-Mass Mapping.
All measurements were performed on a Bruker Reflex III MALDI-TOF MS (Bruker Daltonik GmbH, Bremen, Germany) operating in reflectron mode (32). The obtained peptide-mass fingerprints were analyzed by using the Mascot algorithm (http://www.matrixscience.com) to identify proteins.
Plasmid Construction.
The cDNA library (in vector pGAD10) prepared from green vegetative tissues of A. thaliana ecotype Columbia was a generous gift of L. De Veylder (Ghent University, Ghent, Belgium). Oligonucleotide primers for the amplification of the epimerase cDNA present in the A. thaliana cDNA library were designed with attB1 or attB2 for the insertion into the GATEWAY donor vector pDONR201 (Invitrogen) by homologous recombination. Primers with the following sequences were synthesized by GENSET OLIGOS (Paris): 5′-pENTR_Epim (forward), 5′-GGGG-attB1-CCACCATGGGAACTACCAATGGAACAGAC-3′; and 3′-pENTR_Epim (reverse), 5′-GGGG-attB2-CTCACTCTTTTCCATCAGCCGCGCGGAG-3′. attB1 and attB2 are the minimal 25-bp sequences required for efficient homologous recombination. The PCR product was cloned into the pDONR201 vector, and the resulting plasmid, pENTR_Epim, was used to transfer the gene sequence into pDEST15 [glutathione S-transferase (GST)-tag N-terminal fusion] or pDEST17 (His-tag N-terminal fusion) by means of homologous recombination. The corresponding plasmids, pDEST15_Epim (GST fusion) and pDEST17_Epim (His fusion), were used for expression of the proteins in Escherichia coli BL21(DE3) cells.
Heterologous Expression of the Recombinant Epimerase.
E. coli cells carrying the pDEST15_Epim or pDEST17_Epim plasmids were grown at 37°C to an OD at 600 nm of 0.6 in 300 ml of culture volume and then transferred at 28°C. After a 30-min adaptation, isopropyl-β-d-thiogalactopyranoside was added to a final concentration of 0.4 mM for induction, and the fusion protein was produced overnight. The cells were washed, resuspended in 3 volumes of 50 mM Tris⋅HCl buffer (pH 7.7) containing 1 mM EDTA, 0.5 mM DTT, 1 mM PMSF, 1 mM NAD+ and 20% glycerol, treated for 15 min at 26°C with lysozyme (100 μg/ml), and submitted twice to freezing in liquid nitrogen followed by thawing on ice. Crude extracts and 90% ammonium sulfate fractions were tested for epimerase activity by our HPLC assay (27).
Results
Purification of the GDP-Man 3",5"-Epimerase.
The GDP-Man 3",5"-epimerase was purified from 4-day-old A. thaliana suspension cells (Materials and Methods; Table 1). We observed that the level of total l-AA (l-AA + dehydro-l-AA) and the capacity of A. thaliana cells to convert the exogenous 14C-mannose into the total l-AA pool both decrease after aging of the cell culture (B.A.W., unpublished results). Significantly, this decrease correlates with a drop of the specific activity of GDP-Man 3",5"-epimerase (Fig. 2), suggesting that the enzyme could play a role in the control of l-AA biosynthesis. Because on day 4 of the Arabidopsis cell suspension culture the capacity of cells to synthesize l-AA, their l-AA content as well as the specific activity of GDP-Man 3",5"-epimerase were high, we collected the cells at that specific moment. A high osmolarity buffer containing 0.6 M sucrose was used for cell extraction to prevent disruption of cellular organelles. The epimerase activity could not be measured in crude extracts (Table 1) because of the presence of other enzymes using GDP-Man as substrate (27). The 55–70% ammonium sulfate fraction, which represented 18% of the total protein and contained all of the epimerase activity (Table 1), was separated further by DEAE-Sepharose anion-exchange FPLC. As shown in Fig. 3A, all epimerase activity bound to DEAE-Sepharose and was eluted as two overlapping peaks at ≈75 mM NaCl. Sephacryl S-200 gel filtration chromatography of the pooled epimerase fractions from DEAE-Sepharose (Fig. 3B) resulted in a symmetrical peak of activity corresponding to an 84-kDa protein (data not shown). A 10-fold decrease in the enzymatic activity was observed, probably because of the loss of a cofactor from the enzymatic complex. Further enzyme purification was achieved on a hydroxylapatite column (Fig. 3C); the epimerase eluted at ≈40 mM potassium phosphate buffer.
Figure 2.

Correlation between l-AA content and GDP-Man 3",5"-epimerase activity during the growth of A. thaliana cell suspension. Total l-AA content of cells and specific epimerase activity of the corresponding 55–70% ammonium sulfate fractions were determined as described in Materials and Methods.
Figure 3.
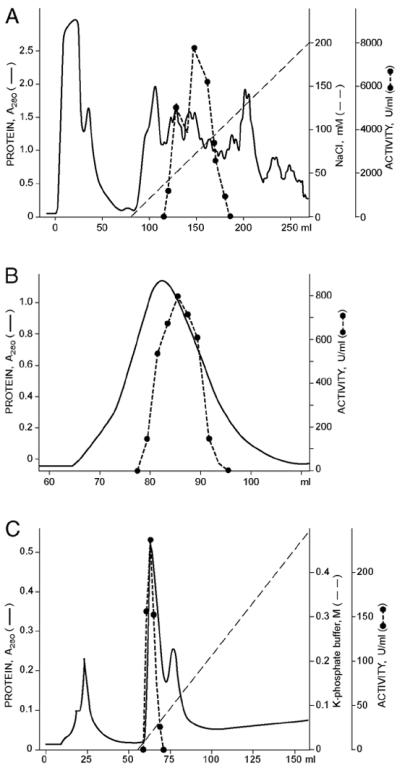
Purification of GDP-Man 3",5"-epimerase from A. thaliana suspension cells. (A) DEAE-Sepharose FPLC of the ammonium sulfate fraction. The enzyme was eluted with a linear gradient of NaCl from 0 to 200 mM (see Materials and Methods). (B) Sephacryl S-200 gel filtration chromatography of epimerase fractions from DEAE-Sepharose. (C) Hydroxylapatite FPLC of epimerase fractions from Sephacryl gel filtration. The proteins were eluted with a linear gradient of 2–500 mM potassium phosphate (pH 7.2) (see Materials and Methods).
The two next affinity steps in the epimerase purification (heparin-agarose and GDP-agarose chromatography; Table 1) were included to remove some persisting and abundant contaminants identified by nano-ESI MS/MS. The nonadsorbed material from GDP-agarose was applied onto a dye-affinity column of Blue-Sepharose; ≈50% of the epimerase activity bound to the Blue-Sepharose column (Table 1). Specific elution with 1 mM NAD+ resulted in the recovery of ≈30% of the applied epimerase activity.
The NAD-eluted Blue-Sepharose fraction contained ≈20 μg of protein in total and showed the highest specific activity of the epimerase (13,600 units/mg of protein) (Table 1). Four protein bands were seen on SDS/PAGE (Fig. 4)—the slowest-migrating 60-kDa band (band 1), a faint 55-kDa band (band 2), and two discrete bands, differing in intensity, of 46 (band 3, major) and 43 kDa (band 4, minor). In-gel tryptic digestion of bands 1 and 2, followed by MALDI-TOF peptide-mass mapping, allowed an unambiguous identification of the proteins as betaine aldehyde dehydrogenase and glutathione synthase, respectively. However, no significant match could be found for bands 3 and 4. Because the molecular mass of the native epimerase was estimated to be ≈84 kDa and no protein bands of high molecular mass (>60 kDa) could be observed on the gel, we deduced that the enzyme had probably a dimeric structure with a subunit of ≈42 kDa. Thus, the two discrete bands of 46 and 43 kDa (Fig. 4) would represent the epimerase subunit(s).
Figure 4.

SDS/PAGE of the partially purified GDP-Man 3",5"-epimerase. Proteins were visualized by Coomassie blue staining. (Left) Molecular mass standards. (Right) NAD-eluted fraction from Blue-Sepharose. Proteins were identified by MS of in-gel tryptic digests. Band 1, betaine aldehyde dehydrogenase; band 2, glutathione synthase; bands 3 and 4, GDP-Man 3",5"-epimerase, with apparent molecular masses of 46 and 43 kDa, respectively.
Identification of GDP-Man 3",5"-Epimerase.
The 46- and 43-kDa protein bands of the NAD-eluted fraction from Blue-Sepharose (bands 3 and 4; Fig. 4) were subjected to in-gel tryptic digestion, followed by nano-ESI MS/MS analysis, as described in Materials and Methods. The MS spectrum of both bands revealed the presence of an abundant doubly charged peptide ion at m/z 785.9, the product ion spectrum of which is shown in Fig. 5. The interpretation of the MS/MS spectrum gave the sequence ISITGAGGFIASHIAR, which matches perfectly with only one protein: a predicted 42.8-kDa protein of A. thaliana annotated as epimerase/dehydratase-like protein, found in the genome database of Arabidopsis (http://www.arabidopsis.org/blast) (European Molecular Biology Laboratory accession no. AF272706; Figs. 5 and 6). To confirm further the identity of the GDP-Man 3",5"-epimerase, the sequence of the hypothetical 42.8-kDa protein of A. thaliana was subjected to an in-silico tryptic digestion using the MS-Digest algorithm (http://prospector.ucsf.edu), and the calculated masses of the theoretical peptides were subsequently compared with those observed in the MALDI-TOF analysis of tryptic digests derived from the 46- and 43-kDa protein bands (Fig. 4; Table 2). As shown in Table 2, 8 of 9 measured peptide masses for the 46-kDa protein (Fig. 4, band 3) digest corresponded exactly (mass difference <20 ppm) to the calculated tryptic peptide masses of the hypothetical 42.8-kDa protein of A. thaliana. Similarly, 14 of the total 16 MALDI-TOF peptides derived from the 43-kDa protein (Fig. 4, band 4) could be identified in the theoretical digest of the predicted 42.8-kDa epimerase/dehydratase-like protein (Table 2). The m/z 785.9 peptide ion identified by the nano-ESI MS/MS (Fig. 5) was also the most abundant ion in the MALDI-TOF analysis (m/z 1570.76; Table 2). The tryptic peptides derived from the 46- and 43-kDa proteins and observed in the MALDI-TOF experiment covered 46% of the epimerase sequence (Fig. 6; Table 2). Obviously, the two discrete epimerase bands of 46 and 43 kDa represent different molecular forms of the same protein. Analysis of the C-terminal peptides observed in the MALDI-TOF of the epimerase bands revealed that the most distal C-terminal peptide present in the 46-kDa band digest (V358VGTQAPVQLGSLR371) was absent from the 43-kDa band-derived sample (Table 2). Instead, the latter showed the presence of two related C-terminal peptides (I331TYFWIK337 and I331TYFWIKEQIEKEK344) located more upstream within the protein sequence (Table 2; Fig. 6), suggesting that the 43-kDa epimerase band represented the product of a proteolytic cleavage at the C terminus of the 46-kDa form. The loss of about 30 amino acids from the C terminus would account for a ≈3-kDa shift in the molecular mass of GDP-Man 3",5"-epimerase and, consequently, the appearance of a minor, faster migrating protein band on the SDS/PAGE (Fig. 4, band 4).
Figure 5.

Nano-ESI MS/MS spectrum of the m/z 785.9 [M + 2H]+2 peptide ion derived from in-gel tryptic digestion of the 46- and 43-kDa bands of GDP-Man 3",5"-epimerase (see Fig. 4).
Figure 6.

Database amino acid sequence of a hypothetical epimerase/dehydratase-like protein of A. thaliana (European Molecular Biology Laboratory accession no. AF272706) corresponding to the purified GDP-Man 3",5"-epimerase. The thick underlined region corresponds to the m/z 785.9 peptide sequence obtained by the nano-ESI MS/MS of the in-gel tryptic digest of the 46- and 43-kDa epimerase bands (see Fig. 5). Peptide ions corresponding to the underlined regions were observed by MALDI-TOF MS (see Table 2).
Table 2.
Comparison of peptide ions observed in the MALDI-TOF MS analysis of the in-gel tryptic digest of the 46- and 43-kDa GDP-Man 3",5"-epimerase bands (see Fig. 5) with those predicted by MS-Digest algorithm (http://prospector.ucsf.edu) for the hypothetical epimerase/dehydratase-like protein of A. thaliana
| Epimerase band | MALDI-TOF- measured mass [M+H]† | MS-Digest-calculated mass [M+H]† | Mass difference, ppm | Missed cleavages | Modifications | Peptide sequence predicted by MS-Digest |
|---|---|---|---|---|---|---|
| 46 kDa | 1424.70 | 1424.82 | −12 | 0 | — | VVGTQAPVQLGSLR |
| 1466.55 | 1466.72 | −17 | 0 | — | FHNIYGPFGTWK | |
| 1549.77 | 1549.90 | −13 | 1 | — | KLPIHHIPGPEGVR | |
| 1570.76 | 1570.87 | −11 | 0 | — | ISITGAGGFIASHIAR† | |
| 1674.86 | * | * | * | * | * | |
| 1820.81 | 1820.88 | −7 | 1 | — | ELEREQYWPSENLK | |
| 1905.75 | 1905.85 | −10 | 0 | — | ESDAWPAEPQDAYGLEK | |
| 2098.97 | 2098.96 | +1 | 1 | — | AQTSTDRFEMWGDGLQTR | |
| 2451.99 | 2452.04 | −5 | 0 | Cys-acrylamide | NEHMTEDMFCDEFHLVDLR | |
| 43 kDa | 944.55 | 944.48 | +7 | 0 | — | LGWAPNMR |
| 970.63 | 970.54 | +7 | 0 | — | ITYFWIK | |
| 1466.63 | 1466.72 | −9 | 0 | — | FHNIYGPFGTWK | |
| 1549.83 | 1549.90 | −7 | 1 | — | KLPIHHIPGPEGVR | |
| 1570.81 | 1570.87 | −6 | 0 | — | ISITGAGGFIASHIAR† | |
| 1614.74 | 1614.78 | −4 | 0 | — | SFTFIDECVEGVLR | |
| 1674.91 | * | * | * | * | * | |
| 1685.81 | 1685.82 | −1 | 0 | Cys-acrylamide | SFTFIDECVEGVLR | |
| 1743.80 | 1743.81 | −1 | 0 | Cys-acrylamide | FFYASSACIYPEFK | |
| 1820.89 | 1820.88 | +1 | 1 | — | ELEREQYWPSENLK | |
| 1855.00 | 1855.00 | 0 | 2 | — | ITYFWIKEQIEKEK | |
| 1905.88 | 1905.85 | +3 | 0 | — | ESDAWPAEPQDAYGLEK | |
| 2099.04 | 2098.96 | +8 | 1 | — | AQTSTDRFEMWGDGLQTR | |
| 2452.09 | 2452.04 | +5 | 0 | Cys-acrylamide | NEHMTEDMFCDEFHLVDLR | |
| 3019.55 | 3019.45 | +10 | 1 | — | QLETTNVSLKESDAWPAEPQDAYGLEK | |
| 3356.69 | * | * | * | * | * |
Therefore, we conclude that the GDP-Man 3",5"-epimerase of A. thaliana is a homodimer composed of apparently two identical 42.8-kDa subunits, the amino acid sequence of which is depicted in Fig. 6. The corresponding gene is located on chromosome 5 of A. thaliana; it contains 6 exons and its GC content is 43.6%. The predicted gene/protein is annotated as epimerase/dehydratase-like protein of 377 amino acids (full length). The polypeptide belongs to the α-β structural class and has neither predicted transmembrane domains nor signal peptide sequence. Its calculated molecular mass is 42,759 Da and the isoelectric point is 5.85.
Heterologous Expression of the Recombinant Epimerase.
The epimerase cDNA was cloned into pDEST15 or pDEST17 vector with the GATEWAY system, and the obtained plasmids, pDEST15_Epim and pDEST17_Epim, were used for expression of the recombinant glutathione S-transferase (GST)- and His-fusion proteins in E. coli, respectively. The epimerase activity was detected in the 90% ammonium sulfate fraction of the induced GST-fusion protein-producing pDEST15_Epim E. coli (353 units/mg of protein) (data not shown). However, the enzyme activity could hardly be measured in the corresponding crude extracts probably as a result of the presence of epimerase inhibitor(s) in the E. coli host. The 90% ammonium sulfate fraction obtained from His-fusion protein-producing pDEST17_Epim E. coli showed only a weak epimerase activity (13 units/mg of protein) (data not shown), suggesting that the soluble His-fusion form of the enzyme is either unstable or catalytically inefficient. No epimerase activity could be detected in noninduced pDEST15_Epim or pDEST17_Epim BL21 E. coli cells.
Discussion
By using combined biochemical and mass spectrometry methods, we obtained a highly purified preparation of GDP-Man 3",5"-epimerase from Arabidopsis thaliana cell suspensions. Mass spectrometric identification of major contaminating proteins present in the enzyme fractions allowed us to develop relatively rapidly a suitable purification scheme for the epimerase. During the purification, an important decrease of the enzymatic activity was observed, probably because of loss of the enzyme-bound cofactor (NAD+). Consequently, low yields were obtained (Table 1), albeit sufficient to identify the epimerase protein (Fig. 6).
The predicted epimerase sequence is homologous (24% identity) to that of a TDP-glucose 4",6"-dehydratase-like protein D18 from A. thaliana (33). Surprisingly, the closest homologue (36% identity) of the epimerase protein that could be found by a blast search is the product of the BmlG gene present in the biosynthetic gene cluster for the antitumor drug bleomycin from Streptomyces verticillus (ref. 34; Fig. 7). The BmlG protein is apparently a 5"-epimerase responsible for the conversion at the sugar nucleotide level of d-mannosyl to l-gulosyl residues, which are then transferred from the sugar nucleotide donor to bleomycin aglycones. The resemblance between GDP-Man 3",5"-epimerase and the Streptomyces 5"-epimerase is a fascinating example of either convergent protein evolution or a horizontal gene transfer between bacteria and plants. The Arabidopsis GDP-Man 3",5"-epimerase shows a significant homology (21% identity) to a hypothetical protein from Synechocystis (Fig. 7). The latter protein has recently been considered as a homolog of GDP-4"-keto,6"-deoxy-d-mannose 3",5"-epimerase/reductase (GER1) from A. thaliana (35). The GDP-Man 3",5"-epimerase shares 21% identity with both GER1 and the corresponding protein, GDP-l-Fuc synthetase (GFS) from E. coli (36). All these proteins are structurally similar to UDP-glucose 4-epimerase (GalE) of E. coli (refs. 36 and 37; Fig. 7). Like other members of the UDP-glucose 4-epimerase family, GDP-Man 3",5"-epimerase is a homodimer and contains a typical dinucleotide-binding Rossmann fold in the N-terminal part of the sequence (amino acids 31–41) (Fig. 7), which represents a variation of the characteristic glycine-rich GXGXXG NAD(P)-binding pattern (38). Additionally, the enzyme shows another characteristic feature of the short-chain dehydrogenase/reductase (SDR) family (39), namely the presence of the conserved serine/threonine residue and of the YXXXK motif, which form the strictly conserved catalytic triad (S143, Y174, and K178 of the epimerase sequence; Fig. 7). Therefore, we conclude that the GDP-Man 3",5"-epimerase of A. thaliana is, to our knowledge, a novel member of the extended SDR family. Furthermore, we have cloned the epimerase gene and shown the enzyme activity in transformed E. coli cells. These results represent unequivocal evidence of the enzymatic function of the product of the epimerase gene identified in the present work.
Figure 7.

Multiple sequence alignment of GDP-Man 3",5"-epimerase from A. thaliana with related proteins detected by PSI-blast. The alignment was created with CLUSTALX (40), using default parameters, and displayed with the POSTSCRIPT VIEWER program. Colors follow CLUSTALW convention (41). EPIM, GDP-Man 3",5"-epimerase from A. thaliana; BlmG, NDP-d-Man 5"-epimerase of S. verticillus; SYNECHOCYSTIS, a hypothetical protein of Synechocystis sp.; GER1, GDP-4"-keto,6"-deoxy-d-mannose 3",5"-epimerase/4"-reductase of A. thaliana; GFS, GDP-l-Fuc synthetase of E. coli; TGD, TDP-d-glucose 4",6"-dehydratase of E. coli; GALE, UDP-d-glucose 4"-epimerase of E. coli. Positions on the ruler: 31–41, dinucleotide binding site; 162, 196, and 200, conserved S (T), Y, and K residues, respectively, of the catalytic triad of the short-chain dehydrogenase/reductase (SDR) protein family. *, conserved residues.
The recognition of the epimerase gene and the expression of the active recombinant protein are prerequisites for further studies on the enzyme structure, mechanism, and regulation to determine a possible role of the plant epimerase in the control of the ascorbate biosynthesis. Moreover, the recombinant enzyme can be used for in vitro enzymatic synthesis of GDP-l-Gal, the substrate for subsequent still unknown steps leading to free l-Gal in the biosynthesis of vitamin C in plants.
Acknowledgments
We thank Dr. Peter Casteels for help with FPLC and Stephane Rombauts for helpful discussions, Magda Puype for technical assistance, Rebecca Verbanck for artwork, and Martine De Cock for preparing the manuscript. This work was supported by a grant from Zeneca (Wilmington, DE).
Abbreviations
- l-AA
l-ascorbic acid
- nano-ESI MS/MS
nano-electrospray ionization tandem mass spectrometry
- MALDI-TOF
matrix-assisted laser desorption ionization–time of flight MS
References
- 1.Smirnoff N, Wheeler G L. Crit Rev Plant Sci. 2000;19:267–290. [Google Scholar]
- 2.Nishikimi M, Fukuyama R, Minoshima S, Shimizu N, Yagi K. J Biol Chem. 1994;269:13685–13688. [PubMed] [Google Scholar]
- 3.Nishikimi M, Yagi K. Subcell Biochem. 1996;25:17–39. doi: 10.1007/978-1-4613-0325-1_2. [DOI] [PubMed] [Google Scholar]
- 4.Davey M W, Van Montagu M, Inzé D, Sanmartin M, Kanellis A, Smirnoff N, Benzie I J J, Strain J J, Favell D, Fletcher J. J Sci Food Agric. 2000;80:825–860. [Google Scholar]
- 5.Mapson L W, Breslow E. Biochem J. 1958;68:395–406. doi: 10.1042/bj0680395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Østergaard J, Persiau G, Davey M W, Bauw G, Van Montagu M. J Biol Chem. 1997;272:30009–30016. doi: 10.1074/jbc.272.48.30009. [DOI] [PubMed] [Google Scholar]
- 7.Imai T, Karita S, Shiratori G-i, Hattori M, Nunome T, Ôba K, Hirai M. Plant Cell Physiol. 1998;39:1350–1358. doi: 10.1093/oxfordjournals.pcp.a029341. [DOI] [PubMed] [Google Scholar]
- 8.Kim S-T, Huh W-K, Kim J-Y, Hwang S-W, Kang S-O. Biochim Biophys Acta. 1996;1297:1–8. doi: 10.1016/0167-4838(96)00077-5. [DOI] [PubMed] [Google Scholar]
- 9.Feingold D S, Avigad G. In: Carbohydrates: Structure and Function. Preiss J, editor. New York: Academic; 1980. pp. 101–170. [Google Scholar]
- 10.Wheeler G L, Jones M A, Smirnoff N. Nature (London) 1998;393:365–369. doi: 10.1038/30728. [DOI] [PubMed] [Google Scholar]
- 11.Conklin P L, Norris S R, Wheeler G L, Williams E H, Smirnoff N, Last R L. Proc Natl Acad Sci USA. 1999;96:4198–4203. doi: 10.1073/pnas.96.7.4198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Keller R, Springer F, Renz A, Kossmann J. Plant J. 1999;19:131–141. doi: 10.1046/j.1365-313x.1999.00507.x. [DOI] [PubMed] [Google Scholar]
- 13.Goudsmit E M, Neufeld E F. Biochem Biophys Res Commun. 1967;26:730–735. doi: 10.1016/s0006-291x(67)80134-7. [DOI] [PubMed] [Google Scholar]
- 14.Mourão P A S, Assreuy A-M. J Biol Chem. 1995;270:3132–3240. doi: 10.1074/jbc.270.7.3132. [DOI] [PubMed] [Google Scholar]
- 15.Su J-S, Hassid W Z. Biochemistry. 1962;1:468–474. doi: 10.1021/bi00909a016. [DOI] [PubMed] [Google Scholar]
- 16.Zablackis E, Huang J, Müller B, Darvill A G, Albersheim P. Plant Physiol. 1995;107:1129–1138. doi: 10.1104/pp.107.4.1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rayon C, Cabanes-Macheteau M, Loutelier-Bourhis C, Salliot-Maire I, Lemoine J, Reiter W-D, Lerouge P, Faye L. Plant Physiol. 1999;119:725–733. doi: 10.1104/pp.119.2.725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zablackis E, York W S, Pauly M, Hantus S, Reiter W-D, Chapple C C S, Albersheim P, Darvill A. Science. 1996;272:1808–1810. doi: 10.1126/science.272.5269.1808. [DOI] [PubMed] [Google Scholar]
- 19.Bonin C P, Potter I, Vanzin G F, Reiter W-D. Proc Natl Acad Sci USA. 1997;94:2085–2090. doi: 10.1073/pnas.94.5.2085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tonetti M, Sturla L, Bisso A, Benatti U, De Flora A. J Biol Chem. 1996;271:27274–27279. doi: 10.1074/jbc.271.44.27274. [DOI] [PubMed] [Google Scholar]
- 21.Giraud M-F, Naismith J H. Curr Opin Struct Biol. 2000;10:687–696. doi: 10.1016/s0959-440x(00)00145-7. [DOI] [PubMed] [Google Scholar]
- 22.Barber G A. Arch Biochem Biophys. 1971;147:619–623. doi: 10.1016/0003-9861(71)90420-6. [DOI] [PubMed] [Google Scholar]
- 23.Barber G A. Arch Biochem Biophys. 1975;167:718–722. doi: 10.1016/0003-9861(75)90516-0. [DOI] [PubMed] [Google Scholar]
- 24.Hebda P A, Behrman E J, Barber G A. Arch Biochem Biophys. 1979;194:496–502. doi: 10.1016/0003-9861(79)90644-1. [DOI] [PubMed] [Google Scholar]
- 25.Barber G A, Hebda P A. Methods Enzymol. 1982;83:522–525. doi: 10.1016/0076-6879(82)83046-2. [DOI] [PubMed] [Google Scholar]
- 26.Barber G A. J Biol Chem. 1979;254:7600–7603. [PubMed] [Google Scholar]
- 27.Wolucka B A, Davey M, Boerjan W. Anal Biochem. 2001;294:161–168. doi: 10.1006/abio.2001.5165. [DOI] [PubMed] [Google Scholar]
- 28.Bradford M M. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 29.Laemmli U K. Nature (London) 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 30.Wilm M, Shevchenko A, Houthaeve T, Breit S, Schweigerer L, Fotsis T, Mann M. Nature (London) 1996;379:466–469. doi: 10.1038/379466a0. [DOI] [PubMed] [Google Scholar]
- 31.Wilm M, Mann M. Anal Chem. 1996;68:1–8. doi: 10.1021/ac9509519. [DOI] [PubMed] [Google Scholar]
- 32.Gevaert K, Demol H, Puype M, Broekaert D, De Boeck S, Houthaeve T, Vandekerckhove J. Electrophoresis. 1997;18:2950–2960. doi: 10.1002/elps.1150181537. [DOI] [PubMed] [Google Scholar]
- 33.Kushnir S, Babiychuk E, Kampfenkel K, Belles-Boix E, Van Montagu M, Inzé D. Proc Natl Acad Sci USA. 1995;92:10580–10584. doi: 10.1073/pnas.92.23.10580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Du L, Sánchez C, Chen M, Edwards D J, Shen B. Chem Biol. 2000;7:623–642. doi: 10.1016/s1074-5521(00)00011-9. [DOI] [PubMed] [Google Scholar]
- 35.Bonin C P, Reiter W-D. Plant J. 2000;21:445–454. doi: 10.1046/j.1365-313x.2000.00698.x. [DOI] [PubMed] [Google Scholar]
- 36.Somers W S, Stahl M L, Sullivan F X. Structure (London) 1998;6:1601–1612. doi: 10.1016/s0969-2126(98)00157-9. [DOI] [PubMed] [Google Scholar]
- 37.Somoza J R, Menon S, Schmidt H, Joseph-McCarthy D, Dessen A, Stahl M L, Somers W S, Sullivan F X. Structure (London) 2000;8:123–135. doi: 10.1016/s0969-2126(00)00088-5. [DOI] [PubMed] [Google Scholar]
- 38.Scrutton N S, Berry A, Perham R N. Nature (London) 1990;343:38–43. doi: 10.1038/343038a0. [DOI] [PubMed] [Google Scholar]
- 39.Jörnvall H, Persson B, Krook M, Atrian S, Gonzàlez-Duarte R, Jeffery J, Ghosh D. Biochemistry. 1995;34:6003–6013. doi: 10.1021/bi00018a001. [DOI] [PubMed] [Google Scholar]
- 40.Thompson J D, Gibson T J, Plewniak F, Jeanmougin F, Higgins D G. Nucleic Acids Res. 1997;25:4876–4882. doi: 10.1093/nar/25.24.4876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Thompson J D, Higgins D G, Gibson T J. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]