

Abstract
Background
Many people with schizophrenia do not reach a satisfactory clinical response with a standard dose of an initially prescribed antipsychotic drug. In such cases, clinicians face the dilemma of increasing the antipsychotic dose in order to enhance antipsychotic efficacy.
Objectives
To examine the efficacy of increasing antipsychotic dose compared to keeping the same dose in the treatment of people with schizophrenia who have not responded (as defined in the individual studies) to an initial antipsychotic drug trial. We also examine the adverse effects associated with such a procedure.
Search methods
We searched the Cochrane Schizophrenia Group Trials Register (10 June 2014, 6 October 2015, and 30 March 2017). We examined references of all included studies for further trials.
Selection criteria
All relevant randomised controlled trials (RCTs), reporting useable data, comparing increasing the antipsychotic dose rather than maintaining the original dose for people with schizophrenia who do not respond to their initial antipsychotic treatment.
Data collection and analysis
At least two review authors independently extracted data . We analysed dichotomous data using relative risks (RR) and the 95% confidence intervals (CI). We analysed continuous data using mean differences (MD) and their 95% CI. We assessed risk of bias for included studies and used GRADE to create a 'Summary of findings' table.
Main results
Ten relevant RCTs with 675 participants are included in this review. All trials were double blind except one single blind. All studies had a run‐in phase to confirm they did not respond to their initial antipsychotic treatment. The trials were published between 1980 and 2016. In most studies the methods of randomisation, allocation and blinding were poorly reported. In addition sample sizes were often small, limiting the overall quality of the evidence. Overall, no clear difference was found between groups in terms of the number of participants who showed clinically relevant response (RR 1.09, 95% CI 0.86 to 1.40, 9 RCTs, N = 533, low‐quality evidence), or left the study early due to adverse effects (RR 1.63, 95% CI 0.52 to 5.07, very low quality evidence), or due to any reason (RR 1.30, 95% CI 0.89 to 1.90, 5 RCTs, N = 353, low‐quality evidence). Similarly, no clear difference was found in general mental state as measured by PANSS total score change (MD −1.44, 95% CI −6.85 to 3.97, 3 RCTs, N = 258, very low quality evidence). At least one adverse effect was equivocal between groups (RR 0.91, 95% CI 0.55 to 1.50, 2 RCTs, N = 191, very low quality evidence). Data were not reported for time in hospital or quality‐of‐life outcomes. Finally, subgroup and sensitivity analyses did not show any effect on the primary outcome but these analyses were clearly underpowered.
Authors' conclusions
Current data do not show any clear differences between increasing or maintaining the antipsychotic dose for people with schizophrenia who do not respond to their initial antipsychotic treatment. Adverse effect reporting was limited and poor. There is an urgent need for further trials in order to determine the optional treatment strategy in such cases.
Plain language summary
Increasing versus maintaining the dose of antipsychotic medication for people with schizophrenia who do not respond to treatment
Review question
If a person with schizophrenia does not initially respond to an antipsychotic medication, is increasing the dose of this antipsychotic effective and safe?
Background
Many people with the serious mental illness schizophrenia do not respond fully (i.e. symptoms such as delusions and hallucinations still remain) with a standard dose of an initially prescribed antipsychotic drug. In such cases, clinicians can consider increasing the antipsychotic dose beyond regular thresholds or switching to a different antipsychotic drug in order to enhance antipsychotic efficacy. The evidence surrounding the optimal treatment strategy is scarce.
Searching for evidence
The Information Specialist of Cochrane Schizophrenia ran an electronic search of their specialised register up to 30 March 2017 for trials that randomised people with schizophrenia who were not responding to their initial antipsychotic treatment to receive either an increased antipsychotic dose or continue on the same dose. The search returned 1919 records, which were checked for eligibility by the review authors.
Evidence found
Ten trials met the review requirements and provided usable data. No clear difference between increasing the dose of the antipsychotic drug and continuing antipsychotic treatment at the same dose was shown for any efficacy (clinical response) or safety (incidence of adverse effects) outcomes. The evidence currently available is limited and of low or very low quality. In particular, very few studies reported adverse effects adequately.
Conclusions
The results of the present review show that there is no good‐quality evidence to support or refute the hypothesis that increasing the antipsychotic dose for patients not responding to their initial antipsychotic treatment differs from continuing antipsychotic treatment at the same dose. No clear evidence regarding safety is available. Therefore, no firm conclusions can be made. Larger, well‐designed trials are needed.
Summary of findings
Summary of findings for the main comparison. Antipsychotic dose increase compared to antipsychotic dose continuation for non response in schizophrenia.
| Antipsychotic dose increase compared to antipsychotic dose continuation for non response in schizophrenia | ||||||
| Patient or population: non response in schizophrenia Setting: inpatients and outpatients Intervention: antipsychotic dose increase Comparison: antipsychotic dose continuation | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | № of participants (studies) | Certainty of the evidence (GRADE) | Comments | |
| Risk with antipsychotic dose continuation | Risk with antipsychotic dose increase | |||||
| Global state: clinically relevant response: Assessed with response ratio follow‐up: range 2 weeks to 12 weeks | Study population | RR 1.09 (0.86 to 1.40) | 533 (9 RCTs) | ⊕⊕⊝⊝ LOW 1 | ||
| 309 per 1000 | 336 per 1000 (265 to 432) | |||||
| Leaving the study early: tolerability ‐ leaving early due to adverse effects. Assessed with risk ratio follow‐up: range 2 weeks to 9 weeks | Study population | RR 1.63 (0.52 to 5.07) | 496 (7 RCTs) | ⊕⊝⊝⊝ VERY LOW 1 2 | ||
| 74 per 1000 | 121 per 1000 (39 to 376) | |||||
| Leaving the study early: acceptability ‐ leaving early due to any reason. Assessed with: Risk ratio follow‐up: range 2 weeks to 9 weeks | Study population | RR 1.30 (0.89 to 1.90) | 353 (5 RCTs) | ⊕⊕⊝⊝ LOW 1 | ||
| 23 per 100 | 30 per 100 (20 to 43) | |||||
| General mental state : PANSS total score change* assessed with: Weighted mean difference follow‐up: range 2 weeks to 9 weeks | The mean general mental state ‐ PANSS total score change ranged from −8.9 to 0.03 points | MD 1.44 points lower (6.85 lower to 3.97 higher) | ‐ | 258 (3 RCTs) | ⊕⊝⊝⊝ VERY LOW 3 4 5 | One other trial used the BPRS total score change and showed no clear difference between the two groups. Pre‐defined outcome: Clinically important change in general mental state not reported. |
| Adverse effects ‐ at least one adverse effect assessed with: Risk ratio follow‐up: range 2 weeks to 9 weeks | Study population | RR 0.91 (0.55 to 1.50) | 191 (2 RCTs) | ⊕⊝⊝⊝ VERY LOW 1 6 | ||
| 716 per 1000 | 652 per 1000 (394 to 1000) | |||||
| Service use: time in hospital | ‐ | see comment | ‐ | (0 studies) | ‐ | No studies reported this outcome. |
| Quality of life ‐ clinically important change in quality of life (defined as at least 50% improvement in HQLS) | Study population | not estimable | 17 (1 RCT) | ⊕⊕⊝⊝ LOW 1 | ||
| 0 per 1000 | 0 per 1000 (0 to 0) | |||||
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk ratio; OR: Odds ratio; | ||||||
| GRADE Working Group grades of evidence High certainty: We are very confident that the true effect lies close to that of the estimate of the effect Moderate certainty: We are moderately confident in the effect estimate: The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different Low certainty: Our confidence in the effect estimate is limited: The true effect may be substantially different from the estimate of the effect Very low certainty: We have very little confidence in the effect estimate: The true effect is likely to be substantially different from the estimate of effect | ||||||
1 Imprecision for dichotomous outcomes: a) sample size should be >800 and/or total number of events>300; in our review, both numbers are much smaller. b) the pooled estimate of effect includes both no effect and an appreciable benefit or appreciable harm.
2 Inconsistency: Heterogeneity: Tau² = 1.03; Chi² = 11.24, df = 5 (P = 0.05); I² = 56%
3 Inconsistency: Heterogeneity: Tau² = 14.71; Chi² = 5.70, df = 2 (P = 0.06); I² = 65%
4 Imprecision for continuous outcomes: a) sample size is lower than 400, b) confidence interval includes no effect and the upper or lower confidence limit crosses the minimal important difference (MID), either for benefit of harm
5 Indirectness: The pre‐specified outcome (clinical important change in mental state) was not reported.
6 Inconsistency: Heterogeneity: Tau² = 0.10; Chi² = 3.62, df = 1 (P = 0.06); I² = 72%
Background
Between one‐fifth and one‐third of people with schizophrenia do not respond to standard antipsychotic treatment adequately (Barnes 2003). One possibility to help these individuals is to increase the dose of the currently prescribed antipsychotic. This approach is based on the premise that increasing dose of an antipsychotic drug would lead to enhanced treatment efficacy (Kinon 2004). In the 1970s and the 1980s there was a trend to use high doses for the treatment of schizophrenia. This strategy, sometimes also called "rapid neuroleptization" (Neborsky 1981), was later given up when a narrative review suggested that high doses were not more efficacious than lower doses (Baldessarini 1988). However, this review was not restricted to non‐responding patients, who might still benefit from a dose increase if standard doses were not effective. The use of high antipsychotic doses is frequent practice, making a systematic review on this question important.
Description of the condition
Schizophrenia is often a chronic and disabling psychiatric disorder. It afflicts approximately 1% of the population world‐wide with little gender differences. Its typical manifestations are 'positive' symptoms such as fixed, false beliefs (delusions) and perceptions without cause (hallucinations), 'negative' symptoms such as apathy and lack of drive, disorganisation of behaviour and thought, and catatonic symptoms such as mannerisms and bizarre posturing (Carpenter 1994). The degree of suffering and disability is considerable with 80% to 90% not working (Marvaha 2004), and approximately 5% dying from suicide (Palmer 2005). Antipsychotic medication is the current treatment of choice in schizophrenia (Kane 1996). Unfortunately, a large number of patients experience no significant improvement despite pharmacological treatment (Lieberman 2005). These patients are often labelled as 'non‐responders'. The exact epidemiology of this phenomenon is not clearly understood, one reason being that the criteria for non response or treatment resistance differ (Howes 2017). But guidelines say that approximately 20% to 30% of patients do not respond to an adequate trial of an antipsychotic drug (Lehmann 2004). There are no clear predictors of non response to antipsychotics, but it is well established that people with a first episode respond better than chronic patients (Jäger 2007).
Description of the intervention
Increasing antipsychotic dose in non‐responsive schizophrenia patients is often done in incremental steps — the dosage is systematically increased until a clinical response is reached or the side effects become too pronounced and no further gain in efficacy is obtained. The effective dose ranges of second‐generation antipsychotics are overall better understood than those of first‐generation antipsychotics (Davis 2004; Baldessarini 1988). But even in their case the effective dose ranges are only based on mean values of many patients. Individual patients might well need higher doses beyond the officially approved ranges. Studies on plasma levels of antipsychotics show that there is a lot of interindividual variability, i.e. two different patients may have very different plasma levels when they receive the same dose of an antipsychotic, for example because of the differing activity of their liver enzymes that metabolise these drugs (mainly of the cytochrome P450 family). This may explain why some patients need much higher doses than others (Hiemke 2011).
How the intervention might work
The idea behind increasing the antipsychotic dose is that for a given non‐responsive schizophrenic patient an individual threshold of sensitivity has to be reached for the drug to be effective. It is well known that considerable individual differences exist in respect to pharmacokinetics antipsychotics, which directly influences their efficacy (Miller 2009). A major factor lies in the differences in the expression of cytochrome P450 enzymes which are responsible for the metabolism of many antipsychotic drugs. Polymorphisms in the genes coding for these enzymes exist which can lead to their excessive expression and thus to too fast elimination of drugs (so‐called 'ultrarapid metabolisers') and subsequent non response. More recent work suggested that some of the individual differences in response to antipsychotic drugs might have their source in the variability of ABCB1 genotypes, meaning that individuals with a favourable genotypic configuration show a lower risk of developing refractoriness to increasing antipsychotic dose (Vijayan 2012).
Why it is important to do this review
One of the major challenges in the pharmacological treatment of schizophrenia is non response to antipsychotics. Increasing antipsychotic dose is one of the major strategies to do so which is frequently applied in clinical practice. Unfortunately, there is a lack of clear evidence to what extent this strategy is effective. This often results in uninformed clinical decisions which may lead to severe side effects, exacerbations of psychosis or relapse in many people with schizophrenia. Our aim is to provide a family of related systematic reviews of this topic in order to contribute to a more evidence‐based clinical practice. The review is also potentially important for policy makers, because the high frequency of non response and treatment resistance lead to high rates of disability and thus costs for society (Vos 2012).
This review is part of three 'sibling' Cochrane Reviews, investigating non response in people with schizophrenia ('Increasing antipsychotic dose versus switching antipsychotic for non response in schizophrenia' (Samara 2015b), and 'Reducing antipsychotic dose for people with schizophrenia' (title only)).
Objectives
To examine the efficacy of increasing antipsychotic dose compared to keeping the same dose in the treatment of people with schizophrenia who have not responded (as defined in the individual studies) to an initial antipsychotic drug trial. We also examine the adverse effects associated with such a procedure.
Methods
Criteria for considering studies for this review
Types of studies
All relevant randomised controlled trials. If a trial was described as 'double blind' but randomisation was implied, we included such trials in a sensitivity analysis (see Sensitivity analysis). We excluded quasi‐randomised studies, such as those allocating by alternate days of the week. When people were given additional treatments, we only included data if the adjunct treatment was evenly distributed between groups and it was only the increasing dose group that was randomised.
Types of participants
Adults, however defined, with schizophrenia or related disorders — including schizophreniform disorder, schizoaffective disorder and delusional disorder, by any means of diagnosis — who were non‐responsive to their current antipsychotic treatment, irrespective of age, gender or race. We accepted any definition of non response that was used in the individual studies. It is a general strategy of the Cochrane Schizophrenia group to also include studies which did not use operationalised diagnostic criteria such as those of the Diagnostic and Statistical Manual of Mental Disorders, 5th Edition (DSM‐5), because in clinical routine practice such criteria are not meticulously used either.
Types of interventions
1. Dose increase
Any increase of the antipsychotic dose, irrespectively of how it was defined. The new doses could be either within recommended target dose ranges as described in the antipsychotics' labels or international recommendations (Gardner 2010), or higher.
2. Dose continuation
Continuation of the current antipsychotic dose.
Types of outcome measures
We divided all outcomes into short term (up to 3 months), medium term (more than 3 months up to 6 months) and long term (more than 6 months).
We endeavoured to report binary outcomes recording clear and clinically meaningful degrees of change (e.g. global impression of much improved, or more than 50% improvement on a rating scale as defined within the trials) before any others. Thereafter we listed other binary outcomes and then those that are continuous.
Primary outcomes
1. Global state: clinically relevant response ‒ as defined by trials *
* We expected that different trials would use different definitions of response. But studies have shown that, as long as relative measures of risk (relative risks, odds ratios) are applied, meta‐analytic results do not differ much depending on the exact cut‐off applied (Furukawa 2011).
2. Leaving the study early
2.1 Tolerability ‒ leaving early due to adverse effects
Secondary outcomes
1. Leaving the study early
1.1 Acceptability ‒ leaving early due to any reason 1.2 Efficacy ‒ leaving early due to inefficacy of treatment
2. Mental state
2.1 General mental state
2.1.1 Clinically important change in general mental state ‒ as defined by each of the studies 2.1.2 Average endpoint general mental state score 2.1.3 Average change in general mental state scores
2.2 Positive symptoms
2.2.1 Clinically important change in positive symptoms ‒ as defined by each of the studies 2.2.2 Average endpoint positive symptom score 2.2.3 Average change in positive symptom scores
2.3 Negative symptoms
2.3.1 Clinically important change in negative symptoms ‒ as defined by each of the studies 2.3.2 Average endpoint negative symptom score 2.3.3 Average change in negative symptom scores
3. Depression
3.1 Clinically important change in depressive symptoms ‒ as defined by each of the studies 3.2 Average endpoint depressive symptom score 3.3 Average change in depressive symptom scores
4. Aggressive behaviour
4.1 Clinically important change in aggressive behaviour ‒ as defined by each of the studies 4.2 Average endpoint aggressive behaviour score 4.3 Average change in aggressive behaviour score
5. Exacerbations of psychosis (as defined by the individual studies)
5.1. Time ill (number of days in exacerbation)
6. Service use
6.1 Hospitalisation ‒ time in hospital (days)
7. Adverse effects
7.1. At least one adverse effect 7.2. Specific side effects (as defined by the original authors, based on any reference values they applied)
8. Quality of life
8.1 Clinically important change in quality of life ‒ as defined by each of the studies 8.2 Average endpoint quality of life 8.3 Average change in quality of life
9. Satisfaction with care
9.1 Clinically important change in satisfaction with care ‒ as defined by each of the studies 9.2 Average endpoint satisfaction with care 9.3 Average change in satisfaction with care
'Summary of findings' table
We used the GRADE approach to interpret findings (Schünemann 2011), and the GRADE profiler (GRADE pro GDT), to export data from this review and create the 'Summary of findings' tables. These tables provide outcome‐specific information concerning the overall quality of evidence from each included study in the comparison, the magnitude of effect of the interventions examined, and the sum of available data on all outcomes that we rated as important to patient care and decision making.
We aimed to select the following main outcomes for inclusion in the 'Summary of findings' table.
Global state: clinically relevant response ‒ as defined by trials.
Leaving the study early: tolerability ‒ leaving early due to adverse effects.
Leaving the study early: acceptability ‒ leaving early due to any reason.
General mental state ‒ clinically important change in general mental state scores.
Adverse effects ‒ at least one adverse effect.
Service use ‒ time in hospital.
Quality of life ‒ clinically important change in quality of life.
If data were not available for these pre‐specified outcomes but were available for ones that are similar, we presented the closest outcome to the pre‐specified one in the table but took this into account when grading the finding.
Search methods for identification of studies
We applied no language restriction within the limitations of the search tools.
Electronic searches
1. Cochrane Schizophrenia Group’s Study‐Based Register of Trials
On 30 March 2017, the Information Specialist searched the register using the following search strategies.
(Dosage Increasing*) in Intervention Field of STUDY
The Cochrane Schizophrenia Group’s Registry of Trials is compiled by systematic searches of major resources (including AMED, BIOSIS, CINAHL, Embase, MEDLINE, PsycINFO, PubMed, and registries of clinical trials) and their monthly updates, handsearches, grey literature, and conference proceedings (see Group Module). There were no language, date, document type, or publication status limitations for inclusion of records into the register.
Searching other resources
1. Reference searching
We inspected references of all included studies for further relevant studies.
2. Personal contact
We contacted the first author of each included study for information regarding unpublished trials. We noted any response in the Characteristics of included studies and thanked the authors in the Acknowledgements.
Data collection and analysis
Selection of studies
At least two authors (EK, MTS) independently inspected citations from the searches to identify relevant abstracts. Where disputes arose, we acquired the full report for more detailed scrutiny. At least two review authors (EK, MTS) obtained and independently inspected full reports of the abstracts meeting the review criteria. We resolved disagreements by discussion with SL. Where it was not possible to resolve disagreement by discussion, we attempted to contact the authors of the study for clarification.
Data extraction and management
1. Extraction
Two review authors (MS, EK) independently extracted data from all included studies. Again, we discussed any disagreement, eventually with SL; documented decisions; and, if necessary, contacted authors of studies for clarification. We extracted data presented only in graphs and figures whenever necessary and possible. We attempted to contact authors through an open‐ended request in order to obtain missing information or for clarification whenever necessary.
2. Management
2.1 Forms
We extracted data onto standard, simple forms.
2.2 Scale‐derived data
We included continuous data from rating scales only if: a) the psychometric properties of the measuring instrument have been described in a peer‐reviewed journal (Marshall 2000); b) the measuring instrument has not been written or modified by one of the trialists for that particular trial; and c) the instrument is a global assessment of an area of functioning and not sub‐scores which are not, in themselves, validated or shown to be reliable. There are exceptions, however: we included sub‐scores from mental state scales measuring positive and negative symptoms of schizophrenia.
Ideally the measuring instrument should either be i. a self‐report or ii. completed by an independent rater or relative (not the therapist). We realise that this is not often reported clearly: we note if this is the case or not in Description of studies.
2.3 Endpoint versus change data
There are advantages of both endpoint and change data. Change data can remove a component of between‐person variability from the analysis. On the other hand calculation of change needs two assessments (baseline and endpoint) which can be difficult in unstable and difficult‐to‐measure conditions such as schizophrenia. We decided to use endpoint data primarily, and only use change data if the latter were not available. We combined endpoint and change data as we preferred to used mean differences (MD) rather than standardised mean differences (Deeks 2011).
2.4 Skewed data
Continuous data on clinical and social outcomes are often not normally distributed. To avoid the pitfall of applying parametric tests to non‐parametric data, we applied the following standards to relevant continuous data before inclusion.
For endpoint data from studies including fewer than 200 participants:
when a scale starts from the finite number zero, we subtracted the lowest possible value from the mean, and divided this by the standard deviation. If this value is lower than 1, it strongly suggests that the data are skewed and we would exclude these data. If this ratio is higher than 1 but less than 2, there is a suggestion that the data are skewed: we would enter these data and test whether their inclusion or exclusion would change the results substantially. If such data changed results we would enter as 'other data'. Finally, if the ratio is larger than 2 we would include these data, because it is less likely that they are skewed (Altman 1996; Higgins 2011).
if a scale starts from a positive value (such as the Positive and Negative Syndrome Scale (PANSS), which can have values from 30 to 210 (Kay 1986)), we would modify the calculation described above to take the scale starting point into account. In these cases skewed data are present if 2 SD > (S − S min), where S is the mean score and 'S min' is the minimum score.
Please note: we would have entered all relevant data from studies of more than 200 participants in the analysis irrespective of the above rules, because skewed data pose less of a problem in large studies. We also entered all relevant change data, as when continuous data are presented on a scale that includes a possibility of negative values (such as change data), it is difficult to tell whether or not data are skewed.
2.5 Common measure
To facilitate comparison between trials, we intended to convert variables that can be reported in different metrics, such as days in hospital (mean days per year, per week or per month) to a common metric (e.g. mean days per month).
2.6 Conversion of continuous to binary
Where possible, we made efforts to convert outcome measures to dichotomous data. This can be done by identifying cut‐off points on rating scales and dividing participants accordingly into 'clinically improved' or 'not clinically improved'. It is generally assumed that if there is a 50% reduction in a scale‐derived score such as the Brief Psychiatric Rating Scale (BPRS, Overall 1962), or the Positive and Negative Syndrome Scale (PANSS, Kay 1986), this could be considered as a clinically significant response (Leucht 2005a; Leucht 2005b). If data based on these thresholds were not available, we used the primary cut‐off presented by the original authors.
2.7 Direction of graphs
Where possible, we entered data in such a way that the area to the left of the line of no effect indicated a favourable outcome for increased dose group. Where keeping to this made it impossible to avoid outcome titles with clumsy double‐negatives (e.g. 'Not un‐improved') we reported data where the left of the line indicated an unfavourable outcome. We noted this in the relevant graphs.
Assessment of risk of bias in included studies
Review authors (MTS and EK) worked independently to assess risk of bias by using criteria described in the Cochrane Handbook for Systematic Reviews of Interventions to assess trial quality (Higgins 2011b). This set of criteria is based on evidence of associations between overestimate of effect and high risk of bias of the article such as sequence generation, allocation concealment, blinding, incomplete outcome data and selective reporting.
If MTS and EK disagreed, they involved SI to make the final rating by consensus. Where inadequate details of randomisation and other characteristics of trials were provided, we contacted authors of the studies in order to obtain further information. We reported non‐concurrence in quality assessment, but if disputes arose as to which category we would allocate a trial then again we resolved by discussion.
The level of risk of bias was noted in the text of the review (Risk of bias in included studies), the Characteristics of included studies table and in the Table 1.
Measures of treatment effect
1. Binary data
For binary outcomes we calculated a standard estimation of the risk ratio (RR) and its 95% confidence interval (CI). It has been shown that RR is more intuitive than odds ratios (Boissel 1999); and that odds ratios tend to be interpreted as RR by clinicians (Deeks 2000). The number needed to treat for an additional beneficial outcome (NNTB) and number needed to treat for an additional harmful outcome (NNTH), with confidence intervals, is intuitively attractive to clinicians but is problematic both in its accurate calculation in meta‐analyses and interpretation (Hutton 2009). For binary data presented in the 'Summary of findings' table, we calculated illustrative comparative risks where possible.
2. Continuous data
For continuous outcomes we estimated mean difference (MD) between groups. We preferred not to calculate effect size measures (standardised mean difference (SMD)). However if scales of very considerable similarity were used, we presumed there was a small difference in measurement, and we calculated effect size and transformed the effect back to the units of one or more of the specific instruments.
Unit of analysis issues
1. Cluster trials
Studies increasingly employ 'cluster randomisation' (such as randomisation by clinician or practice) but analysis and pooling of clustered data poses problems. Firstly, authors often fail to account for intra‐class correlation in clustered studies, leading to a 'unit of analysis' error (Divine 1992), whereby P values are spuriously low, confidence intervals unduly narrow and statistical significance overestimated. This causes type I errors (Bland 1997; Gulliford 1999).
If clustering had not been accounted for in primary studies, we would have presented data in a table, with a (*) symbol to indicate the presence of a probable unit of analysis error. We would have contacted first authors of studies to obtain intra‐class correlation coefficients for their clustered data and to adjust for this by using accepted methods (Gulliford 1999). If clustering had been incorporated into the analysis of primary studies, we presented these data as if from a non‐cluster randomised study, but adjusted for the clustering effect.
We have sought statistical advice and have been advised that the binary data as presented in a report should be divided by a 'design effect'. This is calculated using the mean number of participants per cluster (m) and the intra‐class correlation coefficient (ICC) [Design effect = 1 + (m − 1) * ICC] (Donner 2002). If the ICC was not reported we assumed it to be 0.1 (Ukoumunne 1999).
If cluster studies had been appropriately analysed taking into account intra‐class correlation coefficients and relevant data documented in the report, synthesis with other studies would have been possible using the generic inverse variance technique.
2. Cross‐over trials
A major concern of cross‐over trials is the carry‐over effect. It occurs if an effect (e.g. pharmacological, physiological or psychological) of the treatment in the first phase is carried over to the second phase. As a consequence, on entry to the second phase the participants can differ systematically from their initial state despite a wash‐out phase. For the same reason cross‐over trials are not appropriate if the condition of interest is unstable (Elbourne 2002). As both effects are very likely in people with severe mental illness, we used data of the first phase only of cross‐over studies.
3. Studies with multiple treatment groups
Where a study involves more than two treatment arms, if relevant we presented the additional treatment arms in comparisons. If data were binary we simply added and combined within the two‐by‐two table. If data were continuous we combined data following the formula in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011a). Where the additional treatment arms were not relevant, we did not use these data.
Dealing with missing data
1. Overall loss of credibility
We share the concern that at some degree of loss to follow‐up, data must lose credibility (Xia 2010). However, from which degree of attrition onward this is a problem is unclear. Therefore we did not exclude studies on the basis of degree of attrition, but we took attrition into account in the 'Risk of bias' assessment.
2. Binary
We presented data on a 'once randomised, always analyse' basis (an intention‐to‐treat analysis, ITT). Those leaving the study early were all assumed to have the same rates of outcome as those who completed.
3. Continuous
3.1 Assumptions about participants who left the trials early or who were lost to follow‐up
Various methods are available to account for participants who left the trials early or were lost to follow‐up. Some trials just present the results of study completers, others use the method of 'last observation carried forward' (LOCF, Leucht 2007), while more recently methods such as multiple imputation or mixed‐effects models for repeated measurements (MMRM) have become more of a standard. While the last two methods seem somewhat better than LOCF (Leon 2006), we felt that the high percentage of participants leaving the studies early and differences in the reasons for leaving the studies early between groups is often the core problem in randomised schizophrenia trials. We therefore did not exclude studies based on the statistical approach used. However, for preference we used the more sophisticated approaches. For example, we preferred MMRM or multiple imputation to LOCF and we only presented completer analyses if some kind of ITT data were not available at all. Moreover, we addressed attrition in the 'Risk of bias' assessment.
3.2 Standard deviations
If standard deviations were not reported, we first tried to obtain the missing values from the authors. If not available, where there were missing measures of variance for continuous data, but an exact standard error and confidence intervals available for group means, and either P value or t value available for differences in mean, we calculated them according to the rules described in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011a). When only the standard error (SE) was reported, we calculated standard deviations (SDs) by the formula SD = SE * √(n). The Cochrane Handbook for Systematic Reviews of Interventions presents detailed formulae for estimating SDs from P values, t or F values, confidence intervals, ranges or other statistics (Higgins 2011a). If these formulae did not apply, we calculated the SDs according to a validated imputation method which is based on the SDs of the other included studies (Furukawa 2006). Although some of these imputation strategies can introduce error, the alternative would be to exclude a given study’s outcome and thus to lose information. We nevertheless examined the validity of the imputations in a sensitivity analysis excluding imputed values.
Assessment of heterogeneity
1. Clinical heterogeneity
We considered all included studies initially, without seeing comparison data, to judge clinical heterogeneity. We simply inspected all studies for clearly outlying people or situations which we had not predicted would arise. When such situations or participant groups arose, we discussed these.
2. Methodological heterogeneity
We considered all included studies initially, without seeing comparison data, to judge methodological heterogeneity. We simply inspected all studies for clearly outlying methods which we had not predicted would arise. When such methodological outliers arose, we discussed these.
3. Statistical heterogeneity
3.1 Visual inspection
We visually inspected graphs to investigate the possibility of statistical heterogeneity.
3.2 Employing the I² statistic
We investigated statistical heterogeneity between studies by considering the I² statistic alongside the Chi² P value. The I² statistic provides an estimate of the percentage of inconsistency thought to be due to chance (Higgins 2003). The importance of the observed value of I² depends on i. magnitude and direction of effects and ii. strength of evidence for heterogeneity (e.g. P value from Chi² test, or a confidence interval for I²). We considered an I² estimate greater than or equal to 50% accompanied by a statistically significant Chi² statistic as evidence of substantial levels of heterogeneity (Deeks 2011). When substantial levels of heterogeneity were found in the primary outcome, we explored reasons for heterogeneity (Subgroup analysis and investigation of heterogeneity).
Assessment of reporting biases
Reporting biases arise when the dissemination of research findings is influenced by the nature and direction of results (Egger 1997). These are described in Chapter 10 of the Cochrane Handbook for Systematic Reviews of Interventions (Sterne 2011).
1. Protocol versus full study
We tried to locate protocols of included randomised trials. If the protocol was available, we compared outcomes in the protocol and in the published report . If the protocol was not available, we compared outcomes listed in the Methods section of the trial report with actually reported results.
2. Funnel plot
We are aware that funnel plots may be useful in investigating reporting biases but are of limited power to detect small‐study effects. We planned not to use funnel plots for outcomes where there are 10 or fewer studies, or where all studies are of similar size. In future versions, where funnel plots are possible we will seek statistical advice in their interpretation.
Data synthesis
We understand that there is no closed argument for preference for use of fixed‐effect or random‐effects models. The random‐effects method incorporates an assumption that the different studies are estimating different, yet related, intervention effects. This does seem true to us as we are a priori expecting some clinical heterogeneity between the participants in the different trials. Therefore we chose the random‐effects model for analyses (DerSimonian 1986). There is, however, a disadvantage to the random‐effects model: it puts added weight onto small studies which often are the most biased ones. Depending on the direction of effect, these studies can either inflate or deflate the effect size.
Subgroup analysis and investigation of heterogeneity
1. Subgroup analyses
1.1 Antipsychotic drugs
We performed subgroup analyses based on the antipsychotic drugs included in the selected studies.
1.2 Clinical state, stage or problem
We undertook this review and provided an overview of the effects of dose increase versus dose maintenance for people with schizophrenia in general. In addition, however, we aimed to report data on subgroups of people in the same clinical state, stage and with similar problems.
2. Investigation of heterogeneity
We reported if heterogeneity was high. First, we investigated whether data had been entered correctly. Second, if data were correct we visually inspected the graph and we inspected closely studies outside of the company of the rest to identify reasons that might explain the heterogeneity. Decisions as to whether single studies should be excluded from the analysis, or whether a formal meta‐analysis should not be undertaken at all depend on issues such as whether the heterogeneity was due to differences in direction of effect or only to the degree of the difference between intervention and control (Higgins 2011a). When unanticipated clinical or methodological heterogeneity was obvious we simply stated hypotheses regarding these for future reviews or versions of this review. We did not anticipate undertaking analyses relating to these.
Sensitivity analysis
1. Implication of randomisation
We aimed to include trials in a sensitivity analysis if they were described in some way as to imply randomisation. For the primary outcomes we included these studies; if their inclusion did not result in a substantive difference, they remained in the analyses. If their inclusion did result in important clinically significant but not necessarily statistically significant differences, we did not add the data from these lower quality studies to the results of the better trials, but presented such data within a subcategory.
2. Risk of bias
We analysed the effects of excluding trials that were judged to be at high risk of bias across one or more of the domains described in Assessment of risk of bias in included studies. If the exclusion of trials at high risk of bias did not substantially alter the direction of effect or the precision of the effect estimates, then we included relevant data from these trials in the analysis. Studies with a high risk of bias in terms of randomisation or allocation concealment were excluded right from the start. When randomisation and allocation methods had not been described (and risk of bias was usually rated as unclear) we also entered such trials in a sensitivity analysis.
3. Imputed values
Where we had to make assumptions regarding missing SDs (see Dealing with missing data), we compared the findings of the primary outcomes when we used our assumption/s and when we used data only from studies which provided SDs.
We also undertook a sensitivity analysis to assess the effects of including data from trials where we used imputed values for ICC in calculating the design effect in cluster randomised trials.
4. Fixed and random effects
We synthesised data using a random‐effects model; however, we also synthesised data for the primary outcomes using a fixed‐effect model to evaluate whether this altered the results.
If substantial differences were noted in the direction or precision of effect estimates in any of the sensitivity analyses listed above, we did not pool data from the excluded trials with the other trials contributing to the outcome, but presented them separately.
Results
Description of studies
For substantive description of studies please see Characteristics of included studies and Characteristics of excluded studies tables.
Results of the search
The initial search of the Cochrane Schizophrenia Group Trials Register yielded 1525 reports. We made two later searches of the same Trials Register (using the same search strategy): one in October 2015 which yielded 386 records; and one in March 2017 which yielded 8 records. A total of 1835 records remained after we removed duplicates. After excluding 1778 records based on abstract or title, we obtained and closely inspected 57 full‐text reports. From these, we included 10 studies (referring to 17 full‐text reports); and excluded 37 studies (referring to 40 full‐text reports) (see Figure 1).
1.
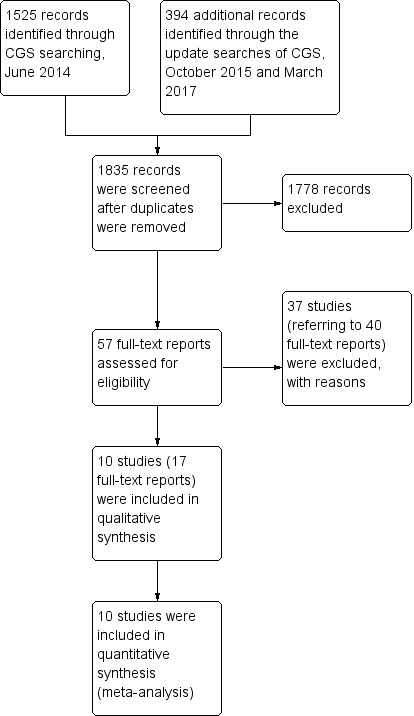
Study flow diagram.
Included studies
We selected 10 studies for inclusion in this review. The studies were published between 1980 and 2016 (for more details, see Characteristics of included studies and the accompanying ‘Risk of bias’ tables).
1. Study design
All studies were randomised; however, only two studies provided an adequate description of randomisation (Honer 2011; Sakurai 2016). All studies had a non‐randomised run‐in phase apart from one trial which had a randomised double‐blind run‐in phase of 2 weeks' duration (Loebel 2014).
2. Length of trials
The included trials varied in duration, both in terms of the run‐in phase and the main phase. The run‐in phase varied from 2 weeks (Huang 1987; Loebel 2014) to 4 weeks (Bjørndal 1980; Honer 2011; Kinon 1993; Lindenmayer 2011; McEvoy 1991; McGorry 2011; Sakurai 2016), whereas the main phase varied from 2 weeks (McEvoy 1991) to 12 weeks (Bjørndal 1980).
3. Participants
There were a total of 675 participants in the 10 included studies. Participants in seven of the included trials appear to have been inpatients (Bjørndal 1980; Huang 1987; Kinon 1993; Lindenmayer 2011; Loebel 2014; McEvoy 1991; McGorry 2011); while in the remaining three studies participants could be either in‐ or outpatients (Goff 2013; Honer 2011; Sakurai 2016). All studies bar one included patients with a diagnosis of schizophrenia according to different operational diagnostic criteria such as Research Diagnostic Criteria (RDC) (McEvoy 1991), DSM‐III (Huang 1987), DSM‐III‐R (Kinon 1993), DSM‐IV (Goff 2013; Honer 2011), DSM‐IV‐TR (Lindenmayer 2011; McGorry 2011), and ICD‐10 (Sakurai 2016) — the exception was Bjørndal 1980, which did not describe any diagnostic criteria. All studies included both male and female participants apart from two: Bjørndal 1980 included only male participants and Huang 1987 did not provide any data on the sex of the participants. The mean age of participants was 36.4 years.
4. Study size
Honer 2011 was the largest study (131 participants) followed by Sakurai 2016 (103 participants) and Loebel 2014 (95 participants) whereas Bjørndal 1980 and McGorry 2011 were the smallest studies (29 and 26 participants respectively). The remaining five studies had 48 to 75 participants.
5. Interventions
Ten studies compared antipsychotic dose increase versus antipsychotic dose continuation. In four studies the dose increase was flexible (Bjørndal 1980; Honer 2011; McEvoy 1991; McGorry 2011), whereas in the remaining studies it was fixed. There was one study on fluphenazine (Kinon 1993), two on haloperidol (Bjørndal 1980; McEvoy 1991), two on quetiapine (Honer 2011; Lindenmayer 2011), one on lurasidone (Loebel 2014), one on risperidone (McGorry 2011), one on thiothixene (Huang 1987), one on ziprasidone (Goff 2013), and one on olanzapine or risperidone (Sakurai 2016).
6. Outcomes
A variety of scales were used to assess clinical response and adverse events. The reporting on efficacy and side effects was incomplete in the original publications. However, we were able to improve this situation by contacting the authors, some of whom agreed to share their data with us (see Acknowledgements).
6.1 Outcome scales
Details of scales that provided usable data are shown below.
6.1.1 Global state
Clinical Global Impression ‒ CGI (Guy 1976)
CGI is a 7‐point rating instrument that is commonly used in studies on schizophrenia. It enables clinicians to quantify severity of illness (CGI‐Severity) and/or overall clinical improvement (CGI‐Improvement) during therapy with low scores indicating decreased severity or greater improvement.
6.1.2 Mental state
Brief Psychiatric Rating Scale ‒ BPRS (Overall 1962)
The BPRS is a scale used to measure the severity of psychiatric symptoms, including psychotic symptoms. The scale usually has 18 items (depending on the version the number of items could vary from 16 to 24), and each item is rated on a seven‐point scoring system varying from 'not present' (1) to 'extremely severe' (7). Higher scores indicate more pronounced symptomatology.
Nurses' Observation Scale for Inpatient Evaluation ‐ NOSIE‐30 (Honigfeld 1965; Honigfeld 1973)
The NOSIE‐30 is a ward behaviour rating scale, especially designed for use by nurses and other subprofessional personnel. This 30‐item scale is quick (can be completed in 5 to 10 minutes) and simple (requires minimum training). The 30 items (behaviours) are rated on a 5‐point scoring system varying from 'never' (0) to 'always' (4) based on their frequency during the three days prior to examination. Higher scores indicate more pronounced symptomatology.
Positive and Negative Symptom Scale ‒ PANSS (Kay 1986)
The PANSS was developed from the BPRS and the Psychopathology Rating Scale. It is used to evaluate the positive, negative and general symptoms in schizophrenia. The scale has 30 items, and each item is rated on a 7‐point scoring system varying from 'absent' (1) to 'extreme' (7). Higher scores indicate more pronounced symptomatology.
Scale for the Assessment of Negative Symptoms ‒ SANS (Andreasen 1982)
The SANS is a scale used to measure the severity of negative symptoms in schizophrenia. The scale is used to evaluate five domains of symptoms: alogia, affective blunting, avolition‒apathy, anhedonia‒asociality and attention impairment. Each symptom is rated on a 6‐point scoring system varying from 'absent' (0) to 'severe' (5). Higher scores indicate more pronounced symptomatology.
6.1.3 Adverse events
Simpson Angus Scale ‒ SAS (Simpson 1970)
This 10‐item scale, with a scoring system of 0 to 4 for each item, measures drug‐induced parkinsonism, a short‐term drug‐induced movement disorder. A low score indicates low levels of parkinsonism.
Abnormal Involuntary Movement Scale ‒ AIMS (NIMH 1970)
The AIMS has been used to assess tardive dyskinesia, a long‐term, drug‐induced movement disorder. However, using this scale in short‐term trials may also be helpful to assess some rapidly occurring abnormal movement disorders such as tremor. A low score indicates low levels of abnormal involuntary movements.
Barnes Akathisia Rating Scale ‒ BAS (Barnes 1989)
This scale comprises items rating the observable, restless movements that characterise akathisia, the subjective awareness of restlessness and any distress associated with the condition. These items are rated from 'normal' (0) to 'severe' (3). In addition, there is an item for rating the global severity (from 'absent' (0) to 'severe' (5)). A low score indicates low levels of akathisia.
6.1.4 Behaviour
Behavioral Activity Rating Scale ‒ BARS (Swift 2002)
The BARS was designed to measure the degree of activity for patients with agitated behaviour rather than to represent the severity of a specific diagnostic entity such as schizophrenia. The BARS describes seven levels of activity, from 'difficult or unable to rouse' (1) to 'violent, requires restraint' (7).
6.1.5 Functioning
Global Assessment of Functioning ‒ GAF (DSM‐IV‐TR 1994)
The GAF has been used to rate the social, occupational, and psychological functioning on a hypothetical continuum of mental health‒illness. The lowest scores are 1 to 10 corresponding to 'Persistent danger of severely hurting self or others OR persistent inability to maintain minimal personal hygiene OR serious suicidal act with clear expectation of death' and the highest scores are 91 to 100 corresponding to 'No symptoms, life's problems never seem to get out of hand, is sought out by others because of his or her many positive qualities'. A score of 0 is given for inadequate information. A high score indicates good functioning.
Social‐Adaptive Functioning Evaluation ‒ SAFE (Harvey 1997)
The SAFE was originally designed to rate severity of impairment in crucial adaptive functioning domains of geriatric patients in a restricted setting, but has now been adapted for use with people of all ages. The scale has 17 items that measure social‒interpersonal, instrumental, and life skills functioning and are rated by observation, caregiver contact, and interaction with the subject if possible. Scoring ranges from 'no impairment' (0) to 'extreme impairment' (4). A low score indicates good functioning.
Social and Occupational Functioning Assessment Scale ‒ SOFAS (DSM‐IV‐TR 1994)
The SOFAS has been used to rate the social and occupational functioning on a hypothetical continuum of mental health‒illness. It takes into account impairments in functioning due to physical limitations or mental impairments, but not due to lack of opportunity and other environmental limitations. The lowest scores are 1 to 10 corresponding to 'Persistent hygiene problems, inability to maintain minimal personal hygiene. Unable to function without harming self or others or without considerable external support' and the highest scores are 91 to 100 corresponding to 'Superior functioning in a wide range of activities'. A score of 0 is given for inadequate information. A high score indicates good functioning.
6.1.6 Quality of Life
Heinrichs‐Carpenter‐Hanlon Quality of Life Scale ‒ HQLS (Heinrichs 1984; Carpenter 1994)
The HQLS is a semi‐structured interviewer‐administered scale containing 21 items divided in four subscales. The structure of the instrument establishes a series of topics to be explored, using specified sample probes. Each item is rated on a 7‐point scale (0 to 6) for which descriptive anchors are provided. High scores reflect normal or unimpaired function, and low scores reflect severe impairment of function.
Excluded studies
We excluded 40 full‐text articles on 37 trials. Three studies were excluded because they were not appropriately randomised (Agid 2013; Bai 2002; DeBuck 1972). Twenty‐three studies were excluded because they did not examine suitable participants (e.g. stable participants or not selected after a run‐in phase to confirm that they had failed to improve with their current antipsychotic treatment (Badgett 1996; Baker 2003; Bastecky 1982; Bondolfi 1995; Branchey 1981; Canuso 2010; Chen 1998; Clerc 1989; Daniel 1997; Ericksen 1978; Gardos 1971; Gulliver 2010; Harris 1997; Hirschowitz 1995; Itil 1970; Kane 1985; Lehmann 1980; McCreadie 1979; Mitchell 2004; NCT00862992; NCT01457339; NCT01569659; Suzuki 1992). Nine studies were excluded because of wrong interventions, most of them did not have a continuation control group (Bitter 1989; CN138032; Coryell 1998; Cookson 1987; de Leon 2007; Hirschowitz 1997; Janicak 1997; Simpson 1999; Volavka 1996). Two studies were excluded since no usable data were presented (NCT00539071; Dencker 1978).
Studies awaiting assessment
None.
Ongoing studies
None.
Risk of bias in included studies
For graphical representations of our judgements of risk of bias please refer to Figure 2 and Figure 3. Full details of judgements are seen in the 'Risk of bias' tables.
2.
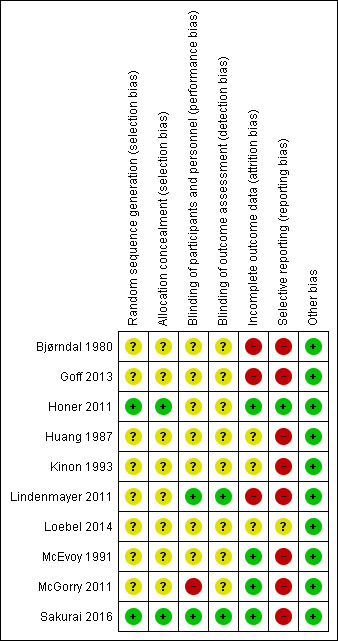
Risk of bias summary: review authors' judgements about each risk of bias item for each included study.
3.
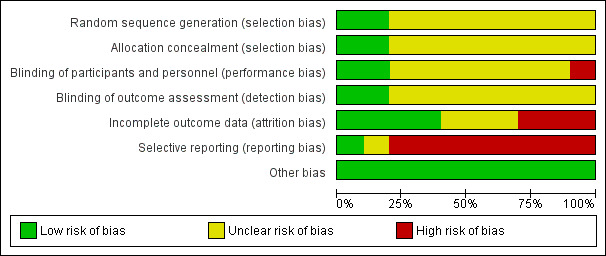
Risk of bias graph: review authors' judgements about each risk of bias item presented as percentages across all included studies.
Allocation
We rated no studies at high risk for allocation bias. In two studies, random sequence generation was adequate (Honer 2011; Sakurai 2016). In the remaining eight studies, this was unclear: they were described as randomised, but none of them provided any further details about random sequence generation. As for allocation concealment, again only two studies provided information and we rated them as adequate (Honer 2011; Sakurai 2016). The remaining eight studies did not provide any details on allocation concealment.
Blinding
Concerning performance bias, we rated two studies at low risk of bias since it was explicitly mentioned that subjects, caregivers and investigators were blinded to the randomisation status (Lindenmayer 2011), and antipsychotics were provided in identical powder form in amount and colour (Sakurai 2016). However we rated one study at high risk of bias since it was described as single‐blind (McGorry 2011). The remaining seven studies we rated with an unclear risk of bias since the term "double‐blind" was the only description provided by the authors.
Concerning detection bias, we rated two studies with a low risk of bias since it was explicitly mentioned that outcome assessors were blinded to the randomisation status (Lindenmayer 2011; Sakurai 2016). All the remaining eight studies we rated with an unclear risk of bias since the term "blind" was the only description provided by the authors.
Incomplete outcome data
The number of participants leaving the studies early was relatively low (< 25%) in four studies which also used an ITT approach to analyse the results (Honer 2011; McEvoy 1991; McGorry 2011; Sakurai 2016); thus we rated these studies at low risk of bias. Three studies had an unclear risk of bias since this outcome was not adequately addressed (Huang 1987; Kinon 1993; Loebel 2014). The remaining three studies we rated with a high risk of bias (Bjørndal 1980; Goff 2013; Lindenmayer 2011).
Selective reporting
One study reported data on all predefined outcomes and we therefore rated it at low risk of bias (Honer 2011). We rated one study with an unclear risk of bias (Loebel 2014). The remaining eight studies we rated with a high risk of bias as not all outcomes were reported; usually, data on adverse effects were missing.
Other potential sources of bias
We judged all studies to be free of other potential sources of bias.
Effects of interventions
See: Table 1
See: Table 1 for the main comparison 'Antipsychotic dose increase versus antipsychotic dose continuation'. We used risk ratios (RR) for dichotomous data and mean differences (MD) for continuous data, with their respective 95% confidence intervals (CIs) throughout.
1. Comparison: Antipsychotic dose increase versus antipsychotic dose maintenance
1.1 Global state: 1a. Clinically relevant response ‒ as defined by trials
Nine studies provided data on the number of responders in the dose increase versus dose maintenance group; only Loebel 2014 did not provide any data on this outcome. There was no clear difference between dose increase versus dose maintenance in any of the nine included studies either when examined separately or pooled (RR 1.09, 95% CI 0.86 to 1.40, 9 RCTs, N = 533, Analysis 1.1). Moreover, no significant heterogeneity was indicated (P = 0.37, I² = 8%). Response was defined as a higher than 0% BPRS total score reduction in Bjørndal 1980; as a 20% or more PANSS total score reduction in Goff 2013, Honer 2011 and Lindenmayer 2011; as a 20% or more BPRS total score reduction in McGorry 2011; and as a 25% or more PANSS total score reduction in Sakurai 2016. In the remaining three studies, response was defined by the score on a global improvement scale (Huang 1987; Kinon 1993); or by a combination of various criteria (McEvoy 1991).
1.1. Analysis.
Comparison 1 Antipsychotic dose increase versus antipsychotic dose maintenance, Outcome 1 Global state: 1a. Clinically relevant response as defined by trials.
1.2 Global state: 1b. Any change (improvement on Global Assessment scale)
There was no clear difference in one single small study by Bjørndal 1980 for this outcome (RR 0.76, 95% CI 0.32 to 1.80, 1 RCT, N = 23, Analysis 1.2).
1.2. Analysis.
Comparison 1 Antipsychotic dose increase versus antipsychotic dose maintenance, Outcome 2 Global state: 1b. Any change (improvement in Global Assesment scale).
1.3 Global state: 2a. Average endpoint score (CGI‐Severity, high = poor)
Three studies reported the average CGI‐Severity score at endpoint (Honer 2011; McEvoy 1991; McGorry 2011). There was no clear difference between dose increase and dose maintenance antipsychotic treatment (MD −0.11, 95% CI −0.40 to 0.19, 3 RCTs, N = 99, Analysis 1.3 ). Moreover, no significant heterogeneity was indicated (P = 0.88, I² = 0%).
1.3. Analysis.
Comparison 1 Antipsychotic dose increase versus antipsychotic dose maintenance, Outcome 3 Global state: 2a. Average endpoint score (CGI‐Severity , high = poor).
1.4 Global state: 2b. Average change score (CGI‐Severity, high = poor)
One study reported the average CGI‐Severity score at change (Loebel 2014). No clear effect between antipsychotic dose increase and dose maintenance was observed (MD −0.40, 95% CI −0.80 to −0.00, 1 RCT, N = 95, Analysis 1.4 ).
1.4. Analysis.
Comparison 1 Antipsychotic dose increase versus antipsychotic dose maintenance, Outcome 4 Global state: 2b. Average change score ( CGI‐Severity, high = poor).
1.5 Global state: 2c. Average endpoint score (CGI‐Improvement, high = poor)
One study reported the average CGI‐Improvement score at endpoint (Sakurai 2016), and showed no difference between the two groups (MD 0.00, 95% CI −0.35 to 0.35, 1 RCT, N = 103, Analysis 1.5).
1.5. Analysis.
Comparison 1 Antipsychotic dose increase versus antipsychotic dose maintenance, Outcome 5 Global state: 2c. Average endpoint score (CGI‐Improvement, high = poor).
1.6 Leaving the study early: 1. Tolerability ‒ due to side effects
Seven studies provided data on the number of participants leaving early due to side effects in the dose increase versus dose maintenance group (Honer 2011; Huang 1987; Lindenmayer 2011; Loebel 2014; McEvoy 1991; McGorry 2011; Sakurai 2016). There was no clear difference between dose increase versus dose maintenance group (RR 1.63, 95% CI 0.52 to 5.07, 7 RCTs, N = 496, Analysis 1.6), but there was some heterogeneity (P = 0.05, I² = 56%). The heterogeneity was due to Sakurai 2016 which had less attrition due to side effects in the dose continuation group compared to the dose increase group. There was no clear difference between the two groups in the remaining six studies.
1.6. Analysis.
Comparison 1 Antipsychotic dose increase versus antipsychotic dose maintenance, Outcome 6 Leaving the study early: 1. Tolerability ‐ due to adverse events.
1.7 Leaving the study early: 2. Acceptability ‒ due to any reason
Five studies provided data on the number of participants leaving early due to any reason in the dose increase versus dose maintenance group (Honer 2011; Huang 1987; Lindenmayer 2011; McGorry 2011; Sakurai 2016). There was no clear difference between dose increase versus dose maintenance group in any of the five included studies either when examined separately or pooled (RR 1.30, 95% CI 0.89 to 1.90, 5 RCTs, N = 353, Analysis 1.7). Moreover, no significant heterogeneity was indicated (P = 0.36, I² = 8%).
1.7. Analysis.
Comparison 1 Antipsychotic dose increase versus antipsychotic dose maintenance, Outcome 7 Leaving the study early: 2. Acceptability of treatment ‐ due to any reason.
1.8 Leaving the study early: 3. Efficacy ‒ due to inefficacy
Four studies provided data on the number of participants leaving early due to inefficacy in the dose increase versus dose maintenance group (Honer 2011; Huang 1987; Lindenmayer 2011; Sakurai 2016). There was no clear difference between dose increase versus dose maintenance group in any of the four included studies either when examined separately or pooled (RR 0.82, 95% CI 0.30 to 2.28, 4 RCTs, N = 336, Analysis 1.8). Moreover, no significant heterogeneity was indicated (P = 0.36, I² = 3%).
1.8. Analysis.
Comparison 1 Antipsychotic dose increase versus antipsychotic dose maintenance, Outcome 8 Leaving the study early: 3. Efficacy ‐ due to inefficacy.
1.9 Mental state: 1a. General ‒ average endpoint score (PANSS total, high = poor)
Two studies reported the average PANSS total score at endpoint (Honer 2011; Lindenmayer 2011). There was no clear difference between dose increase and dose maintenance antipsychotic treatment for this outcome (MD −1.81, 95% CI −7.31 to 3.69, 2 RCTs, N = 191, Analysis 1.9). Moreover, no significant heterogeneity was indicated (P = 0.63, I² = 0%).
1.9. Analysis.
Comparison 1 Antipsychotic dose increase versus antipsychotic dose maintenance, Outcome 9 Mental state: 1a. General ‐ average endpoint score (PANSS total, high = poor).
1.10 Mental state: 1b. General ‒ average change score (PANSS total, high = poor)
Three studies reported the average PANSS total score change (Lindenmayer 2011; Loebel 2014; Sakurai 2016). Lindenmayer 2011 and Sakurai 2016 showed a trend in favour of dose maintenance versus dose increase whereas Loebel 2014 study showed a clear difference in favour of dose increase versus dose maintenance. Overall, there was no clear difference between dose increase and dose maintenance antipsychotic treatment for this outcome (MD −1.44, 95% CI −6.85 to 3.97, 3 RCTs, N = 258, Analysis 1.10) but heterogeneity was considerable, even though not significant (P = 0.06, I² = 65%).
1.10. Analysis.
Comparison 1 Antipsychotic dose increase versus antipsychotic dose maintenance, Outcome 10 Mental state: 1b. General ‐ average change score (PANSS total, high = poor).
1.11 Mental state: 1c. General ‒ average endpoint and/or change score (PANSS total, high = poor)
Four studies reported the average PANSS total score at endpoint or change or both; Honer 2011 reported only endpoint scores, Loebel 2014 and Sakurai 2016 only change scores, whereas Lindenmayer 2011 reported both endpoint and change scores. In this analysis we used endpoint scores for the latter study in accordance with our protocol. There was no clear difference between dose increase and dose maintenance antipsychotic treatment for this outcome (MD −2.13, 95% CI −6.16 to 1.90, 4 RCTs, N = 389, Analysis 1.11). Moreover, no significant heterogeneity was indicated (P = 0.14, I² = 46%).
1.11. Analysis.
Comparison 1 Antipsychotic dose increase versus antipsychotic dose maintenance, Outcome 11 Mental state: 1c. General ‐ average endpoint and/or change score (PANSS total, high = poor).
1.12 Mental state: 1d. General ‒ average endpoint score (BPRS total, high = poor)
Three studies reported the average BPRS total score at endpoint (Kinon 1993; McEvoy 1991; McGorry 2011). There was no clear difference between dose increase and dose maintenance antipsychotic treatment (MD −1.25, 95% CI −4.60 to 2.11, 3 RCTs, N = 99, Analysis 1.12). Moreover, no significant heterogeneity was indicated (P = 0.98, I² = 0%).
1.12. Analysis.
Comparison 1 Antipsychotic dose increase versus antipsychotic dose maintenance, Outcome 12 Mental state: 1d. General ‐ average endpoint score (BPRS total, high = poor).
1.13 Mental state: 1e. General ‒ average change score (BPRS total, high = poor)
Only one study reported the average BPRS total score change (Huang 1987). There was no clear difference between dose increase and dose maintenance antipsychotic treatment (MD −2.38, 95% CI −6.15 to 1.39, 1 RCT, N = 42, Analysis 1.13).
1.13. Analysis.
Comparison 1 Antipsychotic dose increase versus antipsychotic dose maintenance, Outcome 13 Mental state: 1e. General ‐ average change score (BPRS total, high = poor).
1.14 Mental state: 1f. General ‒ average endpoint and/or change score (BPRS total, high = poor)
Four studies reported the average BPRS total score at endpoint or change (Huang 1987; Kinon 1993; McEvoy 1991; McGorry 2011). There was no clear difference between dose increase and dose maintenance antipsychotic treatment for this outcome (MD −1.75, 95% CI −4.25 to 0.76, 4 RCTs, N = 141, Analysis 1.14). Moreover, no significant heterogeneity was indicated (P = 0.97, I² = 0%).
1.14. Analysis.
Comparison 1 Antipsychotic dose increase versus antipsychotic dose maintenance, Outcome 14 Mental state: 1f. General ‐ average endpoint and/or change score (BPRS total, high = poor).
1.15 Mental state: 1g. General average change score (NOSIE total, high = poor)
Only one study reported the average NOSIE total score change (Huang 1987). There was no clear difference between dose increase and dose maintenance antipsychotic treatment (MD 3.70, 95% CI −5.38 to 12.78, 1 RCT, N = 42, Analysis 1.15).
1.15. Analysis.
Comparison 1 Antipsychotic dose increase versus antipsychotic dose maintenance, Outcome 15 Mental state: 1g. General ‐ average change score (NOSIE total, high = poor).
1.16 Mental state: 2a. Positive symptoms ‒ clinically important change
There was no clear difference in one single small study by McGorry 2011 for this outcome (RR 1.33, 95% CI 0.58 to 3.07, 1 RCT, N = 17, Analysis 1.16).
1.16. Analysis.
Comparison 1 Antipsychotic dose increase versus antipsychotic dose maintenance, Outcome 16 Mental state: 2a. Positive symptoms ‐ Clinically important change.
1.17 Mental state: 2b. Positive symptoms ‒ average endpoint subscore (PANSS positive, high = poor)
Two studies reported the average PANSS positive subscore at endpoint (Honer 2011; Lindenmayer 2011). There was no clear difference between dose increase and dose maintenance antipsychotic treatment for this outcome (MD −0.94, 95% CI −2.79 to 0.90, 2 RCTs, N = 191, Analysis 1.17). Moreover, no significant heterogeneity was indicated (P = 0.38, I² = 0%).
1.17. Analysis.
Comparison 1 Antipsychotic dose increase versus antipsychotic dose maintenance, Outcome 17 Mental state: 2b. Positive symptoms ‐ average endpoint subscore (PANSS positive, high = poor).
1.18 Mental state: 2c. Positive symptoms ‒ average change subscore (PANSS positive, high = poor)
Three studies reported the average PANSS positive subscore change (Goff 2013; Lindenmayer 2011; Sakurai 2016). There was no clear difference between dose increase and dose maintenance antipsychotic treatment for this outcome (MD 0.04, 95% CI −1.31 to 1.40, 3 RCTs, N = 238, Analysis 1.18). Moreover, no significant heterogeneity was indicated (P = 0.20, I² = 38%).
1.18. Analysis.
Comparison 1 Antipsychotic dose increase versus antipsychotic dose maintenance, Outcome 18 Mental state: 2c. Positive symptoms ‐ average change subscore (PANSS positive, high = poor).
1.19 Mental state: 2d. Positive symptoms ‒ average endpoint subscore (BPRS positive subscore, high = poor)
There was no clear difference in one single small study by McGorry 2011 for this outcome (MD 0.40, 95% CI −2.94 to 3.74, 1 RCT, N = 17, Analysis 1.19).
1.19. Analysis.
Comparison 1 Antipsychotic dose increase versus antipsychotic dose maintenance, Outcome 19 Mental state: 2d. Positive symptoms ‐ average endpoint subscore (BPRS positive,high = poor).
1.20 Mental state: 3a. Negative symptoms ‒ average endpoint subscore (PANSS negative, high = poor)
Two studies reported the average PANSS negative subscore at endpoint (Honer 2011; Lindenmayer 2011). There was no clear difference between dose increase and dose maintenance antipsychotic treatment for this outcome (MD 0.32, 95% CI −1.48 to 2.11, 2 RCTs, N = 191, Analysis 1.20). Moreover, no significant heterogeneity was indicated (P = 0.64, I² = 0%).
1.20. Analysis.
Comparison 1 Antipsychotic dose increase versus antipsychotic dose maintenance, Outcome 20 Mental state: 3a. Negative symptoms ‐ average endpoint subscore (PANSS negative, high = poor).
1.21 Mental state: 3b. Negative symptoms ‒ average change subscore (PANSS negative, high = poor)
Two studies reported the average PANSS negative subscore change (Lindenmayer 2011; Sakurai 2016). There was no clear difference between dose increase and dose maintenance antipsychotic treatment for this outcome (MD −0.15, 95% CI −0.96 to 0.67, 2 RCTs, N = 163, Analysis 1.21). Moreover, no significant heterogeneity was indicated (P = 0.80, I² = 0%).
1.21. Analysis.
Comparison 1 Antipsychotic dose increase versus antipsychotic dose maintenance, Outcome 21 Mental state: 3b. Negative symptoms ‐ average change subscore (PANSS negative, high = poor).
1.22 Mental state: 3c. Negative symptoms ‒ average endpoint subscore (BPRS negative, high = poor)
There was no clear difference in one single small study by McGorry 2011 for this outcome (MD −0.40, 95% CI −1.97 to 1.17, 1 RCT, N = 17, Analysis 1.22).
1.22. Analysis.
Comparison 1 Antipsychotic dose increase versus antipsychotic dose maintenance, Outcome 22 Mental state: 3c. Negative symptoms ‐ average endpoint subscore (BPRS negative, high = poor).
1.23 Mental state: 3d. Negative symptoms ‒ average endpoint score (SANS, high = poor)
One study reported the average SANS score at endpoint (Kinon 1993). There was no clear difference between dose increase and dose maintenance antipsychotic treatment for this outcome (MD 1.50, 95% CI −14.33 to 17.33, 1 RCT, N = 34, Analysis 1.23).
1.23. Analysis.
Comparison 1 Antipsychotic dose increase versus antipsychotic dose maintenance, Outcome 23 Mental state: 3d. Negative symptoms ‐ average endpoint score (SANS, high = poor).
1.24 Adverse effects ‒ At least one adverse effect
Two studies — Honer 2011 and Lindenmayer 2011 — reported the numbers of participants with at least one adverse effect but did not reveal a clear difference between the antipsychotic dose increase group (75.2%) versus the antipsychotic dose maintenance group (71.6%), Analysis 1.24. The results were considerably but not significantly heterogeneous (P = 0.06, I² = 72%). It should be noted that, particularly in recent trials, efficacy related events such as exacerbation of psychosis can also be considered adverse events which may in part explain the heterogeneity.
1.24. Analysis.
Comparison 1 Antipsychotic dose increase versus antipsychotic dose maintenance, Outcome 24 Adverse effects ‐ At least one adverse effect.
1.25 Adverse effects ‒ Cardiac: QTc prolongation
Two studies reported the numbers of participants with QTc prolongation (Goff 2013; Honer 2011). There was no clear difference between the two groups. In total there were two participants with QTc prolongation in the antipsychotic dose increase group (1.6%) and no events in the dose maintenance group (0%) (RR 2.47, 95% CI 0.12 to 50.39, 2 RCTs, N = 206; heterogeneity test: not applicable, Analysis 1.25).
1.25. Analysis.
Comparison 1 Antipsychotic dose increase versus antipsychotic dose maintenance, Outcome 25 Adverse effects ‐ Cardiac: QTc prolongation.
1.26 Adverse effects ‒ Cardiac: Orthostatic hypotension
Two studies reported the numbers of participants with orthostatic hypotension (Huang 1987; Lindenmayer 2011). There was no clear difference between the two groups. In total, 3 out of 50 participants in the antipsychotic dose increase group and 3 out of 52 participants in the dose maintenance group presented with orthostatic hypotension (RR 1.09, 95% CI 0.25 to 4.82, 2 RCTs, N = 102; heterogeneity test: P = 0.43, I² = 0%, Analysis 1.26).
1.26. Analysis.
Comparison 1 Antipsychotic dose increase versus antipsychotic dose maintenance, Outcome 26 Adverse effects ‐ Cardiac: Orthostatic hypotension.
1.27 Adverse effects ‒ Cardiac: Palpitations
One study reported data in terms of participants reporting palpitations (Huang 1987). There was no clear difference between the antipsychotic dose increase group (4.8%) versus the antipsychotic dose maintenance group (0%) (RR 3.00, 95% CI 0.13 to 69.70, 1 RCT, N = 42, Analysis 1.27).
1.27. Analysis.
Comparison 1 Antipsychotic dose increase versus antipsychotic dose maintenance, Outcome 27 Adverse effects ‐ Cardiac: Palpitations.
1.28 Adverse effects ‒ Cardiac: Premature Ventricular Contractions
One study reported data in terms of participants with premature ventricular contractions (Huang 1987). There was no clear difference between the antipsychotic dose increase group (4.8%) versus the antipsychotic dose maintenance group (0%) (RR 3.00, 95% CI 0.13 to 69.70, 1 RCT, N = 42, Analysis 1.28).
1.28. Analysis.
Comparison 1 Antipsychotic dose increase versus antipsychotic dose maintenance, Outcome 28 Adverse effects ‐ Cardiac: Premature Ventricular Contractions.
1.29 Adverse effects ‒ Constipation
Two studies reported the numbers of participants with constipation (Huang 1987; Lindenmayer 2011). There was no clear difference between the two groups. In total there were five participants with constipation in the antipsychotic dose increase group (10%) and three participants in the dose maintenance group (5.8%) (RR 1.53, 95% CI 0.44 to 5.38, 2 RCTs, N = 102; heterogeneity test: P = 0.62, I² = 0%, Analysis 1.29).
1.29. Analysis.
Comparison 1 Antipsychotic dose increase versus antipsychotic dose maintenance, Outcome 29 Adverse effects ‐ Constipation.
1.30 Adverse effects ‐ Dizziness
Two studies reported the numbers of participants with dizziness (Honer 2011; Huang 1987). There was no clear difference between the two groups. In total there were 18 patients with dizziness in the antipsychotic dose increase group (16.5%) and 15 patients in the dose maintenance group (23.4%) (RR 0.77, 95% CI 0.41 to 1.44, 2 RCTs, N = 173; heterogeneity test: P = 0.56, I² = 0%, Analysis 1.30).
1.30. Analysis.
Comparison 1 Antipsychotic dose increase versus antipsychotic dose maintenance, Outcome 30 Adverse effects ‐ Dizziness.
1.31 Adverse effects ‒ Drooling
One study reported data in terms of participants complaining of drooling (Huang 1987). There was no clear difference between the antipsychotic dose increase group (9.5%) versus the antipsychotic dose maintenance group (4.8%) (RR 2.00, 95% CI 0.20 to 20.41, 1 RCT, N = 42, Analysis 1.31).
1.31. Analysis.
Comparison 1 Antipsychotic dose increase versus antipsychotic dose maintenance, Outcome 31 Adverse effects ‐ Drooling.
1.32 Adverse effects ‒ Death (suicide or natural cause)
One study reported that there were no deaths or suicides during the trial in any of the two groups (Honer 2011).
1.33 Adverse effects ‒ Extrapyramidal: Categorical deterioration (AIMS score)
One study reported data in terms of categorical change (deterioration) in AIMS scores (Honer 2011). There was no clear difference between the antipsychotic dose increase group (19%) versus the antipsychotic dose maintenance group (14%) (RR 1.38, 95% CI 0.59 to 3.26, 1 RCT, N = 131, Analysis 1.33).
1.33. Analysis.
Comparison 1 Antipsychotic dose increase versus antipsychotic dose maintenance, Outcome 33 Adverse effects ‐ Extrapyramidal: Categorical deterioration (AIMS, high = poor).
1.34 Adverse effects ‒ Extrapyramidal: average endpoint score (AIMS, high = poor)
One study reported the average AIMS score at endpoint (Lindenmayer 2011). There was no clear difference between groups (MD 0.70, 95% CI −0.87 to 2.27, 1 RCT, N = 60, Analysis 1.34 ).
1.34. Analysis.
Comparison 1 Antipsychotic dose increase versus antipsychotic dose maintenance, Outcome 34 Adverse effects ‐ Extrapyramidal: average endpoint score (AIMS, high = poor).
1.35 Adverse effects ‐ Extrapyramidal: average change score (AIMS, high = poor)
Two studies reported the average AIMS score change (Lindenmayer 2011; Sakurai 2016). Lindenmayer 2011 favoured dose continuation group whereas Sakurai 2016 favoured dose increase group. Nevertheless, in both studies AIMS scores were very low at baseline. When pooling, there was no clear difference between the two groups (MD 0.41, 95% CI −1.15 to 1.96, 2 RCTs, N = 163, Analysis 1.35), but heterogeneity was significant (P = 0.004, I² = 88%).
1.35. Analysis.
Comparison 1 Antipsychotic dose increase versus antipsychotic dose maintenance, Outcome 35 Adverse effects ‐ Extrapyramidal: average change score (AIMS, high = poor).
1.36 Adverse effects ‒ Extrapyramidal: Akathisia
Two studies reported the numbers of participants with akathisia (Bjørndal 1980; Huang 1987). There was no clear difference between the two groups. In total there were three participants with akathisia in the antipsychotic dose increase group (9.1%) and four participants in the dose maintenance group (12.5%) (RR 0.74, 95% CI 0.04 to 14.02, 2 RCTs, N = 65; heterogeneity test: P = 0.10, I² = 63%, Analysis 1.36).
1.36. Analysis.
Comparison 1 Antipsychotic dose increase versus antipsychotic dose maintenance, Outcome 36 Adverse effects ‐ Extrapyramidal: akathisia.
1.37 Adverse effects ‒ Extrapyramidal: Categorical deterioration (BAS, high = poor)
One study reported data in terms of categorical change (deterioration) in BAS scores (Honer 2011). There was no clear difference between the antipsychotic dose increase group (10%) versus the antipsychotic dose maintenance group (7%) (RR 1.47, 95% CI 0.42 to 5.14, 1 RCT, N = 131, Analysis 1.37).
1.37. Analysis.
Comparison 1 Antipsychotic dose increase versus antipsychotic dose maintenance, Outcome 37 Adverse effects ‐ Extrapyramidal: categorical deterioration (BAS, high = poor).
1.38 Adverse effects ‒ Extrapyramidal: average endpoint (BAS, high = poor)
One study reported the average BAS score at endpoint (Lindenmayer 2011). There was no clear difference between groups (MD −0.20, 95% CI −0.74 to 0.34, 1 RCT, N = 60, Analysis 1.38).
1.38. Analysis.
Comparison 1 Antipsychotic dose increase versus antipsychotic dose maintenance, Outcome 38 Adverse effects ‐ Extrapyramidal: average endpoint score (BAS, high = poor).
1.39 Adverse effects ‒ Extrapyramidal: average change score (BAS, high = poor)
Two studies reported the average BAS score change (Lindenmayer 2011; Sakurai 2016). Lindenmayer 2011 showed a trend in favour of dose continuation group whereas Sakurai 2016 favoured dose increase group. When pooling, there was no clear difference between the two groups (MD −0.11, 95% CI −0.59 to 0.37, 2 RCTs, N = 163, Analysis 1.39), but there was heterogeneity (I² = 69%).
1.39. Analysis.
Comparison 1 Antipsychotic dose increase versus antipsychotic dose maintenance, Outcome 39 Adverse effects ‐ Extrapyramidal: average change score (BAS, high = poor).
1.40 Adverse effect ‒ Extrapyramidal: dystonia and/or dyskinesia
Three studies reported the numbers of participants with dystonia or dyskinesia (Bjørndal 1980; Honer 2011; Huang 1987). There was no clear difference between the two groups. In total there were 13 participants with dystonia or dyskinesia in the antipsychotic dose increase group (10.7 %) and 14 participants in the dose maintenance group (18.7%) (RR 0.48, 95% CI 0.09 to 2.73, 3 RCTs, N = 196; heterogeneity test: P = 0.07, I² = 62%, Analysis 1.40).
1.40. Analysis.
Comparison 1 Antipsychotic dose increase versus antipsychotic dose maintenance, Outcome 40 Adverse effects ‐ Extrapyramidal: dystonia and/or dyskinesia.
1.41 Adverse effects ‒ Extrapyramidal: Categorical deterioration (SAS, high = poor)
One study reported data in terms of categorical change (deterioration) in SAS scores (Honer 2011). There was no clear difference between the antipsychotic dose increase group (17%) versus the antipsychotic dose maintenance group (14%) (RR 1.22, 95% CI 0.51 to 2.93, 1 RCT, N = 131, Analysis 1.41).
1.41. Analysis.
Comparison 1 Antipsychotic dose increase versus antipsychotic dose maintenance, Outcome 41 Adverse effects ‐ Extrapyramidal: categorical deterioration in SAS.
1.42 Adverse effects ‒ Extrapyramidal: average endpoint score (SAS, high = poor)
Two studies reported the average SAS score at endpoint (Honer 2011; Lindenmayer 2011). Overall, there was no clear difference between groups (MD 0.93, 95% CI −1.04 to 2.91, 2 RCTs, N = 191), but the studies' results were significantly heterogeneous (P = 0.006, I² = 87%). Honer 2011 showed no difference in SAS endpoint scores between the two groups (MD −0.02, 95% CI −0.79 to 0.75) whereas Lindenmayer 2011 showed a superiority of the antipsychotic dose maintenance group versus the antipsychotic dose increase group (MD 2.00, 95% CI 0.78 to 3.22). Nevertheless, the Lindenmayer 2011 results favouring the dose maintenance group at endpoint could be attributed to a baseline imbalance in SAS scores between the two groups. This claim is supported by the results of the repeated measures analysis of variance (ANOVA) model with accounted effects for baseline values that showed no clear difference between the two groups at endpoint (P = 0.249) and the analysis of the SAS change scores (see Analysis 1.42).
1.42. Analysis.
Comparison 1 Antipsychotic dose increase versus antipsychotic dose maintenance, Outcome 42 Adverse effects ‐ Extrapyramidal: average endpoint score (SAS, high = poor).
1.43 Adverse effects ‒ Extrapyramidal: average change score (SAS, high = poor)
Two studies reported the SAS score change (Lindenmayer 2011; Sakurai 2016). There was no clear difference between groups (MD 0.21, 95% CI −0.83 to 1.26, 2 RCTs, N = 163; heterogeneity test: P = 0.08, I² = 67%, Analysis 1.43).
1.43. Analysis.
Comparison 1 Antipsychotic dose increase versus antipsychotic dose maintenance, Outcome 43 Adverse effects ‐ Extrapyramidal: average change score (SA, high = poor).
1.44 Adverse effects ‒ Extrapyramidal: Tremor
Two studies reported the numbers of participants with tremor (Honer 2011; Huang 1987). There was no clear difference between the two groups. In total there were 10 participants with headache in the antipsychotic dose increase group (9.2%) and 5 participants in the dose maintenance group (7.8%) (RR 1.59, 95% CI 0.59 to 4.26, 2 RCTs, N = 173; heterogeneity test: P = 0.83, I² = 0%, Analysis 1.44).
1.44. Analysis.
Comparison 1 Antipsychotic dose increase versus antipsychotic dose maintenance, Outcome 44 Adverse effects ‐ Extrapyramidal: tremor.
1.45 Adverse effects ‒ Headache
Two studies reported the numbers of participants with headache (Honer 2011; Huang 1987). There was no clear difference between the two groups. In total there were 12 participants with headache in the antipsychotic dose increase group (11%) and 4 participants in the dose maintenance group (6.3%) (RR 1.46, 95% CI 0.52 to 4.08, 2 RCTs, N = 173; heterogeneity test: P = 0.64, I² = 0%, Analysis 1.45).
1.45. Analysis.
Comparison 1 Antipsychotic dose increase versus antipsychotic dose maintenance, Outcome 45 Adverse effects ‐ Headache.
1.46 Adverse effects ‒ Somnolence and/or drowsiness
Four studies reported the numbers of participants with somnolence or drowsiness (Bjørndal 1980; Honer 2011; Huang 1987; Lindenmayer 2011). There was no clear difference between the two groups. In total there were 25 participants with somnolence or drowsiness in the antipsychotic dose increase group (16.7%) and 11 participants in the dose maintenance group (10.4%) (RR 1.76, 95% CI 0.81 to 3.81, 4 RCTs, N = 256; heterogeneity test: P = 0.32, I² = 13%, Analysis 1.46).
1.46. Analysis.
Comparison 1 Antipsychotic dose increase versus antipsychotic dose maintenance, Outcome 46 Adverse effects ‐ Somnolence and/or drowsiness.
1.47 Adverse effects ‒ Weight at endpoint (high = poor)
Two studies reported data in terms of weight at endpoint (Honer 2011; Lindenmayer 2011). There was no clear difference between groups (MD −1.85, 95% CI −7.09 to 3.39, 2 RCTs, N = 165; heterogeneity test: P = 0.98, I² = 0%, Analysis 1.47).
1.47. Analysis.
Comparison 1 Antipsychotic dose increase versus antipsychotic dose maintenance, Outcome 47 Adverse effects ‐ Weight at endpoint (high = poor).
1.48 Behaviour: Average endpoint score (BARS, high = poor)
One study — Lindenmayer 2011 — reported the average BARS symptom score at endpoint and showed no clear difference between the two groups (MD −0.10, 95% CI −0.50 to 0.30, 1 RCT, N = 60, Analysis 1.48).
1.48. Analysis.
Comparison 1 Antipsychotic dose increase versus antipsychotic dose maintenance, Outcome 48 Behaviour: average endpoint score (BARS, high = good).
1.49 Functioning ‒ Global Assessment of Functioning: average change score (GAF, high = good)
One study — Sakurai 2016 — reported the average GAF score change and showed no clear difference between the two groups (MD −0.60, 95% CI −3.00 to 1.80, 1 RCT, N = 103, Analysis 1.49).
1.49. Analysis.
Comparison 1 Antipsychotic dose increase versus antipsychotic dose maintenance, Outcome 49 Functioning ‐ Global Assessment of Functioning: average change score (GAF, high = good).
1.50 Functioning ‒ Social‐Adaptive Functioning Evaluation: average endpoint score (SAFE, high = poor)
One study — Lindenmayer 2011 — reported the average SAFE score at endpoint and showed no clear difference between the two groups (MD 0.16, 95% CI −0.47 to 0.79, 1 RCT, N = 60, Analysis 1.50).
1.50. Analysis.
Comparison 1 Antipsychotic dose increase versus antipsychotic dose maintenance, Outcome 50 Functioning ‐ Social ‐ Adaptive Functioning Evaluation: average endpoint score (SAFE, high = poor).
1.51 Functioning ‒ Social and Occupational Functioning Assessment: average endpoint score (SOFAS, high = good)
One study — Honer 2011 — reported the average SOFAS symptom score at endpoint and showed no clear difference between the two groups (MD 0.50, 95% CI −3.80 to 4.80, 1 RCT, N = 131, Analysis 1.51).
1.51. Analysis.
Comparison 1 Antipsychotic dose increase versus antipsychotic dose maintenance, Outcome 51 Functioning ‐ Social and Occupational Functioning Assessment: average endpoint score (SOFAS, high = good).
1.52 Quality of life: Clinically important change (at least 50% improvement HQLS, high = good)
One study — McGorry 2011 — reported that there were no participants, from either group, that improved in terms of quality of life, measured by a 50% or more Heinrichs Quality of Life Scale (HQLS) symptom score change, Analysis 1.52
1.52. Analysis.
Comparison 1 Antipsychotic dose increase versus antipsychotic dose maintenance, Outcome 52 Quality of life: Clinically important change (at least 50% improvement HQLS, high = good).
1.53 Quality of life: Average endpoint score (HQLS, high = good)
One study — McGorry 2011 — reported the average HQLS symptom score at endpoint and showed no clear difference between the two groups (MD 5.50, 95% CI −13.66 to 24.66, 1 RCT, N = 17, Analysis 1.53 ).
1.53. Analysis.
Comparison 1 Antipsychotic dose increase versus antipsychotic dose maintenance, Outcome 53 Quality of life: Average endpoint score (HQLS, high = good).
2. Subgroup analyses and investigation of heterogeneity
All subgroup analyses were only performed on the primary outcome "clinically relevant response as defined by authors".
2.1 Single antipsychotic drugs
There were no differences between the single antipsychotic drugs (i.e. fluphenazine, haloperidol, quetiapine, risperidone, thiothixene and ziprasidone) that were included in the present review (test for subgroup differences: Chi² = 6.65, df = 5 (P = 0.25), I² = 24.8%).
2.2 Clinical state, stage or problem
There were no differences between first‐episode and multiple‐episode participants (test for subgroup differences: Chi² = 2.49, df = 1 (P = 0.11), I² = 59.9%).
3. Publication bias
Due to the small number of included studies a funnel plot analysis was not performed.
4. Sensitivity analyses
We only performed sensitivity analyses on the primary outcome "clinically relevant response as defined by authors". As there was no study in which randomisation was implied, this pre‐planned sensitivity analysis did not apply to our review.
4.1 Exclusion of studies with unclear randomisation methods
We judged all but two studies — Honer 2011 and Sakurai 2016 — to have used unclear randomisation methods. Excluding these studies did not change the overall results (RR 1.05, 95% CI 0.77 to 1.44, 2 RCTs, N = 234; heterogeneity test: P = 0.88, I² = 0%).
4.2 Exclusion of studies with unclear allocation concealment methods
We judged all but two studies — Honer 2011 and Sakurai 2016 — to have used unclear allocation concealment methods. Excluding these studies did not change the overall results (RR 1.05, 95% CI 0.77 to 1.44, 2 RCTs, N = 234; heterogeneity test: P = 0.88, I² = 0%).
4.3 Exclusion of studies with high risk of bias regarding blinding
There was only one study with high risk of bias regarding blinding of participants and personnel (McGorry 2011). Excluding this study did not change the overall results (RR 1.07, 95% CI 0.86 to 1.34, 8 RCTs, N = 516; heterogeneity test: P = 0.51, I² = 0%). There was no study with high risk of bias regarding blinding of outcome assessment.
4.4 Exclusion of studies with high risk of bias regarding incomplete outcome data
There were three studies with high risk of bias regarding incomplete outcome data (Bjørndal 1980; Goff 2013; Lindenmayer 2011). Excluding these studies did not change the overall results (RR 1.19, 95% CI 0.92 to 1.54, 6 RCTs, N = 375; heterogeneity test: P = 0.43, I² = 0%).
4.5 Exclusion of studies with imputed values
There was only one study with imputed values for the primary outcome (Goff 2013). In this study, those leaving the study early were all assumed to have the same rates of response as those who completed. Excluding this study did not change the overall results (RR 1.16, 95% CI 0.90 to 1.50, 8 RCTs, N = 458; heterogeneity test: P = 0.40, I² = 4%).
4.6 Fixed‐effect model
When a fixed‐effect model was applied, the overall results did not change (RR 1.10, 95% CI 0.88 to 1.38, 9 RCTs, N = 533; heterogeneity test: P = 0.37, I² = 8%).
Discussion
Summary of main results
1. General
This review included 10 studies with 675 participants that compared antipsychotic dose increase versus antipsychotic dose maintenance in people with schizophrenia or related disorders who were non‐responsive to their initial antipsychotic treatment. The included studies were published over a long period (from 1980 to 2016) and varied in duration both of the run‐in (e.g. 2 to 4 weeks) and the main phase (e.g. 2 to 12 weeks). Despite the comprehensive literature and electronic search and the application of broad inclusion criteria, the total of 10 included studies with 675 participants is a small base upon which to judge the effectiveness of antipsychotic dose increase versus dose maintenance. Trials with small sample sizes lack sufficient power to detect a small to moderate effect, and thus results from such trials are often inconclusive, even when a real effect does exist. A review by Davey Smith and colleagues suggested that meta‐analyses based on summation of small trials should be interpreted as inconclusive, regardless of whether the combined estimate was significant (Davey Smith 1998). The studies included in this review provided therefore no sufficient data on whether increasing the dose of the initial antipsychotic drug brings better results than maintaining the same dose for a longer period of time after an initial period of non response.
2. Treatment effects
2.1 Global state: clinically relevant response ‐ as defined by the authors
Nine out of the 10 included studies presented data on the primary outcome "clinically relevant response as defined by the authors". The meta‐analysis of the data did not show a significant superiority of dose increase versus dose maintenance but since the total number of studies and participants is low, more trials are warranted and no firm conclusions can be drawn.
2.2 Global state: any change ‒ improved
One study reported how many participants improved based on global state, three studies reported the average CGI‐Severity score at endpoint, one study the average CGI‐Severity score change, and one study reported the average CGI‐Improvement score at endpoint. Overall, no clear difference between antipsychotic dose increase and antipsychotic dose maintenance group was shown. Nevertheless, since few data are available in terms of global state, results are again inconclusive and more studies are warranted.
2.3 Leaving the study early: tolerability ‒ due to side effects
Only 47 out of 496 participants left the studies early due to side effects with no difference between groups (30 out of 267 participants in the antipsychotic dose increase group and 17 out of 229 participants in the antipsychotic dose maintenance group). One could suggest that higher antipsychotic dosages seem to be well tolerated in people with schizophrenia who are non‐responsive to their initial antipsychotic treatment. Nevertheless, the data were heterogeneous; most studies showed no difference between groups, but Sakurai 2016 favoured dose continuation group significantly (less attrition due to side effects). In addition, since events such as ‘worsening of psychosis’ are, by definition, recorded as adverse events, especially in modern trials, this outcome may not be an ideal measure of overall tolerability. Moreover, since only seven studies presented data on this outcome, more studies are warranted to draw any solid conclusion.
2.4 Leaving the study early: acceptability ‒ due to any reason
In the five studies that provided data, 90 out of 353 participants left the studies early due to any reason with no difference between groups (55 out of 199 participants in the antipsychotic dose increase group and 35 out of 154 participants in the antipsychotic dose maintenance group). Leaving a study due to any reason is often considered to be a measure of acceptability of treatment. Nevertheless, since only five studies presented data on this outcome, more studies are warranted.
2.5 Leaving the study early: efficacy ‒ due to inefficacy
In the four studies that provided data, 17 out of 336 participants left the studies early due to inefficacy with no difference between groups (8 out of 190 participants in the antipsychotic dose increase group and 9 out of 146 participants in the antipsychotic dose maintenance group). Leaving a study due to inefficacy could be considered as a measure of efficacy of treatment. But again, as only four studies presented data on this outcome, results are inconclusive and more studies are needed.
2.6 Mental state: general
Two studies reported the average PANSS total score at endpoint with no difference between antipsychotic dose increase and antipsychotic dose maintenance group (Honer 2011; Lindenmayer 2011). Regarding the PANSS total score change, three studies were included in the analysis (Lindenmayer 2011; Loebel 2014; Sakurai 2016), and again no difference between the two groups was indicated. Nevertheless, in this analysis there was a considerable, even though not statistically significant, heterogeneity of the study results. Lindenmayer 2011 and Sakurai 2016 showed no difference between groups whereas Loebel 2014 favoured the dose increase group. Apart from the different antipsychotics used, our inspection of the three studies did not reveal an important difference which could explain the considerable heterogeneity; in the Lindenmayer 2011 study quetiapine was employed and exceeded the recommended dose range, in the Sakurai 2016 study olanzapine or risperidone were employed but did not exceed the recommended dose range, and in Loebel 2014 lurasidone was employed but did not exceed the recommended dose range. For lurasidone, there is some evidence from fixed‐dose studies that 160 mg/day had so far been the most efficacious lurasidone dose anyhow (Loebel 2013). When we combined PANSS endpoint and change scores, again no clear difference between the two groups was shown. As for the BPRS scale, three studies reported the average BPRS total score at endpoint and only one study reported the average BPRS total score change. Both analyses showed no difference between groups. When BPRS endpoint and change scores were combined, again no clear difference between the two groups was shown. As for the NOSIE scale, only one study reported the average score change with no difference between groups. Nevertheless, since few studies and participants were included in all the above analyses, results are inconclusive and more studies are needed.
2.7 Mental state: positive and negative symptoms
Specific symptoms of schizophrenia, e.g. positive and negative symptoms, were only reported by few trials (maximum number = 3). No difference between antipsychotic dose increase and antipsychotic dose maintenance group was shown but so few studies were included in the analyses that any meaningful statement is not possible.
2.8 Adverse effects
Adverse effects were often poorly and incompletely reported, and overall data were extremely few. Some outcomes concerning extrapyramidal symptoms were heterogeneous, but since only two or three studies presented relevant data per outcome and scores were very low at baseline, we could make no meaningful clinical interpretation. More studies are warranted.
2.9 Behaviour, functioning and quality of life
Few data were available in terms of behaviour, functioning rating scales and quality of life. No clear difference was found but more studies are warranted. In our opinion, improving patients' functioning and quality of life are important and challenging goals of antipsychotic treatment strategies and should be included in the future research agenda.
3. Subgroup analyses and investigation of heterogeneity
There was no statically significant heterogeneity in the meta‐analysis of the primary outcome "clinically relevant response as defined by authors". Moreover, no subgroup analysis revealed any statistically significant difference, but the power to detect a real difference was very low.
4. Sensitivity analyses
The results of the primary outcome were not much different in a series of pre‐planned sensitivity analyses in which we excluded studies with unclear randomisation, unclear allocation concealment, high risk of bias regarding blinding or incomplete outcome data, and studies with imputed values; or when a fixed‐effect model instead of a random‐effects model was applied. Nevertheless, the statistical power of these analyses was low.
Overall completeness and applicability of evidence
We could include in our review only 10 studies that examined antipsychotic dose increase versus antipsychotic dose maintenance for patients with schizophrenia not responding to their current antipsychotic treatment. The 10 studies were on different antipsychotics, had different lengths of run‐in and follow‐up phases, used different definitions of non response and were conducted in different settings and populations. There was no clear difference between the two groups in any efficacy outcome. In addition, data on most of our secondary outcomes were few (usually one or two included studies per outcome). Moreover, subgroup and sensitivity analyses did not detect any effect but the statistical power of these analyses was low. Therefore, more studies are needed and alternative treatment strategies for non‐responders should be explored.
Quality of the evidence
The quality of the evidence is low to very low based on GRADE (Schünemann 2011). All studies were randomised and all but McGorry 2011 were double blind. Nevertheless, for most of them details were not presented. Therefore it is unclear whether the studies were adequately randomised, whether treatment allocation was really concealed and whether blinding worked. Moreover, many outcomes were assessed in sample sizes too small to detect clear differences if they existed.
Potential biases in the review process
We decided a priori to pool all antipsychotic drugs in this review. We feel that this is justified for efficacy‐related outcomes, because most antipsychotic drugs do not differ in efficacy and if differences exist between some antipsychotic drugs these are not large (Leucht 2009; Leucht 2013). The decision to pool all studies irrespective of the antipsychotic drug used is more problematic for adverse effects, because antipsychotic drugs differ to a large extent in this regard. This could also partly explain the considerable heterogeneity that was present in the meta‐analysis of some adverse effects' outcomes. Furthermore, between‐study heterogeneity is expected to be larger for small studies (IntHout 2015), and it is difficult to estimate when few studies are included in a meta‐analysis (Friede 2017; von Hippel 2015), as in our case.
The search was mainly based on the Cochrane Schizophrenia Group’s register of trials. This is largely made up of searches of published literature. It is possible that there are unpublished studies that we are not aware of and there is a possibility of publication bias, but we were unable to undertake the proposed funnel plot to investigate the presence of publication bias since fewer than 10 studies per outcome were included in analyses. We have chosen to use the random‐effects model for our analyses, which does not assume that the populations from which the different trials are derived are the same. This technique does emphasise the results from smaller trials and it is these studies that are likely to be most prone to bias. Nevertheless, the results of a fixed‐effect model in a sensitivity analysis of the primary outcome were similar.
All in all, we should highlight that few trials were included in the present review and most analyses were clearly underpowered, increasing the possibility of a type II error (i.e. failure to find a difference although it is present).
Agreements and disagreements with other studies or reviews
Schizophrenia treatment guidelines acknowledge that limited evidence exists concerning the treatment strategies in case of non response to the initial antipsychotic treatment; nevertheless, exceeding the antipsychotic dosage outside the recommended range is not recommended (Hasan 2012). Dold 2015, a recent review on the same topic, reached a conclusion similar to that of our review; nevertheless, authors failed to include almost half of the relevant studies.
Authors' conclusions
Implications for practice.
1. For people with schizophrenia
It is well established that many people with schizophrenia do not respond to initial antipsychotic treatment. In such cases, recent evidence suggests that treatment strategy should be revised after just two weeks of treatment since patients not even minimally improved by that point are unlikely to respond later (Samara 2015a). But which alternative strategy should be followed in cases of non response? One could choose to increase the dose of the initial antipsychotic drug, or to switch to another antipsychotic drug, or to augment with a second drug. Increasing the dose is reasonably expected to cause more adverse effects and any clinical decision should balance between the potential benefit in efficacy and the risk of adverse effects. Nevertheless, the results of the current review do not provide evidence that higher antipsychotic dosages increase either the benefit in efficacy or the risk of adverse effects, probably because few data were available and different antipsychotics were combined. For efficacy outcomes, combining different antipsychotics is not expected to be so problematic since differences in efficacy between drugs are small; but for adverse effects, differences between antipsychotics are larger (Leucht 2013), and combining them could blur the picture e.g. combine an haloperidol with an olanzapine study for extrapyramidal side effects. The subgroup analysis examining each drug separately did not reveal any statistically significant difference, but the power to detect a real difference was again very low.
2. For clinicians
All in all, the present review shows that there is not enough evidence to decide on the question whether increasing the antipsychotic dose in general, especially for dosages above the maximal recommended ones, is more effective than maintaining it. In addition, no difference for safety outcomes was shown. If clinicians decide to increase the antipsychotic dose above the maximal approved threshold, informed consent needs to be obtained and the patient should be closely monitored for the risk of developing intolerable side effects. Clinicians should be aware that high‐quality reliable evidence on increasing or not increasing antipsychotic dose is not available at the moment, either in terms of global efficacy or potential harms. Data concerning dose management, comprehending antipsychotic reduction, are thus strongly needed.
3. For managers/policy makers
We found no data for economic outcomes.
Implications for research.
1. General
Outcome reporting remains insufficient in antipsychotic drug trials. Strict adherence to the CONSORT statement (CONsolidated Standards Of Reporting Trials; Moher 2001) would make such studies much more informative. Moreover, outcomes measuring functioning and quality of life are usually missing and, even if they are described, heterogeneous rating scales are employed. In addition, drug adherence and drug plasma levels are not monitored in most antipsychotic trials, but their use could shed light on reasons for non response. Finally, recommended dose ranges are not well established and it seems that for some drugs like risperidone it might be overestimated, whereas for others like olanzapine and quetiapine it might be underestimated.
2. Specific
Further studies are warranted to investigate the optimal treatment strategy when a patient does not respond to the initial antipsychotic treatment since the results of the present review were not conclusive. Large, pragmatic, randomised trials on different antipsychotics, using a widely accepted definition of non response (Leucht 2014), and employing outcomes that measure global functioning as well are needed. Reporting of adverse effects is also insufficient. The focus should be on high dosages, above the maximal recommended ones, since significant variability is expected in optimal dose ranges among individual patients. In addition, the standard recommended dose ranges are derived from clinical trials that include patients meeting strict inclusion and exclusion criteria, but these patients probably differ from those met in everyday clinical practice.
Individualizing antipsychotic treatment should be the goal in schizophrenia research. Evidence suggests that patients with schizophrenia show optimal response to different drugs (Clark 2011). But predicting the optimal drug and dose for individual patients is difficult; decisions are still based on trial and error. Towards personalised antipsychotic treatment, patient and disease characteristics such as socioeconomic status, patient autonomy and disease severity might be useful to differentiate patients and their response to antipsychotics (Correll 2011). Environmental attributes such as diet and smoking should also be taken into account since they can alter drug metabolism and may indicate the need for antipsychotic dosage adjustment (Nebert 2000). Pharmacogenetics, another promising path, is yet in its early stages for use in everyday clinical practice (Eum 2016). Finally, direct linking of drug plasma levels with D2/3R occupancy seems to be feasible based on preliminary data, but replication is needed (Nakajima 2016). Overall, individualizing treatment is still an unmet and challenging need in schizophrenia treatment, but research in this field is ongoing and promising.
For a suggested design of study please see Additional Table 2.
1. Suggested design for future study.
| Methods | Randomisation: random. Allocation: concealed. Blinding: double blind. Duration: at least 2 weeks run‐in phase to confirm non response to initial treatment and at least 4 weeks randomised double‐blind phase. Setting: in‐ or out‐patients. |
| Participants | Diagnosis: patients with schizophrenia, schizoaffective disorder or schizophreniform disorder. N > 300 Gender: male and female patients. Age: mean 30 years (SD = 7.0), range 18 to 65 years. |
| Interventions | All patients firstly receive treatment with one antipsychotic drug for at least 2 weeks. Those patients who do not at least minimally improve after 2 weeks of treatment, are considered non‐responders and are randomised to: 1. Increasing the dose of the antipsychotic drug above the officially recommended dose range. 2. Continuing treatment with the antipsychotic drug at the same, initial dose (within the officially recommended dose range). |
| Outcomes | Response (e.g. defined as PANSS or BPRS decrease ≥ 50%).* Relapse. Leaving the study early due to any reason. Leaving the study early due to side effects. General mental state‐average change in general mental state scores. Adverse effects‐ at least one adverse effect; clinically important general adverse effects; sudden and unexpected death. Service use‐ time in hospital. Quality of life. All outcomes by time ‐ short term (up to 12 weeks), medium term (13 to 26 weeks) and long term (over 26 weeks). |
| Notes | *Primary outcome of interest. |
Acknowledgements
We would like to thank all members of the Cochrane Schizophrenia Group for their editorial assistance, and acknowledge use of the Cochrane Schizophrenia Group’s template for the Methods section which we have adapted for use in this review.
The search term was developed by the Trial Search Co‐ordinator of the Cochrane Schizophrenia Group and the contact author of this protocol. We would like to thank Sergio Covarrubias, Jinmei Qin and Claire Ainsworth for peer reviewing this version of the review.
We would also like to thank Patrick D McGorry and Susy Harrigan for their efforts to provide the original data of their study published in 2011 (McGorry 2011).
Appendices
Appendix 1. Previous searches
Search in 2014 and 2015
Electronic searches
1. Cochrane Schizophrenia Group’s Trials Register
On 10 June 2014, 6 October 2015, the Trials Search Coordinator (TSC) searched the Cochrane Schizophrenia Group’s Registry of Trials using the following search strategies which have been developed based on literature review and consulting the authors of the review:
((("non respon*" or nonrespon* or "not respon*" or "no respon*" or unrespon* or fail* or unsuccess* or *resist* or persist* or residual or untreat* or refract* or ineffective or "not effective" or unchanged) or ((drug* or therap* or treat* or antipsychotic* or neuroleptic* or tranquili* or partial* or incomplete*) NEXT respon*) or ((poor or subsequen*) NEAR3 respon*)) and (dose* or dosage* or dosing)):ti,ab of REFERENCE or (dose* or dosage* or dosing)):sin of STUDY
The Cochrane Schizophrenia Group’s Registry of Trials is compiled by systematic searches of major resources (including AMED, BIOSIS, CINAHL, Embase, MEDLINE, PsycINFO, PubMed, and registries of clinical trials) and their monthly updates, hand‐searches, grey literature, and conference proceedings (see Group Module). There was no language, date, document type, or publication status limitations for inclusion of records into the register.
Searching other resources
1. Reference searching
We inspected references of all included studies for further relevant studies.
2. Personal contact
We contacted the first author of each included study for information regarding unpublished trials. We noted any response in the Characteristics of included studies and thanked the authors in the Acknowledgements.
Data and analyses
Comparison 1. Antipsychotic dose increase versus antipsychotic dose maintenance.
1.32. Analysis.
Comparison 1 Antipsychotic dose increase versus antipsychotic dose maintenance, Outcome 32 Adverse effects ‐ Death (suicide or naturalistic cause).
Comparison 2. Subgroup analysis and investigation of heterogeneity.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Single antipsychotic drugs | 8 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 Fluphenazine | 1 | 34 | Risk Ratio (M‐H, Random, 95% CI) | 2.25 [0.22, 22.53] |
| 1.2 Haloperidol | 2 | 71 | Risk Ratio (M‐H, Random, 95% CI) | 1.10 [0.67, 1.80] |
| 1.3 Quetiapine | 2 | 191 | Risk Ratio (M‐H, Random, 95% CI) | 0.65 [0.15, 2.86] |
| 1.4 Risperidone | 1 | 17 | Risk Ratio (M‐H, Random, 95% CI) | 9.9 [0.63, 155.08] |
| 1.5 Thiothixene | 1 | 42 | Risk Ratio (M‐H, Random, 95% CI) | 1.75 [0.94, 3.26] |
| 1.6 Ziprasidone | 1 | 75 | Risk Ratio (M‐H, Random, 95% CI) | 0.79 [0.45, 1.41] |
| 2 Clinical state, stage or problem | 9 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 First episode patients | 1 | 17 | Risk Ratio (M‐H, Random, 95% CI) | 9.9 [0.63, 155.08] |
| 2.2 Not first episode patients | 8 | 516 | Risk Ratio (M‐H, Random, 95% CI) | 1.07 [0.86, 1.34] |
2.1. Analysis.
Comparison 2 Subgroup analysis and investigation of heterogeneity, Outcome 1 Single antipsychotic drugs.
2.2. Analysis.
Comparison 2 Subgroup analysis and investigation of heterogeneity, Outcome 2 Clinical state, stage or problem.
Comparison 3. Sensitivity analysis.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Excluding studies with unclear randomisation methods | 2 | 234 | Risk Ratio (M‐H, Random, 95% CI) | 1.05 [0.77, 1.44] |
| 2 Excluding studies with unclear allocation concealment methods | 2 | 234 | Risk Ratio (M‐H, Random, 95% CI) | 1.05 [0.77, 1.44] |
| 3 Exclusion of studies with high risk of bias regarding blinding | 8 | 516 | Risk Ratio (M‐H, Random, 95% CI) | 1.07 [0.86, 1.34] |
| 4 Exclusion of studies with high risk of bias regarding incomplete outcome data | 6 | 375 | Risk Ratio (M‐H, Random, 95% CI) | 1.19 [0.92, 1.54] |
| 5 Exclusion of studies with imputed values | 8 | 458 | Risk Ratio (M‐H, Random, 95% CI) | 1.16 [0.90, 1.50] |
| 6 Fixed effects | 9 | 533 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.10 [0.88, 1.38] |
3.1. Analysis.
Comparison 3 Sensitivity analysis, Outcome 1 Excluding studies with unclear randomisation methods.
3.2. Analysis.
Comparison 3 Sensitivity analysis, Outcome 2 Excluding studies with unclear allocation concealment methods.
3.3. Analysis.
Comparison 3 Sensitivity analysis, Outcome 3 Exclusion of studies with high risk of bias regarding blinding.
3.4. Analysis.
Comparison 3 Sensitivity analysis, Outcome 4 Exclusion of studies with high risk of bias regarding incomplete outcome data.
3.5. Analysis.
Comparison 3 Sensitivity analysis, Outcome 5 Exclusion of studies with imputed values.
3.6. Analysis.
Comparison 3 Sensitivity analysis, Outcome 6 Fixed effects.
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Bjørndal 1980.
| Methods | Allocation: randomised, no further details Blinding: double, no further details Duration: 16 weeks; 4 weeks non‐randomised open‐label run‐in phase and 12 weeks randomised double‐blind phase Design: parallel Location: not indicated, probably single centre, Denmark Setting: inpatients | |
| Participants | Diagnosis: chronic schizophrenia, severity of illness was 4 to 6 according to the disability scale used (0 indicated no symptoms, while a score of 7 indicated extremely severe degree of illness), no further details. N = 29, data only on 23 completers Gender: 29 men, 0 women Age: mean 34 years (range 19 to 60 years) for 23 completers History: duration of illness mean 9 years (range 2 to 20), length of stay in hospital mean 5 years (range 0.5 to 17), duration of antipsychotic treatment mean 8 years (range 1 to 18) for 23 completers; no further details. | |
| Interventions | All participants firstly received open‐label 2 mg haloperidol tablets for 4 weeks (maximum dosage being 12 mg/day). Participants were then randomised to either: 1. dose increase: 20 mg haloperidol tablets; mean 103 mg/day at endpoint. N = 12; or 2. dose maintenance: 2 mg haloperidol tablets; mean 15 mg/day at endpoint. N = 11. The starting dose corresponded to the number of tablets reached during the open pretest period (mean 10 mg/day in both groups). During the first 6 weeks, the dose was adjusted according to effect and side effects, while during the following 6 weeks the dose was maintained constant as far as possible. Rescue medication: orphenadrine and chloralodol; no further details. |
|
| Outcomes | Clinically relevant response (defined as > 0% BPRS total score reduction) Global state: improved, possible improved, unchanged, or deteriorated (Global Assessment Scale) Adverse effects Unable to use: Leaving the study early (no separate numbers for the two groups) Mental state: general (BPRS total score, no mean, no SD), Nurses Observation Scale for Inpatient Evaluation (NOSIE total score, no SD) Plasma levels of haloperidol (no mean, no SD) and prolactin (no SD), and not protocol outcomes Other laboratory investigations (no mean, no SD), and not protocol outcomes |
|
| Notes | In this study, there was a 4‐week, open, pretest period during which participants received 2 mg haloperidol tablets (mean 10 mg/day). It is not clearly indicated whether all participants entering the double‐blind phase were non‐responders during the run‐in phase. Nevertheless, based on participant description and BPRS total scores at baseline, we assumed this is the case. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "...patients were randomized..." (pg. 18); no further details |
| Allocation concealment (selection bias) | Unclear risk | No details were presented |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "...12‐week double‐blind phase..." (pg. 18); no further details |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: "...12‐week double‐blind phase..." (pg. 18); no further details |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Quote: "Twenty‐nine male, chronic schizophrenic patients ... consent to participate in the study. Six of the patients dropped out during the study, independent of the drug administered. The 23 patients completing the study ..." (pg. 18); no further details. Only completers were analysed. |
| Selective reporting (reporting bias) | High risk | BPRS total score, NOSIE, plasma levels and other laboratory values were assessed but no usable data were reported in the results. |
| Other bias | Low risk | No obvious risk for other bias |
Goff 2013.
| Methods | Allocation: randomised, no further details Blinding: double, no further details Duration: 11 weeks; 3 weeks non‐randomised open‐label run‐in phase and 8 weeks randomised double‐blind phase. Design: parallel Location: multi‐centre (USA) Setting: inpatients (N = 30) and outpatients (N = 45) | |
| Participants | Diagnosis: schizophrenia or schizoaffective disorder (DSM‐IV) N = 75 Gender: 52 men, 23 women Age: mean 40 years (SD = 11.9), range 16 to 65 years History: age at onset mean 25 years (SD = 9.1), no further details | |
| Interventions | All participants firstly received open‐label ziprasidone treatment, titrated up to 160 mg/day, for a minimum of 3 weeks. Participants with persistent psychotic symptoms defined by a score of 4 (moderate) or greater on any item of PANSS despite ziprasidone treatment were then randomised to either: 1. dose increase: ziprasidone 320 mg/day; dose could be decreased to 240 mg/day. N = 38; or 2. dose maintenance: ziprasidone 160 mg/day. N = 37. Rescue medication: benztropine, propranolol, lorazepam, zolpidem; no further details. |
|
| Outcomes | Global state: clinically relevant response (defined as ≥20% PANSS total score reduction) Mental state: positive symptoms (PANSS positive subscore) Adverse effects: cardiac — QTc prolongation (number of participants with QTc longer than 500 msec) Unable to use: Leaving the study early (numbers not presented) Overall mental state (PANSS total score, no mean, no SD) Global state (change in CGI‐I, no SD; and CGI‐S, no mean, no SD) Negative symptoms (PANSS negative subscore, no mean, no SD) Depression (Calgary Depression Rating Scale (CDRS), no mean, no SD) Functioning (Global Assessment of Functioning, GAF, no mean, no SD) Adverse effects (EPS ‐ SAS (no mean, no SD), Akathisia ‐ BAS (no mean, no SD), tardive dyskinesia ‐ AIMS (no mean, no SD), Side Effect Checklist (no numbers), rate of adverse effects (no SD)) Vital signs (no numbers) Plasma levels (no SD) Cognition (Schizophrenia Cognition Rating Scale, no mean, no SD) |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "...eligible patients were randomly assigned to ziprasidone 40 mg capsules or matching placebo in a 1:1 ratio stratified according to the duration of prior ziprasidone treatment..." (pg. 486); no further details. |
| Allocation concealment (selection bias) | Unclear risk | No details were presented |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: " For the 8‐week double‐blind, placebo‐controlled trial..." (pg. 486); no further details. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: " For the 8‐week double‐blind, placebo‐controlled trial..." (pg. 486); no further details. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | 44% of the participants left the study early. Numbers per group as well as reasons for leaving the study early were not specified. For the primary outcome of clinically relevant response, only completers were analysed. |
| Selective reporting (reporting bias) | High risk | PANSS total score, PANSS negative sbscore, CGI, CDRS, SCoRS, SAS, BAS, AIMS, Side Effect Checklist, vital signs and plasma levels were assessed but no usable data were reported in the results. |
| Other bias | Low risk | No obvious risk for other bias |
Honer 2011.
| Methods | Allocation: randomised, 2:1 with a computerised schedule Blinding: double, no further details Duration: 12 weeks; 4 weeks non‐randomised open‐label run in phase and 8 weeks randomised double‐blind phase. Design: parallel Location: multi‐centre (Canada) Setting: inpatients (N = 27) and outpatients (N = 104) | |
| Participants | Diagnosis: schizophrenia or schizoaffective disorder (DSM IV) N = 131 Gender: 90 men, 41 women Age: mean 39.7 years (SD = 12.1), range 18 to 65 years History: not stated | |
| Interventions | All participants firstly received open‐label quetiapine treatment 800 mg/day for 4 weeks. Participants with persistent positive and negative symptoms and CGI≥4 despite quetiapine treatment were then randomised to either: 1. dose increase: quetiapine 1200 mg/day; dose could be decreased. N = 88; or 2. dose maintenance: quetiapine 800 mg/day. N = 43. Rescue medication: flurazepam, zaleplon, lorazepam, anticholinergic medication. Antidepressant, mood‐stabilizing, or hypnotic medications were continued, if subjects were taking stable doses for a 30‐day period prior to trial entry. |
|
| Outcomes | Global state: clinically relevant response (defined as ≥ 20% PANSS total score reduction) Leaving the study early (due to side effects, any reason, inefficacy) Mental state: general mental state (PANSS total score), positive symptoms (PANSS positive subscore), negative symptoms (PANSS negative subscore) Global state (CGI‐Severity) Adverse effects: (at least one adverse effect, SAS (improved, no change, worsened), BAS (improved, no change, worsened), AIMS (improved, no change, worsened), death/suicide, BMI & weight increase, increase in heart rate, QTcF prolongation (≥450 ms), dizziness, headache, fatigue, somnolence, anxiety, dyskinesia, tremor). Functioning (SOFAS) Unable to use: Plasma levels, not a protocol outcome |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "...randomised (2:1, with a computerized schedule) to supplementation with quetiapine or with placebo. The person who generated the randomization schedule was not involved in determining subject eligibility, administering treatment, or determining outcome." (pg. 14). |
| Allocation concealment (selection bias) | Low risk | Quote: "The person who generated the randomization schedule was not involved in determining subject eligibility, administering treatment, or determining outcome." (pg. 14). |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "...double‐blind, placebo‐controlled trial..." (pg. 13); no further details. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: "...double‐blind, placebo‐controlled trial..." (pg. 13); no further details. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | 21.3% of the participants left the study early. Reasons for leaving the study early were described. An ITT approach was used. |
| Selective reporting (reporting bias) | Low risk | Free from selective reporting |
| Other bias | Low risk | No obvious risk for other bias |
Huang 1987.
| Methods | Randomisation: randomised, no further details Blinding: double, no further details Duration: 11 weeks; 2 weeks non‐randomised single‐blind run in phase and 9 weeks randomised double‐blind phase. Design: parallel Location: single‐centre (USA) Setting: inpatients | |
| Participants | Diagnosis: schizophrenia (DSM III); treatment resistant. N = 50 Gender: not indicated Age: 21 to 55 years History: mentally ill for 2 years or more; treated with 2 or more antipsychotics at usual therapeutic doses (equivalent to thiothixene 60 mg/day) for 6 months or more without appreciable remission. | |
| Interventions | All participants firstly received single‐blind thiothixene 60 mg/day for 2 weeks to confirm treatment resistance e.g. not showing moderate improvement during the first 3 weeks of treatment. Participants were then randomised to either: 1. dose increase: thiothixene up to 400 mg/day. N = 25; or 2. dose maintenance: thiothixene 60 mg/day. N = 25. Rescue medication: benztropine; no further details. |
|
| Outcomes | Global state: clinically relevant response (defined as moderate improvement in Roerig Global Scale (RGS) (Guy 1976)) Leaving the study early (due to adverse events, any reason and inefficacy) Mental state (BPRS total score, BPRS factors such as anxiety‐depression, anergia, thought disturbance, activity, hostility‐suspicion), Nurses Observation Scale for Inpatient Evaluation (NOSIE total score, NOSIE factors such as social competence, social interest, personal neatness, irritability, manifest psychosis, retardation, depression). Adverse effects (side effect check list, dystonia, dry mouth, blurred vision, drowsiness, orthostatic hypotension, tremor, dizziness, drooling, constipation, ataxia, akathisia, palpitations, headache, premature ventricular contractions). Unable to use: Vital signs (CBC, urinalysis, SMA‐12, ECG, blood pressure, pulse rate); no data were presented and not protocol outcomes. |
|
| Notes | 50 participants were randomised. Eight of the 50 participants (4 in each group) showing moderate improvement on the RGS in the first 21 days were eliminated from the study as they were not considered to be treatment‐resistant. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "...randomly assigned..." (pg. 70); no further details |
| Allocation concealment (selection bias) | Unclear risk | No details were presented |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "...under double‐blind control to..." (pg. 70); no further details |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: "...under double‐blind control to..." (pg. 70); no further details |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Unclear whether all those who left early were reported. It may be that 26% (13/50) left the study early. |
| Selective reporting (reporting bias) | High risk | No usable data on vital signs were reported in the results |
| Other bias | Low risk | No obvious risk for other bias |
Kinon 1993.
| Methods | Randomisation: randomised, stratified based on week 3 serum fluphenazine levels Blinding: double, no further details Duration: 8 weeks; 4 weeks non‐randomised open‐label run in phase and 4 weeks randomised double‐blind phase. Design: parallel Location: single‐centre (USA) Setting: inpatients | |
| Participants | Diagnosis: schizophrenia, schizoaffective disorder or schizophreniform disorder (DSM III‐R) N = 58 Gender: 64.1% males (of all participants entering the open label run in phase) (N = 156). Not indicated for randomised participants. Age: mean 29.4 years (SD = 7.0), range 18 to 50 years for all participants entering the open label run in phase (N = 156), not indicated for randomised participants. History: age at first hospitalisation ‒ mean 23.0 years (SD = 6.5); number of previous hospitalisations ‒ mean 2.6 (SD = 2.2); data for all participants entering the open label run in phase (N = 156), not indicated for randomised participants alone. | |
| Interventions | All participants firstly received open label fluphenazine 20 mg/day for 4 weeks (N = 156). Participants who had a rating of worse than mild on each of the four BPRS psychotic items and a rating of less than much improved on the CGI‐I were considered non‐responders (N = 58) and were then randomised to: 1. dose increase: Fluphenazine 80 mg/day. N = 16; or 2. dose maintenance: Fluphenazine 20 mg/day. N = 18; or 3. additional intervention: haloperidol 20 mg/day. N = 13. Rescue medication: benztropine, no further details. |
|
| Outcomes | Global state: clinically relevant response (defined as CGI‐I ≤ 2, at least much improved) Mental state: general (BRPS total score), negative symptoms (modified SANS) Unable to use: Extrapyramidal symptoms (modified SAS, no mean) |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "Non responders were then randomly assigned to double‐blind treatment for...", "Subjects were stratified based on Week 3 serum fluphenazine levels..." (pg. 310); no further details. |
| Allocation concealment (selection bias) | Unclear risk | No details were presented |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "...double‐blind treatment..." (pg.310); no further details |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: "...double‐blind treatment..." (pg.310); no further details |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Outcome not addressed. Data were presented for 81% (47/58) of all randomised participants |
| Selective reporting (reporting bias) | High risk | SAS was used but scores were available only for two items, not total |
| Other bias | Low risk | No obvious risk for other bias |
Lindenmayer 2011.
| Methods | Randomisation: randomised, no further details Blinding: double, no further details Duration: 12 weeks; 4 weeks non‐randomised open‐label run in phase and 8 weeks randomised double‐blind phase. Design: parallel Location: multi‐centre (USA) Setting: inpatients | |
| Participants | Diagnosis: schizophrenia or schizoaffective disorder (DSM‐IV‐R); suboptimal past treatment response N = 60 Gender: 55 men, 5 women Age: mean 40.15 years (SD = 10.2) History: age at onset ‒ mean 20.27 years (SD = 5.88); number of previous hospitalisations ‒ mean = 6.7 (SD = 5.88); suboptimal past treatment response, defined by (i) presence of persistent positive symptoms after at least 6 contiguous weeks of treatment with one or more typical or atypical antipsychotics at dosages ≥ 600 mg/d of chlorpromazine equivalents and (ii) poor level of functioning for the past 2 years defined as lack of competitive employment or enrolment in an academic or vocational program, and not having age‐expected interpersonal relations with someone outside the biological family of origin. | |
| Interventions | All participants firstly received open label quetiapine 600 mg/day for 4 weeks. Participants who did not demonstrate an initial response to quetiapine treatment defined as ≤ 15% PANSS total score reduction were then randomised to either: 1. dose increase: quetiapine 1200 mg/day. N = 29; or 2. dose maintenance: quetiapine 600 mg/day. N = 31. Concomitant mood stabilisers on a stable dose for the past 2 months before trial initiation were allowed to continue. |
|
| Outcomes | Global state: clinically relevant response (defined as ≥ 20% PANSS total score reduction) Leaving the study early (due to side effects, any reason, inefficacy) Mental state: general (PANSS total score), positive symptoms (PANSS positive subscore), negative symptoms (PANSS negative subscore) Adverse effects: (SAS, AIMS, BAS, BMI/weight change, orthostatic hypotension, somnolence, agitation, constipation, weight gain) Functional changes (Social‐Adaptive Functioning Evaluation ‐ SAFE) Aggressive behaviour (BARS total score at endpoint) Unable to use: Global state (CGI no numbers presented) Vital signs and ECG; no data presented and not protocol outcomes Laboratory values (ALT, AST, GGT, glukose, lactate dehydrogenase, cholesterol, triglycerides), not protocol outcomes |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "...patients were randomly assigned to...", "Stratification was done at the time of randomization to avoid imbalances secondary to the presence of concomitant mood stabilizer medications or the use of benztropine (or other antiparkinsonian agents) at baseline." (pg. 161); no further details. |
| Allocation concealment (selection bias) | Unclear risk | No details were presented |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "This was a prospective, randomised, double‐blind, parallel group, 8‐week trial of...", "All clinical and research stuff (subject, caregiver, investigator, outcomes assessor), except for the research pharmacists, were blinded to the randomization status." (pg. 161). |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "This was a prospective, randomised, double‐blind, parallel group, 8‐week trial of...", "All clinical and research stuff (subject, caregiver, investigator, outcomes assessor), except for the research pharmacists, were blinded to the randomization status." (pg. 161). |
| Incomplete outcome data (attrition bias) All outcomes | High risk | 53.3% of the participants left the study early. Numbers per group as well as reasons for leaving the study early were specified. Last observation carried forward (LOCF) methods were used. |
| Selective reporting (reporting bias) | High risk | Data on CGI, vital signs and ECG were not reported in the results. |
| Other bias | Low risk | No obvious risk for other bias |
Loebel 2014.
| Methods | Randomisation: randomised, no further details Blinding: double, no further details Duration: 6 weeks; 2 weeks double‐blind run in phase and 4 weeks randomised double‐blind phase Design: parallel Location: multi‐centre Setting: inpatients | |
| Participants | Diagnosis: acute schizophrenia, no further details N = 95 Gender: 60.1% males (of initially randomised participants to lurasidone 80 mg/day) (N = 198) Age: mean 40.5 years; range 18 to 75 years for all initially randomised participants to lurasidone 80 mg/day History: no details |
|
| Interventions | Participants were firstly randomised to double‐blind treatment with lurasidone 20 mg/day, lurasidone 80 mg/day, or placebo. After 2 weeks, only participants who were randomised to lurasidone 80 mg/day (N = 198) and showed < 20% PANSS total score reduction, were re‐randomised to either: 1. dose increase: lurasidone 160 mg/day. N = 43; or 2. dose maintenance: lurasidone 80 mg/day. N = 52. Rescue medication: no details |
|
| Outcomes | Leaving the study early: due to side effects Mental state: general (PANSS total score) Global state (CGI‐Severity change) Unable to use: Weight gain (not separately presented for the two groups) |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "Eligible patients were randomised to..." (pg. 476); no further details |
| Allocation concealment (selection bias) | Unclear risk | No details are presented |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "...to double‐blind treatment with..." (pg. 476); no further details |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: "...to double‐blind treatment with..." (pg. 476); no further details |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Outcome not addressed |
| Selective reporting (reporting bias) | Unclear risk | Insufficient information to permit judgement |
| Other bias | Low risk | No obvious risk for other bias |
McEvoy 1991.
| Methods | Randomisation: randomised, no further details Blinding: double, no further details Duration: 6 weeks; 4 weeks non‐randomised open‐label run‐in phase and 2 weeks randomised double‐blind phase. Design: parallel Location: single‐centre (USA) Setting: inpatients | |
| Participants | Diagnosis: schizophrenia or schizoaffective disorder (RDC). N = 48 Gender: 53.8% males (of all participants entering the open‐label run in phase) (N = 95). Not indicated for randomised participants. Age: mean 31.5 years (SD = 9.5) for all participants entering the open‐label run in phase, not indicated for randomised participants. History: no details |
|
| Interventions | All participants firstly received open label haloperidol at antipsychotic threshold (NT)* for at least 2 weeks. Non‐responding participants (not ready for discharge) were then randomised to either: 1. dose increase: haloperidol at a dosage two to ten times higher than NT dosage; mean 11.6 mg/day (SD = 4.7). N = 25; or 2. dose maintenance: haloperidol at NT dosage; mean 3.4 mg/day (SD = 2.3). N = 23. Rescue medication: biperiden, lorazepam, diphenhydramine, no further details |
|
| Outcomes | Global state: clinically relevant response (defined as BPRS total score ≤ 32 (16 items), with all psychosis items (conceptual disorganisation, hallucinatory behavior, hostility, suspiciousness. and unusual thought content) rated "mild" or less; their CGl global severity item was rated "mild" or less; and their CGI global change item was rated at least "moderately improved"). Leaving the study early: due to adverse events Mental sate: general (BRPS total score) Global state (CGI‐S) Unable to use: Negative symptoms: Wing negative symptoms scale (no mean, no SD) Mental state; self‐report of perceived medication effects: Medication Response Questionnaire (no mean, no SD) Adverse events: Extrapyramidal Side Effects Scale (no mean, no SD) and anticholonergic side effects (no numbers presented) |
|
| Notes | * Antipsychotic threshold (NT): the lowest antipsychotic dosage at which individual patients develop slight increase in rigidity is hypothesised to correspond to the lowest dosages at which these patients attain maximum antipsychotic benefit (Haase 1961). ** Response criterion: BPRS total score ≤ 32 (16 items), with all psychosis items (conceptual disorganisation, hallucinatory behaviour, hostility, suspiciousness. and unusual thought content) rated "mild" or less; their CGl global severity item was rated "mild" or less; and their CGI global change item was rated at least "moderately improved". |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "...patients were randomly assigned..." (pg. 740); no further details |
| Allocation concealment (selection bias) | Unclear risk | No details are presented |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "...on a double‐blind basis..." (pg. 740); no further details |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: "...on a double‐blind basis..." (pg. 740); no further details |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | An ITT approach was used. 8.3% (4/48) of participants left the study early due to severe extrapyramidal side effects. This is probably the overall attrition rate. |
| Selective reporting (reporting bias) | High risk | Wing negative symptom scale, Medication Response Questionnaire, Extrapyramidal Side Effects Scale and anticholinergic side effects were not presented separately for the group of non‐responders |
| Other bias | Low risk | No obvious risk for other bias |
McGorry 2011.
| Methods | Randomisation: randomised, no further details Blinding: single Duration: 8 weeks; 4 weeks non‐randomised open‐label run in phase and 4 weeks randomised single‐blind phase Design: parallel Location: single‐centre (Australia) Setting: inpatients | |
| Participants | Diagnosis: schizophrenia, schizophreniform, schizoaffective, delusional disorder, psychotic disorder not otherwise specified, or brief psychosis (DSM‐IV); first psychotic episode N = 26 Gender: 18 men, 8 women Age: mean 21.62 years (SD = 3.98) History: age at onset ‒ mean 21 years (SD = 3.8); duration of untreated psychosis ‒ mean 267.1 days (SD = 416.5); a first episode of psychosis | |
| Interventions | All participants firstly received open label risperidone 2 mg/day for 4 weeks. Participants who were considered 'slow responders' defined as a score of > 3 on each of the BPRS psychosis subscale items (i.e. mild), a CGI‐S > 3, and a CGI‐I > 3 were then randomised to: 1. dose increase: risperidone 3 or 4 mg/day (if required). N = 9; or 2. dose maintenance: risperidone 2 mg/day. N = 8; or 3. additional intervention: risperidone 2 mg/day with addition of lithium, titrated up to therapeutic levels (0.6 to 1.2 mmol). N = 9. |
|
| Outcomes | Global state: clinically relevant response (defined as ≥ 20% BPRS total score reduction)
Leaving the study early (due to adverse events and any reason)
Mental state: general (BPRS total score), positive symptoms (BPRS psychosis subscale), negative symptoms (BPRS negative subscale)
Global state (CGI‐S)
Quality of life: number of participants with at least 50% improvement inHQLS and mean endpoint score in HQLS
Unable to use: Global state (CGI‐I, no numbers) Extrapyramidal adverse events (not separately presented for the group of 'slow responders') and weight gain (no SD). |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "...slow responders were randomised..." (pg. 3); no further details |
| Allocation concealment (selection bias) | Unclear risk | No details are presented |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Quote: "...randomised single blind to one of three open treatment groups" (pg. 3); no further details |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: "...randomised single blind to one of three open treatment groups" (pg. 3); no further details |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | 19.2% (5/26) participants left the study early. A LOCF approach was adopted for analysis of Phase II data |
| Selective reporting (reporting bias) | High risk | Original study author sent us data on most outcomes but data on CGI‐I, extrapyramidal side effects and weight gain were not reported. |
| Other bias | Low risk | No obvious risk for other bias |
Sakurai 2016.
| Methods | Randomisation: randomised to 1 of the 2 treatment groups in a 1:1 ratio by simple randomisation stratified by their antipsychotic type and treatment setting; computer‐generated randomisation list. Blinding: double; identical powder form in amount and color; participants blinded to their allocated intervention; assessors blinded to the allocation. Duration: 4 weeks Design: parallel Location: single‐centre (Japan) Setting: inpatients and outpatients | |
| Participants | Diagnosis: schizophrenia, schizoaffective or persistent delusional disorder (ICD‐10) N = 103 Gender: 38 men, 65 women Age: mean 50.7 years (SD = 15.81) History: duration of illness ‒ mean 16.05 years (SD = 14.4); total duration of antipsychotic treatment‐ mean 10.9 years (SD = 14). |
|
| Interventions | All participants had been receiving olanzapine 10 mg/day or risperidone 3 mg/day for at least 4 weeks. Participants who had a total score ≥ 60 on the PANSS, ≥ 3 on the CGI‐S, and ≤ 70 on the GAF were considered non‐responders and were randomised to either: 1. dose increase: olanzapine 20 mg/day or risperidone 6 mg/day (double antipsychotic dose). N = 52; or 2. dose maintenance: olanzapine 10 mg/day or risperidone 3 mg/day. N = 51. |
|
| Outcomes | Global state: clinically relevant response (defined as ≥ 25% PANSS total score reduction)
Leaving the study early (due to adverse events, any reason and inefficacy) Mental state: general (PANSS total score), positive symptoms (PANSS positive subscore), negative symptoms (PANSS negative subscore) Global state (CGI‐I) Functioning: overall (GAF) Adverse effects (EPS ‐ SAS, Akathisia ‐ BAS, tardive dyskinesia ‐ AIMS) Unable to use: Global state: CGI‐S (no mean, no SD) Plasma concentrations, not a protocol outcome |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "...randomly allocated to 1 of the 2 treatment groups in a 1:1 ratio by simple randomization stratified by their antipsychotic type (ie, olanzapine or risperidone) and treatment setting (ie, inpatient or outpatient)....according to a computer‐generated randomization list..." (pg. 1382). |
| Allocation concealment (selection bias) | Low risk | Quote: "The person who was independent of this study in the central office prepared a piece of paper on which 1 of the assigned groups was designated according to a computer‐generated randomization list, inserted it into an envelope on which a participant ID number was written, and sealed it. Upon registration of each participant, 1 of the investigators opened the envelope that corresponded to the participant's ID, and the person who prepared the envelopes confirmed that the envelopes were appropriately opened." (pg. 1382). |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "During the 4‐week observation, all antipsychotic drugs were provided in identical powder form in amount and color with lactose added... Thus, the participants were blinded to their allocated intervention." (pg. 1382). |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "The following assessments were performed by assessors who were blinded to the allocation..." (pg. 1383). |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Significantly more participants in the dose increase group (30.8%) than in the dose continuation group (13.7%) left the study early due to side effects. Reasons for leaving the study early were described. An ITT approach was used. Results between ITT analysis and only competers were similar. |
| Selective reporting (reporting bias) | High risk | CGI‐S was used but scores at endpoint were not available |
| Other bias | Low risk | No obvious risk for other bias |
Scales
AIMS: Abnormal Involuntary Movement Score BAS: Barnes Akathisia Scale
BARS: Behavioral Activity Rating Scale
BAS: Barnes Akathisia Rating Scale
BPRS: Brief Psychiatric Rating Scale
CDRS: Calgary Depression Rating Scale
CGI‐I: Clinical Global impression‐Improvement
CGI‐S: Clinical Global impression‐Severity
CPRS: Comprehensive Psychopathological Rating Scale
GAF: Global Assessment of Functioning
HQLS: Heinrichs‐Carpenter‐Hanlon Quality of Life Scale
NOSIE: Nurse's Observation Scale for Inpatient Evaluation
PANSS: Positive and Negative Syndrome Scale for Schizophrenia
RGS: Roering Global Scale
SAFE: Social‐Adaptive Functioning Evaluation
SANS: Scale for the Assessment of Negative Symptoms
SAS: Simpson Angus Scale
SCoRS: Schizophrenia Cognition Rating Scale
SOFAS: Social and Occupational Functioning Scale
Diagnostic Tools
DSM: Diagnostic and Statistical Manual of Mental Disorders
ICD: International Classification of Diseases
RDC: Research Diagnostic Criteria for schizophrenia or schizoaffective disorders
Others
BMI: Body‐mass‐index
ECG: Electrocardiogram
EPS: Extrapyramidal Symptoms
ITT: Intention‐to‐treat
LOCF: Last observation carried forward
mg: Milligram
msec: Millisecond
N: Number
n.i.: Not indicated
SD: Standard deviation
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Agid 2013 | Allocation: not randomised |
| Badgett 1996 | Allocation: randomised Participants: schizophrenia (DSM‐III‐R) based on the CASH‐interview; at baseline, participants were stabilised on 20 mg/day of haloperidol, not non‐responders |
| Bai 2002 | Allocation: not randomised |
| Baker 2003 | Allocation: randomised Participants: schizophrenia, schizoaffective disorder, schizophreniform disorder, or bipolar I disorder, manic or mixed episode (DSM‐IV); no run‐in phase to confirm that participants have not responded to their current antipsychotic treatment. Interventions: rapid dose escalation or usual clinical practice |
| Bastecky 1982 | Allocation: not indicated Participants: schizophrenia (chronic schizophrenics); no run‐in phase to confirm that participants have not responded to their current antipsychotic treatment |
| Bitter 1989 | Allocation: randomised Participants: schizophrenia or schizoaffective disorder (acutely exacerbating) Interventions: initially 3 plasma levels of haloperidol; if no improvement after 6 weeks, randomly re‐assignment to one of the 3 plasma levels of haloperidol for another 6 weeks. Outcomes: no usable data. |
| Bondolfi 1995 | Allocation: randomised Participants: schizophrenia (chronic according to DSM‐III), treatment‐resistant (defined by unresponsiveness or intolerance to appropriate doses of two different classes of conventional antipsychotics for at least 4 weeks each); no run‐in phase to confirm that participants have not responded to their current antipsychotic treatment. Interventions: no antipsychotic dose increase versus maintenance Comparison; either risperidone or clozapine; drug dosages could be changed after day 14 depending on each participant's response. |
| Branchey 1981 | Allocation: randomised Participants: schizophrenia, chronic type (according to Research Diagnostic Criteria); no run‐in phase to confirm that participants have not responded to their current antipsychotic treatment. Interventions: loxapine continuation versus loxapine decrease; no dose increase group |
| Canuso 2010 | Allocation: randomised Participants: acute exacerbation of schizoaffective disorder (DSM‐IV); no run‐in phase to confirm that participants have not responded to their current antipsychotic treatment. |
| Chen 1998 | Allocation: not indicated Participants: no run‐in phase to confirm that participants have not responded to their current antipsychotic treatment |
| Clerc 1989 | Allocation: distributed by drawing of lots Participants: hospitalised participants all with negative symptoms (ICD‐9 and DSM‐III); no run‐in phase to confirm that participants have not responded to their current antipsychotic treatment. Interventions: 2 ranges of amisulpride doses (20 mg to 120 mg and 100 mg to 600 mg); no dose continuation group |
| CN138032 | Allocation: randomised Participants: treatment‐resistant schizophrenia Interventions: 1. open‐label phase olanzapine or risperidone; 2. single‐blind placebo washout period; 3. double‐blind phase perphenazine vs. aripiprazole; 4. open‐label aripiprazole extended phase. |
| Cookson 1987 | Allocation: randomised Participants: schizophrenia (hebephrenic or paranoid, ICD‐9); treatment‐resistant to low dosages of antipsychotics; improved with higher dosages. Interventions: 50% reduction of dosage or dosage continuation; no dose increase group |
| Coryell 1998 | Allocation: randomised Participants: schizophrenia, acute exacerbation (DSM‐III) Interventions: 1st phase: low‐dose (< 18ng/ml), intermediate or high‐dose (> 25 ng/ml) range of haloperidol plasma levels for 3 weeks; 2nd phase: if no improvement, antipsychotic dose could randomly remain the same, be reduced or increased. |
| Daniel 1997 | Allocation: not indicated Participants: schizophrenia or schizoaffective disorder; no run‐in phase to confirm that participants have not responded to their current antipsychotic treatment. Interventions: ziprasidone 80 mg vs. ziprasidone 160 mg; no dose maintenance group |
| de Leon 2007 | Allocation: randomised Participants: schizophrenia or schizoaffective disorder (DSM‐III); Kane et al criteria (Kane 1988) for defining treatment resistance. Interventions: 1st phase: 16 weeks in one of the following dose‐groups: 100, 300, 600 mg/day clozapine; if no improvement, randomization to one of the two other dosage groups; no dose maintenance group. |
| DeBuck 1972 | Allocation: not randomised for non‐responders Participants: any psychoses with the exception of chronic organic psychosis with superimposed non‐psychotic symptomatology, and endogenous depression with retardation and apathy, and "acute organic psychosis". |
| Dencker 1978 | Allocation: randomised
Participants: schizophrenia; all participants firstly received open‐label 12.5 mg fluphenazine enanthate per week for 2 weeks and those who were proven "refractory to ordinary doses of neuroleptics" were then randomised. Interventions: 250 mg fluphenazine enanthate per week versus 12.5 mg fluphenazine enanthate per week. Outcomes: no usable data due to the cross‐over design of the trial. |
| Ericksen 1978 | Allocation: randomised Participants: schizophrenia (acutely decompensated); no run‐in phase to confirm that participants have not responded to their current antipsychotic treatment. Interventions: haloperidol standard dose versus haloperidol high loading dose; no dose maintenance group. |
| Gardos 1971 | Allocation: not indicated Participants: schizophrenia (treatment resistant, decompensated); no run‐in phase to confirm that participants have not responded to their current antipsychotic treatment. Interventions: thiothixene high dose versus thiothixene low dose Outcomes: no usable data |
| Gulliver 2010 | Allocation: randomised Participants: schizophrenia, participants who maintained stability on open‐label oral olanzapine Interventions: randomised to low, medium or high doses of olanzapine long‐acting injection for 24 weeks |
| Harris 1997 | Allocation: not indicated Participants: people with schizophrenia and related psychotic disorders over the age of 45 who met DSM‐IV criteria for "in remission''; not non‐responders. |
| Hirschowitz 1995 | Allocation: not indicated Participants: participants with schizophrenia, stabilised on 20 mg/day; not non‐responders. |
| Hirschowitz 1997 | Allocation: randomised Participants: schizophrenia (DSM‐III‐R) based on the CASH‐interview; participants free of medication at baseline or stabilised on 20 mg/day. Interventions: no dose continuation or increase group |
| Itil 1970 | Allocation: not indicated. Participants: schizophrenia (chronic); no run‐in phase to confirm that participants have not responded to their current antipsychotic treatment. Interventions: fluphenazine 30 mg versus fluphenazine 800 mg; no dose maintenance or increase group. |
| Janicak 1997 | Allocation: randomised. Participants: schizophrenia or schizoaffective disorder; acutely psychotic. Interventions: low, middle or high plasma level range of haloperidol; after 2 weeks, 50% of the non‐responders were randomly reassigned to the middle plasma level group. |
| Kane 1985 | Allocation: not indicated. Participants: schizophrenia (remitted or stable); no run‐in phase to confirm that participants have not responded to their current antipsychotic treatment. Interventions: low‐dose fluphenazine decanoate versus standard‐dose fluphenazine decanoate; no dose increase group. |
| Lehmann 1980 | Allocation: randomised. Participants: schizophrenia; no run‐in phase to confirm that participants have not responded to their current antipsychotic treatment. Interventions: haloperidol 10 mg/day, 20 mg/day or individual dose of haloperidol; no dose increase or maintenance group. |
| McCreadie 1979 | Allocation: randomised Participants: schizophrenia (female, drug‐resistant, chronic); no run‐in phase to confirm that participants have not responded to their current antipsychotic treatment. Interventions: flupentixol decanoate high‐dose (200 mg/2 weeks) versus standard dose (40 mg/2 weeks); no dose increase or maintenance group. |
| Mitchell 2004 | Allocation: randomised Participants: schizophrenia, schizoaffective disorder, bipolar I disorder (DSM‐IV‐R); stable inpatients; the run‐in phase was not used to identify participants that have not responded to their current antipsychotic treatment. Interventions: run‐in phase of olanzapine 20 mg/day for 10 days; then, randomisation to olanzapine 20 mg/day, 30 mg/day or 40 mg/day for 10 days; finally, for an additional 10 days, 30 mg/day participants received olanzapine 40 mg/day; all other participants remained on the same dose. Outcomes: no usable data |
| NCT00539071 | Allocation: randomised Participants: schizophrenia or schizoaffective disorder; eligible participants would be receiving or had received treatment with risperidone oral or long‐acting, or a combination that did not exceed 50 mg/2 weeks of long‐acting or oral risperidone 8 mg/day for at least 6 weeks within seven years of study entry without satisfactory response. Interventions: long‐acting risperidone 50 mg/week vs 75 mg to 100 mg/2 weeks. Outcomes: no usable data |
| NCT00862992 | Allocation: randomised Participants: schizophrenia (DSM‐IV‐TR); no run‐in phase to confirm that participants have not responded to their current antipsychotic treatment. Interventions: low‐dose versus medium‐dose versus high‐dose of cariprazine; no dose increase or maintenance group. |
| NCT01457339 | Allocation: randomised Participants: clinically stable participants with schizophrenia (on a stable dose of an antipsychotic drug); no run‐in phase to confirm that participants have not responded to their current antipsychotic treatment. Interventions: SPD489 multiple doses versus placebo; no dose increase or maintenance group. |
| NCT01569659 | Allocation: randomised Participants: schizophrenia or schizoaffective disorder (DSM‐IV); Kane et al criteria (Kane 1988) for treatment‐resistance; no run‐in phase to confirm that participants have not responded to their current antipsychotic treatment. Interventions: low dosage of lurasidone (80 mg/day) versus high dose of lurasidone (up to 240 mg/day); no dose increase or maintenance group. |
| Simpson 1999 | Allocation: randomised Participants: any psychoses with the exception of chronic organic psychosis with superimposed non‐psychotic symptomatology, endogenous depression and acute organic psychosis. Interventions: clozapine 100, 300 or 600 mg/d; if no improvement was observed after 16 weeks, participants were randomised to one of the other two dosages, no maintenance group. |
| Suzuki 1992 | Allocation: randomised Participants: schizophrenia (DSM‐III); not non‐responders |
| Volavka 1996 | Allocation: not randomised for the group of non‐responders |
Differences between protocol and review
In the original protocol, we stated that we would apply specific standards to potential skewness of data before inclusion to avoid the pitfall of applying parametric tests to non‐parametric data. However, excluding studies would also lead to loss of valuable information and bias.
In addition, although not stated in the original protocol, we decided to extract and present all available measures of efficacy such as continuous measures of global state, e.g. mean CGI‐Severity (endpoint and change) and mean CGI‐Improvement, and measures of functioning, e.g. GAF, SAFE, and SOFAS.
We have renamed outcomes from 'Clinically significant response' to 'Clinically important change'.
We have now specified 'Summary of findings' table outcomes should be 'Clinically important change' but if data were not available for these pre‐specified outcomes but were available for ones that are similar, we presented the closest outcome to the pre‐specified one in the table but took this into account when grading the finding.
We have updated the Methods template to the latest version provided by Cochrane Schizophrenia. This does not involve a change to the methods but updating of references and rewording of some sections.
We have reworded some of the background to harmonise this review with its 'sibling review' Samara 2015b.
Contributions of authors
Myrto Samara: protocol development, study selection, data extraction, writing the report.
Elisabeth Klupp: study selection, data extraction.
Bartosz Helfer: protocol development.
Philipp Rothe: protocol development.
Johannes Schneider‐Thoma: writing of a summary report for the review.
Stefan Leucht: protocol development, study selection, data extraction, writing the report.
Sources of support
Internal sources
Freistaat Bayern, Germany.
External sources
German Federal Ministry of Education and Research (Bundesministerium für Bildung und Forschung, BMBF) Grant number: 01KG1407, Germany.
Declarations of interest
Myrto Samara ‒ nothing to declare.
Elisabeth Klupp ‒ nothing to declare.
Bartosz Helfer ‒ nothing to declare.
Philipp Rothe ‒ nothing to declare.
Johannes Schneider‐Thoma ‒ nothing to declare.
Stefan Leucht ‐ has received honoraria for consulting from LB Pharma, Lundbeck, Otsuka, TEVA, Geodon Richter, Recordati, LTS Lohmann, and Boehringer Ingelheim; and for lectures from Janssen, Lilly, Lundbeck, Otsuka, SanofiAventis, and Servier
New
References
References to studies included in this review
Bjørndal 1980 {published data only}
- Bjørndal N, Bjerre M, Gerlach J, Kristjansen P, Magelund G, Oestrich IH, et al. High dosage haloperidol therapy in chronic schizophrenic patients: a double‐blind study of clinical response, side effects, serum haloperidol, and serum prolactin. Psychopharmacology 1980;67(1):17‐23. [DOI] [PubMed] [Google Scholar]
Goff 2013 {published data only}
- Goff DC, McEvoy JC, Citrome L, Mech AW, Bustillo JR, Gil R, et al. High‐dose oral ziprasidone versus conventional dosing in schizophrenia patiens with residual symptoms: The ZEBRAS study. Journal of Clinical Psychopharmacology 2013;33(4):485‐90. [DOI] [PubMed] [Google Scholar]
- NCT00403546. High dose oral ziprasidone versus conventional dosing in schizophrenia patients with residual symptoms. clinicaltrials.gov/show/NCT00403546 (accessed 5 December 2015).
Honer 2011 {published data only}
- AstraZeneca. A Canadian, multicentre, double‐blind, randomized, parallel‐group study of the safety, tolerability, and efficacy of treatment with higher doses of quetiapine fumarate (Seroquel) greater than 800mg/day in schizophrenic or schizoaffective subjects. Clinical Study Report CNS.000‐105‐629 30 November 2006.
- Honer WG, MacEwan GW, Gendron A, Stip E, Labelle A, Williams R, et al. A randomized, double‐blind, placebo‐controlled study of the safety and tolerability of high‐dose quetiapine in patients with persistent symptoms of schizophrenia or schizoaffective disorder. Journal of Clinical Psychiatry 2012;73(1):13‐20. [DOI] [PubMed] [Google Scholar]
- Honer WG, MacEwan W, Gendron A, Stip E, Labelle A, Williams R, et al. A double‐blind, placebo‐controlled study of the safety and tolerability of quetiapine 1200mg/d versus 800mg/d in patients with persistent symptoms of schizophrenia or schizoaffective disorder. Schizophrenia Research 2010;117(2‐3):375. [Google Scholar]
- NCT00328978. A Canadian, multicenter, double‐blind, randomized, parallel‐group studyof the safety, tolerability, and efficacy of treatment with higher doses of quetiapine fumarate (Seroquel) greater than 800mg/day in schizophrenic or schizoafective subjects. clinicaltrials.gov/show/NCT00328978 (accessed 5 December 2015).
Huang 1987 {published data only}
- Huang CC, Gerhardstein RP, Kim DY, Hollister L. Treatment‐resistant schizophrenia ‐ controlled study of moderate and high dose thiothixene. International Clinical Psychopharmacology 1987;2(1):69‐75. [DOI] [PubMed] [Google Scholar]
Kinon 1993 {published data only}
- Johns CA, Mayerhoff DI, Lieberman JA, Kane JM. Schizophrenia: alternative neuroleptic strategies. In: Angrist B, Schulz SC editor(s). The Neuroleptic‐Nonresponsive Patient: Characterization and Treatment. Washington, DC, USA: American Psychiatric Press, Inc, 1990:53‐66. [Google Scholar]
- Kinon BJ, Kane JM, Johns C, Perovic R, Ismi M, Koreen A, et al. Treatment of neuroleptic‐resistant schizophrenic relapse. Psychopharmacology Bulletin 1993;29(2):309‐14. [PubMed] [Google Scholar]
Lindenmayer 2011 {published data only}
- Lindenmayer J‐P, Citrome L, Khan A, Kaushik S, Kaushik S. A randomized, double‐blind, parallel‐group, fixed‐dose, clinical trial of quetiapine at 600 versus 1200mg/d for patients with treatment‐resistant schizophrenia or schizoaffective disorder. Journal of Clinical Psychopharmacology 2011;31(April):160‐8. [DOI] [PubMed] [Google Scholar]
- NCT00297947. A randomized, double‐blind, parallel‐group, fixed dose, clinical trial of quetiapine 600mg/day vs 1200mg/day for patients with treatment‐resistant schizophrenia or schizoaffective disorder. clinicaltrials.gov/show/NCT00297947 2006 (accessed 6 December 2015).
Loebel 2014 {published data only}
- Loebel A, Silva R, Goldman R, Watabe K, Cucchiaro J, Kane J. Optimizing Treatment with Lurasidone in Patients with Schizophrenia: Results of a Randomized, Double‐blind, Placebo‐controlled Trial (OPTIMIZE Trial). Neuropsychopharmacology 2014;39(Suppl 1):S473–S647. [Google Scholar]
- Loebel A, Silva R, Goldman R, Watabe K, Pikalov A, Cucchiaro J, et al. Optimizing treatment with lurasidone in patients with schizophrenia: Results of a randomized, double‐blind, placebo‐controlled trial. European Psychiatry 2015;30(Suppl 1):1722. [Google Scholar]
McEvoy 1991 {published data only}
- McEvoy JP, Hogarty GE, Steingard S. Optimal dose of neuroleptic in acute schizophrenia. A controlled study of the neuroleptic threshold and higher haloperidol dose. Archives of General Psychiatry 1991;48(8):739‐45. [DOI] [PubMed] [Google Scholar]
McGorry 2011 {published data only}
- McGorry PD, Cocks J, Power P, Burnett P, Harrigan S, Lambert T. Very low‐dose risperidone in first‐episode psychosis: a safe and effective way to initiate treatment. Schizophrenia Research and Treatment 2011, issue 2011:631690. [DOI: 10.1155/2011/631690] [DOI] [PMC free article] [PubMed]
Sakurai 2016 {published data only}
- Sakurai H, Suzuki T, Bies RR, Pollock BG, Mimura M, Kapur S, et al. Increasing Versus Maintaining the Dose of Olanzapine or Risperidone in Schizophrenia Patients Who Did Not Respond to a Modest Dosage: A Double‐Blind Randomized Controlled Trial. Journal of Clinical Psychiatry 2016;77(10):1381‐90. [DOI] [PubMed] [Google Scholar]
References to studies excluded from this review
Agid 2013 {published data only}
- Agid O, Schulze L, Arenovich T, Sajeev G, McDonald K, Foussias G, et al. Antipsychotic response in first‐episode schizophrenia: efficacy of high doses and switching. European Neuropsychopharmacology 2013;23(9):1017‐22. [DOI] [PubMed] [Google Scholar]
Badgett 1996 {published data only}
- Badgett S, Hitzemann RJ, Burr G, Piscani K, Frecska E, Hirschowitz J. Dose reduction in schizophrenia. Proceedings of the 149th Annual Meeting of the American Psychiatric Association. New York, USA, 1996 May 4‐9.
Bai 2002 {published data only}
- Bai YM, Lin CC, Yu SC. Risperidone for severe tardive dyskinesia: one year follow up study. International Journal of Neuropsychopharmacology 2002;5(Suppl 1):S165. [Google Scholar]
Baker 2003 {published data only}
- Baker RW, Kinon BJ, Maguire GA, Liu H, Hill AL. Effectiveness of rapid initial dose escalation of up to forty milligrams per day of oral olanzapine in acute agitation. Journal of Clinical Psychopharmacology 2003;23(4):342‐8. [DOI] [PubMed] [Google Scholar]
Bastecky 1982 {published data only}
- Bastecky J, Kalvach Z, Halkova E. Longterm application of high and medium doses of clorotepin to chronic schizophrenics: an open comparative study. Activitas Nervosa Superior 1982;24(4):230‐2. [Google Scholar]
Bitter 1989 {published data only}
- Bitter I, Volavka J, Cooper J, Scheurer, Camus L, Bakall R. Haloperidol blood levels and clinical effects in schizophrenia. Psychiatry Today: Accomplishments and Promises. Excerpta Medica International Congress Series 1989;899:350. [Google Scholar]
Bondolfi 1995 {published data only}
- Bondolfi G, Baumann P, Patris M, May J, Billeter U, Dufour H, et al. A randomized double‐bind trial of risperidone versus clozapine for treatment‐resistant chronic schizophrenia. European Neuropsychopharmacology 1995;5(3):349. [Google Scholar]
Branchey 1981 {published data only}
- Branchey MH, Branchey LB, Richardson MA. Effects of neuroleptic adjustment on clinical condition and tardive dyskinesia in schizophrenic patients. American Journal of Psychiatry 1981;138(5):608‐12. [DOI] [PubMed] [Google Scholar]
- Branchey MH, Branehey LB, Richardson MA. Effects of gradual decrease and discontinuation of neuroleptics on clinical condition and tardive dyskinesia. Psychopharmacology Bulletin 1981;17(1):118‐20. [PubMed] [Google Scholar]
Canuso 2010 {published data only}
- Canuso CM, Lindenmayer JP, Kosik‐Gonzalez C, Turkoz I, Carothers J, Bossie CA, et al. A randomized, double‐blind, placebo‐controlled study of 2 dose ranges of paliperidone extended‐release in the treatment of subjects with schizoaffective disorder. Journal of Clinical Psychiatry 2010;71(5):587‐98. [DOI] [PubMed] [Google Scholar]
Chen 1998 {published data only}
- Chen YG, Zhao JP, Xie G. A study on serum concentration and clinical response of clozapine with different dose administration for treatment of schizophrenia. Chinese Journal of Psychiatry 1998;31(2):104‐7. [Google Scholar]
Clerc 1989 {published data only}
- Clerc G. Double‐blind study of amisulpride at different dosages in negative schizophrenic patients. In: Borenstein P, Boyer P, Braconnier A, Carnoy P, Clerc G, Costa e Silva JA, et al. editor(s). Amisulpride. Paris: Expansion Scientifique Francaise, 1989:105‐10. [Google Scholar]
CN138032 {published data only}
- Bristol‐Myers Squibb. A multicenter, randomized, double‐blind study of flexible doses of aripiprazole versus perphenazine in the treatment of patients with treatment‐resistant schizophrenia [Clinical Study Report CN138032]. ctr.bms.com/pdf//CN138‐032%20ST.pdf (accessed 7 December 2015).
Cookson 1987 {published data only}
- Cookson IB. The effects of a 50% reduction of cis(z)‐flupenthixol decanoate in chronic schizophrenic patients maintained on a high dose regime. International Clinical Psychopharmacology 1987;2(2):141‐9. [DOI] [PubMed] [Google Scholar]
Coryell 1998 {published data only}
- Coryell W, Miller DD, Perry PJ. Haloperidol plasma levels and dose optimization. American Journal of Psychiatry 1998;155(1):48‐53. [DOI] [PubMed] [Google Scholar]
Daniel 1997 {published data only}
- Daniel D, Reeves K, Harrigan EP. The efficacy and safety of ziprasidone 80 mg‐day and 160 mg‐day in schizophrenia and schizoaffective disorder. Schizophrenia Research 1997;24(1‐2):204. [Google Scholar]
DeBuck 1972 {published data only}
- DeBuck RP. Relative safety and efficacy of high and low dose administration of fluphenazine‐hcl to psychotic patients. Proceedings of the 8th International Congress of the C.I.N.P. Copenhagen, Denmark, 1972:265‐72.
de Leon 2007 {published data only}
- Leon J, Diaz FJ, Josiassen RC, Cooper TB, Simpson GM. Weight gain during a double‐blind multidosage clozapine study. Journal of Clinical Psychopharmacology 2007;27(1):22‐7. [DOI] [PubMed] [Google Scholar]
Dencker 1978 {published data only}
- Dencker SJ, Johansson R, Lundin L, Malm U. High doses of fluphenazine enanthate in schizophrenia. A controlled study. Acta Psychiatrica Scandinavica 1978;57(5):405‐14. [DOI] [PubMed] [Google Scholar]
Ericksen 1978 {published data only}
- Ericksen SE, Hurt SW, Chang S. Haloperidol dose, plasma levels, and clinical response: a double‐blind study. Psychopharmacology Bulletin 1978;14(2):15‐6. [PubMed] [Google Scholar]
Gardos 1971 {published data only}
- Gardos G, Cole JO, Orzack MH, Weinberger E, McCraith D. Differential effects of high and low doses of thiothixene in schizophrenics. Proceedings of the 5th World Congress of Psychiatry. Ciudad de Mexico, Mexico, 1971:306‐7.
Gulliver 2010 {published data only}
- Gulliver A, Sun B, Karagianis JL, Zhao F, Watson SB, McDonnell DP. Dose‐associated changes in safety and efficacy parameters observed in a 24‐week maintenance trial of olanzapine long‐acting injection in patients with schizophrenia. Proceedings of the 163rd Annual Meeting of the American Psychiatric Association. New Orleans, LA, 2010.
Harris 1997 {published data only}
- Harris MJ, Paulsen JS, Lacro JoP, Rockwell E, Jeste DV. Neuroleptic maintenance treatment in the elderly. Proceedings of the 150th Annual Meeting of the American Psychiatric Association; 1997 May 17‐22; San Diego, California, USA. 1997.
Hirschowitz 1995 {published data only}
- Hirschowitz J, Hitzemann R, Curtis C, Piscani K. Dose reduction in schizophrenia. Proceedings of the 34th Annual Meeting of the American College of Neuropsychopharmacology; 1995 Dec 11‐15; San Juan, Puerto Rico. 1995.
Hirschowitz 1997 {published data only}
- Hirschowitz J, Hitzemann R, Piscani K, Burr G, Frecska E, Culliton D, et al. The Dose Reduction in Schizophrenia (DORIS) study: a final report. Schizophrenia Research 1997;23(1):31‐43. [DOI] [PubMed] [Google Scholar]
Itil 1970 {published data only}
- Itil T. Fluphenazine ‐ Hi versus Lo dose. Psychopharmacology Bulletin 1970;7(2):52‐5. [Google Scholar]
- Itil T, Keskiner A, Heinemann L, Han T, Gannon P, Hsu W. Treatment of resistant schizophrenics with extreme high dosage fluphenazine hydrochloride. Psychosomatics 1970;11(5):456‐63. [DOI] [PubMed] [Google Scholar]
Janicak 1997 {published data only}
- Janicak PG, Javaid JI, Sharma RP, Leach A, Dowd S, Davis JM. A two‐phase, double‐blind randomized study of three haloperidol plasma levels for acute psychosis with reassignment of initial non‐responders. Acta Psychiatrica Scandinavica 1997;95(4):343‐50. [DOI] [PubMed] [Google Scholar]
Kane 1985 {published data only}
- Kane JM, Rifkin A, Woerner M, Reardon G, Kreisman D, Blumenthal R, et al. High‐dose versus low‐dose strategies in the treatment of schizophrenia. Psychopharmacology Bulletin 1985;21(3):533‐7. [PubMed] [Google Scholar]
Lehmann 1980 {published data only}
- Lehmann E, Quadbeck H, Tegeler J, Fararuni M, Heinrich K. Drug‐response differences of high and standard dosage of fluphenazine‐decanoate in relation to schizophrenic symptoms. Pharmakopsychiatrie und Neuropsychopharmakologie 1980;13(3):117‐29. [DOI] [PubMed] [Google Scholar]
McCreadie 1979 {published data only}
- McCreadie RG, Flanagan WL, McKnight J, Jorgensen A. High dose flupenthixol decanoate in chronic schizophrenia. British Journal of Psychiatry 1979;135:175‐9. [DOI] [PubMed] [Google Scholar]
Mitchell 2004 {published data only}
- Mitchell M, Earley W, Bari M, Riesenberg R, Marquez E, Kurtz D, et al. Preliminary pharmacokinetic and tolerability profiles of olanzapine 20, 30, and 40 mg/day. Schizophrenia Research 2004;67(1):164. [Google Scholar]
- Mitchell M, Riesenberg R, Bari MA, Marquez E, Kurtz D, Falk D, et al. A double‐blind, randomized trial to evaluate the pharmacokinetics and tolerability of 30 or 40 mg/d oral olanzapine relative to 20 mg/d oral olanzapine in stable psychiatric subjects. Clinical Therapeutics 2006;28(6):881‐92. [DOI] [PubMed] [Google Scholar]
NCT00539071 {published data only}
- NCT00539071. High dose risperidone consta for patients with schizophrenia with unsatisfactory response to standard dose risperidone or long‐acting injectable. clinicaltrials.gov/ct2/show/NCT00539071 (accessed 6 December 2015).
NCT00862992 {published data only}
- NCT00862992. Safety, pharmacokinetics and efficacy study of MP‐214 in patients with schizophrenia. clinicaltrials.gov/ct2/show/NCT00862992 (accessed 8 December 2015).
NCT01457339 {published data only}
- NCT01457339. Assess the safety and pharmacokinetics of ascending, multiple oral doses of spd489 in adults with schizophrenia. clinicaltrials.gov/ct2/show/NCT01457339 (accessed 8 December 2015).
NCT01569659 {published data only}
- NCT01569659. High dose lurasidone for patients with treatment resistant schizophrenia. clinicaltrials.gov/ct2/show/NCT01569659 (accessed 8 December 2015).
Simpson 1999 {published data only}
- Simpson GM, Josiassen RC, Stanilla JK, Leon J, Nair C, Abraham G, et al. Double‐blind study of clozapine dose response in chronic schizophrenia. American Journal of Psychiatry 1999;156(11):1744‐50. [DOI] [PubMed] [Google Scholar]
Suzuki 1992 {published data only}
- Suzuki E, Kanba S, Nibuya M, Koshikawa H, Nakaki T, Yagi G. Plasma homovanillic acid, plasma anti‐D1 and ‐D2 dopamine‐receptor activity, and negative symptoms in chronically mediated schizophrenia. Biological Psychiatry 1992;31(4):357‐64. [DOI] [PubMed] [Google Scholar]
Volavka 1996 {published data only}
- Volavka J, Cooper TB, Czobor P, Meisner M. Effect of varying haloperidol plasma levels on negative symptoms in schizophrenia and schizoaffective disorder. Psychopharmacology Bulletin 1996;32(1):75‐9. [PubMed] [Google Scholar]
Additional references
Andreasen 1982
- Andreasen NC, Olsen S. Negative v positive schizophrenia: Definition and validation. Archives of General Psychiatry 1982;39(7):789‐94. [DOI] [PubMed] [Google Scholar]
Baldessarini 1988
- Baldessarini RJ, Cohen BM, Teicher MH. Significance of neuroleptic dose and plasma level in the treatment of psychoses. Archives of General Psychiatry 1988;45(1):79‐91. [DOI] [PubMed] [Google Scholar]
Barnes 1989
- Barnes TR. A rating scale for drug‐induced akathisia. British Journal of Medicine 1989;154:672‐6. [DOI] [PubMed] [Google Scholar]
Barnes 2003
- Barnes TR, Buckley P, Schulz, SC. Treatment‐resistant schizophrenia. In: Hirsch SR, Weinberger D editor(s). Schizophrenia. 2nd Edition. Oxford: Blackwell Science Ltd, 2003:489–516. [Google Scholar]
Bland 1997
- Bland JM. Statistics notes. Trials randomised in clusters. BMJ 1997;315(7108):600. [DOI] [PMC free article] [PubMed] [Google Scholar]
Boissel 1999
- Boissel JP, Cucherat M, Li W, Chatellier G, Gueyffier F, Buyse M, et al. The problem of therapeutic efficacy indices. 3. Comparison of the indices and their use [Apercu sur la problematique des indices d'efficacite therapeutique, 3: comparaison des indices et utilisation. Groupe d'Etude des Indices D'efficacite]. Therapie 1999;54(4):405‐11. [PUBMED: 10667106] [PubMed] [Google Scholar]
Carpenter 1994
- Carpenter WT, Buchanan RW. Schizophrenia. New England Journal of Medicine 1994;330(10):681‐90. [DOI] [PubMed] [Google Scholar]
Clark 2011
- Clark SL, Adkins DE, Oord EJ. Analysis of efficacy and side effects in CATIE demonstrates drug response subgroups and potential for personalized medicine. Schizophrenia Research 2011;132(2‐3):114‐20. [DOI] [PMC free article] [PubMed] [Google Scholar]
Correll 2011
- Correll CU, Cañas F, Larmo I, Levy P, Montes JM, Fagiolini A, et al. Individualizing antipsychotic treatment selection in schizophrenia: characteristics of empirically derived patient subgroups. European Psychiatry 2011;26(Suppl 1):3‐16. [DOI] [PubMed] [Google Scholar]
Davey Smith 1998
- Davey Smith G, Egger M. Meta‐analysis: Unresolved issues and future developments. BMJ 1998;316(7126):221‐5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Davis 2004
- Davis JM, Chen N. Dose‐response and dose equivalence of antipsychotics. Journal of Clinical Psychopharmacology 2004;24(2):192‐208. [DOI] [PubMed] [Google Scholar]
Deeks 2000
- Deeks J. Issues in the selection for meta‐analyses of binary data. Proceedings of the 8th International Cochrane Colloquium. Cape Town: The Cochrane Collaboration, 2000.
Deeks 2011
- Deeks JJ, Higgins JP, Altman DG. Chapter 9: Analysing data and undertaking meta‐analyses. In: Higgins JP, Green S, editor (s). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from handbook.cochrane.org.
DerSimonian 1986
- DerSimonian R, Laird N. Meta‐analysis in clinical trials. Controlled Clinical Trials 1986;7(3):177‐88. [DOI] [PubMed] [Google Scholar]
Divine 1992
- Divine GW, Brown JT, Frazier LM. The unit of analysis error in studies about physicians' patient care behavior. Journal of General Internal Medicine 1992;7(6):623‐9. [DOI] [PubMed] [Google Scholar]
Dold 2015
- Dold M, Fugger G, Aigner M, Lanzenberger R, Kasper S. Dose escalation of antipsychotic drugs in schizophrenia: a meta‐analysis of randomized controlled trials. Schizophrenia Research 2015;166(1‐3):187‐93. [DOI] [PubMed] [Google Scholar]
Donner 2002
- Donner A, Klar N. Issues in the meta‐analysis of cluster randomized trials. Statistics in Medicine 2002;21(19):2971‐80. [DOI] [PubMed] [Google Scholar]
DSM‐IV‐TR 1994
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders. Fourth. Washington DC: American Psychiatric Association, 1994:34. [Google Scholar]
Egger 1997
- Egger M, Davey Smith G, Schneider M, Minder C. Bias in meta‐analysis detected by a simple, graphical test. BMJ 1997;315(7109):629‐34. [DOI] [PMC free article] [PubMed] [Google Scholar]
Elbourne 2002
- Elbourne D, Altman DG, Higgins JP, Curtina F, Worthingtond HV, Vaile A. Meta‐analyses involving cross‐over trials: methodological issues. International Journal of Epidemiology 2002;31(1):140‐9. [DOI] [PubMed] [Google Scholar]
Eum 2016
- Eum S, Lee AM, Bishop JR. Pharmacogenetic tests for antipsychotic medications: clinical implications and considerations. Dialogues in Clinical Neuroscience 2016;18(3):323‐37. [DOI] [PMC free article] [PubMed] [Google Scholar]
Friede 2017
- Friede T, Röver C, Wandel S, Neuenschwander B. Meta‐analysis of few small studies in orphan diseases. Research Synthesis Methods 2017;8(1):79‐91. [DOI] [PMC free article] [PubMed] [Google Scholar]
Furukawa 2006
- Furukawa TA, Barbui C, Cipriani A, Brambilla P, Watanabe N. Imputing missing standard deviations in meta‐analyses can provide accurate results. Journal of Clinical Epidemiology 2006;59(7):7‐10. [DOI] [PubMed] [Google Scholar]
Furukawa 2011
- Furukawa TA, Akechi T, Wagenpfeil S, Leucht S. Relative indices of treatment effect may be constant across different definitions of response in schizophrenia trials. Schizophrenia Research 2011;126(1‐3):212‐9. [DOI] [PubMed] [Google Scholar]
Gardner 2010
- Gardner DM, Murphy AL, O'Donnell H, Centorrino F, Baldessarini RJ. International consensus study of antipsychotic dosing. American Journal of Psychiatry 2010;167(6):686‐93. [DOI] [PubMed] [Google Scholar]
Gulliford 1999
- Gulliford MC. Components of variance and intraclass correlations for the design of community‐based surveys and intervention studies: data from the Health Survey for England 1994. American Journal of Epidemiology 1999;149(9):876‐83. [DOI] [PubMed] [Google Scholar]
Guy 1976
- Guy U. ECDEU assessment manual for psychopharmacology. Revised. Rockville, Maryland, USA: National Institute of Mental Health, 1976. [Google Scholar]
Haase 1961
- Haase H. Extrapyramidal modification of fine movements. A “conditio sine qua non” of the fundamental therapeutic action of neuroleptic drugs. Revue Canadienne de Biologie 1961;20:425‐49. [PubMed] [Google Scholar]
Harvey 1997
- Harvey PD, Davidson M, Mueser KT, Parrella M, White L, Powchik P. Social‐Adaptive Functioning Evaluation (SAFE): a rating scale for geriatric psychiatric patients. Schizophrenia Bulletin 1997;23(1):131‐45. [DOI] [PubMed] [Google Scholar]
Hasan 2012
- Hasan A, Falkai P, Wobrock T, Lieberman J, Glenthoj B, Gattaz WF, et al. World federation of societies of biological psychiatry (WFSBP) guidelines for the biological treatment of schizophrenia, part 1: update 2012 on the acute treatment of schizophrenia and the management of treatment resistance. World Journal of Biological Psychiatry 2012;13(5):318‐78. [DOI] [PubMed] [Google Scholar]
Heinrichs 1984
- Heinrichs DW, Hanlon TE, Carpenter WT. The Quality of life scale: an instrument for rating the schizophrenic deficit sndrome. Schizophrenia Bulletin 1984;10(3):388‐98. [DOI] [PubMed] [Google Scholar]
Hiemke 2011
- Hiemke C, Baumann P, Bergemann N, Conca A, Dietmaier O, Egberts K, et al. AGNP consensus guidelines for therapeutic drug monitoring in psychiatry: update 2011. Pharmacopsychiatry 2011;44(6):195‐235. [DOI] [PubMed] [Google Scholar]
Higgins 2003
- Higgins JP, Thompson SG, Deeks JJ, Altman DG. Measuring inconsistency in meta‐analyses. BMJ 2003;327(7414):557‐60. [DOI] [PMC free article] [PubMed] [Google Scholar]
Higgins 2011a
- Higgins JP, Green S. Chapter 7: Selecting studies and collecting data. In: Higgins JP, Green S, editor(s). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from handbook.cochrane.org.
Higgins 2011b
- Higgins JP, Altman DG, Sterne JA. Chapter 8: Assessing risk of bias in included studies. In: Higgins JP, Green S, editor(s). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from handbook.cochrane.org.
Honigfeld 1965
- Honigfeld G, Klett CJ. The nurses' observation scale for inpatient evaluation: a new scale for measuring improvement in chronic schizophrenia. Journal of Clinical Psychology 1965;21:65‐71. [DOI] [PubMed] [Google Scholar]
Honigfeld 1973
- Honigfeld G. NOSIE‐30: History and current status of its use in pharmacopsychiatric research. In: P. Pichot, R. Olivier‐Martin editor(s). Modem Problems in Pharmacopsychiatry: Psychological Measurement. Basel: Karger, 1973. [DOI] [PubMed] [Google Scholar]
Howes 2017
- Howes OD, McCutcheon R, Agid O, Bartolomeis A, Beveren NJ, Birnbaum ML, et al. Treatment‐Resistant Schizophrenia: Treatment Response and Resistance in Psychosis (TRRIP) Working Group Consensus Guidelines on Diagnosis and Terminology. American Journal of Psychiatry 2017;174(3):216‐29. [DOI] [PMC free article] [PubMed] [Google Scholar]
Hutton 2009
- Hutton JL. Number needed to treat and number needed to harm are not the best way to report and assess the results of randomised clinical trials. British Journal of Haematology 2009;146(1):27‐30. [DOI] [PubMed] [Google Scholar]
IntHout 2015
- IntHout J, Ioannidis JP, Borm GF, Goeman JJ. Small studies are more heterogeneous than large ones: a meta‐meta‐analysis. Journal of Clinical Epidemiology 2015;68(8):860‐9. [DOI] [PubMed] [Google Scholar]
Jäger 2007
- Jäger M, Riedel M, Messer T, Laux G, Pfeiffer H, Naber D, et al. Psychopathological characteristics and treatment response of first episode compared with multiple episode schizophrenic disorders. European Archives of Psychiatry and Clinical Neuroscience 2007;257(1):47‐53. [DOI] [PMC free article] [PubMed] [Google Scholar]
Kane 1988
- Kane J, Honigfeld G, Singer J, Meltzer H. Clozapine for the treatment‐resistant schizophrenic. A double‐blind comparison with chlorpromazine. Archives of General Psychiatry 1988;45(9):789‐96. [DOI] [PubMed] [Google Scholar]
Kane 1996
- Kane JM. Pharmacologic treatment of schizophrenia. Biological Psychiatry 1999;46(10):1396–1408. [DOI] [PubMed] [Google Scholar]
Kay 1986
- Kay SR, Opler LA, Fiszbein A. Positive and Negative Syndrome Scale (PANSS) Manual. North Tonawanda, NY: Multi‐Health Systems, 1986. [Google Scholar]
Kinon 2004
- Kinon BJ, Ahl J, Stauffer VL, Hill AL, Buckley PF. Dose Response and Atypical Antipsychotics in Schizophrenia. CNS Drugs 2004;18(9):597‐616. [DOI] [PubMed] [Google Scholar]
Lehmann 2004
- Lehman AF, Lieberman JA, Dixon LB, McGlashan TH, Miller AL, Perkins DO, et al. Practice guideline for the treatment of patients with schizophrenia, second edition. American Journal of Psychiatry 2004;161(2 Suppl):1‐56. [PubMed] [Google Scholar]
Leon 2006
- Leon AC, Mallinckrodt CH, Chuang‐Stein C, Archibald DG, Archer GE, Chartier K. Attrition in randomized controlled clinical trials: methodological issues in psychopharmacology. Biological Psychiatry 2006;59(11):1001‐5. [DOI] [PubMed] [Google Scholar]
Leucht 2005a
- Leucht S, Kane JM, Kissling W, Hamann J, Etschel E, Engel R. Clinical implications of brief psychiatric rating scale scores. British Journal of Psychiatry 2005;187:366‐71. [PUBMED: 16199797] [DOI] [PubMed] [Google Scholar]
Leucht 2005b
- Leucht S, Kane JM, Kissling W, Hamann J, Etschel E, Engel RR. What does the PANSS mean?. Schizophrenia Research 2005;79(2‐3):231‐8. [PUBMED: 15982856] [DOI] [PubMed] [Google Scholar]
Leucht 2007
- Leucht S, Engel RR, Bauml J, Davis JM. Is the superior efficacy of new generation antipsychotics an artifact of LOCF?. Schizophrenia Bulletin 2007;33(1):183‐91. [PUBMED: 16905632] [DOI] [PMC free article] [PubMed] [Google Scholar]
Leucht 2009
- Leucht S, Corves C, Arbter D, Engel R, Li C, Davis JM. Second‐generation versus first‐generation antipsychotic drugs for schizophrenia: a meta‐analysis. Lancet 2009;373(9657):31‐41. [DOI] [PubMed] [Google Scholar]
Leucht 2013
- Leucht S, Cipriani A, Spineli L, Mavridis D, Orey D, Richter F, et al. Comparative efficacy and tolerability of 15 antipsychotic drugs in schizophrenia: a multiple‐treatments meta‐analysis. Lancet 2013;382(9896):951‐62. [DOI] [PubMed] [Google Scholar]
Leucht 2014
- Leucht S. Measurements of response, remission, and recovery in schizophrenia and examples for their clinical application. Journal of Clinical Psychiatry 2014;75(Suppl 1):8‐14. [DOI] [PubMed] [Google Scholar]
Lieberman 2005
- Lieberman JA, Stroup TS, McEvoy JP, Swartz MS, Rosenheck RA, Perkins DO, et al. Effectiveness of antipsychotic drugs in patients with chronic schizophrenia. New England Journal of Medicine 2005;353(12):1209–23. [DOI] [PubMed] [Google Scholar]
Loebel 2013
- Loebel A, Cucchiaro J, Sarma K, Xu L, Hsu C, Kalali AH, et al. Efficacy and safety of lurasidone 80 mg/day and 160 mg/day in the treatment of schizophrenia: a randomized, double‐blind, placebo‐ and active‐controlled trial. Schizophrenia Research 2013;145(1‐3):101‐9. [DOI] [PubMed] [Google Scholar]
Marshall 2000
- Marshall M, Lockwood A, Bradley C, Adams C, Joy C, Fenton M. Unpublished rating scales: a major source of bias in randomised controlled trials of treatments for schizophrenia. British Journal of Psychiatry 2000;176:249‐52. [DOI] [PubMed] [Google Scholar]
Marvaha 2004
- Marwaha S, Johnson S. Schizophrenia and employment ? A review. Social Psychiatry and Psychiatric Epidemiology 2004;39(5):337‐49. [DOI] [PubMed] [Google Scholar]
Miller 2009
- Miller R. Mechanisms of Action of Antipsychotic Drugs of Different Classes, Refractoriness to Therapeutic Effects of Classical Neuroleptics, and Individual Variation in Sensitivity to their Actions: PART I. Current Neuropharmacology 2009;7(4):302‐14. [DOI] [PMC free article] [PubMed] [Google Scholar]
Moher 2001
- Moher D, Schulz KF, Altman D. The CONSORT statement: revised recommendations for improving the quality of reports of parallel‐group randomised trials. Journal of the American Medical Association 2001;285(15):1987–91. [DOI] [PubMed] [Google Scholar]
Nakajima 2016
- Nakajima S, Uchida H, Bies RR, Caravaggio F, Suzuki T, Plitman E, et al. Dopamine D2/3 Receptor Occupancy Following Dose Reduction Is Predictable With Minimal Plasma Antipsychotic Concentrations: An Open‐Label Clinical Trial. Schizophrenia Bulletin 2016;42(1):212‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Nebert 2000
- Nebert DW, Dieter MZ. The evolution of drug metabolism. Pharmacology 2000;61(3):124‐35. [DOI] [PubMed] [Google Scholar]
Neborsky 1981
- Neborsky R, Janowsky D, Munson E, Depry D. Rapid treatment of acute psychotic symptoms with high‐ and low‐dose haloperidol. Behavioral considerations. Archives of General Psychiatry 1981;38(2):195‐9. [DOI] [PubMed] [Google Scholar]
NIMH 1970
- National Institute of Mental Health. Clinical global impression. In: Guy W, Bonato RR editor(s). Manual for the ECDEU Assessment Battery. Washington DC: NIMH, 1970. [Google Scholar]
Overall 1962
- Overall JE, Gorham DR. The brief psychiatric rating scale. Psychological Reports 1962;10:799‐812. [Google Scholar]
Palmer 2005
- Palmer BA, Pankratz VS, Bostwick JM. The lifetime risk of suicide in schizophrenia: a reexamination. Archives of General Psychiatry 2005;62(3):247‐53. [DOI] [PubMed] [Google Scholar]
Samara 2015a
- Samara MT, Leucht C, Leeflang MM, Anghelescu IG, Chung YC, Crespo‐Facorro B, et al. Early Improvement As a Predictor of Later Response to Antipsychotics in Schizophrenia: A Diagnostic Test Review. American Journal of Psychiatry 2015;172(7):617‐29. [DOI] [PubMed] [Google Scholar]
Samara 2015b
- Samara MT, Helfer B, Rothe PB, Leucht S. Increasing antipsychotic dose versus switching antipsychotic for non response in schizophrenia. Cochrane Database of Systematic Reviews 2015, Issue 10. [DOI: 10.1002/14651858.CD011884] [DOI] [PMC free article] [PubMed] [Google Scholar]
Schünemann 2011
- Schünemann HJ, Oxman AD, Vist GE, Higgins JP, Deeks JJ, Glasziou P, et al. Chapter 12: Interpreting results and drawing conclusions. In Higgins JP, Green S, editor(s). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from handbook.cochrane.org.
Simpson 1970
- Simpson M, Angus JW. A rating scale for extrapyramidal side effects. Acta Psychiatrica Scandinavica 1970;212:11‐9. [DOI] [PubMed] [Google Scholar]
Sterne 2011
- Sterne JA, Egger M, Moher D. Chapter 10: Addressing reporting biases. In: Higgins JP, Green S, editor(s). Cochrane Handbook for Systematic Reviews of Intervention. Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from handbook.cochrane.org.
Swift 2002
- Swift RH, Harrigan EP, Cappelleri JC, Kramer D, Chandler LP. Validation of the behavioural activity rating scale (BARS): a novel measure of activity in agitated patients. Journal of Psychiatric Research 2002;36(2):87‐95. [DOI] [PubMed] [Google Scholar]
Ukoumunne 1999
- Ukoumunne OC, Gulliford MC, Chinn S, Sterne JA, Burney PG. Methods for evaluating area‐wide and organistation‐based intervention in health and health care: a systematic review. Health Technology Assessment 1999;3(5):1‐75. [PubMed] [Google Scholar]
Vijayan 2012
- Vijayan NN, Mathew A, Balan S, Natarajan C, Nair CM, Allencherry PM, et al. Antipsychotic drug dosage and therapeutic response in schizophrenia is influenced by ABCB1 genotypes: a study from a south Indian perspective. Pharmacogenomics 2012;13(10):1119‐27. [DOI] [PubMed] [Google Scholar]
von Hippel 2015
- Hippel PT. The heterogeneity statistic I2 can be biased in small meta‐analyses. BMC Medical Research Methodology 2015;15(1):35. [DOI] [PMC free article] [PubMed] [Google Scholar]
Vos 2012
- Vos T, Flaxman AD, Naghavi M, Lozano R, Michaud C, Ezzati M, et al. Years lived with disability (YLDs) for 1160 sequelae of 289 diseases and injuries 1990‐2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet 2012;380(9859):2163‐96. [DOI] [PMC free article] [PubMed] [Google Scholar]
Xia 2010
- Xia J, Adams CE, Bhagat N, Bhagat V, Bhoopathi P, El‐Sayeh H, et al. Loss to outcomes stakeholder survey: the LOSS study. Schizophrenia Research 2010;117(2‐3):278. [Google Scholar]
References to other published versions of this review
Helfer 2015
- Helfer B, Leucht S, Rothe PH, Samara MT. Increasing antipsychotic dose for non response in schizophrenia. Cochrane Database of Systematic Reviews 2015, Issue 10. [DOI: 10.1002/14651858.CD011883] [DOI] [PMC free article] [PubMed] [Google Scholar]