

Abstract
Many breakthroughs have been made in the past decade regarding our knowledge of the biological basis of the diffuse gliomas, the most common primary central nervous system (CNS) tumors. These tumors as a group are aggressive, associated with high mortality and have a predilection for adults. However, a subset of CNS glial and glioneuronal tumors are characterized by a more circumscribed pattern of growth and occur more commonly in children and young adults. They tend to be indolent, but our understanding of anaplastic changes in these tumors continues to improve as diagnostic classifications evolve in the era of molecular pathology and more integrated and easily-accessible clinical databases. The presence of anaplasia in pleomorphic xanthoastrocytomas and gangliogliomas is assigned a WHO grade III under the current classification, while the significance of anaplasia in pilocytic astrocytomas remains controversial. However, recent data highlight the association of the latter with aggressive clinical behavior, as well as the presence of molecular genetic features of both pilocytic and diffuse gliomas, with the recognition that the precise terminology remains to be defined. We review current concepts and advances regarding histopathology and molecular understanding of pilocytic astrocytomas, pleomorphic xanthoastrocytomas and gangliogliomas, with a focus on their anaplastic counterparts.
Keywords: pilocytic astrocytoma, ganglioglioma, pleomorphic xanthoastrocytoma, anaplasia, BRAF
Introduction
Diagnostic neuropathology continues to evolve due to the contributions of the now fairly mature field of molecular pathology and the increasing digitalization of clinical information. In comparing WHO classifications of Central Nervous System tumors from 2007 and 2016, many entities are now defined by means of specific molecular alterations, such as IDH wild-type or mutant in the group of diffuse gliomas. This new classification advances important conceptual discussions regarding the relationship between histopathology/morphology and molecular alterations and the degree to which they provide orthogonal or redundant information. As a result, categories that long represented an umbrella of heterogeneous entities are now being progressively refined, partially along molecular fault lines, but also more generally. An important example, which we explore here, is that of the officialization of distinct diagnostic categories for anaplastic subsets of otherwise generally benign tumors.
Anaplasia, from the Greek ana and plasis, which translates directly to “backward formation” or “to form backwards”, refers to the loss of mature or specialized features of differentiated cells or tissues. Anaplasia can therefore be thought of as dedifferentiation and generally labels a more aggressive neoplasm. Because of the nature of anaplastic transformation, meaning the loss of unique, clearly identifiable features, anaplastic transformation in tumors can be hard to define. Even so, this remains an important task due to the prognostic importance of these changes, and one that is likely to become more feasible with the proliferation of molecular profiling of larger series of (sometimes rare) tumors. Particularly in the context discussed here, that of otherwise rather well-behaved neoplasms, anaplasia can lead to the recommendation of additional treatments beyond surgery and thus have a profound clinical impact. Anaplastic variants have been well studied primarily in the diffuse gliomas, where they represent distinct categories with specified outcomes requiring therapies other than surgery (e.g. anaplastic astrocytoma and anaplastic oligodendroglioma, both WHO grade III). Anaplasia has also been studied in other primary intraparenchymal (e.g. ependymoma) and extra-axial (meningioma) CNS tumors.
Here, we focus on two circumscribed gliomas and a glioneuronal tumor: pilocytic astrocytoma (PA, WHO grade I), pleomorphic xanthoastrocytoma (PXA, WHO grade II) and ganglioglioma (GG, WHO grade I), along with their anaplastic counterparts. We review important pathologic features of each tumor as well as summarize current knowledge of their molecular alterations, paying special attention to distinctive aspects of anaplasia in each of these entities. Throughout, we emphasize the need to further characterize these emerging categories of prognostic significance. Lastly, we highlight common themes across all three tumors and highlight remaining gray zones.
1. Pilocytic Astrocytoma, WHO Grade I
Epidemiologically, pilocytic astrocytoma (PA) is the most common brain tumor in children, affecting young adults as well. The 2007-2011 Central Brain Tumor Registry of the United States (CBTRUS) report estimates its incidence at 0.84 cases per 100,000 individuals under 19 years of age [1]. PAs are usually found in the cerebellum, often showing a strong and diffusely enhancing, sometimes cystic, calcified appearance on MRI, but can occur anywhere in the neuraxis, including the spinal cord, and usually favoring midline structures. PAs are most often sporadic but can occur in association with neurofibromatosis type 1 (NF1), often affecting the optic pathways and the hypothalamus when they occur in this setting. They are, in addition, the most common primary brain tumor in patients with this syndrome.
As a whole, pilocytic astrocytomas have a 10-year survival over 90% even after surgical resection without additional treatment. Certain locations may be difficult to access surgically, resulting in subtotal resections and a greater likelihood of local recurrence. PA is defined as a WHO grade I, circumscribed (non-diffuse) glioma of low-to-moderate cellularity. Histologically, it is characterized by a biphasic entity featuring compact piloid areas of bipolar astrocytes with hair-like processes and Rosenthal fibers alongside more discohesiveareas of myxoid stroma featuring microcysts and eosinophilic granular bodies (EGBs). Cytology is generally bland, with piloid areas having elongated nuclei and microcystic areas having more round or oval nuclei. Additional features may include glomeruloid vessels and hyalinized vessels, infarct-like, generally non-palisading necrosis, leptomeningeal involvement with a desmoplastic reaction, as well as pleomorphic nuclei and hyperchromasia with rare mitoses. Immunohistochemistry typically documents a glial phenotype, with near uniform expression of GFAP and OLIG2.
Molecular features
After initial evaluations revealed what appeared to be grossly normal cytogenetics in pilocytic astrocytoma, a number of independent studies published in 2008 employing the improved array comparative genomic hybridization (aCGH) analyses ushered in a far better understanding of the molecular changes underlying PAs.
A study by Pfister et al. uncovered duplication of the proto-oncogene BRAF as the most common abnormality in pediatric low-grade astrocytomas by means of array-based genome-wide assessment of DNA copy number [2]. They also identified activating BRAF mutations in some of the patients, as well as duplications in adult patients. These alterations led to activation of the MAPK pathway and increased proliferation [2]. A similar study by Bar et al. identified a 1.9Mb region in chromosome 7q34 including the BRAF kinase domain that was mutated in 18 of 20 posterior fossa pilocytic astrocytomas with concomitant activation of downstream MEK and ERK [3].
Work by Jones et al. not only replicated the tandem duplication in 7q34 (seen in 66% of PAs in their series and none of the 244 higher-grade astrocytomas), but also note that the rearrangement leads to an oncogenic fusion gene, KIAA1549:BRAF, featuring a constitutively active BRAF kinase domain that can result in transformation of NIH3T3 cells [4].
A more recent study of whole-genome sequencing of 96 pilocytic astrocytomas described recurrent alterations in FGFR1 and PTPN11 and reaffirmed the key role played by the MAPK pathway, claiming that PA “is predominantly a single-pathway disease” [5]. They describe additional changes, such as NTRK2 fusion genes in non-cerebellar tumors and additional examples of genomic alterations leading to BRAF activation [5].
1.1. Pilocytic Astrocytoma with Anaplasia, no WHO grade
Pilocytic astrocytomas will, on rare occasions, demonstrate anaplastic features (Figure 1 A, B). Until recently, this anaplastic transformation was only addressed in the literature in the form of case reports or retrospective case series [6 and references within]. Furthermore, studies would employ different criteria and nomenclature for defining anaplastic (malignant) pilocytic astrocytomas (A-PAs) given that official consensus criteria do not yet exist. A 2010 study defining A-PAs as harboring at least 4 mitoses / 10 hpf (but usually more than 5), demonstrating hypercellularity and moderate-to-severe cytologic atypia in addition to typical PA features estimated the frequency of A-PAs at 1.7%, or 34 out of 2200 consultation cases [6].
Figure 1. Pilocytic astrocytoma with anaplasia: pathology and molecular genetic features.
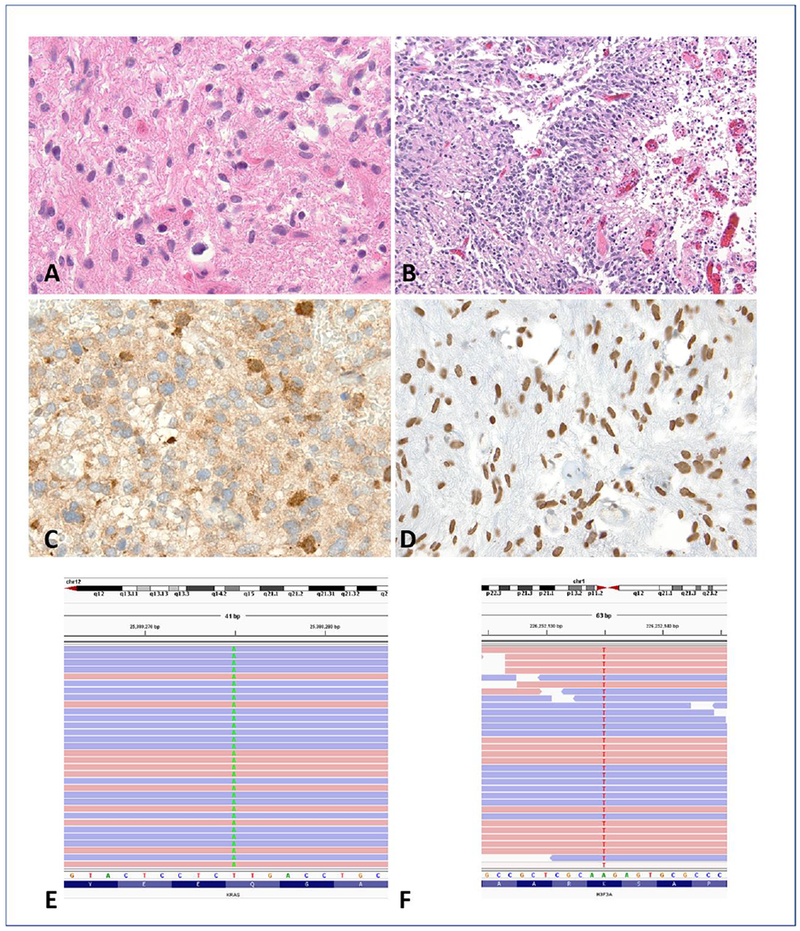
This tumor developed in the posterior fossa in a 4 year old child and demonstrated a well differentiated piloid component with Rosenthal fibers (A). In addition there were sharply circumscribed cellular regions with brisk mitotic activity and pseudopallisading necrosis consistent with anaplasia (B). Immunohistochemical stains demonstrated ATRX loss (C) and expression of H3 K27M mutant protein (D), a feature of a subset of pilocytic asrocytomas with anaplasia, histologically defined. Next-generation sequencing detected KRAS p.Q61H (c.183A>T) (E) and H3F3A p.K27M (c.83A>T) mutations (F).
This study also provided evidence that anaplastic features, when present over greater than one low power field (20×), convey prognostic significance. Median overall survival was found to be 24 months (95% CI: 17-29) and progression-free survival of 14 months (95% CI: 11-29), with prior radiation, PA precursor, mitoses/10 hpf as a continuous variable and necrosis were associated with significantly decreased overall and progression free-survival [6]. A-PAs are thus associated with poorer survival than classic PA, but still portending a far better prognosis than glioblastoma, with tumors without necrosis being more analogous to St Anne-Mayo grade 2 astrocytomas, and those with necrosis being more analogous to grade 3 astrocytomas. This emphasizes the importance of distinguishing A-PA from classic WHO grade I pilocytic astrocytomas, given the clinical ramifications.
Indeed, follow up studies have demonstrated unique molecular features of A-PAs. For example, a series including 43 conventional PAs, 24 clinically aggressive / recurrent PAs and 25 histologically anaplastic PAs was assessed with respect to its molecular genetics, gene expression and immunohistochemistry [7]. This study uncovered heterozygous PTEN/10q and homozygous p16 deletions in about a third and a fifth, respectively, of A-PAs, but in none of the other categories [7]. Consistent with previous molecular studies in standard PAs, BRAF duplication was identified in a third of sporadic A-PAs and two thirds of cerebellar examples. In addition, activation of PI3K/AKT and mTOR may play a role in the increased proliferation characterizing anaplastic PAs [7].
In 2018, Reinhardt and colleagues carried out DNA methylation profiling of 102 A-PAs, arriving at the conclusion that 83 of these tumors had a shared methylation signature that was different from reference cases [8]. They referred to this group as a “methylation class anaplastic astrocytoma with piloid features”, or MC AAP for short. The rest of the samples clustered with the reference classes, with subsequent molecular studies being consistent with this classification. The most frequent molecular alterations reported included CDKN2A/B (64/73, 87%), alterations of the MAPK pathway (19/24, 79%, mostly NF1 mutations and BRAF fusions) and ATRX mutations (31/69, 45%) [8]. These tumors as a group were associated with worse overall survival than PA and other low grade tumors, similar to glioblastoma IDH mutant, but worse than glioblastoma IDH wild-type.
Another study focusing on histologically defined pilocytic astrocytoma with anaplasia documented the presence of ATRX and H3-K27M alterations, as well as alternative lengthening of telomeres (ALT), in 57 A-PA resections from 36 patients [9] (Figure 1 C-F). ALT was present in 25 cases and 9 of 11 benign PA precursors. ATRX loss was found in over half of the cases: ALT+/ATRX− (20/24, 83%) and ALT−/ATRX+ (11/11, 100%). BRAF duplications/fusions were present in 8/26 cases tested, with no BRAF p.V600E mutations identified. Interestingly, H3-K27M was present in 5 of 32 (16%) cases along with ATRX loss and ALT suggesting that A-PAs are heterogeneous neoplasms harboring genetic alterations usually associated with both PA and diffuse gliomas [9]. Loss of ATRX expression in A-PA has been reported by others [10]. Other rarer MAPK activating alterations have been reported in A-PA, for example FGFR1 duplications [11].
2. Pleomorphic Xanthoastrocytoma (PXA), WHO Grade II
Pleomorphic xanthoastrocytoma (PXA) is a rather rare tumor, comprising <1% of all primary brain tumors [1]. It tends to occur in children and young adults with a reported median age of presentation of 22 [12]. The 2014 CBTRUS report estimates its incidence at 0.3 cases per 100,000 individuals [1]. As a whole, PXAs have a 5-year recurrence-free rate of 70.9% and overall survival rate of 90.4%, with extent of resection being the most significant predictor of recurrence [13].
PXAs are nearly exclusively supratentorial, superficially located within the cerebral hemispheres, most commonly in the temporal lobe and often involving the leptomeninges. Indeed, PXAs are most commonly brought to medical attention due to recurring, intractable seizures [14]. On imaging, PXAs are often peripherally located, superficial, enhancing lesions without significant edema presenting as either a cystic mass containing a mural nodule (70%) or a predominantly solid mass with cystic changes (30%) [15].
PXA is a WHO grade II, circumscribed (non-diffuse) astrocytic glioma that is minimally mitotically active (< 5 mitoses / hpf). The Greek xanthos means ‘yellow’ and it is used a descriptor for PXA in reference to the accumulation of vast amounts of lipid in a subset of neoplastic cells, often termed “xanthomatous” cells, that are usually large, multinucleated cells. Histologically, PXA is characterized by a combination of large, pleomorphic, often multinucleated astrocytes that can have xanthomatous changes, with intermingled spindle cells, a dense pericellular reticulin network, and granular bodies (usually eosinophilic but sometimes pale). Intranuclear inclusions, prominent nucleoli and lymphocytic infiltrates are also common findings in PXA.
Staining patterns can be variable, though usually involve positive GFAP in the large pleomorphic and xanthomatous cells. Interestingly, despite being astrocytic tumors, PXAs can show features of neuronal differentiation, such as synaptophysin positivity as well as neurofilament, class III beta-tubulin or MAP2 [16,17]. In addition, S100 and CD34 can be positive, leading some to favor a multipotent neuroectodermal precursor as the cell of origin over the more classically postulated subpial astrocytes suggested in the first description of this entity in 1979 by Kepes, Rubinstein and Eng [18].
Molecular features
Early molecular characterization of PXAs revealed complex karyotypes involving gains of chr3 and chr7 and alterations in the long arm of chrl [19–21]. These alterations, however, are not specific to PXA. A subsequent study utilizing comparative genomic hybridization revealed the most common recurrent alteration (50%) to be loss of chr9 corresponding to homozygous deletion of CDKN2A/B in 9p21.3 [22].
BRAF point mutations occur in approximately 60% of PXAs [23] and can be effectively assessed using immunochemistry [24]. BRAF p.V600E, however, is a very common primary CNS tumor mutation, especially in pilocytic astrocytoma and ganglioglioma [25–27]. Importantly, IDH sequencing [28] or immunohistochemistry against IDH R132H [13], have failed to reveal any abnormalities in IDH in the vast majority of PXA, making this entity clearly distinct from diffuse, infiltrating astrocytomas. However, at least one exceptional case combining features of infiltrating IDH mutant gliomas and BRAF mutant PXA have been reported [29].
A series of 62 PXAs (46 WHO grade II and 16 with anaplastic features) was evaluated for alterations in frequently aberrant targets in infiltrating gliomas, namely TP53, CDKN2A, CDK4, MDM2, and EGFR [30]. The rate of p53 mutations was very low at 5%, consistent with previous studies estimating it at 2% [31]. Furthermore, no MDM2, EGFR and CDK4 gene amplifications could be found and none of the tumors showed homozygous loss of CDKN2A [30]. Additional studies point to examples of alterations in TSC2 and NF1, and an ETV6-NTRK3 fusion [32,33]. These results indicate that PXAs are molecularly distinct from diffuse astrocytomas and emphasize the support of molecular markers in rendering more accurate diagnoses.
2.1. Anaplastic pleomorphic xanthoastrocytoma, WHO Grade III
Unlike the pilocytic astrocytomas described above, PXAs do have a specific, official definition for their anaplastic counterpart, albeit only since the most recent edition of the WHO classification in 2016 [34]. These changes followed multiple publications providing evidence, importantly, of differences in prognosis [12,13]. Anaplastic PXAs (A-PXAs) are defined as PXAs with >= 5 mitoses / 10 hpf [13] and are assigned a WHO grade III, in contrast with non-anaplastic PXAs, which are grade II. A-PXAs usually have a recognizable PXA focus [12] and are otherwise similar to PXA, including in their location, imaging features, histology and immunophenotype (Figure 2A-E). Increased mitotic activity will frequently be associated with necrosis, however this is not a requirement for diagnosing A-PXA. Pleomorphism will at times be less pronounced than in regular PXA (Figure 2C), and neoplastic cells will more often infiltrate diffusely. Some reports mention other morphologies, including small-cell, fibrillary and epithelioid/rhabdoid transformation [35]. A-PXAs portend a significantly worse prognosis, with a 5-year overall survival of 89.4% for less than 5 mitoses per 10 hpf compared to 55.6% for 5 or more mitoses per 10 hpf [13]. Similarly, 5-year overall survival rate for patients with tumor necrosis was significantly worse (42.2%) when compared to patients without tumor necrosis (90.2%).
Figure 2. Anaplastic pleomorphic xanthastrocytoma (A-PXA) (WHO grade III): pathology and molecular genetic features.
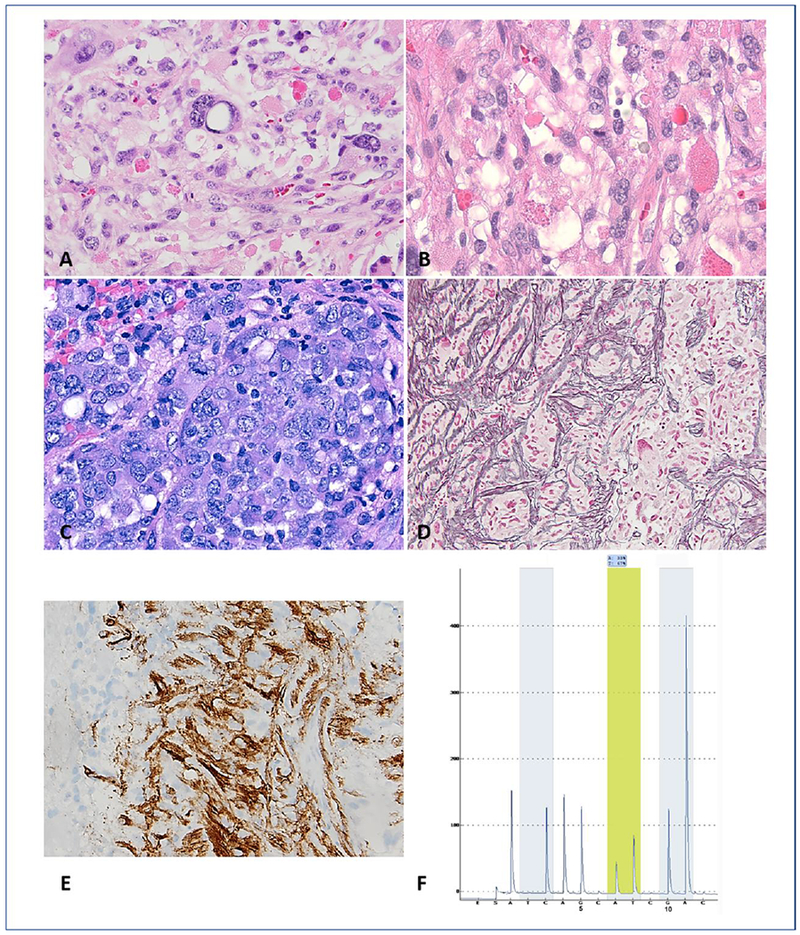
A 40-year-old man presented with a temporal lobe mass. Histologic features included moderate cellularity, pleomorphism and numerous eosinophilic granular bodies (A). In addition, mitotic activity was easy to find (B). Cellular areas with a paradoxic decrease in pleomorphism and epithelioid morphology is a feature of some A-PXA (C). There is an increased in reticulin deposition (D) and CD34 expression (E) in this A-PXA. Detection of V600E by pyrosequencing assay (F). Analysis of sequence flanking the T>A hotspot (yellow shading) within codon 600 allows for the detection of V600E mutation. Nucleotide dispensation order and the numerical positions 5 and 10 are shown below the pyrogram.
Initial efforts to identify unique molecular features of PXA with anaplastic features did not immediately meet with success. Indeed, an observation was made that the frequency of BRAF p.V600E mutation was is fact lower in cases showing anaplastic features (9/19 cases, 47.4%) than in PXA without such anaplasia (30/40 cases, 75%)[13,36]. A more recent study of 24 PXA and 14 A-PXA primary resection specimens using the OncoScan chromosomal microarray revealed that the rate of CDKN2A/B deletion in PXAs (83%) was similar to that in A-PXAs (93%), and equivalent regardless of BRAF mutation status, with BRAF p.V600E (87%) and BRAF wild-type (87%) (Figure 2F) [37]. Histologic grade correlated best with overall survival (p=0.003), unlike specific molecular features. While whole chromosome gains/losses were frequent, chromosomal losses and copy-neutral LOH involving chr22 and chr14 were significantly more common in A-PXA. These changes, however, ranged from identical profiles before and after anaplastic transformation, to numerous copy number changes, highlighting the complexity of molecularly separating anaplastic tumors from their regular counterparts [37].
There is still no molecular marker uniquely or specifically associated with anaplastic features in PXA. A study that retrospectively reviewed 55 PXAs and found that the TERT promoter was mutated more frequently in PXA than in A-PXA, but this failed to reach statistical significance, and would alter likelihoods rather than serve as a specific marker [38]. Another study examined tissue from 15 patients with A-PXA, including 4 patients with multiple recurrences (2 of which showed anaplastic transformation) confirmed that CDKN2A homozygous deletion in combination with RAF alterations is a defining genetic feature of both PXA and anaplastic PXA [39]. Like the studies above, alterations in TERT were frequent (47%) and chromosomal copy number alterations are widespread, but ultimately cannot, by themselves, separate anaplastic tumors from the rest [39]. Rare alterations in the MAPK pathway in the absence of BRAF p.V600E have been reported in A-PXA, for instance NRF1-BRAF and ATG7-RAF1 fusions [40].
3. Ganglioglioma, WHO Grade I
While gangliogliomas (GG) can occur anywhere in the CNS, they are most commonly present in the temporal lobe (79%) and frontal lobe (12%) of children and young adults [41]. GG is the tumor most commonly associated with chronic, treatment-resistant temporal lobe epilepsy, and therefore is a part of the entities grouped under ‘long-term epilepsy associated tumors’ (LEATs) [42]. On imaging, about two thirds of tumors have a cystic, sometimes septated appearance with a mural nodule, or may be a solid mass isointense to the gray matter on T1-WI. There is no mass effect. Calcification can often be seen on CT [43]. GGs are indolent tumors, with a recurrence-free survival rate estimated at 97% over a 7.5 year-period [41].
Immunophenotype: GFAP will stain the glial component, while the neuronal component may stain with MAP2, neurofilaments, chromogranin-A (more robust in dysplastic neurons than normal ones) and synaptophysin. CD34 will stain most gangliogliomas, especially in the temporal lobe, including neural cells in peritumoral cortex [44]. Antibodies against the BRAF pV600E mutation can be used to identify ganglion cells in tumors bearing this genetic abnormality.
Gangliogliomas (GG) are WHO grade I, well-differentiated, slow-growing glioneuronal tumors comprised of neoplastic ganglion cells and neoplastic glial cells. Neoplastic ganglion cells will have a lack of cytoarchitectural organization with clustering and cytomegaly, with binucleated forms in less than half of the cases. Perimembranous aggregation of Nissl substance may be seen, along with eosinophilic granular bodies and, more rarely, Rosenthal fibers. The fibrillary matrix surrounding these elements is generally conspicuous and may include myxoid degeneration, microcystic changes and dystrophic calcification. Mitoses and necrosis are rare, but not incompatible with the diagnosis. Extensive lymphoid infiltrates may be present within the parenchyma or along perivascular spaces, and a malformative capillary network component may be seen.
Molecular features
A series of 61 GGs screened with array-based comparative genomic hybridization indicate that 66% of gangliogliomas have chromosomal abnormalities, with frequent gains of chr7 (21%) and frequent losses of chr22 (16%)[45].
BRAF p.V600E mutation is identified in ~20-60% GGs [25,27,46,47]. Again, BRAF p.V600E is not specific to gangliogliomas. Mutant BRAF protein is detected for the most part in ganglion cells, but can also be found in the other tumor components [46]. A small study examined 3 gangliogliomas that did not have a BRAF V600E mutation with the goal of uncovering alternative genetic aberrations. They identified BRAF-KIAA1549 and MACF1 as parallel means to activate the MAPK pathway [33]. IDH mutations should not be present in ganglioglioma and, if found, should prompt strong consideration of diffuse glioma [47,48], which may contain neoplastic ganglion cells on occasion [49]. Likewise, combined loss of 1p and 19q should exclude a diagnosis of GG.
A recent series of 54 pediatric midline GG examined the co-ocurrence of BRAF p.V600E mutations and H3 K27M mutations. Half of the patients had a BRAF p.V600E mutation and 9.3% had combined H3F3A/BRAF mutations at diagnosis. There were no H3 K27M mutations in the absence of BRAF mutations in this cohort. Importantly, the authors caution against using H3 K27M mutation alone as a marker for grade IV diffuse gliomas [50]. A case report examining a ganglioglioma originating in the posterior fossa uncovered a deletion in 9q21 by aCGH that resulted in a novel molecular fusion TLE4-NTRK2 that made the patient eligible to being treated by entrectinib, a selective tyrosine kinase inhibitor [51].
A recent study used targeted next-generation sequencing on a cohort of 40 gangliogliomas to try to uncover mutations beyond the well-known BRAF p.V600E, as well as interacting mutations and additional copy number alterations [52]. While only 18 tumors had the BRAF V600E mutation, 36/40 had genetic alterations targeting the MAPK pathway, including different BRAF mutations and fusions, KRAS and RAF1 mutations, FGFR1/2 alterations and a case of biallelic NF1 mutation. A few of these tumors additionally had a CDKN2A homozygous deletion and one also had a subclonal PTEN mutation. Of the 4 tumors without MAPK alterations, one was found to have a novel ABL2-GAB2 gene fusion [52].
3.1. Anaplastic Ganglioglioma, WHO Grade III
Anaplastic Ganglioglioma (AGG) refers to a subset of GGs with elevated mitotic activity in the anaplastic glial component of this mixed glioneuronal tumor and will often additionally demonstrate increased cellularity and pleomorphism, microvascular proliferation and necrosis (Figure 3A-D)[34]. The WHO states that the “exclusion of diffuse glioma with entrapped pre-existing neuronal cells is obligatory for the diagnosis of anaplastic ganglioglioma and may be facilitated by additional genetic analysis” [48].
Figure 3. Anaplastic ganglioglioma (A-GG) (WHO grade III): pathology and molecular genetic features.
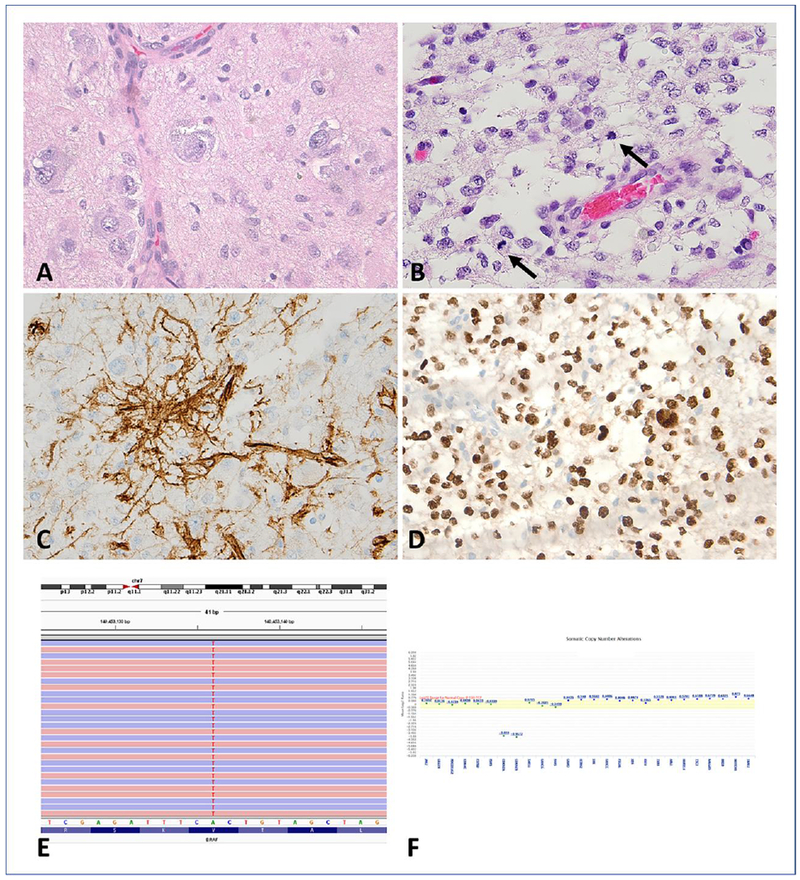
A-GG with a neoplastic ganglion cell component (A) and a cellular astrocytic components with brisk mitotic activity (arrows)(B). CD34 (C) and OLIG2 (D) expression in A-GG. Next- generation sequencing detected c.1799T>A substitution resulting in BRAF V600E mutation (E) and homozygous loss of CDKN2A (mean Log2 ratio: −3.818) and CDKN2B (mean Log2 ratio: −3.957)(F).
A small case study focusing on 2 gangliogliomas and their anaplastic recurrences identified losses of CDKN2A/B and DMBT1 or amplification of CDK4 [45]. However, these changes were already present in the original lesion and are therefore not specific to the anaplastic process. A CDKN2A deletion was also identified in 2/3 AGG [53]. Yet another small series examined BRAF p.V600E mutations and found a frequency of 3/6 [27](Figure 3E,F).
Until recently, only small series were available to try to address the difference in prognosis afforded by this anaplastic transformation, leading to results that were hard to interpret or extrapolate from. However, its persistence as an independent entity in the most recent WHO classification (2016) has motivated larger analyses. For example, a study by Zanello and colleagues assessed AGGs in a retrospective series including 18 patients (13 adults and 5 children) with AGGs (14 de novo and 4 secondary) representing 8% of the larger GG cohort of 222 [54]. They report a median progression-free survival of 10 months and a median overall survival of 27 months, which renders AGGs a far more aggressive entity than their GG counterpart [54]. From a molecular point of view, this cohort shared the BRAF p.V600E mutations (39%) seen in GG, but also hTERT promoter mutations (61%), p53 accumulation (39%), ATRX loss (17%) and p.K27M H3F3A mutations (17%) [54].
Another multicenter French group retrospectively evaluated 43 adult cases to gain insight into the natural course and prognosis of anaplastic gangliogliomas [55]. Their results support a challenging pathological diagnosis (needing to exclude a fifth of the originally available cases due to misclassification) and very poor survival that was nearly analogous to glioblastoma. Indeed, median progression-free survival was reported at 8.0 months, median overall survival at 24.7 months and 3-year and 5-year survival rates at 38.4% and 24.9%, respectively [55]. In addition, this group suggests survival benefits are associated with complete resection and adjuvant chemoradiotherapy [55].
Lastly, a systematic review of pediatric anaplastic gangliogliomas in 2018 included 24 studies following 34 patients [56]. Mean patient age was 9.18 years, and all underwent surgery. Mean overall survival was 43 months, with 1- and 3-year survival being 76.6% and 45.5%, respectively [56]. These numbers, while not as dire as those reported by the Zanello et al. study, still reflect the far poorer prognosis of AGGs as compared to GGs.
Miscellaneous Tumors
Although anaplastic changes are best recognized in the circumscribed tumors described above, they also may rarely develop in other tumors. These include dysembryoplastic neuroepithelial tumor [57–59]. Desmoplastic infantile ganglioglioma is another circumscribed WHO grade I tumor that may contain high grade histologic areas resembling glioblastoma or embryonal tumors [60]. However, these should not lead to up grading of these tumors, since they are not necessarily associated with a worse outcome.
Conclusions
In reviewing two circumscribed gliomas (Pilocytic Astrocytoma and Pleomorphic Xanthoastrocytoma) and the glioneuronal tumor Ganglioglioma, as well as their anaplastic counterparts, some general themes emerge. First and foremost, case series, while often limited by the rarity of these tumors and therefore statistical power, do support the notion that anaplastic features portend a significantly worse prognosis, thus justifying and emphasizing the need to better understand and identify anaplastic variants of known tumors.
Anaplastic PXAs only earned their official WHO designation within the last revision in 2016. Anaplastic PAs remains an unofficially handled entity, although at this point with equivalent evidence to support a difference in prognosis and similar classification systems (i.e. mitotic counts), it is reasonable to expect anaplastic pilocytic astrocytoma to be included in the next revision of the WHO classification. Indeed, disease categories within CNS tumors continue to evolve despite the maturity in the field in large part due to the success of molecular diagnoses and the proliferation of better clinical searchable databases.
Surprisingly, these anaplastic entities have, by and large, the same genetic aberrations as their classic counterparts. For example, they all have BRAF alterations in a substantial proportion of cases, and it even appears that the frequency may be lower in some anaplastic tumors. In some cases, there is evidence of an increased number of copy number alterations in anaplastic recurrences, but this is hard to separate from the natural tendency to accumulate further changes over time, even in the absence of transformation. In the case of anaplastic changes in PA, ATRX alterations and ALT are frequent events, and in our experience may be identifiable in putative non-anaplastic precursors years prior to the identification of histologic anaplasia. However, in rare instances they are clearly identifiable only during tumor progression.
Further studies will reveal additional genetic aberrations in these tumors, and it stands to reason that we will be able to identify alterations that help differentiate anaplastic transformation more reliably. Even so, the genetic diversity uncovered so far, and the lack of clear recurring alterations that are specific to anaplasia (or to individual non-anaplastic tumor types, for that matter) underscores the importance of continued integrated diagnoses with strong emphasis on histopathologic morphology, immunohistochemistry, and the addition of emerging areas of discovery, such as epigenetics and regulatory RNAs, to our molecular diagnostic tumor panels.
Acknowledgments
This work was supported in part by the Pilocytic/Pilomyxoid Fund, including Lauren’s First and Goal, and the Stick it to Brain Tumors Annual Women’s Ice Hockey Tournament (F.J.R.); Department of Defense grant W81XWH-18-1-0496 (FJR); NIGMS Medical Scientist Training Program Award T32-GM007309 (E.P.), and NIH grant P30 CA006973 to the Sidney Kimmel Comprehensive Cancer Center (PI: W. Nelson).
References
- 1.Ostrom QT, Gittleman H, Liao P, Rouse C, Chen Y, Dowling J, Wolinsky Y, Kruchko C, Barnholtz-Sloan J (2014) CBTRUS statistical report: primary brain and central nervous system tumors diagnosed in the United States in 2007-2011. Neuro Oncol 16 Suppl 4: iv1–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pfister S, Janzarik WG, Remke M, Ernst A, Werft W, Becker N, Toedt G, Wittmann A, Kratz C, Olbrich H, Ahmadi R, Thieme B, Joos S, Radlwimmer B, Kulozik A, Pietsch T, Herold-Mende C, Gnekow A, Reifenberger G, Korshunov A, Scheurlen W, Omran H, Lichter P (2008) BRAF gene duplication constitutes a mechanism of MAPK pathway activation in low-grade astrocytomas. J Clin Invest 118: 1739–1749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bar EE, Lin A, Tihan T, Burger PC, Eberhart CG (2008) Frequent gains at chromosome 7q34 involving BRAF in pilocytic astrocytoma. J Neuropathol Exp Neurol 67: 878–887. [DOI] [PubMed] [Google Scholar]
- 4.Jones DT, Kocialkowski S, Liu L, Pearson DM, Backlund LM, Ichimura K, Collins VP (2008) Tandem duplication producing a novel oncogenic BRAF fusion gene defines the majority of pilocytic astrocytomas. Cancer Res 68: 8673–8677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jones DT, Hutter B, Jager N, Korshunov A, Kool M, Warnatz HJ, Zichner T, Lambert SR, Ryzhova M, Quang DA, Fontebasso AM, Stutz AM, Hutter S, Zuckermann M, Sturm D, Gronych J, Lasitschka B, Schmidt S, Seker-Cin H, Witt H, Sultan M, Ralser M, Northcott PA, Hovestadt V, Bender S, Pfaff E, Stark S, Faury D, Schwartzentruber J, Majewski J, Weber UD, Zapatka M, Raeder B, Schlesner M, Worth CL, Bartholomae CC, von Kalle C, Imbusch CD, Radomski S, Lawerenz C, van Sluis P, Koster J, Volckmann R, Versteeg R, Lehrach H, Monoranu C, Winkler B, Unterberg A, Herold-Mende C, Milde T, Kulozik AE, Ebinger M, Schuhmann MU, Cho YJ, Pomeroy SL, von Deimling A, Witt O, Taylor MD, Wolf S, Karajannis MA, Eberhart CG, Scheurlen W, Hasselblatt M, Ligon KL, Kieran MW, Korbel JO, Yaspo ML, Brors B, Felsberg J, Reifenberger G, Collins VP, Jabado N, Eils R, Lichter P, Pfister SM, International Cancer Genome Consortium PedBrain Tumor P (2013) Recurrent somatic alterations of FGFR1 and NTRK2 in pilocytic astrocytoma. Nat Genet 45: 927–932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rodriguez FJ, Scheithauer BW, Burger PC, Jenkins S, Giannini C (2010) Anaplasia in pilocytic astrocytoma predicts aggressive behavior. Am J Surg Pathol 34: 147–160. [DOI] [PubMed] [Google Scholar]
- 7.Rodriguez EF, Scheithauer BW, Giannini C, Rynearson A, Cen L, Hoesley B, Gilmer-Flynn H, Sarkaria JN, Jenkins S, Long J, Rodriguez FJ (2011) PI3K/AKT pathway alterations are associated with clinically aggressive and histologically anaplastic subsets of pilocytic astrocytoma. Acta Neuropathol 121: 407–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Reinhardt A, Stichel D, Schrimpf D, Sahm F, Korshunov A, Reuss DE, Koelsche C, Huang K, Wefers AK, Hovestadt V, Sill M, Gramatzki D, Felsberg J, Reifenberger G, Koch A, Thomale UW, Becker A, Hans VH, Prinz M, Staszewski O, Acker T, Dohmen H, Hartmann C, Mueller W, Tuffaha MSA, Paulus W, Hess K, Brokinkel B, Schittenhelm J, Monoranu CM, Kessler AF, Loehr M, Buslei R, Deckert M, Mawrin C, Kohlhof P, Hewer E, Olar A, Rodriguez FJ, Giannini C, NageswaraRao AA, Tabori U, Nunes NM, Weller M, Pohl U, Jaunmuktane Z, Brandner S, Unterberg A, Hanggi D, Platten M, Pfister SM, Wick W, Herold-Mende C, Jones DTW, von Deimling A, Capper D (2018) Anaplastic astrocytoma with piloid features, a novel molecular class of IDH wildtype glioma with recurrent MAPK pathway, CDKN2A/B and ATRX alterations. Acta Neuropathol 136: 273–291. [DOI] [PubMed] [Google Scholar]
- 9.Rodriguez FJ, Brosnan-Cashman JA, Allen SJ, Vizcaino MA, Giannini C, Camelo-Piragua S, Webb M, Matsushita M, Wadhwani N, Tabbarah A, Hamideh D, Jiang L, Chen L, Arvanitis LD, Alnajar HH, Barber JR, Rodriguez-Velasco A, Orr B, Heaphy CM (2019) Alternative lengthening of telomeres, ATRX loss and H3-K27M mutations in histologically defined pilocytic astrocytoma with anaplasia. Brain Pathol 29: 126–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Olar A, Tran D, Mehta VP, Reinhardt A, Manekia JH, Garnovskaya M, Ellezam B, Luthra R, Sulman EP, Mohila CA, Campbell GA, Powell SZ, Fuller GN, Aldape KD, Adesina AM (2018) ATRX protein loss and deregulation of PI3K/AKT pathway is frequent in pilocytic astrocytoma with anaplastic features. Clin Neuropathol, 10.5414/NP301105. [DOI] [PubMed] [Google Scholar]
- 11.Ballester LY, Penas-Prado M, Leeds NE, Huse JT, Fuller GN (2018) FGFR1 tyrosine kinase domain duplication in pilocytic astrocytoma with anaplasia. Cold Spring Harb Mol Case Stud 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Giannini C, Scheithauer BW, Burger PC, Brat DJ, Wollan PC, Lach B, O’Neill BP (1999) Pleomorphic xanthoastrocytoma: what do we really know about it? Cancer 85: 2033–2045. [PubMed] [Google Scholar]
- 13.Ida CM, Rodriguez FJ, Burger PC, Caron AA, Jenkins SM, Spears GM, Aranguren DL, Lachance DH, Giannini C (2015) Pleomorphic Xanthoastrocytoma: Natural History and Long-Term Follow-Up. Brain Pathol 25: 575–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Thom M, Blumcke I, Aronica E (2012) Long-term epilepsy-associated tumors. Brain Pathol 22: 350–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Crespo-Rodriguez AM, Smirniotopoulos JG, Rushing EJ (2007) MR and CT imaging of 24 pleomorphic xanthoastrocytomas (PXA) and a review of the literature. Neuroradiology 49: 307–315. [DOI] [PubMed] [Google Scholar]
- 16.Giannini C, Scheithauer BW, Lopes MB, Hirose T, Kros JM, VandenBerg SR (2002) Immunophenotype of pleomorphic xanthoastrocytoma. Am J Surg Pathol 26: 479–485. [DOI] [PubMed] [Google Scholar]
- 17.Powell SZ, Yachnis AT, Rorke LB, Rojiani AM, Eskin TA (1996) Divergent differentiation in pleomorphic xanthoastrocytoma. Evidence for a neuronal element and possible relationship to ganglion cell tumors. Am J Surg Pathol 20: 80–85. [DOI] [PubMed] [Google Scholar]
- 18.Kepes JJ, Rubinstein LJ, Eng LF (1979) Pleomorphic xanthoastrocytoma: a distinctive meningocerebral glioma of young subjects with relatively favorable prognosis. A study of 12 cases. Cancer 44: 1839–1852. [DOI] [PubMed] [Google Scholar]
- 19.Li YS, Ramsay DA, Fan YS, Armstrong RF, Del Maestro RF (1995) Cytogenetic evidence that a tumor suppressor gene in the long arm of chromosome 1 contributes to glioma growth. Cancer Genet Cytogenet 84: 46–50. [DOI] [PubMed] [Google Scholar]
- 20.Sawyer JR, Roloson GJ, Chadduck WM, Boop FA (1991) Cytogenetic findings in a pleomorphic xanthoastrocytoma. Cancer Genet Cytogenet 55: 225–230. [DOI] [PubMed] [Google Scholar]
- 21.Sawyer JR, Thomas EL, Roloson GJ, Chadduck WM, Boop FA (1992) Telomeric associations evolving to ring chromosomes in a recurrent pleomorphic xanthoastrocytoma. Cancer Genet Cytogenet 60: 152–157. [DOI] [PubMed] [Google Scholar]
- 22.Weber RG, Hoischen A, Ehrler M, Zipper P, Kaulich K, Blaschke B, Becker AJ, Weber-Mangal S, Jauch A, Radlwimmer B, Schramm J, Wiestler OD, Lichter P, Reifenberger G (2007) Frequent loss of chromosome 9, homozygous CDKN2A/p14(ARF)/CDKN2B deletion and low TSC1 mRNA expression in pleomorphic xanthoastrocytomas. Oncogene 26: 1088–1097. [DOI] [PubMed] [Google Scholar]
- 23.Dias-Santagata D, Lam Q, Vernovsky K, Vena N, Lennerz JK, Borger DR, Batchelor TT, Ligon KL, Iafrate AJ, Ligon AH, Louis DN, Santagata S (2011) BRAF V600E mutations are common in pleomorphic xanthoastrocytoma: diagnostic and therapeutic implications. PLoS One 6: e17948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ida CM, Vrana JA, Rodriguez FJ, Jentoft ME, Caron AA, Jenkins SM, Giannini C (2013) Immunohistochemistry is highly sensitive and specific for detection of BRAF V600E mutation in pleomorphic xanthoastrocytoma. Acta Neuropathol Commun 1: 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chappe C, Padovani L, Scavarda D, Forest F, Nanni-Metellus I, Loundou A, Mercurio S, Fina F, Lena G, Colin C, Figarella-Branger D (2013) Dysembryoplastic neuroepithelial tumors share with pleomorphic xanthoastrocytomas and gangliogliomas BRAF(V600E) mutation and expression. Brain Pathol 23: 574–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Donson AM, Kleinschmidt-DeMasters BK, Aisner DL, Bemis LT, Birks DK, Levy JM, Smith AA, Handler MH, Foreman NK, Rush SZ (2014) Pediatric brainstem gangliogliomas show BRAF(V600E) mutation in a high percentage of cases. Brain Pathol 24: 173–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schindler G, Capper D, Meyer J, Janzarik W, Omran H, Herold-Mende C, Schmieder K, Wesseling P, Mawrin C, Hasselblatt M, Louis DN, Korshunov A, Pfister S, Hartmann C, Paulus W, Reifenberger G, von Deimling A (2011) Analysis of BRAF V600E mutation in 1,320 nervous system tumors reveals high mutation frequencies in pleomorphic xanthoastrocytoma, ganglioglioma and extra-cerebellar pilocytic astrocytoma. Acta Neuropathol 121: 397–405. [DOI] [PubMed] [Google Scholar]
- 28.Balss J, Meyer J, Mueller W, Korshunov A, Hartmann C, von Deimling A (2008) Analysis of the IDH1 codon 132 mutation in brain tumors. Acta Neuropathol 116: 597–602. [DOI] [PubMed] [Google Scholar]
- 29.Yamada S, Kipp BR, Voss JS, Giannini C, Raghunathan A (2016) Combined “Infiltrating Astrocytoma/Pleomorphic Xanthoastrocytoma” Harboring IDH1 R132H and BRAF V600E Mutations. Am J Surg Pathol 40: 279–284. [DOI] [PubMed] [Google Scholar]
- 30.Kaulich K, Blaschke B, Numann A, von Deimling A, Wiestler OD, Weber RG, Reifenberger G (2002) Genetic alterations commonly found in diffusely infiltrating cerebral gliomas are rare or absent in pleomorphic xanthoastrocytomas. J Neuropathol Exp Neurol 61: 1092–1099. [DOI] [PubMed] [Google Scholar]
- 31.Giannini C, Hebrink D, Scheithauer BW, Dei Tos AP, James CD (2001) Analysis of p53 mutation and expression in pleomorphic xanthoastrocytoma. Neurogenetics 3: 159–162. [DOI] [PubMed] [Google Scholar]
- 32.Bettegowda C, Agrawal N, Jiao Y, Wang Y, Wood LD, Rodriguez FJ, Hruban RH, Gallia GL, Binder ZA, Riggins CJ, Salmasi V, Riggins GJ, Reitman ZJ, Rasheed A, Keir S, Shinjo S, Marie S, McLendon R, Jallo G, Vogelstein B, Bigner D, Yan H, Kinzler KW, Papadopoulos N (2013) Exomic sequencing of four rare central nervous system tumor types. Oncotarget 4: 572–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang J, Wu G, Miller CP, Tatevossian RG, Dalton JD, Tang B, Orisme W, Punchihewa C, Parker M, Qaddoumi I, Boop FA, Lu C, Kandoth C, Ding L, Lee R, Huether R, Chen X, Hedlund E, Nagahawatte P, Rusch M, Boggs K, Cheng J, Becksfort J, Ma J, Song G, Li Y, Wei L, Wang J, Shurtleff S, Easton J, Zhao D, Fulton RS, Fulton LL, Dooling DJ, Vadodaria B, Mulder HL, Tang C, Ochoa K, Mullighan CG, Gajjar A, Kriwacki R, Sheer D, Gilbertson RJ, Mardis ER, Wilson RK, Downing JR, Baker SJ, Ellison DW, St. Jude Children’s Research Hospital-Washington University Pediatric Cancer Genome P (2013) Whole-genome sequencing identifies genetic alterations in pediatric low-grade gliomas. Nat Genet 45: 602–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Louis DN, Ohgaki H, Wiestler OD, Cavenee WK (Editors) (2016) World Health Organization Histological Classification of Tumours of the Central Nervous System. International Agency for Research on Cancer, France: (Revised 4th edition). [Google Scholar]
- 35.Kepes JJ (1993) Pleomorphic xanthoastrocytoma: the birth of a diagnosis and a concept. Brain Pathol 3: 269–274. [DOI] [PubMed] [Google Scholar]
- 36.Schmidt Y, Kleinschmidt-DeMasters BK, Aisner DL, Lillehei KO, Damek D (2013) Anaplastic PXA in adults: case series with clinicopathologic and molecular features. J Neurooncol 111: 59–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vaubel RA, Caron AA, Yamada S, Decker PA, Eckel Passow JE, Rodriguez FJ, Nageswara Rao AA, Lachance D, Parney I, Jenkins R, Giannini C (2018) Recurrent copy number alterations in low-grade and anaplastic pleomorphic xanthoastrocytoma with and without BRAF V600E mutation. Brain Pathol 28: 172–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ma C, Feng R, Chen H, Hameed NUF, Aibaidula A, Song Y, Wu J (2018) BRAF V600E, TERT, and IDH2 Mutations in Pleomorphic Xanthoastrocytoma: Observations from a Large Case-Series Study. World Neurosurg 120: e1225–e1233. [DOI] [PubMed] [Google Scholar]
- 39.Phillips JJ, Gong H, Chen K, Joseph NM, van Ziffle J, Bastian BC, Grenert JP, Kline CN, Mueller S, Banerjee A, Nicolaides T, Gupta N, Berger MS, Lee HS, Pekmezci M, Tihan T, Bollen AW, Perry A, Shieh JTC, Solomon DA (2019) The genetic landscape of anaplastic pleomorphic xanthoastrocytoma. Brain Pathol 29: 85–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Phillips JJ, Gong H, Chen K, Joseph NM, van Ziffle J, Jin LW, Bastian BC, Bollen AW, Perry A, Nicolaides T, Solomon DA, Shieh JT (2016) Activating NRF1-BRAF and ATG7-RAF1 fusions in anaplastic pleomorphic xanthoastrocytoma without BRAF p.V600E mutation. Acta Neuropathol 132: 757–760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Luyken C, Blumcke I, Fimmers R, Urbach H, Wiestler OD, Schramm J (2004) Supratentorial gangliogliomas: histopathologic grading and tumor recurrence in 184 patients with a median follow-up of 8 years. Cancer 101: 146–155. [DOI] [PubMed] [Google Scholar]
- 42.Blumcke I, Aronica E, Urbach H, Alexopoulos A, Gonzalez-Martinez JA (2014) A neuropathology-based approach to epilepsy surgery in brain tumors and proposal for a new terminology use for long-term epilepsy-associated brain tumors. Acta Neuropathol 128: 39–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Adachi Y, Yagishita A (2008) Gangliogliomas: Characteristic imaging findings and role in the temporal lobe epilepsy. Neuroradiology 50: 829–834. [DOI] [PubMed] [Google Scholar]
- 44.Blumcke I, Giencke K, Wardelmann E, Beyenburg S, Kral T, Sarioglu N, Pietsch T, Wolf HK, Schramm J, Elger CE, Wiestler OD (1999) The CD34 epitope is expressed in neoplastic and malformative lesions associated with chronic, focal epilepsies. Acta Neuropathol 97: 481–490. [DOI] [PubMed] [Google Scholar]
- 45.Hoischen A, Ehrler M, Fassunke J, Simon M, Baudis M, Landwehr C, Radlwimmer B, Lichter P, Schramm J, Becker AJ, Weber RG (2008) Comprehensive characterization of genomic aberrations in gangliogliomas by CGH, array-based CGH and interphase FISH. Brain Pathol 18: 326–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Koelsche C, Wohrer A, Jeibmann A, Schittenhelm J, Schindler G, Preusser M, Lasitschka F, von Deimling A, Capper D (2013) Mutant BRAF V600E protein in ganglioglioma is predominantly expressed by neuronal tumor cells. Acta Neuropathol 125: 891–900. [DOI] [PubMed] [Google Scholar]
- 47.Dougherty MJ, Santi M, Brose MS, Ma C, Resnick AC, Sievert AJ, Storm PB, Biegel JA (2010) Activating mutations in BRAF characterize a spectrum of pediatric low-grade gliomas. Neuro Oncol 12: 621–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Horbinski C, Kofler J, Yeaney G, Camelo-Piragua S, Venneti S, Louis DN, Perry A, Murdoch G, Nikiforova M (2011) Isocitrate dehydrogenase 1 analysis differentiates gangliogliomas from infiltrative gliomas. Brain Pathol 21: 564–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Perry A, Burton SS, Fuller GN, Robinson CA, Palmer CA, Resch L, Bigio EH, Gujrati M, Rosenblum MK (2010) Oligodendroglial neoplasms with ganglioglioma-like maturation: a diagnostic pitfall. Acta Neuropathol 120: 237–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pages M, Beccaria K, Boddaert N, Saffroy R, Besnard A, Castel D, Fina F, Barets D, Barret E, Lacroix L, Bielle F, Andreiuolo F, Tauziede-Espariat A, Figarella-Branger D, Puget S, Grill J, Chretien F, Varlet P (2018) Co-occurrence of histone H3 K27M and BRAF V600E mutations in paediatric midline grade I ganglioglioma. Brain Pathol 28: 103–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Prabhakaran N, Guzman MA, Navalkele P, Chow-Maneval E, Batanian JR (2018) Novel TLE4-NTRK2 fusion in a ganglioglioma identified by array-CGH and confirmed by NGS: Potential for a gene targeted therapy. Neuropathology, 10.1111/neup.12458. [DOI] [PubMed] [Google Scholar]
- 52.Pekmezci M, Villanueva-Meyer JE, Goode B, Van Ziffle J, Onodera C, Grenert JP, Bastian BC, Chamyan G, Maher OM, Khatib Z, Kleinschmidt-DeMasters BK, Samuel D, Mueller S, Banerjee A, Clarke JL, Cooney T, Torkildson J, Gupta N, Theodosopoulos P, Chang EF, Berger M, Bollen AW, Perry A, Tihan T, Solomon DA (2018) The genetic landscape of ganglioglioma. Acta Neuropathol Commun 6: 47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.von Deimling A, Fimmers R, Schmidt MC, Bender B, Fassbender F, Nagel J, Jahnke R, Kaskel P, Duerr EM, Koopmann J, Maintz D, Steinbeck S, Wick W, Platten M, Muller DJ, Przkora R, Waha A, Blumcke B, Wellenreuther R, Meyer-Puttlitz B, Schmidt O, Mollenhauer J, Poustka A, Stangl AP, Lenartz D, von Ammon K (2000) Comprehensive allelotype and genetic anaysis of 466 human nervous system tumors. J Neuropathol Exp Neurol 59: 544–558. [DOI] [PubMed] [Google Scholar]
- 54.Zanello M, Pages M, Tauziede-Espariat A, Saffroy R, Puget S, Lacroix L, Dezamis E, Devaux B, Chretien F, Andreiuolo F, Sainte-Rose C, Zerah M, Dhermain F, Dumont S, Louvel G, Meder JF, Grill J, Dufour C, Pallud J, Varlet P (2016) Clinical, Imaging, Histopathological and Molecular Characterization of Anaplastic Ganglioglioma. J Neuropathol Exp Neurol 75: 971–980. [DOI] [PubMed] [Google Scholar]
- 55.Terrier LM, Bauchet L, Rigau V, Amelot A, Zouaoui S, Filipiak I, Caille A, Almairac F, Aubriot-Lorton MH, Bergemer-Fouquet AM, Bord E, Cornu P, Czorny A, Dam Hieu P, Debono B, Delisle MB, Emery E, Farah W, Gauchotte G, Godfraind C, Guyotat J, Irthum B, Janot K, Le Reste PJ, Liguoro D, Loiseau H, Lot G, Lubrano V, Mandonnet E, Menei P, Metellus P, Milin S, Muckenstrum B, Roche PH, Rousseau A, Uro-Coste E, Vital A, Voirin J, Wager M, Zanello M, Francois P, Velut S, Varlet P, Figarella-Branger D, Pallud J, Zemmoura I, Club de Neuro-Oncologie of the Societe Francaise de N (2017) Natural course and prognosis of anaplastic gangliogliomas: a multicenter retrospective study of 43 cases from the French Brain Tumor Database. Neuro Oncol 19: 678–688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bouali S, Ben Said I, Zehani A, Drissi C, Bouhoula A, Kallel J, Jemel H (2018) Pediatric Intracranial Anaplastic Gangliogliomas: Illustrative Case and Systematic Review. World Neurosurg 119: 220–231. [DOI] [PubMed] [Google Scholar]
- 57.Rushing EJ, Thompson LD, Mena H (2003) Malignant transformation of a dysembryoplastic neuroepithelial tumor after radiation and chemotherapy. Ann Diagn Pathol 7: 240–244. [DOI] [PubMed] [Google Scholar]
- 58.Ray WZ, Blackburn SL, Casavilca-Zambrano S, Barrionuevo C, Orrego JE, Heinicke H, Dowling JL, Perry A (2009) Clinicopathologic features of recurrent dysembryoplastic neuroepithelial tumor and rare malignant transformation: a report of 5 cases and review of the literature. J Neurooncol 94: 283–292. [DOI] [PubMed] [Google Scholar]
- 59.Chao L, Tao XB, Jun YK, Xia HH, Wan WK, Tao QS (2013) Recurrence and histological evolution of dysembryoplastic neuroepithelial tumor: A case report and review of the literature. Oncol Lett 6: 907–914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.VandenBerg SR, May EE, Rubinstein LJ, Herman MM, Perentes E, Vinores SA, Collins VP, Park TS (1987) Desmoplastic supratentorial neuroepithelial tumors of infancy with divergent differentiation potential (“desmoplastic infantile gangliogliomas”). Report on 11 cases of a distinctive embryonal tumor with favorable prognosis. J Neurosurg 66: 58–71. [DOI] [PubMed] [Google Scholar]