

Abstract
Background.
Trauma-induced coagulopathy (TIC) is a common and deadly bleeding disorder. Platelet dysfunction is present during TIC, but its mechanisms remain unclear. Platelets are currently thought to become “exhausted”, a state in which they have released their granule contents and can no longer aggregate or contract.
Methods.
This prospective observational cohort study tested the hypothesis that platelet exhaustion is present during TIC and characterized the early time course of platelet dysfunction. Blood was collected from 95 adult trauma patients at a Level I trauma center at time of Emergency Department arrival and several time points over 72 hours. Platelet activation state and function were characterized using CD62P (P-selectin) and PAC-1 surface membrane staining, PFA-100, aggregometry, viscoelastic platelet mapping, and, to test for exhaustion, their ability to express CD62P after ex vivo adenosine diphosphate (ADP) agonism. Platelet function was compared between patients with and without TIC, defined by prothrombin time ≥ 18 seconds.
Results.
Platelets in TIC showed no initial increase in their level of surface activation markers or impairment of their capacity to express CD62P in response to ADP stimulation. However, TIC platelets were impaired in nearly all functional assays, spanning adhesion, aggregation, and contraction. These effects largely remained after controlling for platelet count and fibrinogen concentration and resolved after 8 hours.
Conclusion.
TIC platelets exhibit early impairment of adhesion, aggregation, and contraction with retained alpha granule secretion ability, suggesting a specific mechanism of cytoskeletal or integrin dysfunction that is not a result of more general platelet exhaustion.
Keywords: Blood platelet disorders, Hemorrhagic disorders, Platelet activation, Platelet aggregation, Trauma
Introduction
Trauma is a leading cause of death and disability, both in the United States and worldwide.(1) Bleeding is the second leading cause of death and the leading cause of preventable death in injured patients.(2, 3) Trauma-induced coagulopathy (TIC) is a common and deadly complication of injury, occurring in 25–35% of severely injured patients and associated with a 3- to 4-fold increase in mortality.(4, 5) TIC manifests as a complex, multifactorial derangement of hemostasis.
The importance of platelets in hemostasis is difficult to overstate. They serve as the primary substrate for clot initiation, the staging ground for the coagulation cascade and thrombin generation, and a key contributor to mechanical clot strength. In fact, platelet-mediated clot contraction contributes over 80% of overall clot stiffness and is the primary regulator of clot lysis after injury.(6–9)
The platelet lesion in TIC is complex and poorly understood. Platelet adhesion, measured using the PFA-100, has trended towards shorter aperture closure times after trauma, suggesting enhanced platelet adhesive function.(10) However, decreased platelet aggregation in response to common agonists is common, present in as many as 45% of patients with major trauma, and is associated with a remarkable 10-fold higher early mortality.(11–13) Further examination of platelet-mediated clot contraction using thrombelastography platelet mapping has also detected significantly decreased contractile responsiveness to ADP, arachidonic, and, to a lesser extent, thrombin receptor stimulation in trauma patients.(14)
Despite this appearance of platelet hypofunction, platelets tend to appear activated after injury, as determined by membrane markers of platelet activation. Trauma patients tend to have either normal or increased levels of platelet-derived microparticles and increased membrane markers of platelet activation (CD62P and PAC-1).(10, 13) These increased levels of activation markers combined with decreased function has spawned a theory in which platelets quickly become “exhausted” after major trauma, meaning they have become activated and released the contents of their granules and consequently lose their ability to contribute mechanically to clot formation by their aggregation and contraction. Platelet exhaustion was originally described in a small group of patients with a heterogeneous collection of disease processes: renal allograft rejection, hemolytic uremic syndrome or thrombotic thrombocytopenic purpura, disseminated intravascular coagulation due to incompatible transfusion, and systemic lupus erythematosus.(15) They collectively exhibited a particular platelet phenotype of decreased aggregation, reduced adenine nucleotide and serotonin content, and bleeding time prolonged beyond what the platelet count would predict. Supporting this theory for trauma, platelet CD62P (P-selectin) expression, an indicator of alpha granule release, after ex vivo ADP stimulation has been found to be modestly decreased in platelets from trauma patients.(13)
We previously demonstrated that significant platelet dysfunction contributed to distinct hemostatic responses in Emergency Department patients when identified using hierarchical clustering analysis.(16) Platelet-induced clot contraction was also found to be strongly associated with thromboelastography (TEG) clot formation when measured in the same Emergency Department cohort(7), suggesting that platelet dysfunction was an important component of TIC in this cohort of trauma patients. In the present study, we look more specifically at platelet function in this cohort to elucidate the mechanism of the noted platelet dysfunction. Because trauma patients’ coagulability status can be dynamic over the early days after injury, we also examined changes in platelet function over the first 3 days of hospital admission. We hypothesized that platelets from those subjects meeting criteria for TIC would display platelet exhaustion, identified by increased surface membrane markers of activation (CD62P and PAC-1), decreased secretion response to further ex vivo agonism, and decreased adhesion, aggregation, and clot contraction. Our secondary hypothesis was that this phenotype would gradually normalize over the first three days, with any differences becoming non-significant at 72 hours. To test these hypotheses, we prospectively measured platelet phenotypes in this cohort at Emergency Department arrival and serially over the first 3 days of hospital admission.
Methods
Study Design and Patient Population
This was a prospective cohort study of trauma patients presenting to an urban Level I trauma center, from which some of the non-platelet-specific findings have been previously reported.(7) In brief, trauma patients were identified from trauma team activations, which are triggered by criteria meant to capture patients with serious injury. Subjects were screened, and consent for study participation was obtained from either the patient or a legally authorized representative. Blood was collected into standard vacutainers at the time of ED arrival and before the patients received any blood products. Additional blood samples were collected at 8, 24, 48, and 72 hours after arrival. To minimize confounding influences of blood transfusion, minor injury, or survival bias, subjects were excluded if they received any blood products prior to study sample blood draw, were not expected to survive for 3 days after injury, or were minimally injured and either did not require admission or were expected to be discharged from the hospital before 3 days of hospitalization. Vital signs, injury profiles, and outcomes were collected from the medical record, and the injury severity score (ISS) and sequential organ failure assessment (SOFA) were calculated for each patient. The study was approved by the Virginia Commonwealth University institutional human subjects review board.
Assays
All samples were processed in the Virginia Commonwealth University coagulation advancement lab, Virginia Commonwealth University School of Pharmacy. All measurements were carried out within 2 hours of sample collection, as previously described.(7) Briefly, platelet adhesion and aggregation in response to collagen/ADP and collagen/epinephrine stimulation were measured by the PFA-100 (Dade International, Deerfield, IL). Isolated platelet aggregation in response to ADP (10 μM) and collagen (2 μg/mL) was measured using a whole blood impedance aggregometer (Chronolog, Havertown, PA). Platelet contractile force (PCF) and clot elastic modulus (CEM) were measured in re-calcified whole blood using the Hemostasis Analysis System (HAS) (Hemodyne Inc., Richmond, VA). Viscoelastic clot formation was measured in whole blood using TEG (TEG 5000, Haemonetics Corp., Braintree, MA) using recalcification (10mM Ca2+) and kaolin stimulation, as well as standard TEG platelet mapping (TEG-PM). Platelet flow cytometry was performed using an Accuri™ C6 flow cytometer (BD Biosciences, San Jose, CA, USA) on whole blood samples stained with antibodies specific to the activated form of αIIbβ3 (monoclonal antibody PAC-1 conjugated with fluorescein isothiocyanate conjugate [FITC], BD Biosciences, San Jose, CA, USA) and CD62P (mouse anti-human CD62P monoclonal antibody conjugated with phycoerythrin [PE], BD Pharmingen, Franklin Lakes, NJ). CD62P staining was also performed on whole blood samples after ex vivo incubation with ADP (5μM) for 2 minutes to detect platelet responsiveness to further stimulation. The Accuri C6 fluidics was set to the medium flow rate and a forward scatter threshold of 30,000. Twenty thousand events were collected into a pre-set platelet gate using standard methods including anti-CD41a conjugated with PE-Cy5 (mouse anti-human, BD Pharmingen, Franklin Lakes, NJ) as a platelet marker. Collected data were analyzed using the FlowJo software package version 7.6.5 (FlowJo, Ashland, OR).
Definitions and Statistical Approach
In the primary analysis, patients were divided into two groups according to the presence or absence of TIC at Emergency Department arrival, which was originally defined as PT ≥ 18.(17) This definition was chosen for consistency with previous literature and because this plasma-based definition is independent of any specific type of platelet dysfunction. We have used a similar definition in previous studies, thus facilitating comparisons.(7) Demographics and test results were compared using the chi square test for dichotomous variables, Student’s t-test for normally distributed variables, and Mann-Whitney U test for non-normally distributed variables at each individual time point.
To test whether any platelet function defects were independent of platelet count and fibrinogen concentration, we fit the data to a multivariate linear regression model. The model included as independent variables platelet count, fibrinogen concentration, and a dichotomous variable representing the presence or absence of TIC and as the dependent variable the numerical assay result of interest. The Wald test was performed on the TIC term to determine statistical significance in predicting an independent difference in assay result.
A secondary analysis was also performed to examine the time course of platelet function in TIC. This analysis included only the patients with a complete set of data for the duration of the 72-hour study. Patients were characterized as either having or not having TIC according to their results at hour 0. To determine the overall and group-specific effects of time, a generalized estimating equation (GEE) was fit to the data with terms for time, the presence of TIC, and the interaction between time and the presence of TIC. Robust standard errors were used to account for heteroskedasticity. Lab values at individual time points were compared by Wilcoxon rank-sum test.
Several lab values were censored beyond certain detection limits. These include PT > 70 sec, PTT > 120 sec, fibrinogen < 100 mg/dL, and PFA-100 collagen/ADP and collagen/epinephrine closure times > 300 sec.
For all tests, a p-value less than 0.05 was considered significant. All statistics were run using Stata SE 13.1 (StataCorp, College Station, TX).
Results
One hundred seventy three trauma patients were screened for inclusion, of which 95 met inclusion criteria and were included in the primary analysis with 15 exhibiting TIC, as previously described.(7) Descriptive characteristics are included in Table 1. In brief, at the time of ED arrival TIC patients had lower systolic and diastolic blood pressure, higher heart rate, higher Injury Severity Score, lower Glasgow Coma Score, higher base deficit, and lower hemoglobin and hematocrit. The coagulation data that are not platelet-specific were reported previously.(7)
Table 1. Descriptive characteristics of included patients, stratified by the presence of TIC.
(7) Vital signs and laboratory values are at time of ED arrival. Data are presented as median (interquartile range). P-values obtained by Wilcoxon rank-sum test.
| All Included Patients (n=95) |
No TIC (n=80) | TIC (n=15) | P | |
|---|---|---|---|---|
| Demographics/Vital Signs | ||||
| Age, years | 33 (24–50) | 33 (24–52) | 36 (22–45) | 0.353 |
| Heart Rate, beats per min | 98 (78–115) | 93 (78–111) | 110 (101–136) | 0.006 |
| Systolic Blood Pressure, mmHg | 126 (112–149) | 129 (116–152) | 112 (89–127) | 0.010 |
| Diastolic Blood Pressure, mmHg | 80 (67–90) | 80 (68–91) | 71 (53–81) | 0.025 |
| Respiratory Rate, breaths per min | 18 (15–22) | 18 (15–21) | 20 (17–26) | 0.133 |
| Oxygen Saturation, % | 99 (97–100) | 99 (97–100) | 98 (95–100) | 0.522 |
| Temperature, °C | 36.4 (36.1–36.9) | 36.5 (36.1–36.9) | 36.3 (35.7–36.5) | 0.182 |
| Injury Severity Score | 17 (10–27) | 15 (9–25) | 30 (21–43) | <0.001 |
| Glasgow Coma Score | 15 (6–15) | 15 (9–15) | 9 (3–15) | 0.048 |
| Laboratory Values | ||||
| [Lactate], mmol/L | 3.6 (2.4–5.2) | 3.4 (2.4–5.0) | 4.5 (3.1–5.5) | 0.088 |
| Base Deficit, mEq/L | 2.1 (−0.4–5.4) | 1.6 (−0.8–4.9) | 5.4 (2.1–14.5) | 0.001 |
| Hemoglobin, g/dL | 12.9 (11.4–14.0) | 13.3 (11.8–14.5) | 10.5 (8.6–11.9) | <0.001 |
| Hematocrit, % | 37.0 (32.7–41.9) | 37.9 (33.7–43.1) | 32.4 (29.4–36.4) | <0.001 |
| Platelet Count, x109/L | 229 (183–273) | 231 (187–279) | 211 (156–250) | 0.162 |
| PT, sec | 14.7 (13.5–16.1) | 14.4 (13.4–15.5) | 19.5 (18.5–21.2) | <0.001 |
| aPTT, sec | 32.2 (29.3–35.7) | 31.3 (28.8–34.2) | 39.7 (34.4–49.4) | <0.001 |
| Fluid Resuscitation, mL | ||||
| Total Crystalloid at Hour 0 | 1300 (400–2000) | 1000 (300–2000) | 2000 (1400–2600) | 0.037 |
| Total Blood Products at Hour 0 | 0 (0–0) | 0 (0–0) | 0 (0–0) | 0.212 |
Platelet test results at the time of ED arrival are summarized in Table 2. TIC patients showed longer PFA-100 closure times in response to collagen and epinephrine, though not in response to collagen and ADP. They also showed impaired aggregation in response to both ADP and collagen. Few platelets stained positive for PAC-1 or CD62P at ED arrival, and there were no differences between groups. Platelets from both groups also responded briskly to ex vivo ADP stimulation with similarly increased expression of CD62P (Figure 1).
Table 2. Platelet-specific data for all trauma patients at hour 0, stratified by the presence of TIC.
Data are presented as median (interquartile range). P-values obtained by Wilcoxon rank-sum test. REF, reference range; AUC, area under the curve.
| Lab Value | No TIC (n=80) | TIC (n=15) | P |
|---|---|---|---|
| Platelet count, x109/L (REF 150–450) | 231 (187–279) | 211 (156–250) | 0.162 |
| PFA-100 collagen/epinephrine closure time, sec (REF <180) |
97 (84–126) | 155 (119–300) | 0.002 |
| PFA-100 collagen/ADP closure time, sec (REF <120) | 64 (54–81) | 70 (49–129) | 0.613 |
| Platelet ADP aggregation, ohms (REF 6–24) | 11.0 (8.5–13.0) | 6.0 (3.5–8.5) | 0.001 |
| Platelet collagen aggregation, ohms (REF 14–28) | 14.5 (12.0–18.0) | 12.0 (6.5–13.5) | 0.002 |
| Platelet Contractile Force, Kdynes | 8.0 (6.6–9.4) | 6.0 (5.0–8.7) | 0.029 |
| TEG-PM ADP inhibition, % | 78.7 (57.8–97.0) | 96.1 (65.2–97.3) | 0.292 |
| TEG-PM AA inhibition, % | 38.9 (19.6–65.8) | 35.1 (13.7–52.0) | 0.356 |
| Percent PAC-1-positive | 6.0 (1.8–12.6) | 3.5 (0.5–10.2) | 0.265 |
| Percent CD62P-positive | 3.0 (1.4–7.8) | 2.6 (0.8–5.7) | 0.268 |
| Percent that became CD62P-positive after ADP stimulation |
70.1 (57.5–76.8) | 72.5 (63.2–77.2) | 0.914 |
Figure 1. Platelets showed no difference in activation patterns between the TIC and non-TIC groups.
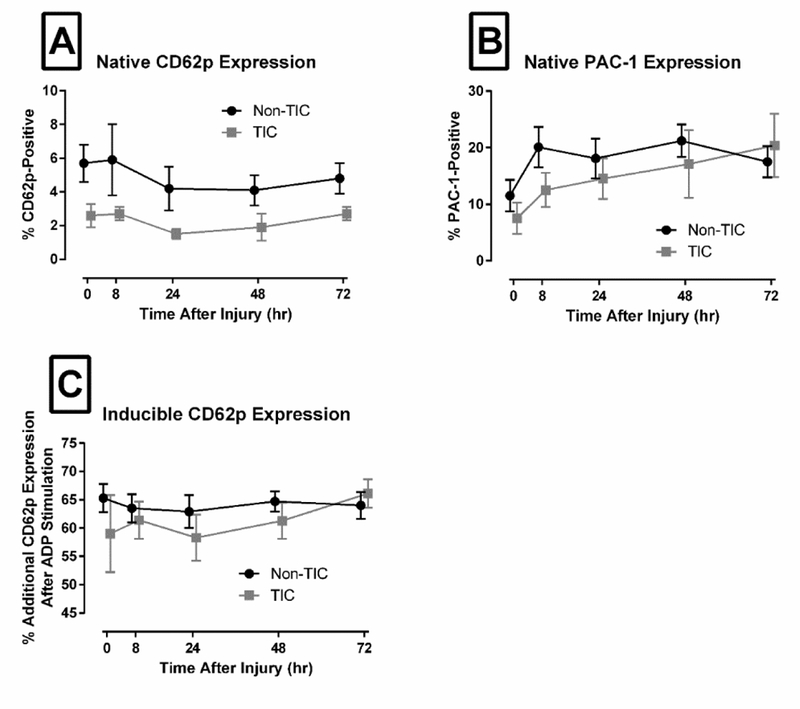
Plots show mean ± standard error. (A) CD62P expression in unstimulated patient sample was not significantly different between groups at any time point. (B) PAC-1 expression in unstimulated sample also showed no significant difference between groups at any time point. (C) Additional CD62P expression that is inducible by ex vivo addition of ADP also showed no significant difference between groups at any time point.
As previously described, TIC patients had decreased platelet counts and fibrinogen concentrations.(7) After controlling for these variables, the effect of TIC on PFA-100 closure time in response to collagen/epinephrine remained significant (p=0.010) and in response to PFA-100 collagen/ADP remained non-significant (p=0.362). The association between TIC and impaired platelet aggregation in response to both ADP and collagen also remained significant (p=0.007 and 0.010, respectively). The associations between TIC, TEG maximum amplitude (MA), and percent lysis at 30 minutes after onset (LY30) became non-significant after adjusting for platelet count and fibrinogen concentration (p=0.149 and 0.133, respectively). The association of TIC with PCF was also no longer significant (p=0.841). The effect of TIC on TEG-PM in response to ADP and AA remained non-significant (p=0.997 and 0.070, respectively).
Fifty subjects had a complete data set and were included in the longitudinal analysis. Their descriptive characteristics are summarized in Table 3, and their platelet function test results are summarized in Table 4 (GEE analysis) and Table 5 (individual time point data). Both aggregation and TEG MA were decreased in the TIC group across all time points. The platelet count decreased in both groups over time but more quickly in the TIC group. The fibrinogen concentration was decreased initially in the TIC group, but increased over time and more quickly for the TIC group. Remarkably, many of the early platelet defects seen in the TIC group at ED arrival were no longer significant by Hour 8. This response was seen in aggregation in responses to both ADP and collagen, TEG MA, and PCF. There were also general pro-hemostatic trends with time in many assays, including ADP aggregation, TEG MA, PCF, CEM, and TEG-PM ADP and AA inhibition. These values sometimes reached and exceeded the pro-hemostatic limit of the reference range. Collagen aggregation improved with time only in the TIC group. One notable exception to this trend is PFA-100, which prolonged with time in both groups. Individual time point comparisons revealed decreased platelet adhesion in the TIC group at early and late time points, though they remained mostly within the reference range. CD62P expression was lower across time in the TIC group, though no individual time point was different enough to reach significance. There were no differences in PAC-1 expression or ADP-induced CD62P expression across time or at any single time point.
Table 3. Descriptive characteristics of the 50 patients included in the longitudinal data analysis.
Data are presented as median (interquartile range). P-values obtained by Wilcoxon rank-sum test.
| All Included Patients (n=50) |
No TIC (n=38) | TIC (n=12) | P | |
|---|---|---|---|---|
| Demographics/Vital Signs | ||||
| Age, years | 37 (24–53) | 44 (25–54) | 27.5 (23.5–42.5) | 0.149 |
| Heart Rate, beats per min | 100 (84–116) | 98 (80–108) | 117 (104–135) | 0.011 |
| Systolic Blood Pressure, mmHg | 127.5 (112–151) | 132 (118–153) | 112 (94–116) | 0.005 |
| Diastolic Blood Pressure, mmHg | 76.5 (61–92) | 81 (64–96) | 61 (49.5–77) | 0.006 |
| Respiratory Rate, breaths per min | 18.5 (15–24) | 18 (14–24) | 20 (17.5–25) | 0.487 |
| Oxygen Saturation, % | 99 (97–100) | 99 (97–100) | 99 (96–100) | 0.812 |
| Temperature, °C | 36.3 (35.9–36.7) | 36.3 (36.0–36.8) | 36.0 (35.5–36.5) | 0.260 |
| Injury Severity Score | 22 (14–30) | 19 (13–29) | 28.5 (21–42) | 0.012 |
| Glasgow Coma Score | 13.5 (3–15) | 14.5 (3–15) | 7 (3–14) | 0.144 |
| Laboratory Values | ||||
| [Lactate], mmol/L | 4.4 (2.8–5.2) | 3.8 (2.7–4.9) | 5.2 (4.5–11.2) | 0.007 |
| Base Deficit, mEq/L | 3.1 (1.0–6.5) | 2.2 (0.7–4.8) | 7.8 (3.9–13.6) | <0.001 |
| Hemoglobin, g/dL | 13.3 (11.8–14.4) | 13.9 (12.5–14.9) | 11.0 (10.2–13.0) | <0.001 |
| Hematocrit, % | 36.5 (32.5–41.0) | 38.6 (35.3–43.6) | 32.3 (30.4–35.9) | 0.004 |
| Platelet Count, x109/L | 236 (177–279) | 243 (179–284) | 217 (151–265) | 0.156 |
| PT, sec | 15.1 (14.0–17.7) | 14.7 (13.7–15.3) | 20.2 (19.3–21.2) | <0.001 |
| aPTT, sec | 33.5 (29.5–38.2) | 32.3 (29.0–35.7) | 39.0 (32.6–46.1) | 0.007 |
| Crystalloid Fluid Resuscitation, mL | ||||
| Total Crystalloid Given at Hour 0 | 1500 (500–2000) | 1200 (350–2000) | 2100 (2000–3000) | 0.003 |
| Total Crystalloid Given at Hour 8 | 550 (0–2700) | 547 (0–2660) | 1668 (253–4090) | 0.520 |
| Blood Products at Hour 8, mL | ||||
| Total Blood Products | 0 (0–620) | 0 (0–310) | 1400 (310–2555) | <0.001 |
| Packed Red Blood Cells | 0 (0–620) | 0 (0–310) | 930 (310–1550) | <0.001 |
| Fresh Frozen Plasma | 0 (0–0) | 0 (0–0) | 225 (0–788) | <0.001 |
| Pooled Platelets | 0 (0–0) | 0 (0–0) | 20 (0–200) | <0.001 |
| Apheresis Platelets | 0 (0–0) | 0 (0–0) | 0 (0–0) | 0.011 |
| Cryoprecipitate | 0 (0–0) | 0 (0–0) | 20 (0–200) | 0.015 |
Table 4. Generalized estimating equation results for longitudinal data.
The coefficient represents the change in the lab value associated with either the presence of TIC (TIC), a 1-unit increase in time (Time), or a 1-unit increase in time in the presence of TIC (TIC*Time). GEE, generalized estimating equation; CI, confidence interval.
| Lab Value | GEE Coefficient [95% CI] |
P |
|---|---|---|
| Platelet count, x109/L: | ||
| TIC | −28.2 [−63.8, 7.3] | 0.119 |
| Time | −0.64 [−0.92, −0.36] | <0.001 |
| TIC*Time | −0.52 [−1.03, −0.02] | 0.042 |
| PT, sec: | ||
| TIC | 4.0 [3.3, 4.7] | <0.001 |
| Time | 0.01 [0, 0.02] | 0.156 |
| TIC*Time | −0.04 [−0.07, −0.02] | 0.001 |
| aPTT, sec: | ||
| TIC | 2.0 [−3.3, 7.3] | 0.455 |
| Time | 0.13 [0.06, 0.20] | <0.001 |
| TIC*Time | 0.04 [−0.13, 0.21] | 0.623 |
| Fibrinogen concentration, mg/dL: | ||
| TIC | −93.0 [−139.8, −46.2] | <0.001 |
| Time | 6.62 [5.66, 7.59] | <0.001 |
| TIC*Time | 1.97 [0.36, 3.58] | 0.016 |
| PFA-100 collagen/epinephrine closure time, sec: | ||
| TIC | 11.9 [−27.3, 51.0] | 0.553 |
| Time | 0.07 [−0.38, 0.53] | 0.747 |
| TIC*Time | 0.31 [−0.50, 1.12] | 0.459 |
| PFA-100 collagen/ADP closure time, sec: | ||
| TIC | 14.5 [−16.5, 45.5] | 0.360 |
| Time | 0.56 [0.18, 0.95] | 0.004 |
| TIC*Time | 0.95 [−0.12, 2.02] | 0.083 |
| Platelet ADP aggregation, ohms: | ||
| TIC | −2.4 [−4.3, −0.5] | 0.014 |
| Time | 0.02 [0, 0.04] | 0.014 |
| TIC*Time | 0.04 [−0.01, 0.09] | 0.103 |
| Platelet collagen aggregation, ohms: | ||
| TIC | −2.8 [−4.6, −1.1] | 0.002 |
| Time | 0.01 [−0.02, 0.03] | 0.619 |
| TIC*Time | 0.05 [0, 0.10] | 0.036 |
| HAS platelet contractile force, kdynes: | ||
| TIC | −1.2 [−2.4, 0] | 0.053 |
| Time | 0.04 [0.02, 0.07] | 0.001 |
| TIC*Time | 0.03 [−0.02, 0.08] | 0.277 |
| HAS clot elastic modulus, kdynes/cm2: | ||
| TIC | −2.6 [−7.0, 1.8] | 0.250 |
| Time | 0.17 [0.07, 0.27] | 0.001 |
| TIC*Time | −0.01 [−0.19, 0.17] | 0.926 |
| TEG MA, mm: | ||
| TIC | −3.5 [−6.7, −0.4] | 0.028 |
| Time | 0.11 [0.08, 0.15] | <0.001 |
| TIC*Time | 0.05 [−0.02, 0.12] | 0.145 |
| TEG LY30, %: | ||
| TIC | 2.7 [−0.4, 5.8] | 0.087 |
| Time | 0.02 [0, 0.04] | 0.113 |
| TIC*Time | −0.06 [−0.13, 0] | 0.051 |
| TEG-PM ADP inhibition, %: | ||
| TIC | −1.7 [−16.8, 13.4] | 0.822 |
| Time | −0.43 [−0.56, −0.30] | <0.001 |
| TIC*Time | −0.02 [−0.28, 0.25] | 0.905 |
| TEG-PM AA inhibition, %: | ||
| TIC | −9.3 [−22.8, 4.2] | 0.177 |
| Time | −0.23 [−0.39, −0.08] | 0.003 |
| TIC*Time | −0.06 [−0.42, 0.31] | 0.768 |
| Percent PAC-1-positive: | ||
| TIC | −6.7 [−13.7, 0.2] | 0.058 |
| Time | 0.05 [0.03, 0.13] | 0.261 |
| TIC*Time | 0.11 [−0.06, 0.29] | 0.207 |
| Percent CD62P-positive: | ||
| TIC | −3.1 [−5.7, −0.5] | 0.019 |
| Time | −0.02 [−0.05, 0.02] | 0.305 |
| TIC*Time | 0.02 [−0.02, 0.05] | 0.397 |
| Percent that became CD62P-positive after ADP stimulation: |
||
| TIC | −5.4 [−14.8, 4.0] | 0.256 |
| Time | 0 [−0.06, 0.06] | 0.940 |
| TIC*Time | 0.08 [−0.04, 0.21] | 0.180 |
Table 5. Longitudinal laboratory value data for subset of patients with complete longitudinal data, stratified by the presence of TIC at hour 0.
Data are presented as median (interquartile range). P-values obtained by Wilcoxon rank-sum test.
| Lab Value | Non-TIC (n=38) | TIC (n=12) | P |
|---|---|---|---|
| Platelet count, x109/L (REF 150–450): | |||
| Hour 0 | 243 (179–284) | 217 (151–265) | 0.156 |
| Hour 8 | 208 (174–256) | 181 (115–248) | 0.251 |
| Hour 24 | 190 (155–240) | 142 (115–185) | 0.044 |
| Hour 48 | 176 (139–231) | 132 (107–145) | 0.005 |
| Hour 72 | 186 (147–237) | 122 (109–151) | 0.001 |
| PT, sec (REF 11–15): | |||
| Hour 0 | 14.7 (13.7–15.3) | 20.2 (19.3–21.2) | <0.001 |
| Hour 8 | 14.4 (13.8–15.6) | 16.7 (16.0–18.1) | <0.001 |
| Hour 24 | 15.2 (14.0–17.2) | 17.8 (17.2–18.6) | <0.001 |
| Hour 48 | 15.6 (14.2–16.9) | 17.2 (15.2–17.9) | 0.071 |
| Hour 72 | 14.8 (13.8–16.6) | 16.2 (15.1–17.6) | 0.030 |
| aPTT, sec (REF 25–40): | |||
| Hour 0 | 32.3 (29.0–35.7) | 39.0 (32.6–46.1) | 0.007 |
| Hour 8 | 33.9 (30.4–36.8) | 34.6 (32.3–39.4) | 0.570 |
| Hour 24 | 39.9 (34.8–44.7) | 41.1 (38.1–47.2) | 0.394 |
| Hour 48 | 41.1 (35.9–47.0) | 42.1 (36.6–52.3) | 0.733 |
| Hour 72 | 39.7 (36.4–44.6) | 45.7 (39.9–55.8) | 0.058 |
| Fibrinogen concentration, mg/dL (REF 250–400): | |||
| Hour 0 | 223 (194–326) | 132 (113–172) | <0.001 |
| Hour 8 | 245 (209–307) | 202 (153–269) | 0.073 |
| Hour 24 | 418 (352–489) | 389 (305–474) | 0.264 |
| Hour 48 | 629 (516–717) | 574 (465–714) | 0.577 |
| Hour 72 | 694 (612–819) | 746 (642–864) | 0.411 |
| PFA-100 collagen/epinephrine closure time, sec (REF <180): | |||
| Hour 0 | 98 (85–152) | 130 (108–214) | 0.090 |
| Hour 8 | 107 (84–143) | 108 (78–127) | 0.768 |
| Hour 24 | 102 (86–171) | 120 (95–157) | 0.715 |
| Hour 48 | 111 (86–140) | 144 (123–296) | 0.029 |
| Hour 72 | 103 (92–142) | 144 (103–217) | 0.222 |
| PFA-100 collagen/ADP closure time, sec (REF <120): | |||
| Hour 0 | 64 (53–82) | 70 (52–79) | 0.955 |
| Hour 8 | 66 (56–78) | 87 (59–211) | 0.039 |
| Hour 24 | 83 (67–96) | 100 (71–112) | 0.122 |
| Hour 48 | 99 (77–136) | 157 (114–236) | 0.005 |
| Hour 72 | 100 (81–147) | 279 (99–300) | 0.018 |
| Platelet ADP aggregation, ohms (REF 6–24): | |||
| Hour 0 | 10.8 (7.5–13.0) | 7.3 (4.5–8.0) | 0.002 |
| Hour 8 | 9.5 (7.0–13.0) | 9.8 (5.5–12.3) | 0.609 |
| Hour 24 | 10.5 (7–15) | 10.3 (6–12) | 0.593 |
| Hour 48 | 10.5 (8.5–13) | 11 (9.3–13.3) | 0.501 |
| Hour 72 | 11.3 (9–15) | 10.5 (7.5–16.3) | 0.524 |
| Platelet collagen aggregation, ohms (REF 14–28): | |||
| Hour 0 | 14.5 (11.5–17.5) | 11.0 (6.8–12.5) | 0.001 |
| Hour 8 | 12.5 (10.0–16.0) | 11.8 (8.0–14.0) | 0.363 |
| Hour 24 | 13.8 (11.0–15.5) | 15.0 (11.3–16.5) | 0.481 |
| Hour 48 | 13.5 (11.5–15.5) | 13.8 (12.3–16.3) | 0.759 |
| Hour 72 | 14.3 (11.0–18.0) | 13.0 (11.8–17.3) | 0.955 |
| HAS platelet contractile force, kdynes (REF 4.5–9.5): | |||
| Hour 0 | 8.4 (6.8–9.6) | 5.8 (3.8–8.1) | 0.002 |
| Hour 8 | 6.7 (5.2–9.3) | 6.4 (5.6–7.7) | 0.388 |
| Hour 24 | 6.9 (5.0–8.6) | 8.2 (7.2–11.0) | 0.122 |
| Hour 48 | 9.1 (5.9–13.1) | 9.5 (6.5–11.8) | 0.867 |
| Hour 72 | 10.2 (7.6–14.4) | 8.0 (7.7–13.8) | 0.668 |
| HAS clot elastic modulus, kdynes/cm2 (REF 14–35): | |||
| Hour 0 | 29.2 (22.9–37.0) | 23.6 (17.9–30.0) | 0.051 |
| Hour 8 | 23.6 (18.6–30.9) | 20.7 (16.4–26.3) | 0.388 |
| Hour 24 | 24.7 (17.6–31.9) | 26.9 (21.2–40.5) | 0.273 |
| Hour 48 | 41.8 (17.4–47.3) | 26.3 (17.1–42.0) | 0.403 |
| Hour 72 | 37.8 (21.7–52.0) | 21.5 (16.0–56.4) | 0.384 |
| TEG MA, mm (REF 50–70): | |||
| Hour 0 | 63.0 (58.8–65.1) | 54.6 (50.9–59.1) | <0.001 |
| Hour 8 | 61.7 (58.3–66.0) | 62.6 (58.2–64.1) | 0.586 |
| Hour 24 | 64.0 (61.9–67.1) | 64.9 (61.4–66.2) | 0.901 |
| Hour 48 | 67.3 (65.1–70.4) | 65.2 (64.2–69.1) | 0.303 |
| Hour 72 | 68.9 (66.6–72.0) | 69.4 (64.8–71.6) | 0.650 |
| TEG LY30, % (REF <7.5): | |||
| Hour 0 | 0.2 (0–1.4) | 2.3 (0.1–5.8) | 0.074 |
| Hour 8 | 0.1 (0–0.6) | 0 (0–0.3) | 0.446 |
| Hour 24 | 0.7 (0–1.6) | 0.7 (0.1–1.6) | 0.836 |
| Hour 48 | 1.1 (0.2–2.6) | 1.3 (0.2–2.1) | 0.980 |
| Hour 72 | 0.6 (0–2.1) | 0.7 (0.1–1.3) | 0.605 |
| TEG-PM ADP inhibition, %: | |||
| Hour 0 | 81.4 (61.4–94.7) | 94.6 (64.4–99.8) | 0.291 |
| Hour 8 | 57.4 (38.0–96.4) | 67.5 (28.4–96.7) | 0.706 |
| Hour 24 | 37.9 (30.0–61.5) | 27.9 (19.4–84.4) | 0.484 |
| Hour 48 | 35.7 (19.4–63.8) | 26.3 (10.0–54.3) | 0.443 |
| Hour 72 | 42.2 (27.2–61.0) | 44.4 (7.1–61.7) | 0.886 |
| TEG-PM AA inhibition, %: | |||
| Hour 0 | 40.7 (21.8–62.9) | 37.2 (24.7–54.5) | 0.706 |
| Hour 8 | 36.8 (17.6–86.0) | 40.2 (1.2–61.5) | 0.641 |
| Hour 24 | 23.3 (6.3–62.8) | 16.8 (9.4–27.3) | 0.511 |
| Hour 48 | 24.9 (1.6–59.1) | 6.0 (0–24.4) | 0.206 |
| Hour 72 | 21.4 (0–55.7) | 7.5 (0–28.1) | 0.569 |
| Percent PAC-1-positive: | |||
| Hour 0 | 3.7 (2.1–12.5) | 3.1 (0.5–13.1) | 0.417 |
| Hour 8 | 10.5 (0.3–35.7) | 11.0 (4.7–18.9) | 0.980 |
| Hour 24 | 12.4 (2.2–25.9) | 14.7 (4.7–19.0) | 0.979 |
| Hour 48 | 18.6 (8.5–30.6) | 12.7 (5.0–17.8) | 0.341 |
| Hour 72 | 14.4 (3.4–28.1) | 19.3 (1.8–30.9) | 0.716 |
| Percent CD62P-positive: | |||
| Hour 0 | 3.2 (1.3–7.9) | 2.4 (0.9–2.6) | 0.180 |
| Hour 8 | 2.8 (0.8–5.1) | 2.4 (1.5–3.8) | 0.910 |
| Hour 24 | 1.6 (0.4–3.8) | 1.1 (0.9–2.0) | 0.577 |
| Hour 48 | 1.6 (0.6–4.8) | 0.8 (0.6–2.3) | 0.458 |
| Hour 72 | 2.0 (0.8–8.5) | 2.8 (1.3–3.4) | 0.874 |
| Percent that became CD62P-positive after ADP stimulation: | |||
| Hour 0 | 69.6 (59.6–75.8) | 65.8 (49.5–75.1) | 0.585 |
| Hour 8 | 64.9 (57.6–72.6) | 64.1 (55.9–67.9) | 0.471 |
| Hour 24 | 69.6 (55.3–75.1) | 60.6 (53.2–67.5) | 0.178 |
| Hour 48 | 68.2 (58.2–73.1) | 58.3 (54.0–69.7) | 0.270 |
| Hour 72 | 67.6 (55.3–74.0) | 66.6 (60.3–73.9) | 0.803 |
Discussion
Our results identify a platelet dysfunction in TIC that primarily involves aggregation, manifests during the critical early hours after injury, and does not include alpha granule secretory dysfunction. This suggests a central mechanism of specific cytoskeletal or integrin dysfunction underlying the TIC platelet phenotype and does not support generalized platelet exhaustion as previously described.
We found no difference in levels of platelet activation state on arrival to the ED, as evidenced by the percentage of platelets that bound the PAC-1 antibody (specific for the activated form of αIIbβ3) or the anti-CD62P antibody (specific for exocytosed alpha granule membrane). Furthermore, TIC platelets were not different in their ability to release granules upon ex vivo activation by ADP stimulation. Prior studies have shown mixed results in this area. One found a modest but statistically significant decrease in inducible CD62P expression in trauma patients(13), and another found that dense granule content was maintained in trauma patient platelets.(18) The current study is larger and more comprehensive than previous work on this subject, and it suggests that platelets remain activatable and are thus not fully exhausted during TIC.
We also found that platelet dysfunction that was relatively common and phenotypically consistent with several prior studies. Most consistent was the loss of adhesive and aggregation function during TIC. Previous work has found similar functional phenotypes where platelet aggregation, measured by impedance aggregometry, was decreased and its decrease was significantly associated with increased mortality.(11–13) Of note, each of these prior studies showed decreased responses to different agonists, which could suggest that the impairment is occurring in a downstream intracellular activation pathway common to all agonists. Our study took this analysis one step further and found that these impairments remained after statistically controlling for fibrinogen concentration and platelet count as possible confounders.
The presence of both normal secretory activity and impaired aggregation during TIC in this study suggests that platelet dysfunction in TIC is more likely to arise either from an abnormal intracellular signaling pathway common to numerous platelet stimuli that is downstream from granule secretion or from integrin dysfunction (Figure 2). In fact, the TIC platelets in this study are phenotypically similar to the αIIbβ3 dysfunction seen in Glanzmann thrombasthenia (GT). The defining phenotypic characteristic of GT is globally impaired platelet aggregation, which was present in our TIC population.(19) Viscoelastic thrombus strength, elastic modulus, and contractile force are also all decreased in GT, all of which was seen in the TIC group.(20, 21) Impaired αIIbβ3 adhesive function also results in decreased platelet contraction.(22) GT patients typically exhibit prolonged PFA-100 closure times,(23) which was partially seen in TIC patients. GPIb and αIIbβ3 receptors are also known to be susceptible to cleavage and inactivation by plasmin proteolytic activity, which is increased during TIC.(24–26) Increased plasmin activity in this cohort is also supported by our previous work showing increased fibrinolysis during TIC in this cohort.7 Therefore, derangement of αIIbβ3 function during TIC could result from its abnormal intracellular activation, cleavage of receptors, or binding to hypofunctional ligands.
Figure 2. Graphic representation of platelet dysfunction in TIC.
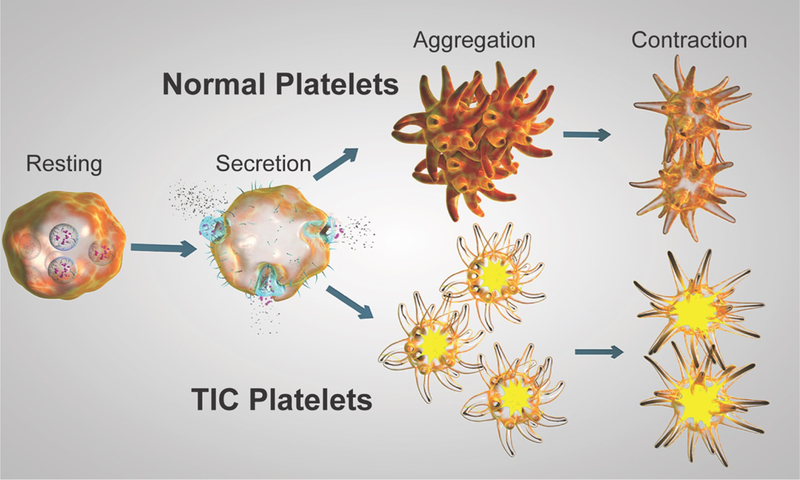
Granule secretion in response to agonist exposure is preserved in TIC platelets. However, compared to normal platelets, TIC platelets exhibit impaired aggregation and contraction. The combination of normal activatability and impaired aggregation suggests that platelet dysfunction in TIC likely arises from either intrinsic platelet or integrin dysfunction.
Another important finding of our study is that the duration of platelet impairment was short-lived. Most assays trended toward becoming pro-hemostatic over time, sometimes exceeding the limit of the reference range. The deficiencies seen at the time of ED arrival were mostly resolved by the next time point 8 hours later (ADP and collagen aggregation, TEG MA, and PCF). This dysfunction occurs in the critical early hours after injury, during which most mortality attributable to hemorrhage after trauma takes place.(27) The recovery time is not due to the loss of data from non-survivors in later time points, as all patients who did not survive the full 72 hours were excluded from this analysis. It is possible that the rapid recovery is due to transfusion of healthy platelets, because there was a significantly higher volume of platelets transfused to the TIC group. However, this is made less likely by the fact that the actual amounts transfused to both groups were quite small at 8 hours (medians of 0 and 20 mL of pooled platelets in the non-TIC and TIC groups, respectively). It is also somewhat unclear whether the population included in the longitudinal analysis accurately represent the larger population included in the primary analysis. While the defects seen in the TIC group in the larger primary analysis were consistently reproduced in the smaller longitudinal population at Hour 0 (decreased ADP aggregation, collagen aggregation, and PCF), one test was not (prolonged collagen/epinephrine PFA-100 closure, which lost significance at p=0.090). It is unclear whether this lost significance is from a loss of power in the smaller longitudinal population (50 vs. 95 patients in the primary analysis) or selection bias towards healthier patients having complete longitudinal data, but both effects may have certainly contributed. There are several longitudinal studies published on TIC, and only a handful on platelet dysfunction after trauma. One study showed prolongation of PFA-100 closure times through the entire 72-hour follow-up period(10), which we have replicated, and another showed a decline in aggregation function that occurred early and persisted throughout the 24-hour follow-up period(28). There has been a broad range of reported durations of TIC, ranging from 6 hours to 5 days after injury, and the duration appears to be highly dependent on the resuscitation methods employed.(29, 30) The reason for our discrepant results could be that the resuscitation strategy employed at this trauma center rapidly corrects coagulopathy, or that the current cohort was less injured than others (mean Injury Severity Score of 20).(7) The persistence of PFA-100 abnormalities in TIC suggests a lasting defect specific to the adhesion pathway that requires further investigation.
Our study has several limitations. First, it is an observational study that should be generalized with caution. Second, our longitudinal analysis cohort excluded patients who were not expected to survive 72 hours. This removed the effect of survival bias on our data. However, patients who die in the early period after injury also tend to be the ones who develop the most severe TIC. Since the longitudinal cohort survived for at least 72 hours after injury, they were capable of achieving hemostasis, suggesting that platelets remained capable of making important contributions to hemostasis in this less-severe TIC phenotype. Third, our study sample included only a relatively small number of patients with TIC (n=15). Fourth, as with any similarly designed study, the analysis of circulating platelets is subject to some selection bias, as these platelets likely do not accurately represent those that are participating in thrombus formation. As it is difficult to sample platelets actively participating in thrombus formation, this bias was unavoidable within the constraints of our study design, but it should be remembered when interpreting our findings. Fifth, our study was not designed or powered to examine any phenotypically distinct subtypes of TIC. The coagulopathy associated with isolated brain injury is perhaps the best-described example of this. A set of simple regression analyses (not shown) found no significant correlation between TBI severity and the abnormalities seen in PFA-100 or aggregometry, but this could be due to a lack of power for this exploratory analysis. Finally, several variables that would have been helpful to further define the platelet abnormality observed (e.g., VWF concentration, VWF multimer profile, PAC-1 and anti-CD62P ligand density) were not collected. Future studies should investigate in greater detail the underlying cause of functional platelet impairment after trauma. A more focused and detailed approach to the specific lesion, focusing on integrin expression and cellular function taking place downstream from initial activation events, is needed to improve future clinical management of this disorder.
In conclusion, our data indicate that circulating TIC platelets are dysfunctional primarily in their ability to mechanically aggregate and contract but maintain normal alpha granule secretory function. Their aggregatory and contractile dysfunction resolves within 8 hours in surviving trauma patients. Platelet integrin dysfunction or inhibition is a clear target for further investigation into the mechanism of platelet dysfunction during TIC.
Essentials.
Platelets in trauma-induced coagulopathy (TIC) are impaired, but the mechanism is not known.
We performed comprehensive longitudinal platelet function testing in trauma patient samples.
Platelets in TIC are widely impaired early after injury, but platelet activatability is intact.
This suggests a mechanism of transient platelet cytoskeletal/integrin dysfunction during TIC.
Acknowledgments
This study was supported by the U.S. Defense Health Program, U.S. Army Medical Research and Materiel Command grant W81XWH-11–2-0089. A. St. John was supported in part by the National Heart, Lung, and Blood Institute (NHLBI) (grant 4 K12 HL 087165). N. White was supported in part by the National Center for Advancing Translational Sciences (NCATS) (grant KL2 TR000421), a component of the National Institutes of Health (NIH). The views expressed in this article are those of the authors and do not necessarily reflect the official policy or position of the Departments of the Army and Defense or the U.S. Government and do not necessarily represent the official view of the NCATS or NIH. Figure 2 was generated for a fee by Ella Maru Studio, Charleston, SC. Biostatistics consultation was provided by the University of Washington Departments of Biostatistics and Statistics.
N. White reports grants from NIH-NCATS during the conduct of the study, grants from U.S. Department of Defense, and personal fees from Stasys Medical Corp outside the submitted work. In addition, Dr. White has a patent through the University of Washington licensed to Stasys Medical Corp.
Footnotes
Disclosure of Conflict of Interests
The authors state that they have no conflict interests.
Addendum
A. St. John, J. Newton, E. Martin, B. Mohammed, D. Contaifer, J. Saunders, G. Brophy, B. Spiess, K. Ward, D. Brophy, and N. White designed the research; J. Newton, E. Martin, B. Mohammed, D. Contaifer, J. Saunders, G. Brophy, B. Spiess, K. Ward, and D. Brophy collected data; A. St. John, J. López, and N. White analyzed data; A. St. John, N. White, and Ella Maru Studio generated the figures; A. St. John, J. Newton, E. Martin, B. Mohammed, D. Contaifer, J. Saunders, G. Brophy, B. Spiess, K. Ward, D. Brophy, J. López, and N. White wrote the paper.
References
- 1.Murray CJ, Vos T, Lozano R, Naghavi M, Flaxman AD, Michaud C, Ezzati M, Shibuya K, Salomon JA, Abdalla S, Aboyans V, Abraham J, Ackerman I, Aggarwal R, Ahn SY, Ali MK, Alvarado M, Anderson HR, Anderson LM, Andrews KG, et al. Disability-adjusted life years (DALYs) for 291 diseases and injuries in 21 regions, 1990–2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet. 2012;380:2197–223. [DOI] [PubMed] [Google Scholar]
- 2.Kauvar DS, Lefering R, Wade CE. Impact of hemorrhage on trauma outcome: an overview of epidemiology, clinical presentations, and therapeutic considerations. The Journal of trauma. 2006;60:S3–11. [DOI] [PubMed] [Google Scholar]
- 3.Eastridge BJ, Mabry RL, Seguin P, Cantrell J, Tops T, Uribe P, Mallett O, Zubko T, Oetjen-Gerdes L, Rasmussen TE, Butler FK, Kotwal RS, Holcomb JB, Wade C, Champion H, Lawnick M, Moores L, Blackbourne LH. Death on the battlefield (2001–2011): implications for the future of combat casualty care. The journal of trauma and acute care surgery. 2012;73:S431–7. [DOI] [PubMed] [Google Scholar]
- 4.Cap A, Hunt B. Acute traumatic coagulopathy. Current opinion in critical care. 2014;20:638–45. [DOI] [PubMed] [Google Scholar]
- 5.Brohi K, Singh J, Heron M, Coats T. Acute traumatic coagulopathy. The Journal of trauma. 2003;54:1127–30. [DOI] [PubMed] [Google Scholar]
- 6.Hoffman M, Monroe DM 3rd., A cell-based model of hemostasis. Thrombosis and haemostasis. 2001;85:958–65. [PubMed] [Google Scholar]
- 7.White NJ, Newton JC, Martin EJ, Mohammed BM, Contaifer D Jr., , Bostic JL, Brophy GM, Spiess BD, Pusateri AE, Ward KR, Brophy DF. Clot Formation Is Associated With Fibrinogen and Platelet Forces in a Cohort of Severely Injured Emergency Department Trauma Patients. Shock (Augusta, Ga). 2015;44 Suppl 1:39–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Harr JN, Moore EE, Chin TL, Ghasabyan A, Gonzalez E, Wohlauer MV, Banerjee A, Silliman CC, Sauaia A. Platelets are dominant contributors to hypercoagulability after injury. The journal of trauma and acute care surgery. 2013;74:756–62; discussion 62–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Moore HB, Moore EE, Chapman MP, Gonzalez E, Slaughter AL, Morton AP, D’Alessandro A, Hansen KC, Sauaia A, Banerjee A, Silliman CC. Viscoelastic measurements of platelet function, not fibrinogen function, predicts sensitivity to tissue-type plasminogen activator in trauma patients. Journal of thrombosis and haemostasis : JTH. 2015;13:1878–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jacoby RC, Owings JT, Holmes J, Battistella FD, Gosselin RC, Paglieroni TG. Platelet activation and function after trauma. The Journal of trauma. 2001;51:639–47. [DOI] [PubMed] [Google Scholar]
- 11.Kutcher ME, Redick BJ, McCreery RC, Crane IM, Greenberg MD, Cachola LM, Nelson MF, Cohen MJ. Characterization of platelet dysfunction after trauma. The journal of trauma and acute care surgery. 2012;73:13–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Solomon C, Traintinger S, Ziegler B, Hanke A, Rahe-Meyer N, Voelckel W, Schochl H. Platelet function following trauma. A multiple electrode aggregometry study. Thrombosis and haemostasis. 2011;106:322–30. [DOI] [PubMed] [Google Scholar]
- 13.Ramsey MT, Fabian TC, Shahan CP, Sharpe JP, Mabry SE, Weinberg JA, Croce MA, Jennings LK. A prospective study of platelet function in trauma patients. The journal of trauma and acute care surgery. 2016;80:726–33. [DOI] [PubMed] [Google Scholar]
- 14.Wohlauer MV, Moore EE, Thomas S, Sauaia A, Evans E, Harr J, Silliman CC, Ploplis V, Castellino FJ, Walsh M. Early platelet dysfunction: an unrecognized role in the acute coagulopathy of trauma. Journal of the American College of Surgeons. 2012;214:739–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pareti FI, Capitanio A, Mannucci L, Ponticelli C, Mannucci PM. Acquired dysfunction due to the circulation of “exhausted” platelets. The American journal of medicine. 1980;69:235–40. [DOI] [PubMed] [Google Scholar]
- 16.White NJ, Contaifer D Jr., , Martin EJ, Newton JC, Mohammed BM, Bostic JL, Brophy GM, Spiess BD, Pusateri AE, Ward KR, Brophy DF. Early hemostatic responses to trauma identified with hierarchical clustering analysis. Journal of thrombosis and haemostasis : JTH. 2015;13:978–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brohi K, Cohen MJ, Ganter MT, Schultz MJ, Levi M, Mackersie RC, Pittet JF. Acute coagulopathy of trauma: hypoperfusion induces systemic anticoagulation and hyperfibrinolysis. The Journal of trauma. 2008;64:1211–7; discussion 7. [DOI] [PubMed] [Google Scholar]
- 18.Bartels AN, Johnson C, Lewis J, Clevenger JW, Barnes SL, Hammer RD, Ahmad S. Platelet adenosine diphosphate inhibition in trauma patients by thromboelastography correlates with paradoxical increase in platelet dense granule content by flow cytometry. Surgery. 2016;160:954–9. [DOI] [PubMed] [Google Scholar]
- 19.Albanyan A, Al-Musa A, AlNounou R, Al Zahrani H, Nasr R, AlJefri A, Saleh M, Malik A, Masmali H, Owaidah T. Diagnosis of Glanzmann thrombasthenia by whole blood impedance analyzer (MEA) vs. light transmission aggregometry. International journal of laboratory hematology. 2015;37:503–8. [DOI] [PubMed] [Google Scholar]
- 20.Castellino FJ, Liang Z, Davis PK, Balsara RD, Musunuru H, Donahue DL, Smith DL, Sandoval-Cooper MJ, Ploplis VA, Walsh M. Abnormal whole blood thrombi in humans with inherited platelet receptor defects. PloS one. 2012;7:e52878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jen CJ, McIntire LV. The structural properties and contractile force of a clot. Cell motility. 1982;2:445–55. [DOI] [PubMed] [Google Scholar]
- 22.Kiyoi T, Tomiyama Y, Honda S, Tadokoro S, Arai M, Kashiwagi H, Kosugi S, Kato H, Kurata Y, Matsuzawa Y. A naturally occurring Tyr143His alpha IIb mutation abolishes alpha IIb beta 3 function for soluble ligands but retains its ability for mediating cell adhesion and clot retraction: comparison with other mutations causing ligand-binding defects. Blood. 2003;101:3485–91. [DOI] [PubMed] [Google Scholar]
- 23.Harrison P, Robinson MS, Mackie IJ, Joseph J, McDonald SJ, Liesner R, Savidge GF, Pasi J, Machin SJ. Performance of the platelet function analyser PFA-100 in testing abnormalities of primary haemostasis. Blood coagulation & fibrinolysis : an international journal in haemostasis and thrombosis. 1999;10:25–31. [DOI] [PubMed] [Google Scholar]
- 24.Pasche B, Ouimet H, Francis S, Loscalzo J. Structural changes in platelet glycoprotein IIb/IIIa by plasmin: determinants and functional consequences. Blood. 1994;83:404–14. [PubMed] [Google Scholar]
- 25.Raza I, Davenport R, Rourke C, Platton S, Manson J, Spoors C, Khan S, De’Ath HD, Allard S, Hart DP, Pasi KJ, Hunt BJ, Stanworth S, MacCallum PK, Brohi K. The incidence and magnitude of fibrinolytic activation in trauma patients. Journal of thrombosis and haemostasis : JTH. 2013;11:307–14. [DOI] [PubMed] [Google Scholar]
- 26.Adelman B, Michelson AD, Loscalzo J, Greenberg J, Handin RI. Plasmin effect on platelet glycoprotein Ib-von Willebrand factor interactions. Blood. 1985;65:32–40. [PubMed] [Google Scholar]
- 27.Bardes JM, Inaba K, Schellenberg M, Grabo D, Strumwasser A, Matsushima K, Clark D, Brown N, Demetriades D. The Contemporary Timing of Trauma Deaths. The journal of trauma and acute care surgery. 2018. [DOI] [PubMed] [Google Scholar]
- 28.Rahbar E, Cardenas JC, Matijevic N, Del Junco D, Podbielski J, Cohen MJ, Cotton BA, Holcomb JB, Wade CE. Trauma, Time, and Transfusions: A Longitudinal Analysis of Coagulation Markers in Severely Injured Trauma Patients Receiving Modified Whole Blood or Component Blood Products. Shock (Augusta, Ga). 2015;44:417–25. [DOI] [PubMed] [Google Scholar]
- 29.Hayakawa M, Sawamura A, Gando S, Kubota N, Uegaki S, Shimojima H, Sugano M, Ieko M. Disseminated intravascular coagulation at an early phase of trauma is associated with consumption coagulopathy and excessive fibrinolysis both by plasmin and neutrophil elastase. Surgery. 2011;149:221–30. [DOI] [PubMed] [Google Scholar]
- 30.Kutcher ME, Kornblith LZ, Vilardi RF, Redick BJ, Nelson MF, Cohen MJ. The natural history and effect of resuscitation ratio on coagulation after trauma: a prospective cohort study. Annals of surgery. 2014;260:1103–11. [DOI] [PMC free article] [PubMed] [Google Scholar]