

Abstract
Domestication of a transposon to give rise to the RAG1/RAG2 recombinase and V(D)J recombination was a pivotal event in the evolution of the jawed vertebrate adaptive immune system. The evolutionary adaptations that transformed the ancestral RAG transposase into a RAG recombinase with appropriately regulated DNA cleavage and transposition activities are not understood. Here, beginning with cryo-electron microscopy structures of RAG’s evolutionary relative, the ProtoRAG transposase from amphioxus, we identify amino acid residues and domains whose acquisition or loss underpins RAG’s propensity for coupled cleavage, preference for asymmetric DNA substrates, and inability to perform transposition in cells. In particular, we identify two jawed-vertebrate-specific adaptations—arginine 848 in RAG1 and an acidic region in RAG2—that together suppress RAG-mediated transposition more than 1000-fold. Our findings reveal a two-tiered mechanism for suppression of RAG-mediated transposition, illuminate the forces at work during the evolution of V(D)J recombination, and provide insight into the principles governing transposon molecular domestication.
Keywords: RAG, V(D)J recombination, evolution, ProtoRAG, DNA transposition, exaptation, cryo-electron microscopy
Jawed vertebrates have evolved a sophisticated adaptive immune system that relies on assembly of immunoglobulin and T-cell receptor genes from arrays of V, D, and J gene segments in developing B and T lymphocytes. The assembly reaction, known as V(D)J recombination, is initiated when the RAG1/RAG2 endonuclease (RAG) cleaves adjacent to the gene segments at recombination signal sequences (RSSs) composed of conserved heptamer and nonamer elements separated by a 12 or 23 bp spacer (12RSS and 23RSS)1 (Fig. 1a). DNA cleavage by RAG occurs by a nick-hairpin mechanism with hairpin formation occurring in a coordinated (coupled) manner in a synaptic complex containing one 12RSS and one 23RSS, a restriction known as the 12/23 rule (Fig. 1b). The 12/23 rule and coupled cleavage are fundamental features of RAG that are thought to contribute to the proper orchestration of V(D)J recombination and protection of genome integrity1–3.
Fig. 1 ∣. Uncoupled DNA cleavage by BbRAGL.

a, RSS and TIR substrates. Underlining indicates sequence identity in TR2 and arrows the site of cleavage. The TIR heptamer sequence can be found in endogenous human RSS sequences38.
b, Schematic of DNA cleavage, recombination, and transposition by RAG/BbRAGL. Inset, Nick-hairpin mechanism of DNA cleavage. Triangle indicates an RSS or TIR in this and other figures (wide side of triangle is the heptamer end).
c, Domain diagrams of the RAG and BbRAGL proteins. CC, catalytic core; NBD, nonamer binding domain; CTT, C-terminal tail; PHD, plant homeodomain finger. Numbers indicate amino acid domain boundaries (mouse RAG is depicted and used in all experiments except where indicated). NBD* is named for consistency and not to imply function.
d, Time course of DNA cleavage by cRAG and cBbRAGL with substrates containing a pair of TIRs or RSSs. Red asterisk, double cleavage band; black asterisk, single 3’TIR cleavage band. Cleavage products were resolved on acrylamide gels and are indicated schematically (circles indicating hairpin ends). For gel source data, see Supplementary Figure 1.
e, f, Cleavage of substrates containing two or one TIRs or RSSs as indicated above the lanes, by cBbRAGL, cRAG, or complexes in which cBbRAG1L/cRAG1 lack the indicated domain. Black asterisks mark the two single cleavage products. Reaction time of 60 min was used here and in other cleavage reactions unless otherwise specified.
g, Transposition frequency measured in HEK293T cells using the assay of Extended Data Fig. 8a.
Transposon “molecular domestication” has contributed broadly to the evolution of new proteins and activities4–6, with RAG and V(D)J recombination representing a paradigmatic example of this process. Current evidence supports a model in which RAG1 and RAG2 evolved from the transposase genes of an ancient “RAG transposon” while disassembled (“split”) immunoglobulin and T-cell receptor genes arose from transposon insertion into a receptor gene, with the inserted terminal inverted repeats (TIRs) of the transposon becoming the RSSs7–9. This model received strong support from the discovery in the cephalochordate amphioxus of ProtoRAG, a transposon possessing numerous features that implicate it as a descendent of the RAG transposon10.
The RAG transposon domestication model predicts a critical divergence during chordate evolution in which, in jawed vertebrates, the RAG transposase acquired properties of a recombinase, while in amphioxus (and likely other invertebrate chordate lineages11), transposase functions were retained. Particularly pivotal would have been a divergence in post-cleavage reaction steps (Fig. 1b), with RAG-generated DNA ends preferentially undergoing end joining (recombination) instead of transposition and ProtoRAG retaining a strong preference for transposition over end-joining10. Indeed, RAG is strikingly poor at performing transposition in living cells12–14, with only a single bone fide transposition event thus far identified in mice or humans15,16. How the ancestral RAG transposon was domesticated to yield a RAG recombinase with minimal in vivo transposition activity and a strong propensity for coupled cleavage of asymmetric substrates stands as a central mystery in the evolution of V(D)J recombination and jawed vertebrate adaptive immunity. Here, we use the structure of ProtoRAG transposase as a lens through which to view this evolutionary transformation.
Uncoupled DNA cleavage by ProtoRAG
ProtoRAG from Branchiostoma belcheri (Extended Data Fig. 1a) is composed of convergently transcribed RAG1-like (BbRAG1L) and RAG2-like (BbRAG2L) genes flanked by 5’ and 3’ TIRs composed of a heptamer similar to the RSS heptamer, an adjacent, conserved 9–10 bp element referred to as TIR region 2 (TR2), and additional flanking sequences10 (Fig. 1a). The BbRAG1L protein contains a “core” region (cBbRAG1L; aa 468–1136) with sequence similarity (33% aa identity) to the core region of RAG1 (cRAG1; aa 384–1008 in mouse) (Fig. 1c). Within cRAG1 and cBbRAG1L, we define catalytic cores (CC and CC*, respectively) that lack one or more DNA binding elements (Fig. 1c). BbRAG2L resembles only core RAG2 (aa 1–350 in mouse; 22% aa identity) and lacks all RAG2 C-terminal elements including an acidic hinge and plant homeodomain finger (Fig. 1c).
With a cleavage substrate containing a 5’/3’TIR pair, both “core” BbRAGL (cBbRAG1L with BbRAG2L) (Fig. 1d) and full length BbRAG1L/BbRAG2L (Extended Data Fig. 1b), generate a strong band corresponding to single cleavage at the 3’TIR (black asterisk) comparable in intensity to the 5’/3’TIR double cleavage band (red asterisk). In contrast, core RAG (cRAG) predominantly generates the 12/23RSS double cleavage product (Fig. 1d). Furthermore, core and full length BbRAGL robustly cleave substrates containing either a single 5’TIR or a single 3’TIR, (Fig. 1e and Extended Data Fig. 1b) while cRAG cleaves single RSS substrates poorly (Fig. 1f). These results indicate that DNA cleavage by BbRAGL is less tightly coupled than by RAG.
Deletion of the nonamer binding domain (NBD) from cRAG1 eliminates activity (Fig. 1f), while cBbRAGL lacking its corresponding NBD* domain (which has limited sequence similarity to NBD but, like NBD17, forms a dimer in solution (Extended Data Fig. 1c, d)) retains substantial activity (Fig. 1e). In addition, while the C-terminal tail (CTT) of RAG1 is dispensable for activity2, the C-terminal tail of BbRAG1L (CTT*; Fig. 1c) is important for BbRAGL cleavage activity10 (Fig. 1e). Hence, RAG1 and BbRAG1L have evolved opposite dependencies on the N- and C-terminal portions of their core regions.
RAG and BbRAGL exhibit a third important difference: while both are active transposases in vitro10,18,19, only BbRAGL exhibits substantial transposition activity in cells10,12–16 (Fig. 1g). We note that BbRAGL activity is being assessed in a heterologous (mammalian) cell context.
Structure of the ProtoRAG Transposase
To better understand these functional differences, we determined the structure of cBbRAGL together with HMGB1 (a DNA bending cofactor that stimulates RAG1 and BbRAGL10 cleavage) bound to the 3’TIR (which is bound more efficiently by the cBbRAGL tetramer than the 5’TIR (Extended Data Fig. 2a–c)). Single particle cryo-EM analysis yielded structures for cBbRAGL/HMGB1 bound to intact and nicked 3’TIRs (designed to mimic the first step of cleavage as in Fig. 1b, inset), with resolution of 4.3 Å for the nicked 3’TIR structure after application of 2-fold symmetry (Fig. 2a, Extended Data Fig. 2d–f and Extended Data Table 1).
Fig. 2 ∣. Cryo-EM structure of cBbRAGL-nicked 3’TIR complex.

a, Symmetrized cryo-EM structure at 4.3 Å resolution of the cBbRAGL/HMGB1-nicked 3’TIR complex.
b, Superimposition of one subunit of cBbRAG1L (nicked-3’TIR complex) with cRAG1 (nicked RSS complex; PDB 5ZDZ).
c, Superimposition of nicked RSS (PDB 5ZDZ) with nicked 3’TIR showing flipped bases and three catalytic residues and calcium ions (spheres) in the RAG/BbRAGL active site.
d, e, The additional density at the C-terminus of BbRAG1L is in close proximity to TR2 (orange) (d) and together with the opposite subunit of BbRAG1L largely encircles the DNA (e).
f, Sequence alignment of CTT* from deuterostome invertebrate RAG1L and cnidarian (hydra) Transib proteins. Species name abbreviations are defined in the legend of Extended Data Fig. 5b. Asterisks, conserved residues with Zn2+ coordination potential.
g, Cleavage reactions using CTT* mutants of BbRAG1L. Red asterisk, double cleavage band; black asterisks, single TIR cleavage bands.
cBbRAGL-3’TIR complexes contain a central cBbRAG1L dimer capped by two monomers of BbRAG2L and two DNA duplexes (Fig. 2a and Extended Data Fig. 2g, h). Rather than the Y shape adopted by cRAG complexes20–23 (Extended Data Fig. 3a), cBbRAGL complexes were roughly V-shaped because no density was discernable for NBD*, HMGB1, or the heptamer-distal 25 bp of the TIRs. Despite an estimated 700 million years of evolutionary divergence, ProtoRAG and RAG exhibit a remarkable degree of structural similarity. cBbRAG1L recapitulates the structural domains of the RAG1 catalytic core while BbRAG2L, like RAG2, adopts a structure consistent with a 6-bladed β-propeller fold (Fig. 2b and Extended Data Fig. 3b–d). Structural similarity is clear in the vicinity of the active site and heptamer, with the nicked 3’TIR exhibiting two flipped (extrahelical) bases similar to those of nicked RSSs bound by RAG21,22 (Fig. 2c and Extended Data Fig. 3e). Like RAG21–23, BbRAGL undergoes an “open” to “closed” conformation upon TIR nicking and is particularly flexible in the BbRAGL-intact TIR complex (Extended Data Fig. 3f–h), with both molecules of BbRAG1L making extensive contacts with both DNA molecules (Extended Data Fig. 4). These striking structural parallels support the hypothesis that RAG and BbRAGL evolved from a common RAG transposon ancestor.
A novel ProtoRAG DNA binding domain
The cBbRAGL-nicked 3’TIR cryo-EM map contained unaccounted-for density at the C-terminus of the BbRAG1L catalytic core that could readily accommodate the Cα backbone of BbRAG1L CTT* (Extended Data Fig. 5a) and was in close proximity to the TR2 element of the 3’TIR (Fig. 2d). This suggested that CTT* is a DNA binding domain that together with residues from the opposite subunit of BbRAG1L forms a clamp to bind TR2 (Fig. 2e). CTT* exhibits sequence conservation among RAG1-like proteins from invertebrates (Fig. 2f) but not with vertebrate RAG1 CTT (Extended Data Fig. 5b). Mutation of residues C1114, H1222, or C1227 in the highly conserved Cx2Cx3GHx4C motif of CTT* reduced activity, while mutation of two less well-conserved residues had no discernable effect on activity (Fig. 2g), indicating that integrity of the conserved CCHC motif is important for CTT* function. Consistent with the hypothesis that BbRAGL makes important contacts with TR2, the heptamer and TR2 are the only portions of the TIR essential for cleavage (Extended Data Fig. 5c–g).
Modular domain function and the 12/23 rule
To investigate how the distinct functional properties of RAG and BbRAGL relate to structural domains, we generated chimeric RAG1-BbRAG1L proteins in which the BbRAG1L catalytic core with or without CTT*, was fused with the RAG1 NBD (Fig. 3a, b), and reciprocally, the RAG1 catalytic core was fused to NBD* and/or CTT* of BbRAG1L (Fig. 3c). Corresponding hybrid RSS/TIR DNA targets were used as cleavage substrates (Fig. 3b, c).
Fig. 3 ∣. DNA cleavage properties of chimeric RAG1-BbRAG1L proteins.

a-c, Schematic diagrams of cBbRAG1L and cRAG1 (a), and chimeric proteins containing the catalytic core of BbRAG1L (b) or RAG1 (c) with matching chimeric cleavage targets. In targets T1 and T2, the heptamer derives from the TIR and the remainder from the 12/23RSS while in T3 and T4, the heptamer derives from the RSS and the remainder from the 5’/3’TIR.
d-h, Cleavage reactions using chimeric proteins and substrates containing one or two targets as indicated above and below the lanes. ΔNon, nonamer region deleted; T1*, T1 with a C-to-A mutation of heptamer position 1 which renders the target uncleavable; mTR2, scrambling of TR2 in both target sites. Asterisks as in Fig. 2g.
When supplied with the RAG1 NBD, the BbRAG1L catalytic core no longer requires CTT* (Fig. 3d) and becomes dependent on the RSS nonamer for activity (Fig. 3e), with spacer length requirements (12±1 bp or 23±1 bp) identical to that of RAG (Extended Data Fig. 6a, b). Reciprocally, when deprived of its NBD, the RAG1 catalytic core becomes dependent on CTT* and TR2 for activity and is active without NBD* or any portion of the DNA substrate except the heptamer and TR2 (Fig. 3f–h). Thus, CTT* renders the RAG1 catalytic core independent of a nonamer binding domain, the RSS nonamer, substrate asymmetry, and hence the 12/23 rule. Notably, proteins containing the BbRAG1L catalytic core exhibit uncoupled cleavage (Fig. 3d) while those containing the RAG1 catalytic core display coupled cleavage (Extended Data Fig. 6c, d). We conclude that the catalytic cores of RAG1 and BbRAG1L dictate the propensities of these enzymes for coupled versus uncoupled cleavage and that the functional organization of ProtoRAG TIRs is different from that of RSSs because of a dependency on different DNA binding domains (Extended Data Fig. 6e). Furthermore, our findings argue that the choice of dominant DNA binding domain was pivotal for the evolution of the 12/23 rule since CTT* would need to have been eliminated to allow dependency on the rule.
Residues controlling coupled cleavage
While searching for features that might explain the intrinsic functional differences between the catalytic cores of RAG and BbRAGL, we observed that S963, which flanks the RAG1 catalytic glutamate E962, is positioned to form a hydrogen bond with E649 in apo RAG (Fig. 4a) and RAG bound to intact RSSs (Fig. 4b) but not when RAG is bound to nicked RSSs and poised for hairpin formation (Fig. 4c). BbRAG1L cannot form this hydrogen bond because E649/S963 have been replaced by V751/A1064 (Fig. 4d). Whether bound to intact or nicked TIRs, BbRAG1L adopts a structure similar to that of RAG1 bound to nicked DNA (Fig. 4e, f and Extended Data Fig. 6f) and hence appears to be constitutively poised for hairpin formation. Notably, the E649-S963 aa pair, while strictly conserved in jawed vertebrate RAG1, is absent from known invertebrate RAG1-like proteins (Fig. 4g).
Fig. 4 ∣. Residues that control coupled cleavage.

a-c, Structure of region surrounding RAG1 E649/S963 before RSS binding (PDB 4WWX) (a), bound to intact RSS (PDB 6CIK) (b), and bound to two nicked RSSs with base flipping (PDB 5ZE1) (c). In (c), E649-S963 hydrogen bond potential is disrupted due to a change in the relative orientation of the residues and acquisition of a potassium ion (K+)
d, Structure of region surrounding BbRAG1L V751/A1064 bound to nicked TIR.
e, f, Superimposition of protein structural elements containing RAG1 E649/S963 (e) or BbRAG1L V751/A1064 (f) bound to intact or nicked DNA. E649, S963, V751, and A1064 are highlighted with dark colors. In (e), the intact DNA structure was obtained with a RAG1 E962Q mutant22.
g, Sequence alignments of RAG1, RAG1-like, and Transib proteins in the vicinity of RAG1 E649 and S963. Species name abbreviations are defined in the legend of Extended Data Fig. 5b.
h, Cleavage reactions using cRAG with RAG1 mutations and DNA substrates containing one or two RSSs as indicated above the lanes. Asterisks as in Fig. 2g.
Incorporating residues of BbRAG1L into RAG1 revealed that E649V, S963A, and E649V/S963A RAG1 mutants display increased uncoupled cleavage activity compared to WT (Fig. 4h and Extended Data Fig. 6g). In contrast, Y994F had no effect and N961A consistently decreased uncoupled cleavage compared to WT RAG1 (Fig. 4h and Extended Data Fig. 6g, h). Reciprocal mutations in BbRAG1L revealed that the V751E but not A1064S mutant displays decreased uncoupled cleavage, while the V751E/A1064S double mutation almost abolished cleavage (Extended Data Fig. 6i). We propose that RAG1 E649 helps dictate coupled cleavage by mechanisms that are partially dependent on hydrogen bond formation with S963 and that lacking E649, BbRAG1L is more likely than RAG to adopt an active site configuration that is “hairpin-competent”. Notably, E649A mutant RAG1 was previously found to exhibit increased uncoupled cleavage activity in vitro and in vivo24.
Two-tiered control of RAG transposition
We reasoned that structural comparisons of RAG and BbRAGL might shed light on their dramatically different capacities to perform transposition in cells. In the RAG post-cleavage complex, RAG1 R848 is near the RSS 3’-OH that attacks target DNA during transposition (Fig. 5a). R848 is strictly conserved in jawed vertebrate RAG1 but is replaced by methionine in BbRAG1L and other invertebrate RAG1-like proteins (Fig. 5b, c). R848M mutant RAG1 cleaves DNA at WT levels and exhibits a striking (~8-fold) increase in transposition activity in vitro relative to WT, manifest as efficient generation of a slow-mobility band that represents inversion-circle intramolecular transposition products18 (Fig. 5d and Extended Data Fig. 7a, b) and enhanced transposition of an RSS-flanked antibiotic resistance gene into a target plasmid (Fig. 5e and Extended Data Fig. 7c). Hence, methionine at RAG1 position 848 stimulates RAG-mediated transposition at a post-cleavage step. Several different amino acids at position 848 support cleavage, with alanine stimulating and glutamate suppressing transposition relative to WT (Extended Data Fig. 7d, e).
Fig. 5 ∣. Reawakening the RAG transposon in vivo.

a, Structure of region surrounding RAG1 R848 after hairpin formation (PDB 5ZE2).
b, Structure of region surrounding RAG1 R848 (PDB 5ZDZ) or BbRAG1L M949 after nicking.
c, Sequence alignments of RAG1, RAG1-like, and Transib proteins in the vicinity of RAG1 R848. Red-shaded residues, highly-conserved binding surface for adenine base of heptamer adjacent to flipped C +1.
d, Cleavage reactions comparing intramolecular transposition by WT and R848M RAG1. The intramolecular transposition product was confirmed to contain inversion circles by inverse PCR DNA sequencing18.
e, Results of in vitro transposition reactions with WT or R848M RAG1 (mean ± SEM). Two-tailed t-test: **, p<0.01.
f, g, Results of in vivo plasmid-to-plasmid transposition assays with RAG2 1–383 (f) or 1–350 (g) and the indicated full length WT or mutant RAG1 protein, and with full length BbRAGL (mean ± SEM). Total antibiotic resistant colony numbers (gray bars) were corrected (black bars) for the fraction of colonies found to harbor plasmids with bone fide transposition events. Two-tailed t-test: *, p<0.05; **, p<0.01; ***, p<0.005 compared to WT RAG1.
h, Number of bone fide transposition events (3–7 bp target site duplication) identified in plasmid-to-genome transposition experiment.
Importantly, in an in vivo “plasmid-to-plasmid” transposition assay (Extended Data Fig. 8a), the R848M RAG1 mutation increased activity to detectable levels (Fig. 5f) while a reciprocal M949R mutation in BbRAG1L decreased activity relative to WT (Extended Data Fig. 8b, c). R848M RAG1 was, however, still ~100-fold less active than BbRAGL (Fig. 5f), raising the possibility that additional mechanisms suppress RAG-mediated transposition in vivo.
The RAG2 protein used in the assays of Fig. 5f (aa 1–383) contains part of the RAG2 acidic hinge (Fig. 1c), a domain present in jawed vertebrate RAG2 but absent from BbRAG2L10 and other known invertebrate RAG2-like proteins11. Strikingly, complete removal of the acidic hinge (RAG2 1–350) increased in vivo transposition activity ~100-fold (Fig. 5g), a result recapitulated in a second cell line and with human RAG proteins (Extended Data Fig. 8d, e). Transposition stimulation depended strongly on the RAG1 R848M mutation, as WT RAG1 lacked detectable transposition activity when paired with RAG2 1–350 (Fig. 5g). Together, RAG1 R848 and the RAG2 acidic hinge suppress RAG-mediated transposition in vivo more than 1000-fold. Transposition products generated in vitro and in vivo exhibited predominantly 5 bp target site duplications, as expected18,19 (Extended Data Fig. 7f). RAG2 acidic hinge deletion does not increase RAG-mediated transposition or DNA cleavage in vitro (Extended Data Fig. 7g, h) or substantially alter protein expression or V(D)J recombination activity in vivo (Extended Data Fig. 8f, g). Hence, the RAG2 acidic hinge suppresses transposition specifically at a post-cleavage step and only in cells. Mapping experiments revealed that aa 362–383 play a critical role in suppressing in vivo transposition by RAG2 1–383 (Extended Data Fig. 8h, i). In assays using RAG2 1–350, a RAG1 E649V mutation boosted transposition while S963A had little effect (Fig. 5g). We conclude that evolutionary adaptations arose early in jawed vertebrate evolution in RAG1 and RAG2 to provide two-tiered protection against RAG-mediated transposition.
To test whether this conclusion extends to RAG-mediated transposition into the genome, we employed a “plasmid-to-genome” transposition assay, with transposition target sites identified by high-throughput sequencing (Extended Data Fig. 9a–c). When paired with RAG2 1–350, E649V/R848M RAG1, WT RAG1, and no RAG1 yielded 930, 16, and zero independent transposition events, respectively (Fig. 5h and Extended Data Fig. 9d). Insertion sites were found on all chromosomes (Extended Data Fig. 9e) and were strongly biased to active genes, particularly in the vicinity of the transcription start site (Extended Data Fig. 9f–h). 180 of the 930 E649V/R848M RAG1-mediated insertions (19%) occurred in protein-coding exons (p=4e-82), which is noteworthy given that the primordial split antigen receptor gene of jawed vertebrates is believed to have been generated by RAG transposon insertion into an exon7,25,26. These data demonstrate that reversal of the protective adaptations acquired by jawed vertebrate RAG1 and RAG2 “reawakens” the RAG transposase and enables widespread transposition into genes and exons in the human genome.
Molecular Domestication of the RAG Transposon
The evolutionary adaptations that protect jawed vertebrate lymphocytes from insertional mutagenesis caused by RAG-mediated transposition have been a long-standing target of investigation27,28 and, a priori, could have involved changes in the RAG proteins, changes in the host cellular milieu, or both. Efficient RSS ligation was unlikely to suffice as a protective mechanism because signal joints can be re-cleaved and transposed by RAG29. Our findings reveal two critical adaptations, intrinsic to the RAG proteins and found only in jawed vertebrates, that each potently suppress RAG-mediated transposition in vivo and that together render the reaction almost undetectable. Like RAG1 R848, the RAG2 acidic hinge suppresses transposition at a post-cleavage step of the reaction, but unlike R848, these suppressive effects are detectable only in the context of living cells. The RAG2 acidic hinge has been implicated in the regulation of RAG catalytic activity30, chromatin targeting31, repair pathway choice32,33, and stability of the RAG-signal end complex33. It remains to be determined whether these activities are relevant to the suppression of RAG-mediated transposition in vivo and whether other proteins contribute to this suppression.
Accumulating evidence supports a model for RAG evolution (Extended Data Fig. 10) in which a Transib transposon34 captured a RAG2-like open reading frame in an early deuterostome to give rise to the original RAG transposon, which in turn gave rise to RAG1/RAG2 and RSSs in jawed vertebrates and RAG1L/RAG2L transposable elements and gene pairs in invertebrates9. We propose that the modular design of the RAG complex—with largely autonomous catalytic cores, swappable DNA binding modules, and a RAG2 accessory subunit—facilitated adaptation of RAG family enzymes to changing host environments and functional demands, including the adaptations in jawed vertebrate that led to a “tamed” RAG recombinase possessing coupled cleavage activity, adherence to the 12/23 rule, and suppressed transposition activity (Extended Data Fig. 10). Our findings contribute to the paradigm of transposon molecular domestication4,6, which is now recognized to encompass elements in almost all branches of life ranging from CRISPR in bacteria35 to active transposases encoded in the human genome whose function, and process of domestication, remain unknown36,37.
METHODS
Statistical methods were not used to predetermine sample size and experiments were not randomized. Investigators were not blinded to allocation during experiments and outcome assessment.
Plasmid generation.
pTT5M, a derivative of pTT5 containing a maltose binding protein (MBP) open reading frame (ORF), described previously10, was modified by inserting an in frame PreScission Protease cleavage site at the C-terminus of MBP, to create pTT5MP. Codon optimized BbRAG1L core (aa 468–1136) and full length BbRAG2L were cloned into pTT5MP at a NotI restriction site that lies downstream of the protease cleavage site by In-Fusion cloning. Truncated BbRAG1L open reading frames (ORFs) (aa 468–1136, aa 484–1136 and ∆NBD (aa 547–1136)) were cloned into pTT5M, as were mouse RAG1 core (aa 384–1008), RAG1 core ∆NBD (aa 462–1008) and RAG2 core (aa 1–383) ORFs. Chimera protein ORFs and point mutants thereof were cloned into pTT5MP. No difference in expression levels were noted between pTT5M and pTT5MP vectors.
A 5’TIR and a 3’TIR, each with 3’ flanking ProtoRAG sequences, were inserted together into the BamHI site of pUC19 by In-Fusion, creating a substrate with 402 bp between the tips of the TIRs. This vector was further modified to eliminate all instances of 5’-CAC in the DNA between the TIRs and in the ~130 bp of pUC19 flanking the 5’TIR and ~280 bp of pUC19 flanking the 3’TIR. This CAC-free region containing the TIRs was then subcloned into the EcoRV/NruI sites of pBR322 to create pB-5’/3’TIR. pB-5’/3’TIR was modified by deletion of the 5’TIR or the 3’TIR using PCR and In-Fusion cloning to create pB-5’TIR and pB-3’TIR. Other alterations to replace or modify the TIRs of pB-5’/3’TIR, pB-5’TIR, or pB-3’TIR, using In-Fusion cloning, resulted in plasmids containing the needed combinations of RSS, chimeric TIR/RSS, and scrambled TIR mutant sequences. The mutations that scrambled portions of the TIR were made by changing A to C, T to G, C to A, and G to T.
Protein expression and purification.
pTT5MP-BbRAG1L core and pTT5MP-BbRAG2L plasmids were cotransfected into expi293F™ cells using the ExpiFectamine™ 293 Transfection Kit. Cells (30–200 ml culture) containing co-expressed proteins were harvested 5 days after transfection by centrifugation (500g) and frozen at −80°C. Cells were re-suspended in lysis buffer (25 mM Tris, pH7.5, 1M KCl, 1 mM DTT) and disrupted by 3 cycles of freezing in liquid nitrogen and thawing in a room temperature water bath. Cell lysates were further disrupted by dounce homogenization, centrifuged at 45,000 r.p.m. (Beckman Coulter Optima LE-80K Ultracentrifuge, Type 50.2 Ti rotor ) for 1 hr at 4°C (all subsequent steps at 4°C), and the supernatant mixed with pre-equilibrated amylose resin and incubated for 2 hr with continual rotation. The beads were loaded onto a gravity flow column and washed with 50–100 ml of lysis buffer and protein eluted with 5–10 ml of elution buffer (25 mM Tris, pH7.5, 0.5M KCl, 1 mM DTT, 40 mM Maltose) depending on the initial cell culture volume. The eluate was further purified by size exclusion chromatography (SEC) on a Superdex™ 200 Increase 10/300 GL column in 20 mM HEPES pH7.6, 0.5 mM TCEP, 150 mM KCl and 5 mM MgCl2 or 5 mM Ca(OAc)2 (Ca2+ buffer used when protein was purified for assembling protein/DNA complexes for cryo-EM because Ca2+ supports DNA binding but not cleavage by RAG1 and BbRAGL10). SEC peak fractions were collected and pooled and protein concentrated to 4–10 µM using an Amicon centrifugal concentrator and frozen at −80°C. Other proteins were expressed and purified following a similar procedure. In all cases, the RAG1 core, BbRAG1L, or chimeric protein was co-expressed with the appropriate RAG2 core or BbRAG2L protein.
Full length (FL) His6-hHMGB1 and His6-hHMGB1∆C (aa 1–165 lacking the acidic C-terminal region) were expressed and purified as described previously10,39.
HEK293T cells were obtained from ATCC, expi293F™ cells were obtained from Thermo Fisher Scientific, and HCT116 cells were obtained from Eric Hendrickson, University of Minnesota. Cell lines used were not authenticated or tested for mycoplasma contamination.
DNA cleavage and cryo-EM substrates.
Linear substrate DNA used in cleavage experiments (e.g., Fig. 1d) was generated by PCR using the pBR322-based vectors as template, purified by agarose gel electrophoresis, and diluted to 100 nM concentration as a working stock. Unmutated TIR sequences are shown in Extended Data Fig. 5c. Unmutated RSS sequences are: 12RSS; 5’-CACAGTGGTAGTAGGCTGTACAAAAACC and 23RSS; 5’-CACAGTGGTAGTA CTCCACTGTCTGGCTGTACAAAAACC. The 3’TIR and 5’TIR DNA substrates, intact or nicked, used in SEC and for synaptic complex purification were assembled by annealing two complementary oligonucleotides:
3’TIR oligo sequence:
5’-CTTGGCAGCGCGCTGCACTATGATACTTACGCTATACCCAGCAGTGTCTGGTCGCCA
TCTTG
5’TIR oligo sequence:
5’-AACTTAGTACATACGCACTATGAAAACTTACGTGTGCATAAGGTCGGCGGCCATCTTG
In vitro DNA cleavage.
WT BbRAGL or RAG proteins (25 nM final concentration of each protein), substrate DNA (final concentration 10 nM) and 175 ng His6-hHMGB1 were incubated in reaction buffer (25 mM HOPS, pH7.0, 50 mM KCl, 2 mM DTT, 1.5 mM MgCl2; 16 μl final reaction volume) at 37°C for 1 hr or for the indicated time period. For reactions with chimeric proteins containing the RAG1 catalytic core, the final concentration of each protein was 50 nM. For reactions with chimeric proteins containing the BbRAG1L catalytic core, the final concentration of each protein was 50 nM, the Mg2+ concentration was 5 mM, and reaction time was 2 hr. In these experiments, control reactions for each experiment used the same conditions as the reactions with the chimeric proteins. Reactions were stopped by adding 1.25 μl 2.5% SDS, 5 μl proteinase K (150 μg/ml), 2 μl 0.5 M EDTA followed by incubation at 55°C for at least 3 hr. Samples were briefly centrifuged and the supernatant mixed with 1.7 μl 80% glycerol and loaded on a non-denaturing 1X TBE (Tris-borate-EDTA buffer) 6% polyacrylamide gel. After 1 hr electrophoresis at 100 V, gels were stained with SYBR GOLD in 1X TBE buffer for 20 min and imaged using a PharosFX™ Plus (Bio-Rad).
Confirmation of intramolecular transposition band.
The slow mobility band (as in Fig. 5d, arrow) was excised and DNA purified and subject to inverse PCR using primers F and R. The major PCR product band (Extended Data Fig. 7a, arrow) was excised and the DNA purified, cloned, and sequenced. Inversion circle transposition products were identified as described previously18.
F: TATTATGAGGCCCTTTCGTCTTC
R: CGCCTATTTTTATAGGTTAATGTCATG
BbRAGL/3’TIR synaptic complex assembly for cryo-EM.
Purified MBP-BbRAGL complex was mixed with 3’TIR DNA substrate and His6-hHMGB1∆C at a ratio of 1:2.5:2.5 in 20 mM HEPES pH7.6, 0.5 mM TCEP, 10 mM CaCl2 and 150 mM KCl and incubated at room temperature for 30 min. After incubation, 5% (v/v) PreScission Protease was added and the sample was incubated at room temperature for 1 hr to cleave off the MBP tags. The mixture was loaded on a Superdex™ 200 Increase 10/300 GL column and purified by SEC in 20 mM HEPES pH7.6, 0.5 mM TCEP, 150 mM KCl and 5 mM Ca(OAc)2. The peak column fractions were collected and concentrated (if necessary) to a protein concentration of ~0.4 mg/mL. The sample was immediately used to prepare cryo-EM grids.
Cryo-EM data acquisition.
3 μl of purified complex was applied to freshly glow discharged C-flat™ 400 mesh, R2/1 and R1.2/1.3 holey grids for intact DNA and nicked DNA complexes, respectively. Grids were blotted for 4 seconds in 100% humidity and plunge frozen in liquid ethane using a Vitrobot Mark 3 (FEI Company). A Titan Krios electron microscope (Janelia Research Campus, HHMI) operated at 300 kV, with a spherical aberration corrector and a Gatan Image Filter (slit width of ~20 eV), was used to acquire images with a K2 Summit direct electron detector (Gatan) in super-resolution mode. The image stacks were collected at a nominal magnification of 81,000X, corresponding to 0.675 Å per super-resolution pixel, at a dose rate of ~10.2 electron per physical pixel per second. The total exposure was 80 and 54 electrons per Å2, fractionated into 50 and 40 frames, for intact DNA complex and nicked DNA complex, respectively. All images were acquired in a defocus range from −1.0 to −2.5 μm. The statistics of data acquisition are summarized in Extended Data Table 1.
Image processing.
A total of 5164 and 4429 LZW-compressed TIFF image stacks were collected for intact DNA complex and nicked DNA complex, respectively. MotionCor2 1.140 was used for beam-induced motion correction and dose weighting. The first 2 frames were discarded, and the output aligned images were binned 2X in Fourier space, resulting in a pixel size of 1.35 Å for further processing. The non-dose weighted aligned images were used for ctf estimation by Gctf 1.0641. The dose-weighted images were used for autopicking, classifications and reconstruction. Roughly 3000 particles were manually picked, followed by a round of 2D classification to generate templates for RELION 1.4 autopicking. The autopicked particles were subjected to 2D classification in RELION-2.142,43 to remove junk particles. Particle coordinates in good classes were extracted for further manual inspection such that bad particles and images were discarded. A previously published cryo-EM map (EMD-6488)21 was low-pass filtered to 60 Å to serve as a starting reference for multiple rounds of 3D classification in RELION-2.1 without imposing symmetry. Good 3D classes were combined and used for gold standard refinement in RELION-2.1 with either C1 or C2 symmetry. Resolution estimation was based on the Fourier shell correlation cutoff at 0.143 (FSC0.143) between the 2 half maps, after a soft mask was applied to mask out solvent region (Extended Data Table 1). The final maps were corrected for K2 detector modulation and sharpened by their corresponding negative B-factor within RELION-2.1. Local resolution variation was estimated by the local resolution module in RELION-2.1.
Modeling and refinement.
An initial model was obtained by structural profiling of cBbRAG1L and BbRAG2L sequence propensities as previously described44–46. In brief, separate models of cBbRAG1L (aa 473–1110) and BbRAG2L were built accounting for accessibility, charge, hydropathy, consensus secondary structure, consensus intrinsic disorder profiles and fold recognition assessment using the Discovery Studio software suite 3.0 (Accelrys). The models were refined by remote homology techniques starting from mouse RAG1 structural templates in PDB: 3GNA17 for NBD* of BbRAG1L and PDB: 4WWX20 for cBbRAG1L and BbRAG2L. To eliminate steric conflicts and further minimize energy, these models were iteratively refined until convergence by repeated cycles of Generalized Born simulated annealing molecular dynamics for implicit solvent using NAMD 2.12 with CHARMM36 force field47 followed by model assessment of the Global Distance Test Total Score (GDT_TS) with QA-RecombineIT48 and local loop remodeling in regions showing the highest divergence. Annealing simulations were performed with harmonic restraints on the backbone protein atom positions in regions of regular secondary structure, while irregular loop regions were left to move freely. This brought the cBbRAG1L and BbRAG2L models to GDT_TS 60 and >67, and RMSD of 2.9 Å and 2.3 Å, respectively. Finally, the assembled cBbRAG1L/BbRAG2L structure was subjected to molecular dynamics simulation in explicit solvent to confirm robustness and stability and to assess configuration dynamics of cBbRAG1L and BbRAG2L domains relative to one another.
The BbRAG1L (aa 545–1104) and BbRAG2L (aa 1–366) model thus derived was flexibly fitted into the C2 symmetrized map of the nicked 3’TIR complex (4.3 Å) by Molecular Dynamics Flexible Fitting (MDFF)49. The flexibly fitted model was able to account for most density, except that BbRAG1L loops 640–650, 704–720, 732–740, 1046–1053 and BbRAG2L loops 11–22, 34–49, 67–74, 85–108, 179–190 300–314, which were adjusted and rebuilt in COOT 0.850. The density for loop 603–630 in BbRAG1L was insufficient for model building. An all-alanine chain was built to fit the density for the C-terminal tail (CTT*) of BbRAG1L (1105–1125). The DNA chains from the previously published model (PDB: 3JBY)21 were fit into the map and then changed to the correct DNA sequence in COOT 0.8. The model was adjusted in COOT 0.8 manually with iterative cycles of automatic rebuilding using Rosetta FastRelax protocol51. The model was further refined using the phenix.real_space_refine module in PHENIX with secondary structure restraints and Ramachandran restraints52. The final model was validated using MolProbity53 and EMringer54 (Extended Data Table 1). All molecular representations were generated in PyMol (http://www.pymol.org) and Chimera55.
In vitro transposition assay.
The in vitro intermolecular transposition reaction (Extended Data Fig. 7b) was performed as described under in vitro DNA cleavage. The 12/23RSS substrate was replaced by 10 nM linear donor fragment with tetracycline resistant marker and 10 nM pECFP-1 target plasmid. The final concentrations of RAG protein, Mg2+, and DTT were 50 nM, 1.5 mM, and 2 mM respectively. After proteinase K digestion, DNA was ethanol precipitated. 50 ng DNA was transformed into electrocompetent MC1061 bacterial cells and spread on plates containing kanamycin or tetracycline/kanamycin/streptomycin. Transposition efficiency was calculated by dividing the number of colonies obtained on double antibiotic plates by the number of colonies obtained on the kanamycin-alone plate10,18.
In vivo plasmid-to-plasmid transposition assay.
The in vivo plasmid-to-plasmid transposition assay (Extended Data Fig. 8a) was performed as described previously12. In brief, 293T cells were transfected with 4 µg each of the pEBB-RAG1 or mutant and pTT5M-RAG2 truncations or pEBB-FL RAG2, 6 µg of donor plasmid (pTetRSS), and 10 µg of target plasmid (pECFP-1) using polyethylenimine. The medium was changed 24 hr after transfection and cells were harvested after 48 hr. Plasmid DNA was precipitated and 300 ng DNA was transformed into electrocompetent MC1061 bacterial cells and plated on kanamycin or kanamycin/tetracycline/streptomycin (KTS) plates. For each protein combination assayed in Fig. 5f, g, plasmids from 30 colonies (except for very low efficiency reactions) from KTS plates were sequenced to determine if they contained a bone fide transposition event (3–7 bp TSD). Total transposition efficiency was calculated as described under in vitro transposition assay and a corrected value was calculated from the fraction of plasmids that contained a transposition event.
Western blotting.
Cells were harvested 48 hr after transfection of protein expression vectors, resuspended in lysis buffer (50 mM Tris, pH 7.5; 150 mM NaCl; 1% N-P40; cocktail protease inhibitor) on ice, and further disrupt by sonication. After centrifugation to remove insoluble debris, samples were mixed with loading buffer, subjected to SDS-PAGE, and transferred to a PVDF membrane, which were incubated separately with anti-RAG156, anti-RAG256, and mouse monoclonal anti-β-actin (Sigma #A1978) antibody.
In vivo recombination assay.
1 μg of RAG or BbRAGL expression vectors were co-transfected with 2 μg of pCJGFP32 or pTIRG810, respectively, into expi293F™ cells using polyethylenimine (DNA:PEI ratio of 1:3). Cells were harvested 72 h post-transfection, washed twice with PBS containing 1% FBS, stained with DAPI (4′,6-diamidino-2-phenylindole) and percent live cells expressing GFP was determined by flow cytometry as shown in Supplementary Data 2.
In vivo plasmid-to-genome transposition assay.
The in vivo plasmid-to-genome transposition assay (Extended Data Fig. 9a) was performed by transfecting 293T cells with 4 µg each of the pEBB-RAG1 or pEBB-RAG1-E649V/R848M or empty vector and pTT5M-RAG2 (1–350), and 6 µg of donor pBSK-12puro2312. 48 hr after transfection, 5×106 cells were split into medium containing 0.8 µg/mL puromycin. After 2–3 weeks of culture, colonies (many hundred from each experiment; colony formation was dependent on inclusion of the donor plasmid) were digested with trypsin, pooled, and re-seeded into new medium containing puromycin and cultured further to obtain sufficient cells. For each experiment, 107 cells were harvested and the genomic DNA was precipitated. Transposition insertion targets from three independent experiments (no RAG1, WT-RAG1, RAG1 E649V/R848M) were amplified using 12RSS and 23RSS LAM-PCR primers with six different barcodes (12v, 23v, 12wt, 23wt, 12m, 23m) as described previously57. Equal amounts of LAM-PCR product from the six groups were mixed and diluted as the library for high-throughput sequencing.
LAM-PCR primers: 12RSS: 5’-Biotin-ctttattgaggcttaagcagtgggttc
23RSS: 5’-Biotin-actgacactcgacctcgacaggattg
Nested-PCR primers:
12RSS: 5’-acactctttccctacacgacgctcttccgatctXXXXXXgcaaaaagcagatcttattttcgtt
23RSS: 5’-acactctttccctacacgacgctcttccgatctXXXXXXcttatcatgtctggatcgctTtatatacg
Where XXXXX represents the barcode.
High throughput sequencing and data analysis.
High-throughput sequencing was performed on an Illumina NextSeq 500. Cutadapt version 1.16 was used to identify barcodes with the adapter-matching error rate set to allow one mismatch in a 7–8 bp barcode. 95.9% of reads were successfully matched to a barcode and unmatched reads were discarded. Next, cutadapt was used to trim the barcode sequences, primer sequences, and 12RSS and 23RSS sequences. The resulting trimmed sequencing data contain only vector sequence or genomic DNA sequence from transposition events and other random integration events. Overall, 60.0% of barcoded sequencing reads contained identifiable RSS sequences and other reads were discarded. Trimmed sequences were aligned to human genome GRCh38 using Bowtie2 (version 2.2.9) using “very sensitive” end-to-end alignment mode. High-quality alignments (MAPQ >= 30; identified with Samtools 1.5) were converted to bed intervals using the bedtools bamToBed utility (bedtools version 2.27.1). Overlapping same-stranded events were merged for each of the six libraries. Bone fide transposition events give rise to 12RSS- and 23RSS-flanking genomic sequences that map to the same site in the genome but in opposite directions and with short overlaps (the target site duplication), a signature that was readily distinguished from random integration of the donor plasmid or excised RSS fragments (Extended Data Fig. 9b). To accomplish this, Bedtools intersect was used to identify loci where corresponding 12RSS and 23RSS libraries showed evidence of transposition events on opposite strands. All intersecting intervals with a 3–7 bp overlap were judged to be transposition events. Gene, exon, and transcription start site (TSS) definitions were downloaded from Ensembl gene v93, dataset Human genes (GRCh38.p12). Active TSSs and active genes/exons were defined based on H3K4me3 (experiment ENCSR000DTU) and H3K36me3 (experiment ENCSR910LIE) ChIP-seq datasets, respectively, from HEK293 cells from ENCODE (https://www.encodeproject.org).
Statistics and reproducibility.
DNA cleavage experiments were typically performed 3 or more times. Exceptions are: Fig. 3i, Fig. 4i, Extended Data Fig. 6a, 6c, and 6f (lanes 10 and 11), all n=2. Statistical analyses were performed using a two-tailed t-test (e.g., Fig. 5e–g)) or a one-tailed Fisher’s exact test (Fig. 5i).
Data availability statement.
The model of the cBbRAGL-nicked 3’TIR synaptic complex has been deposited in the Protein Data Bank with accession code PDB: 6B40. The cryo-EM maps of cBbRAGL in complex with intact or nicked 3’TIRs have been deposited in EMDataBank with accession codes EMD-7043, 7044, 7045, and 7046. High-throughput DNA sequence data to identify transposition events in the human genome have been deposited in the NCBI Sequence Read Archive with accession codes SRR8430227-SRR8430233 (Project PRJNA514369).
Extended Data
Extended Data Fig. 1. ProtoRAG transposon and analysis of the BbRAG1L NBD* domain.

a, Schematic diagram of the ProtoRAG transposon, and below it, the jawed-vertebrate RAG locus and prototypical antigen receptor gene (IGH).
b, Schematic diagram of full length and truncated BbRAG1L proteins (top), and cleavage reactions performed with those proteins (plus BbRAG2L) and TIR substrates, as indicated above and below the lanes. Core BbRAG1L (aa 468–1136) retains the cleavage pattern of full length BbRAG1L, while full length BbRAG1L exhibits strong single TIR cleavage (lanes, 6, 7). Closed and open arrowheads, single 5’TIR and single 3’TIR cleavage products, respectively. For gel source data, see Supplementary Figure 1.
c, Sequence alignment of BbRAG1L NBD* with RAG1 NBD showing divergent sequences with similar predicted secondary structure elements (alpha helices 1, 2 and 3).
d, Size-exclusion chromatography-multiple angle light scattering (SEC-MALS) analysis of the purified NBD* protein, indicating that the protein is a dimer in solution.
Extended Data Fig. 2. Biochemical properties and cryo-EM structure of cBbRAGL-3’TIR synaptic complexes.

a, SEC-MALS of MBP-cBbRAGL, indicating that the complex is a heterotetramer with two subunits each of cBbRAG1L and BbRAG2L.
b, c, SEC profiles of cBbRAGL incubated with intact (b) or nicked (c) 3’TIR, 5’TIR or 3’/5’TIRs showing resolution of protein-DNA complex from free DNA. Gels display the components of pooled column fractions containing the protein-DNA complex.
d, Representative 2D class averages of cryo-EM particles of cBbRAGL bound to intact or nicked 3’TIRs.
e, (Left) Fourier shell correlation (FSC) curves of the half maps from gold standard refinements of cBbRAGL-nicked 3’TIR complex with no symmetry applied (blue), cBbRAGL-intact 3’TIR complex with no symmetry applied (red), and with C2 symmetry applied (green). (Right) FSC curves of the gold standard refinement of cBbRAGL-nicked 3’TIR complex with C2 symmetry applied (blue) and of the C2 symmetrized map and model (green). Resolution of the maps are read by the cutoff values at FSC = 0.143.
f, Color coded local resolution estimation of the C2 symmetrized map of cBbRAGL in complex with nicked 3’TIR, viewed from a perspective similar (with a 30 degree rotation) to that of (g). Resolution is in general better for cBbRAG1L than for BbRAG2L.
g, h, Cryo-EM maps of cBbRAGL bound to intact 3’TIRs (5.3 Å overall resolution) (g) or nicked 3’TIRs (5.0 Å overall resolution) (h). One BbRAG1L subunit (gray) has been rendered partially transparent to allow visualization of DNAs inside the protein. Continuous DNA density running through the protein core is visible with nicked but not intact TIRs, arguing that the DNA in the vicinity of the active site becomes more rigidly constrained upon nicking. This is notable in light of the recent finding that DNA in the RAG active site melts and swivels in preparation for nicking23. Clear differences between the two DNAs are visible in the bottom half of the structures, with 3’TIR-a (orange) protruding below the protein and density for 3’TIR-b (red) dissipating before the DNA emerges from the protein core. This argues that the two identical DNA molecules are engaged differently by cBbRAGL, with one (3’TIR-b) less rigidly constrained by its interactions with protein.
Extended Data Fig. 3. Structural features of cBbRAGL.

a, Comparison of the models of cBbRAGL and cRAG (PDB 5ZDZ) bound to nicked DNA but with DNA removed, illustrating the absence of NBD* from the cBbRAGL structure. NBD is a dimer that can pivot on a flexible hinge to accommodate the different spacer lengths of a 12RSS and 23RSS, providing a structural explanation for the 12/23 rule20–22,58. We speculate that NBD*, HMGB1, and distal TIR sequences constitute a flexible domain located below the main complex, by analogy with RAG-RSS complexes.
b, Superimposition of cBbRAGL-nicked-3’TIR synaptic complex with RAG-nicked RSS synaptic complex (PDB 5ZDZ).
c, BbRAG2L adopts a doughnut-shaped structure consistent with that of a 6-bladed β-propeller. Because of low resolution, some elements cannot be unambiguously modeled as β-strands. Putative β-propellers I-VI are labeled, as are the N- and C-termini of the protein, showing that, as with RAG2, propeller I is composed of both N- and C-terminal sequences.
d, Color coded linear diagram of cBbRAG1L subdomains (top) and cartoon of the BbRAG1L dimer (bottom) with the subdomains of one subunit color coded as in the linear diagram. The other subunit is gray except for the preR subdomain. Stars indicate a gap in the BbRAG1L model spanning aa 603–630. Nomenclature and figure layout as in20. DDBD, dimerization and DNA binding domain; PreR, pre-RNase H domain; RNH, RNase H domain; ZnC2 and ZnH2, domains that contribute two cysteines and two histidines, respectively, for zinc coordination; CTD, C-terminal domain; CTT*, C-terminal tail.
e, Superimposition of cryo-EM map on the model of the nicked 3’TIR in the vicinity the flipped bases near the site of nicking.
f, g, 3D classes of cryo-EM maps of cBbRAGL bound to intact (f) or nicked (g) 3’TIRs (DNA omitted). One class is enlarged and shown from two vantage points below. The arrow points to the cleft that narrows in the open-to-closed transition. With intact DNA, three distinct 3D classes are distinguishable that vary in the degree of closure of the two arms of the V.
h, Superimposition of three forms of cBbRAGL illustrating the movement of a 3’TIR and BbRAG2L subunit (color coded as in e, f) that takes place during the open-to-closed transition. One cBbRAG1L/2L dimer has been aligned and movement is visualized in the other dimer.
Extended Data Fig. 4. Protein-DNA interactions in the cBbRAGL-nicked 3’TIR synaptic complex.

a, Schematic diagram of the detailed interactions between BbRAG1L and nicked 3’TIR DNA. Bold/underlined text, main chain interactions; regular text, side chain interactions; purple text, interactions involving BbRAG1L subunit a (defined as the subunit whose active site engages the TIR depicted); blue text, interactions involving symmetric BbRAG1L subunit b. BbRAG2L-DNA interactions could not be unambiguously assigned and are not depicted.
b, c, Orthogonal views of the nicked 3’TIR-BbRAG1L subunit a interaction (b) and the nicked RSS-RAG1 subunit a interaction (c). Protein electrostatic surface potential is indicated with blue (positive charge) and red (negative charge) using the scale (KT/e) below panels d, e.
d, BbRAG1L subunit b-nicked 3’TIR interaction.
e, RAG1 subunit b-nicked RSS interaction.
Extended Data Fig. 5. CTT, CTT*, and mutational analysis of ProtoRAG TIRs.

a, Superimposition showing CTT* extending from a structurally conserved region at the C-terminus of the catalytic core regions of mouse RAG1 (mRAG1) , zebra fish RAG1 (zRAG1), and BbRAG1L.
b, Sequence alignment of CTT from six vertebrate RAG1 proteins. Species name abbreviations used in this paper: Mmu, mus musculus (mouse); Hsa, homo sapien (man); Gag, Gallus gallus (chicken); Xla, Xenopus tropicalis (frog); Dre, Danio rerio (zebrafish); Bb, Branchiostoma belcheri (amphioxus); Pfl, Ptychodera flava (acorn worm); Spu, Strongylocentrotus purpuratus (purple sea urchin); Afo, Asterias forbesi (sea star); Etr, Eucidaris tribuloides (pencil urchin); Aga, Anopheles gambiae (mosquito); Aae, Aedes aegypti (mosquito); Dps, Drosophila pseudoobscura (fruit fly); Hze, Helicoverpa zea (corn earworm); Hvu, Hydra vulgaris (hydra).
c, Schematic indicating sub-regions of TIRs. Region 1 contains the heptamer and one additional bp, which in Fig. 1a and throughout the paper is defined as part of TR2. Otherwise, region 2 (broken up into 2a and 2b for the 5’ TIR) corresponds to TR2. Poorly conserved regions 3 and 4 separate TR2 from a distal conserved 9 bp element (region 5).
d-g, Cleavage of substrates containing a single 5’TIR (d, e) or a single 3’TIR (f, g), either intact (WT) or with the indicated region scrambled, by cBbRAGL (d, f) or the ΔNBD* cBbRAGL complex (e, g). Closed and open arrowheads, 5’TIR and 3’TIR cleavage products, respectively. Region 5 is completely dispensable for cleavage and regions 3 and 4 contribute modestly to 3’TIR but not 5’TIR cleavage. Upon deletion of NBD* from cBbRAG1L, 3’TIR cleavage loses all dependency on regions 3 and 4, consistent with the possibility that NBD* engages in functionally important interactions with regions 3 and 4 of the 3’TIR.
Extended Data Fig. 6. Activities of chimeric RAG1-BbRAG1L proteins and residues that influence coupled cleavage.

a, b, Cleavage by NBD-CC* is dependent on the length of the spacer between the TIR heptamer and the RSS nonamer. Substrates depicted schematically above the gel images. In (a), the substrates contain a single target based on T1 (Fig. 3b) whose spacer ranges in length from 10–14 bp. In (b), the substrate contains target T1 and a partner target based on T2 (Fig. 3b) whose spacer ranges in length from 20–25 bp. Dark arrowheads, T1 cleavage products; open arrowheads, T2 cleavage products.
c, d, Cleavage reactions using the NBD*-CC-CTT* and CC-CTT* proteins and T3 and T4 substrates (all depicted schematically in Fig. 3c), as indicated above the lanes. T3* and T4*, T3 and T4 targets with a C-to-A mutation of heptamer position 1 which renders the target uncleavable; [T4/T4]H+TR2 and [T3/T3]H+TR2, substrates in which both targets have had all substrate sequences except the heptamer and TR2 deleted. Asterisks as in Fig. 2g.
e, Cartoon depicting differences in the major protein-DNA interactions of BbRAGL and RAG.
f, Superposition of RAG1 and BbRAG1L in the region containing E649 and S963 in complexes bound to nicked DNA substrates illustrating the similarity of positioning of the active site residues E962 and E1063 and flanking residues N961 and N1062. h, RAG1 N961 and BbRAG1L N1062 have the potential to participate in hydrogen bond networks after nicking and could thereby stabilize the hairpin-competent configuration of the enzyme. This is notable in light of the fact that N961A mutant RAG1 displays enhanced coupled cleavage compared to WT RAG1.
i, Cleavage reactions using WT and mutant cBbRAG1L proteins (with BbRAG2L) and substrates containing one or two TIRs as indicated above and below the lanes (left). V751E cBbRAG1L, but not A1064S, reduces uncoupled single 3’TIR cleavage (lower black asterisk, lane 2; reduction also seen in lane 8) and single 5’TIR cleavage (seen most clearly in lane 5). The strong reduction in cleavage seen with the V751E/A1064S BbRAG1L double mutant suggests the possibility that hydrogen bonding between these two residues holds the active site in an inactive configuration. At right: quantitation of uncoupled cleavage as the ratio of the intensity of the 3’TIR single cleavage band (lower black asterisk) to that of the double cleavage band (red asterisk) as in lanes 1–3. Mean ± SEM. Two-tailed t-test: **, p<0.01, compared to WT cBbRAG1L.
Extended Data Fig. 7. In vitro transposition by WT and mutant RAG proteins.

a, Schematic of intramolecular transposition. If the 3’ OH nucleophiles attack the same strand as they are located on, the products are two deletion circles (top), but if they attack the opposite strands, a single inversion circle product is generated (bottom). Staggered attack on the target DNA backbone yields single stranded gaps in the products, represented as five short vertical lines.
b, Inverse PCR reaction to amplify inversion circles from purified intramolecular transposition product as in Fig. 5d, third lane. The band indicated with an arrow was excised, cloned, and sequenced, yielding sites at which intramolecular transposition occurred to yield inversion circles, indicated in the map of the excised 12/23RSS central fragment (below). Half arrows indicate approximate location of PCR primers. The location of deletion circle joints detected by sequencing are not indicated.
c, Schematic of intermolecular in vitro transposition assay. An RSS-flanked Tet gene is mobilized from a linear donor by RAG-mediated DNA cleavage and can transpose into a target plasmid, which is detected after bacterial transformation by the appearance of colonies on Kan/Tet/Str (KTS) plates (Streptomycin (Str) is not relevant in this assay).
d, In vitro DNA cleavage and intramolecular transposition by position 848-mutant cRAG1 (with RAG2 1–383). Increased transposition compared to WT cRAG1 is revealed by diminished intensity of the double cleavage band and increased intensity of the slow-migrating intramolecular inversion circle transposition product band (red arrow). Note, however, that the intensity of the inversion circle band underestimates the efficiency of transposition because deletion circle transposition products, which are of heterogeneous size and hence not visible as a discrete band, are also produced18.
e, Quantitation of intramolecular transposition efficiency from three independent experiments as in (d), measured by ratio of double cleavage band to 23RSS cleavage band (the latter serving as an internal control for the total amount of cleavage). The ratio decreases as intramolecular transposition increases in efficiency, consuming the double cleavage band. Mean, with data range indicated by box. Two tailed t-test; p-values are indicated. f, Distribution of transposition target site duplication lengths determined by sequencing of plasmid transposition products or from high-throughput sequencing of plasmid-to-genome transposition products (Extended Data Fig. 9d), as indicated above the bars. The RAG1 protein used is indicated below the bars. In vitro reactions as in Fig. 5e using RAG2 1–383; in vivo plasmid target reactions as in Fig. 5g using RAG2 1–350; genome transposition products generated using RAG2 1–350. In a small fraction of plasmids, sequencing revealed deletions at the site of insertion of the RSSs (red; deletion).
g, In vitro cleavage and intramolecular transposition reactions using RAG2 1–352 and RAG2 1–383 (as indicated above the lanes) and WT or mutant cRAG1 (as indicated below the lanes). Transposition is readily detected with both forms of RAG2 and is increased by the RAG1 R848M mutation.
h, In vitro intermolecular transposition assays using RAG2 1–383 and RAG2 1–352 and WT or mutant cRAG1 (as indicated below the lanes). Deleting the RAG2 acidic hinge does not increase the efficiency of intermolecular transposition in vitro.
Extended Data Fig. 8. In vivo transposition by RAG and BbRAGL proteins.

a, Schematic of plasmid-to-plasmid in vivo transposition assay. An RSS-flanked Tet gene is mobilized from a donor plasmid by RAG-mediated DNA cleavage and can transpose into a target plasmid, which is detected after bacterial transformation by the appearance of colonies on Kan/Tet/Str (KTS) plates (Streptomycin (Str) reduces background in the assay by selecting against bacteria harboring the rpsL gene, present in the donor plasmid).
b, Schematic of in vivo GFP fluorescence recombination assay, used to generate data of panels (c) (right), (e) (right) and (g). Excision of the polyadenylation sequence (Poly-A) together with its flanking RSSs or TIRs (triangles) by RAG or BbRAGL and resealing of the plasmid allows for expression of GFP.
c, In vivo transposition (left) and recombination (right) activity in HEK293T cells of WT and M949R BbRAG1L (together with BbRAG2L). Mean ± SEM. Two-tailed t-test: ***, p<0.005, compared to WT BbRAG1L.
d, In vivo transposition activity assayed in human colon cancer cell line HCT116 with full length RAG1 R848M/E649V and either RAG2 1–350 or 1–383. As in HEK293T cells, transposition is strongly inhibited by the RAG2 acidic hinge. Mean ± SEM.
e, In vivo transposition (left) and recombination (right) activity in HEK293T cells of WT and R851M human RAG1 together with different forms of human RAG2, beginning at amino acid 1 and ending with the amino acid indicated below the bars. Mean ± SEM. Two-tailed t-test: ***, p<0.005; ****, p<0.001 compared to WT human RAG1.
f, g, Protein expression (f) and recombination activity (g) in HEK293T cells of WT and mutant mouse RAG1 and RAG2 proteins used in the in vivo transposition assays in this study. The data demonstrate that the large increases in transposition activity observed with some proteins (e.g., RAG2 1–350 and 1–352, and RAG1 R848M) are not due to large increases in protein expression or cleavage/recombination activity.
h, i, In vivo transposition activity assayed in HEK293T cells with full length RAG1 R848M (h) or R848M/E649V (i) and various forms of RAG2, beginning at amino acid 1 and ending with the amino acid indicated below the bars. FL, full length RAG2.
Extended Data Fig. 9. Transposition into the human genome by mutant RAG proteins.

a, Schematic of plasmid-to-genome in vivo transposition assay. An RSS-flanked Puro expression cassette is mobilized from a plasmid donor by RAG-mediated DNA cleavage and can transpose into the genome, which is detected by selection with puromycin and high-throughput sequencing.
b, Schematic illustrating detection of bone fide transposition events into the genome by LAM-PCR and high-throughput sequencing. LAM-PCR is performed on genomic DNA with biotinylated primers (half arrows) that extend into the DNA flanking either the 12RSS or 23RSS; thereafter, independent libraries are prepared and sequenced for the 12RSS and 23RSS flanks. If the donor plasmid randomly inserts into the genome (i), then the RSS is flanked by donor plasmid sequences. If the RSS fragment is cleaved at one or both RSSs and randomly inserted into genome (ii), then a match with an appropriate sequence duplication (indicative of a TSD) will not be found between the 12RSS and 23RSS libraries. Finally, if the RSS fragment is inserted into the genome by transposition (iii), a match with a 3–7 bp TSD will be found in the 12RSS and 23RSS libraries.
c, Tissue culture plates stained with crystal violet showing puromycin-resistant colonies for experiments using RAG2 1–350 and either WT or R848M/E649V RAG1. Colony numbers increase about two-fold with the mutant RAG1 protein but many colonies are seen with WT RAG1 due to random integration of the donor plasmid. Essentially no colonies are seen if the donor plasmid is omitted (first column of plates)
d, Summary of sequence data obtained from the plasmid-to-genome transposition experiments. For each of the six libraries, column 1 shows the total number of reads with a barcode and RSS, columns 2 and 3 show a breakdown of number of reads in which RSS flanking sequences map to the human genome or the donor plasmid (a small fraction of reads do map to either genome or plasmid due to poor read quality), column 4 shows the number of unique reads that map to the genome (after elimination of duplicates), and column 5 shows the number of bone fide transposition events detected.
e, Rainfall circos plot of transposition events into chromosomes of HEK293T cells.
f-h, Genome features of transposon integration sites mediated by R848M/E649V RAG1 and RAG2 1–350. f, Number (percent) of transposition events into the genome features indicated. TSS, transcription start site. One-tailed Fisher’s exact test was used to determine whether the frequency of transposition events was greater than that expected by chance: genes (p=9e-30); protein coding genes (p=5e-35); exons (p=6e-86); protein coding exons (p=4e-82) and within 2 kb of a TSSs (p=5e-180). g, h, Meta-analysis of integration sites within gene bodies (g) and flanking TSSs (h).
Extended Data Fig. 10. Model for RAG evolution in metazoans.

Steps leading from the ancestral Transib transposon, consisting of a RAG1-like open reading frame flanked by RSS-like TIRs34, to the RAG recombinase and “split” antigen receptor genes of jawed vertebrates. Box I. Capture of a RAG2-like open reading frame by a Transib transposon to generate the ancestral RAG transposon in an early deuterostome. Box II. Key events in the evolution of RAG1/RAG2 and antigen receptor genes of jawed vertebrates: (A) Insertion of the RAG transposon into the exon of a gene encoding an immunoglobulin-domain receptor protein to generate the ancestral antigen receptor gene and (B) Loss of CTT* and acquisition of E649 and S963 by RAG1 facilitated evolution of the 12/23 rule and coupled cleavage, respectively, while acquisition of RAG1 R848 and the RAG2 acidic hinge powerfully suppressed RAG transposition activity. The order of events depicted in box II is not known. RAG-related elements, if found in members of a given lineage, are indicated at right, as is the presence of the CTT* domain. Protostome lineages have been collapsed into a single branch. While vertical transmission is consistent with the distribution of RAG1/RAG2 transposon/recombinase elements in deuterostomes11, horizontal transmission might have contributed to the spread of Transib elements.
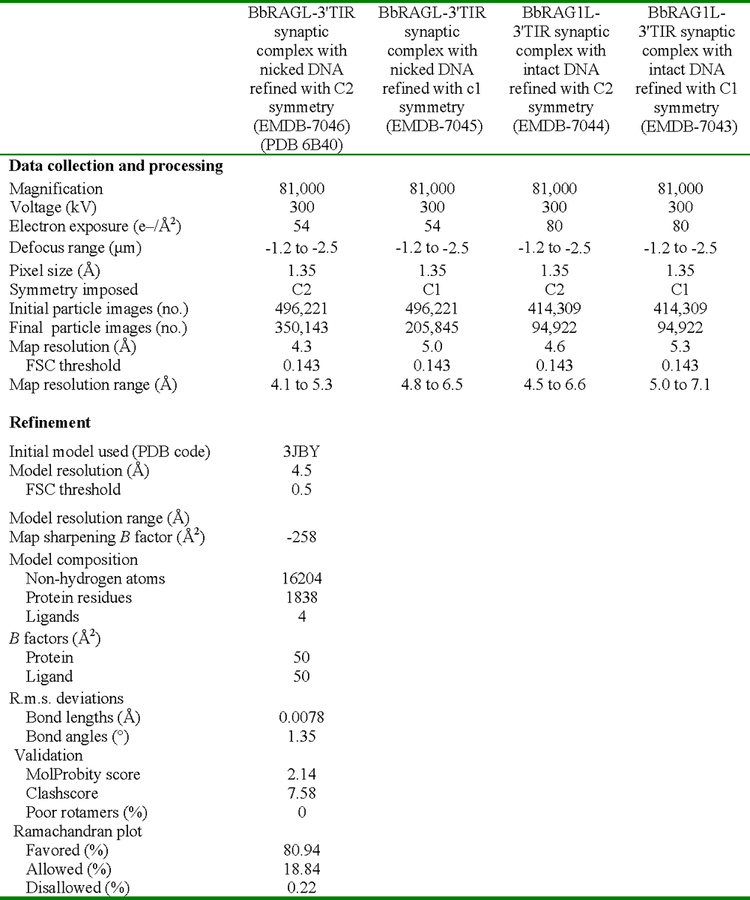
Extended Data Table 1. Cryo-EM data collection, refinement and validation statistics.
Summary of relevant parameters used during cryo-EM data collection and processing. Refinement and validation statistics are provided for the molecular model of the BbRAGL-3’TIR synaptic complex with nicked DNA with C2 symmetry.
 |
Supplementary Material
ACKNOWLEDGMENTS
The authors thank Y. Kong for computational analysis of genome transposition data, W. Eliason for assistance with SEC-MALS, C. Akey for help with freezing of the grids for BbRAGL-intact 3’TIR, R. Huang and H. Chuan for help with cryo-EM data collection, J. Wang for advice and insight in structural analysis, E. Hendrickson for HCT116 cells, XEGEN for NBD phylogenetic sequence analysis, Z. Chou, X. Liu, H. Zhang for an insertion-site mapping script, and M. Ciubotaru for advice, and members of the Schatz lab for comments on the manuscript. This work was supported in part by R01 AI32524 and R01 AI137079 (D.G.S.), R01 AI102778 (Y. X.), 2013CB917800 from the Ministry of Science and Technology of China and 91231206 from the National Natural Science Foundation of China (A. X.), UEFISCDI grant PN-III-ID-PCE-2016–0650 and Romanian Academy programs 1 & 3 of IBAR (M.D.S. and A.J.P.) and grants from Centre National de la Recherche Scientifique and Aix-Marseille Université (P.P.).
Footnotes
End notes
Supplementary information is linked to the online version of the paper at www.nature.com/nature.
The authors declare no competing interests.
REFERENCES
- 1.Gellert M V(D)J recombination: RAG proteins, repair factors, and regulation. Annu. Rev. Biochem 71, 101–132, (2002). [DOI] [PubMed] [Google Scholar]
- 2.Schatz DG & Swanson PC V(D)J recombination: mechanisms of initiation. Annu. Rev. Genet 45, 167–202, (2011). [DOI] [PubMed] [Google Scholar]
- 3.Lewis SM The mechanism of V(D)J joining: lessons from molecular, immunological, and comparative analyses. Adv. Immunol 56, 27–150, (1994). [DOI] [PubMed] [Google Scholar]
- 4.Sinzelle L, Izsvak Z & Ivics Z Molecular domestication of transposable elements: from detrimental parasites to useful host genes. Cell. Mol. Life Sci 66, 1073–1093, (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Levin HL & Moran JV Dynamic interactions between transposable elements and their hosts. Nat. Rev. Genet 12, 615–627, (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jangam D, Feschotte C & Betran E Transposable Element Domestication As an Adaptation to Evolutionary Conflicts. Trends Genet 33, 817–831, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Thompson CB New insights into V(D)J recombination and its role in the evolution of the immune system. Immunity 3, 531–539, (1995). [DOI] [PubMed] [Google Scholar]
- 8.Fugmann SD The origins of the Rag genes--from transposition to V(D)J recombination. Semin. Immunol 22, 10–16, (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Carmona LM & Schatz DG New insights into the evolutionary origins of the recombination-activating gene proteins and V(D)J recombination. FEBS J 284, 1590–1605, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Huang S et al. Discovery of an Active RAG Transposon Illuminates the Origins of V(D)J Recombination. Cell 166, 102–114, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Morales Poole JR, Huang SF, Xu A, Bayet J & Pontarotti P The RAG transposon is active through the deuterostome evolution and domesticated in jawed vertebrates. Immunogenetics 69, 391–400, (2017). [DOI] [PubMed] [Google Scholar]
- 12.Chatterji M, Tsai CL & Schatz DG Mobilization of RAG-generated signal ends by transposition and insertion in vivo. Mol. Cell Biol 26, 1558–1568, (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Reddy YV, Perkins EJ & Ramsden DA Genomic instability due to V(D)J recombination-associated transposition. Genes Dev 20, 1575–1582, (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Curry JD et al. Chromosomal reinsertion of broken RSS ends during T cell development. J. Exp. Med 204, 2293–2303, (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Messier TL, O’Neill JP, Hou SM, Nicklas JA & Finette BA In vivo transposition mediated by V(D)J recombinase in human T lymphocytes. EMBO J 22, 1381–1388, (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Little AJ, Matthews AG, Oettinger MA, Roth DB & Schatz DG in Molecular Biology of B cells (eds Alt FW, Honjo T, Radbruch A, & Reth M) Ch. 2, 13–34 (Academic Press/Elsevier Limited, 2015). [Google Scholar]
- 17.Yin FF et al. Structure of the RAG1 nonamer binding domain with DNA reveals a dimer that mediates DNA synapsis. Nat. Struct. Mol. Biol 16, 499–508, (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Agrawal A, Eastman QM & Schatz DG Transposition mediated by RAG1 and RAG2 and its implications for the evolution of the immune system. Nature 394, 744–751, (1998). [DOI] [PubMed] [Google Scholar]
- 19.Hiom K, Melek M & Gellert M DNA transposition by the RAG1 and RAG2 proteins: a possible source of oncogenic translocations. Cell 94, 463–470, (1998). [DOI] [PubMed] [Google Scholar]
- 20.Kim MS, Lapkouski M, Yang W & Gellert M Crystal structure of the V(D)J recombinase RAG1-RAG2. Nature 518, 507–511, (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ru H et al. Molecular Mechanism of V(D)J Recombination from Synaptic RAG1-RAG2 Complex Structures. Cell 163, 1138–1152, (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kim MS et al. Cracking the DNA Code for V(D)J Recombination. Mol. Cell 70, 358–370 e354, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ru H et al. DNA melting initiates the RAG catalytic pathway. Nat. Struct. Mol. Biol 25, 732–742, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kriatchko AN, Anderson DK & Swanson PC Identification and characterization of a gain-of-function RAG-1 mutant. Mol. Cell Biol 26, 4712–4728, (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sakano H, Hppi K, Heinrich G & Tonegawa S Sequences at the somatic recombination sites of immunoglobulin light-chain genes. Nature 280, 288–294, (1979). [DOI] [PubMed] [Google Scholar]
- 26.Hsu E & Lewis SM in Molecular Biology of B Cells (eds Alt FW, Honjo T, Radbruch A, & Reth M) Ch. 9, 133–149 (Academic Press, 2015). [Google Scholar]
- 27.Jones JM & Gellert M The taming of a transposon: V(D)J recombination and the immune system. Immuno. Rev 200, 233–248, (2004). [DOI] [PubMed] [Google Scholar]
- 28.Chatterji M, Tsai CL & Schatz DG New concepts in the regulation of an ancient reaction: transposition by RAG1/RAG2. Immuno. Rev 200, 261–271, (2004). [DOI] [PubMed] [Google Scholar]
- 29.Neiditch MB, Lee GS, Huye LE, Brandt VL & Roth DB The V(D)J recombinase efficiently cleaves and transposes signal joints. Mol. Cell 9, 871–878, (2002). [DOI] [PubMed] [Google Scholar]
- 30.Lu C, Ward A, Bettridge J, Liu Y & Desiderio S An autoregulatory mechanism imposes allosteric control on the V(D)J recombinase by histone H3 methylation. Cell Rep 10, 29–38, (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ward A, Kumari G, Sen R & Desiderio S The RAG-2 Inhibitory Domain Gates Accessibility Of The V(D)J Recombinase To Chromatin. Mol. Cell Biol 38, e00159, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Corneo B et al. Rag mutations reveal robust alternative end joining. Nature 449, 483–486, (2007). [DOI] [PubMed] [Google Scholar]
- 33.Coussens MA et al. RAG2’s acidic hinge restricts repair-pathway choice and promotes genomic stability. Cell Rep 4, 870–878, (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kapitonov VV & Jurka J RAG1 core and V(D)J recombination signal sequences were derived from Transib transposons. PLoS Biol 3, e181, (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Krupovic M, Beguin P & Koonin EV Casposons: mobile genetic elements that gave rise to the CRISPR-Cas adaptation machinery. Curr. Opin. Microbiol 38, 36–43, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Majumdar S, Singh A & Rio DC The human THAP9 gene encodes an active P-element DNA transposase. Science 339, 446–448, (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Henssen AG et al. Genomic DNA transposition induced by human PGBD5. eLife 4, e10565, (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lefranc MP & Lefranc G The T cell receptor FactsBook (Academic Press, 2001). [Google Scholar]
- 39.Bergeron S, Anderson DK & Swanson PC RAG and HMGB1 proteins: purification and biochemical analysis of recombination signal complexes. Methods Enzymol 408, 511–528, (2006). [DOI] [PubMed] [Google Scholar]
- 40.Zheng SQ et al. MotionCor2: anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat. Methods 14, 331–332, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhang K Gctf: Real-time CTF determination and correction. J. Struct. Biol 193, 1–12, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Scheres SH RELION: implementation of a Bayesian approach to cryo-EM structure determination. J. Struct. Biol 180, 519–530, (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kimanius D, Forsberg BO, Scheres SH & Lindahl E Accelerated cryo-EM structure determination with parallelisation using GPUs in RELION-2. eLife 5, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Slootweg EJ et al. Structural determinants at the interface of the ARC2 and leucine-rich repeat domains control the activation of the plant immune receptors Rx1 and Gpa2. Plant Physiol 162, 1510–1528, (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhang YH, Shetty K, Surleac MD, Petrescu AJ & Schatz DG Mapping and Quantitation of the Interaction between the Recombination Activating Gene Proteins RAG1 and RAG2. J. Biol. Chem 290, 11802–11817, (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kozuki T et al. Roles of the C-terminal domains of topoisomerase IIalpha and topoisomerase IIbeta in regulation of the decatenation checkpoint. Nucl. Acids Res 45, 5995–6010, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Phillips JC et al. Scalable molecular dynamics with NAMD. J. Comput. Chem 26, 1781–1802, (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pawlowski M, Bogdanowicz A & Bujnicki JM QA-RecombineIt: a server for quality assessment and recombination of protein models. Nucl. Acids Res 41, W389–397, (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Trabuco LG, Villa E, Mitra K, Frank J & Schulten K Flexible fitting of atomic structures into electron microscopy maps using molecular dynamics. Structure 16, 673–683, (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Emsley P, Lohkamp B, Scott WG & Cowtan K Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr 66, 486–501, (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.DiMaio F et al. Atomic-accuracy models from 4.5-A cryo-electron microscopy data with density-guided iterative local refinement. Nat. Methods 12, 361–365, (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Adams PD et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr 66, 213–221, (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chen VB et al. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr 66, 12–21, (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Barad BA et al. EMRinger: side chain-directed model and map validation for 3D cryo-electron microscopy. Nat. Methods 12, 943–946, (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pettersen EF et al. UCSF Chimera--a visualization system for exploratory research and analysis. J. Comput. Chem 25, 1605–1612, (2004). [DOI] [PubMed] [Google Scholar]
- 56.Coster G, Gold A, Chen D, Schatz DG & Goldberg M A dual interaction between the DNA damage response protein MDC1 and the RAG1 subunit of the V(D)J recombinase. J. Biol. Chem 287, 36488–36498, (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hu J et al. Detecting DNA double-stranded breaks in mammalian genomes by linear amplification-mediated high-throughput genome-wide translocation sequencing. Nat. Protoc 11, 853–871, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lapkouski M, Chuenchor W, Kim MS, Gellert M & Yang W Assembly Pathway and Characterization of the RAG1/2-DNA Paired and Signal-end Complexes. J. Biol. Chem 290, 14618–14625, (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The model of the cBbRAGL-nicked 3’TIR synaptic complex has been deposited in the Protein Data Bank with accession code PDB: 6B40. The cryo-EM maps of cBbRAGL in complex with intact or nicked 3’TIRs have been deposited in EMDataBank with accession codes EMD-7043, 7044, 7045, and 7046. High-throughput DNA sequence data to identify transposition events in the human genome have been deposited in the NCBI Sequence Read Archive with accession codes SRR8430227-SRR8430233 (Project PRJNA514369).