

Abstract
Chronic low-grade neuroinflammation is increasingly implicated in organ damage caused by alcohol abuse. Purinergic P2X7 receptors (P2X7Rs) play an important role in the generation of inflammatory responses during a number of CNS pathologies as evidenced from studies using pharmacological inhibition approach. P2X7Rs antagonism has not been tested during chronic alcohol abuse. In the present study, we tested the potential of P2X7R antagonist A804598 to reduce/abolish alcohol-induced neuroinflammation using chronic intragastric ethanol infusion and high-fat diet (Hybrid) in C57BL/6J mice. We have previously demonstrated an increase in neuroinflammatory response in 8 weeks of Hybrid paradigm. In the present study, we found neuroinflammatory response to 4 weeks of Hybrid exposure. A804598 treatment reversed the changes in microglia and astrocytes, reduced/abolished increases in mRNA levels of number of inflammatory markers, including IL-1β, iNOS, CXCR2, and components of inflammatory signaling pathways, such as TLR2, CASP1, NF-kB1 and CREB1, as well in the protein levels of pro-IL-1β and Nf-kB1. The P2X7R antagonist did not affect the increase in mRNA levels of fraktalkine (CX3CL1) and its receptor CX3CR1, an interaction that plays a neuroprotective role in neuron-glia communication. P2X7R antagonism also resulted in reduction of the inflammatory markers but did not alter steatosis in the liver. Taken together, these findings demonstrate how P2X7R antagonism suppresses inflammatory response in brain and liver but does not alter the neuroprotective response caused by Hybrid exposure. Overall, these findings support an important role of P2X7Rs in inflammation in brain and liver caused by combined chronic alcohol and high-fat diet.
Keywords: Intragastric ethanol and high fat diet exposure, Purinergic P2X7 receptor pharmacological inhibition, Neuroinflammation, Liver inflammation and steatosis, Immunofluorescence, Taqman custom gene expression
Introduction
Alcohol abuse continues to be a major concern worldwide as the most popular consumed mind-altering substance. Chronic alcohol abuse leads to an enormous economic cost as well as produces significant health related issues including organ damage (Sacks et al. 2015; Stahre et al. 2014). Presently, the pharmaco-social treatment options for alcohol use disorder are limited due, in part, to multiple targets in the brain and the lack of understanding of complex neurobiological mechanisms of alcohol action. One area of developing interest related to the effects of alcohol is the inflammatory processes induced by ethanol as a potential mechanism for organ/tissue damage. Unfortunately, the inflammatory response to ethanol is a complex process that involves a myriad of proteins and many different signaling pathways (Alfonso-Loeches and Guerri 2011; Vetreno and Crews 2014).
Adenosine triphosphate (ATP)-activated purinergic P2X7 receptors (P2X7Rs) belong to the P2X receptor superfamily consisting of 7 family members, P2X1-P2X7 (Khakh and North 2006; North and Jarvis 2013). Among the family members, P2X7Rs are unique in that: 1) They are activated by high micromolar to millimolar rather than low micromolar (P2X1-P2X6) concentrations of ATP; 2) Activation results in the formation of a homotrimer receptor with a large pore allowing transmembrane fluxes of small molecules up to 900 Da, including ATP (Skaper et al. 2010; Volonte et al. 2012). In addition, P2X7Rs interact with many intracellular adaptor and signaling proteins due to their unusually long C-terminus. P2X7Rs have been identified most frequently on immune cells such as mast cells, macrophages, microglia, and dendritic cells (Monif et al. 2010).
P2X7Rs play an important role in the generation of inflammatory responses and as such have been implicated in a number of CNS disorders. For example, P2X7Rs are upregulated during Alzheimer’s disease (McLarnon et al. 2006), amyotrophic lateral sclerosis (Volonte et al. 2011), Huntington’s disease (Diaz-Hernandez et al. 2009), behavioral and mood disorders (Skaper et al. 2010). Pharmacological blockade of the P2X7R and deletion of p2xr7 gene were shown to ameliorate neuropathology in animal models of neurodegenerative diseases (Chessell et al. 2005; Takenouchi et al. 2010; Tewari and Seth 2015), spinal cord injury and multiple sclerosis (Matute et al. 2007; Peng et al. 2009).
We have recently started testing the role of P2X7Rs in alcohol-induced neuroinflammation using a model of chronic ethanol (alcohol molecule) exposure in mildly obese C57BL/6J mice, developed at the Southern California Research Center for Alcoholic Liver and Pancreatic Diseases (ALPD) and Cirrhosis. This model results in dramatic blood ethanol concentrations (BECs), severe hepatic steatosis and elevated alanine aminotransferase levels (Ueno et al. 2012). In our initial studies, we demonstrated that 8 weeks of iG ethanol exposure combined with high-fat diet in C57BL/6J mice caused an increase in pro-inflammatory markers in the brain along with activation of microglia and astrocytes, two important glial cells in regulating CNS immune responses (Asatryan et al. 2015). These changes paralleled increased P2X7R expression in similar ethanol-sensitive brain regions (Asatryan et al. 2015). In addition, our recent work suggested functional involvement of P2X7Rs in alcohol effects. We found that ethanol potentiated the pore activity of P2X7Rs in an allosteric manner in cultured BV2 microglial cells (Asatryan et al. 2018). On the other hand, high fat diet was shown to increase the expression of P2X7Rs in corneal epithelium (Kneer et al. 2018). Furthermore, P2X7Rs mediated obesity-induced inflammatory Th17 response in visceral adipose tissues in humans, (Pandolfi et al. 2016) suggesting a functional role for P2X7Rs.
In the present study, we used a pharmacological inhibition approach to further test the role of P2X7Rs in neuroinflammation caused by exposure to chronic alcohol and high-fat diet. Among the number of commercially available antagonists, we selected to use A804598 in these experiments due to its high potency and selectivity towards P2X7Rs from different species with the resultant block of agonist-stimulated release of IL-1β (Donnelly-Roberts et al. 2009) and ability to readily cross the blood-brain barrier (Able et al. 2011). This antagonist has been tested on neuroimmune signaling and behavioral consequences in vivo in experimental animal models of stress (Catanzaro et al. 2014), neuropathic pain (Jung et al. 2017), traumatic brain injury (Liu et al. 2017), and visual processing in retina (Chavda et al. 2016). We tested the effect of A804598 on the inflammatory responses in the brain and liver caused by exposure to chronic alcohol and high fat diet. Specifically, we studied the effects on astrocytes, microglia, neuroinflammatory, neuroprotective markers, components of signaling and neurotransmission pathways in the brain as well as on steatosis and inflammation in liver.
Methods
Hybrid Model, P2X7 Antagonist Treatments
Age matched (8–10 weeks), male C57BL/6J mice obtained from Jackson laboratories (Bar Harbor, ME) were used in the experiments. All animals were treated in accordance with the National Institutes of Health Guide for Care and Use of Laboratory Animals and protocols approved by the USC Institutional Animal Care and Use Committee.
Mice were single-housed at the Animal Core facility during all the procedures including surgeries, housing and treatments. The experimental strategy was based on the Hybrid model of chronic ethanol exposure developed at the Southern California Center for ALPD and Cirrhosis. The model combines an established intragastric (iG) ethanol infusion with Western high fat diet in mice and was developed to study alcoholic liver disease (Ueno et al. 2012; Xu et al. 2011). Briefly, mice underwent surgery to place a catheter in their stomachs. After 1 week of recovery from the surgery, mice were fed ad libitum high-fat solid diet (HCFD pellets- 1% w/w cholesterol, 20%Cal lard, 17% corn oil:HCFD, Dyets Inc. #180724) for 2 weeks. Mice were then divided into 2 groups and were exposed via iG catheters to liquid high fat diet (HFD - 35%Cal corn oil) plus either ethanol (~27 g/kg/day) at 60% of total required calories (denoted as Hybrid throughout the manuscript) or isocaloric dextrose (denoted as Control). All mice continued to consume HCFD for the remaining 40% calories. In addition, once per week, mice in the Hybrid group were given an additional binge bolus dose of ethanol (3.5~5 g/kg) during the dark cycle. Starting the 2nd week, mice in the Control (n = 10) and half of the Hybrid (n = 10) groups were given vehicle, consisting of 2% dimethyl sulfoxide, 30% polyethylene glycol 300, and 68% (2-Hydroxypropyl)-β-cyclodextrin (Sigma-Aldrich (Sigma), St. Louis, MO). At the same time, the other half of mice in the Hybrid group received P2X7 antagonist N-cyano-N″-[(1S)-1-phenylethyl]-N′-5-quinolinyl-guanidine (A804598,5 mg/kg, Tocris Bioscience, Bristol, UK) freshly dissolved in the vehicle (denoted as Hybrid + A804598, n = 13). The iG route of administration and lower dose was selected based on our pilot study, which demonstrated low tolerability of mice to intraperitoneal injection of higher doses of the antagonist (5–25 mg/kg), as well as to avoid potential toxicities and interaction with ethanol. Both vehicle and antagonist were given 3 times a week through the iG catheter. The treatments continued for 3 weeks for the total duration of 4 weeks. Mice were harvested 2 days after last infusion of either vehicle or antagonist, with ethanol treatment continuing until sacrifice.
Harvesting Brain and Liver Tissues
Shortly after the exposures, the iG tubes were removed, and mice were euthanized by CO2. Mice were then perfused transcardially with PBS after the blood sample collection from the posterior vena cava, decapitated, and livers removed. Brains were removed from the skull and either segmented coronally into three general sections (front, mid, rear) for immunohistochemistry (IHC) or hippocampi were dissected for reverse transcription-quantitative real-time polymerase chain reaction (RT-qPCR) or for Western blotting. Whole livers were weighed at the time of collection, and then small 1 cm pieces were cut from the median lobes for later histological evaluations and RT-qPCR. Brain and liver tissues collected for RT-qPCR were snap frozen and kept at −80 °C, whereas tissues for IHC were placed in 4% paraformaldehyde (Sigma-Aldrich, St. Louis, MO) for two days, then transferred to 70% ethanol before further processing.
Blood Ethanol Concentration (BEC) Measurement
Blood was collected using heparin-treated syringes, centrifuged and BECs in plasma measured using ANALOX GM7 Analyzer (Analox Instruments USA, MA).
Histology and Immunofluorescence
All the histological procedures were performed at the USC School of Pharmacy Histology Core Lab. Fixed brain and liver tissues were embedded in paraffin and 5 μm sections were prepared using a rotary Microtome (MicroHM310, Microm International GmbH, Wolldorf, Germany). Liver tissue slices were deparaffinized, rehydrated and stained for hematoxylin and eosin (H&E). These slides were imaged using a Nikon Eclipse 80i microscope and Optronics MicroFire True Color 4MP digital CCD camera (Liver Core, USC School of Medicine). Matched coronal microtome sections (5–6 μm) of the hippocampi from different treatment groups were obtained using visualization under a histological microscope in conjunction with the Mouse Atlas (Paxinos and Franklin, 4th Edition, 2012). Hippocampal brain sections were deparaffinized, antigen retrieved by heating in a common pressure cooker in citric acid (10 mM sodium citrate, 0.05% Tween 20, pH 6.0), permeabilized using 1% Triton X100, blocked using 5% goat serum and 1% BSA. For immunofluorescence (IF) staining, the sections were incubated with rabbit polyclonal anti-GFAP (1:2000, 2 h at room temperature, Millipore, Temecula, CA) or rabbit polyclonal anti-Iba1 (1:250, 24–48 h at 4 °C, Wako Chemicals, Richmond, VA). After 3 washes in PBS-Tween, the slices were incubated with secondary anti-rabbit antibody containing DyLight 550 (Thermo Fisher Scientific (Thermo), Waltham, MA). The slides were then cover-slipped using Vectashield hard set mounting medium with DAPI (Vector Labs, Burlingame, CA) and visualized using the Nikon Diaphot 300 Inverted Microscope and X-Cite 120LED for fluorescent illumination (Nikon Inc., Melville, NY).
Reverse Transcription and Quantitative Real-Time Polymerase Chain Reaction (RT-qPCR)
Dissected frozen hippocampal or liver tissue samples were thawed in Trizol (Invitrogen, Carlsbad, CA) and homogenized using 400 μm silica beads (OPS Diagnostics LLC, Lebanon, NJ) in Bullet Blender (Next Advance Inc., Averill Park, NY). Following homogenization, RNA was isolated using RNeasy Mini Kit (Qiagen, Valencia, CA) and then converted to cDNA using High-Capacity RNA-to-cDNA Kit (Life Technologies, Grand Island, NY). qPCR was conducted using SYBR Master mix (Thermo) and primer sets in a ABI 7900 fast real-time system (Applied Biosystems, Houston, TX). GAPDH was used as normalization control. The sequences of target gene primer sets are presented in Table 1.
Table 1.
Primer sequences for marker genes used in PCR reactions
| Gene | Primer sequence |
|---|---|
| IL-1β | F - 5′-TCGCTCAGGGTCACAAGAAA-3′ R - 5′-CATCAGAGGCAAGGAGGAAAAC-3′ |
| TNFα | F - 5′-CATCTTCTCAAAATTCGAGTGACAA-3′ R - 5′-TGGGAGTAGACAAGGTACAACCC-3′ |
| Ccl2 (Mcp1) | F - 5′-CCACTCACCTGCTGCTACTCAT-3′ R - 5′-TGGTGATCCTCTTGTAGCTCTCC-3′ |
| Nos2 (iNos) | F - 5′-CCTGGTACGGGCATTGCT-3′ R - 5′-GCTCATGCGGCCTCCTT-3′ |
| Cx3cl1 | F – 5′-GTGCTGACCCGAAGGAGAAA-3′ R – 5′-CGCTTCTCAAACTTGCCACC-3′ |
| Cx3cr1 | F – 5′-ATTGGCTTCTTTGGGGGCAT-3′ R – 5′-CGGTTGTTCATGGAGTTGGC-3′ |
| Cxcl5 | F – 5′-CGGTTCCATCTCGCCATTCA-3′ R – 5′-GCTATGACTGAGGAAGGGGC-3′ |
| Cxcr2 | F – 5′-TAGTGTTGACTGGAGGCTGG-3′ R – 5′-CCTTCAGGGCTTTCCCATCA-3′ |
| Gapdh | F – 5′- GCATGGCCTTCCGTGTTC-3′ R – 5′- GATGTCATCATACTTGGCAGGTTT-3′ |
F, forward; R, reverse
Taqman Custom Gene Expression
Hippocampal samples were prepared in the same manner as in RT-qPCR. Taqman 45-mer 384-well plates pre-loaded with custom gene primers and special matrix were obtained from Invitrogen (Thermo). Samples were mixed with Taqman master mix (Thermo) and water in a total volume of 10 μl/well and loaded onto the plates. PCR reactions were performed using ABI Quant Studio OpenArray Real-Time PCR System (Applied Biosystems, Houston, TX). See Table 2 for the full list of genes analyzed in the assay.
Table 2.
List of target genes in Taqman custom gene expression qPCR
| Neuroinflammation | Signaling/Transcription | Neurotoxicity/Apoptosis | Neurochemical/Neuromodulation |
|---|---|---|---|
| Il1β | Akt1 | Bax | P2xr4 |
| Il6 | Pik3ca | Bcl2 | P2xr7 |
| Il10^ | Mapk1 | Casp3 | P2yr1 |
| Il18 | Mapk14 | Rbfox3 (NeuN) | P2yr6 |
| Ccl2 (Mcp1) | Tlr2 | Trp53 | Adora1 |
| Ccl5 | Tlr4 | Adora2a | |
| Tnfα | Myd88 | Gabra1 | |
| Csf2^ | Ilr1 | Gabra4 | |
| Ifng^ | Hmgb1 | Grin1 | |
| Nos2 | Nfkb1 | Grin2a | |
| Aif1 | Creb1 | Drd1 | |
| Gfap | Nlrp3 | Drd2 | |
| Cxcr2 | Casp1 | ||
| Panx1 |
, Expression of genes for Il10; Csf2 and Ifng were under detection limit
Western Blotting
Dissected hippocampal regions were fast frozen on dry ice and kept in −80 °C until processed. Protein was obtained by brief sonication in RIPA buffer (10 mM Tris, 140 mM NaCl, 1 mM EDTA, 1% Triton X-100, 0.1% sodium deoxycholate, pH 8.0 with addition of protease inhibitor cocktail (Sigma). Protein was measured using BCA assay (Thermo) and separation performed on 10% SDS-PAGE using 25 μg of each sample per lane. Protein was then transferred to PVDF membranes and blotted against IL-1β, p65 NF-kB1 and β-Actin. The following antibodies and dilutions were used: biotinylated anti-mouse IL-1β antibody (1:1000, overnight at 4 °C, R&D Systems, Biotechne, Minneapolis, MN), rabbit polyclonal p65 NF-kB1 (1:1000, overnight at 4 °C, Cell Signaling Technology, Danvers, MA), mouse monoclonal β-Actin (1:10000, 10–15 min room temperature, Sigma). Membranes were incubated for 1–2 h at room temperature with streptavidin-HRP (1:2000, Cell Signaling) for IL-1β, anti-rabbit or anti-mouse HRP conjugated antibodies (1:10000, Bio-Rad, Hercules, CA) for p65 NF-kB1 and β-Actin, respectively. Protein bands were visualized using Chemic-Doc Touch digital Imaging system (Bio-Rad) after subsequent treatment with enhanced chemiluminescence substrate (Clarity Western ECL substrate, Bio-Rad).
Data Analysis
IHC data were analyzed using Image J software (NIH). Fat accumulation in H&E liver staining was analyzed using threshold adjustments in Image J to automatically count fat deposits in liver tissues.
For quantitative analyses of immunostained sections, we obtained images of hippocampus regions at 10× magnification and then focused on the areas around the dentate gyrus at 40× magnification. Cells positively stained for Iba1 or GFAP were identified by typical cellular morphology, and the number of cells were counted manually from 5 to 7 images per mouse (n = 3/group). Cell bodies for microglia and astrocytes were identified and manually outlined. Microglial cell bodies were clearly visible and easy to identify. Cell bodies of astrocytes were identified using respective GFAP images without DAPI and simultaneously controlled with images with DAPI. Cell body areas as well as fluorescence intensity (fluorescence unit per cell body area) were quantified using ImageJ software capabilities. Fluorescence intensity on the images were corrected for background noise which was determined from sections stained with secondary antibody only and set as threshold. Cell body area and fluorescence intensity data were normalized using Control data which were set at “1”.
Densitometry data for Western blots were analyzed using Image J software. Integrated densities for IL-1β and p65 NF-kB1 bands were normalized over the corresponding β-Actin bands. Obtained data for Hybrid and Hybrid + A804598 are presented as fold differences from the Control values, which were averaged and arbitrarily adjusted to “1”.
2(–Delta Ct) method was used for the analyses of the PCR data (Schmittgen and Livak 2008).
All data presented as mean ± SEM using GraphPAD Prism (San Diego, CA). Differences between mouse groups were determined using one-way ANOVA and significance set at P ≤ 0.05.
Results
P2X7R Antagonist Did Not Affect Hybrid-induced changes on Body and Liver Weights, Liver/Body Ratio and BECs
As illustrated in Fig. 1a, body weights of mice in the Control group increased over the course of the experiment. Body weights in mice exposed to Hybrid did not increase and, as a result, were significantly lower (by ~12%) compared to the Control group during the period of 22–30 days (Fig. 1a). Treatment with P2X7R antagonist A804598 did not significantly alter body weights caused by Hybrid (Fig. 1a).
Fig. 1.

a Body weights (in grams) during the course of the study; b Liver weights (in grams), c liver to body weight ratios and d BECs (mM) at the end of the study. A804598 did not affect changes in body and liver weights and BECs caused by Hybrid exposure. Values reported are mean ± SEM. *** P < 0.001 compared to Control; n = 10/group for a, b, c; n = 6/group for d
Hybrid exposure caused a 60% increase in liver weights as compared to the Controls and this effect was similar during P2X7R antagonist treatment (Fig. 1b). Liver/body weight ratio was 2-fold (~100%) higher for mice in Hybrid and Hybrid + A804598 groups compared to the Control group (Fig. 1c). This larger difference was most probably due to the observed difference in the body weights between the Control and Hybrid or Hybrid + A804598 groups towards the end of the exposure period (Fig. 1a).
Hybrid treatment resulted in BECs of ≥50 mM (Fig. 1d). Treatment with A804598 did not significantly affect BEC levels induced by Hybrid (Fig. 1d).
P2X7R Antagonist Reversed Hybrid-Induced Changes in Microglia and Astrocytes in Hippocampus
Immunofluorescence staining of hippocampal slices with an antibody to microglial marker Iba1 demonstrated morphological changes in microglia in the Hybrid group (Fig. 2a, upper & middle panels). There was some rounding of the cell bodies and less prominent extensions with Hybrid compared to Control. P2X7R antagonist inclusion during the Hybrid + A804598 exposure resulted in reversal of the morphological changes in microglia (Fig. 2a). However, these changes did not translate into a quantitative difference in the cell body area (Fig. 2a, bar graph, left). There were also no significant changes in Iba1-positive cell number with Hybrid (Fig. 2a, bar graph, middle). Additional analyses demonstrated a significant decrease in Iba1 fluorescence intensity in the Hybrid group and reversal of this effect in the presence of A804598 (Fig. 2a, bar graph, right).
Fig. 2. Effects of P2X7R antagonist A804598 on Hybrid-induced changes in microglia and astrocytes in hippocampus.
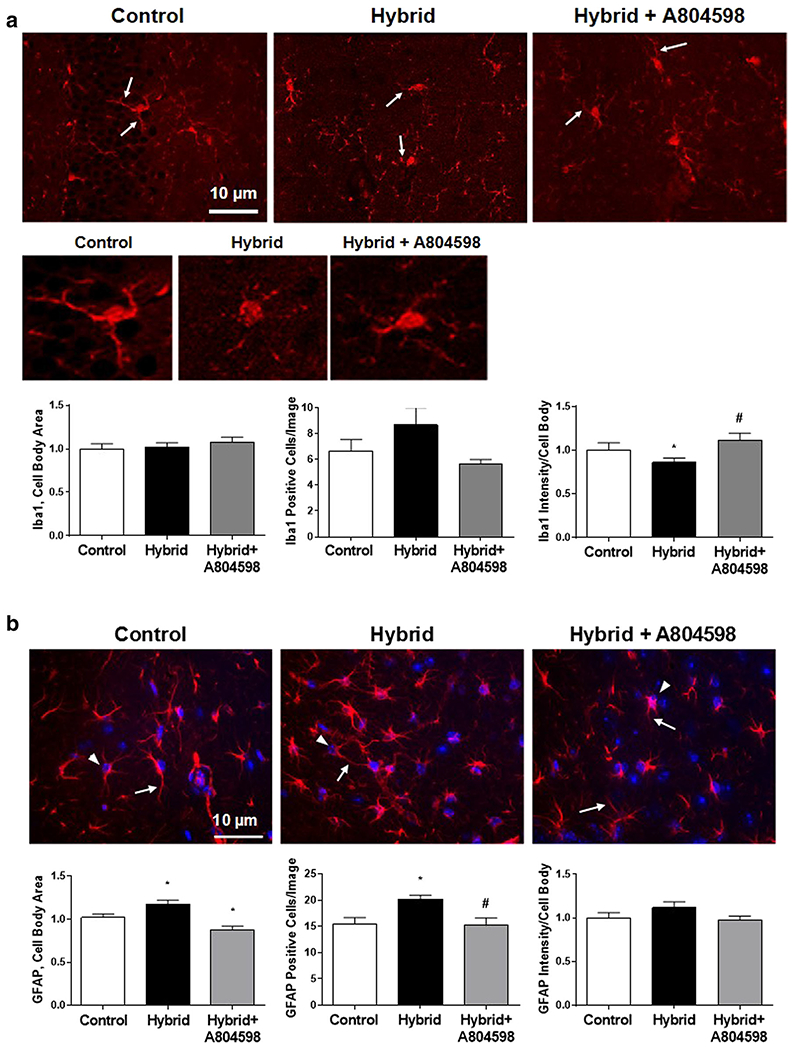
a Iba1 staining of microglia. Upper and middle panels – representative IF images of microglia; middle panel, zoom in for single cells. Bottom panel – bar graphs for quantitative analysis for cell body area, Iba1-positive cells and Iba1 fluorescence intensity. b GFAP staining of astrocytes; DAPI nuclear counter stain is also shown. Upper panel – representative IF images. Lower panel – bar graphs for quantitative analysis for cell body area, GFAP-positive cells and GFAP fluorescence intensity. Arrows on both a and b point to cell extensions and arrowheads on b indicate cell bodies. P2X7R antagonist reversed the changes in both microglia and astrocytes induced by Hybrid exposure. All values are reported as mean ± SEM. * P < 0.05 compared to Control, # P < 0.05 compared to Hybrid, (5–7 images/each mouse, n = 3/group)
IF staining of hippocampal slices with GFAP antibody demonstrated some morphological changes in astrocytes. Hybrid exposure resulted in shorter astrocytic processes (Fig. 2b, upper panel) and a small but significant increase in the cell body area (Fig. 2b, upper panel and bar graph, left) compared to the Controls. (Fig. 2b, upper panel). Furthermore, astrocytes underwent proliferation as demonstrated by an increase in the number of GFAP-positive cells during Hybrid exposure (Fig. 2b, upper panel & bar graph, middle) with no apparent change in the averaged GFAP fluorescence intensity calculated from cellular bodies (Fig. 2b, bar graph, right). Administration of P2X7R antagonist reversed the morphological changes and inhibited astrocyte proliferation as demonstrated by reversal of the increase in cell body areas and reduced GFAP-positive cell numbers (Fig. 2b).
P2X7R Antagonist Reversed the Neuroinflammatory Response in Hippocampus Caused by Hybrid Exposure
Taqman custom plates were used to test for the expression of a wide variety of genes in the hippocampal samples. Focusing on the inflammatory panel of genes, we found that Hybrid exposure caused a neuroinflammatory response (Fig. 3). Hybrid caused significant increases in pro-inflammatory markers Il1β, Tnfα, Ccl2 (Mcp1), Nos2 (iNos) and Cxcr2. The increases in all indicated markers were attenuated by P2X7R antagonist treatment. Significant effects of the antagonist treatment were found for Il1β, Nos2 and Cxcr2 (Fig. 3). Hybrid did not cause changes in pro-inflammatory markers such as Il6, Il18 or Ccl5. In addition, Hybrid induced an increase in the astrocytic marker Gfap gene but did not cause changes in the marker gene for microglia, Iba1. There was an apparent reduction in Gfap levels with P2X7R antagonist treatment, however, this effect was not significant.
Fig. 3. P2X7R antagonist effects on Hybrid-induced changes in gene expression of inflammatory markers in hippocampus.
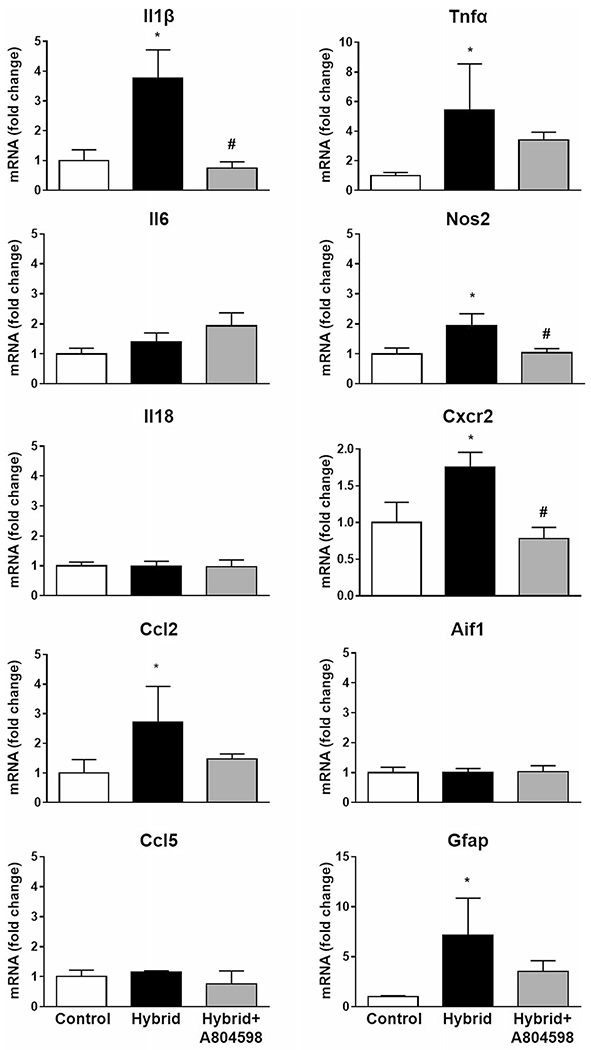
Hybrid induced a neuroinflammatory response as evident by increases in pro-inflammatory markers Il1β, Tnfα, Ccl2, Nos2, Cxcr2 and Gfap. A804598 treatment significantly abolished increases in Il1β, Nos2 and Cxcr2. Data were obtained using Taqman custom gene expression plates. Gapdh was used as a normalization control in the data analyses. All values reported as fold changes compared to Control; mean ± SEM. * P < 0.05, # P < 0.05 compared to Hybrid (n = 4/group for Control and Hybrid and n = 5 for Hybrid + A804598)
Hybrid exposure also caused changes in a few components of the inflammatory signaling pathways (Fig. 4). We found increases in the gene expression for pattern recognition receptor Tlr2, component of the inflammasome pathway such as Casp1, transcription factors Nfkb1 and Creb1, with Hybrid exposure. Changes in Casp1, Nfkb1 and Creb1 were statistically significant. Administration of A804598 abolished the increases in all the indicated markers. We did not find changes in genes for Tlr4, Nlrp3, Myd88, Hmgb1, Il1r, Panx1 or components of intracellular signaling pathways such as Akt, Pi3k, Mapk1, Mapk14 (Table 3).
Fig. 4. Effects of the P2X7R antagonist A804598 on changes in expression of genes related to inflammatory signaling pathways in hippocampus caused by Hybrid exposure.

Increases in mRNA levels of Tlr2, Casp1, Nfkb1, Creb1 were noted. A804598 treatment eliminated these increases. Data were obtained using Taqman custom gene expression plates. Gapdh was used as a normalization control in the data analyses. All values reported as fold changes compared to Control; mean ± SEM. * P < 0.05, # P < 0.05 compared to Hybrid (n = 4/group for Control and Hybrid and n = 5 for Hybrid + A804598)
Table 3.
Changes in gene expression in hippocampal tissues
| Gene | Control | Hybrid | Hybrid + A804598 |
|---|---|---|---|
| Signaling | |||
| Akt | 1.0 ± 0.10 | 0.9. ± 0.08 | 1.04 ± 0.09 |
| Pi3k | 1.0 ± 0.18 | 1.08 ± 0.19 | 0.96 ± 0.10 |
| Mapk1 | 1.0 ± 0.23 | 0.97 ± 0.17 | 1.06 ± 0.10 |
| Mapk14 | 1.0 ± 0.17 | 1.04 ± 0.09 | 1.11 ± 0.10 |
| Neurotoxicity/Apoptosis | |||
| Rbfox/NeuN | 1.0 ± 0.12 | 1.42 ± 0.34 | 1.13 ± 0.12 |
| Bax | 1.0 ± 0.10 | 1.19 ± 0.13 | 1.16 ± 0.05 |
| Bcl2 | 1.0 ± 0.10 | 1.20 ± 0.10 | 1.08 ± 0.17 |
| Trp53 | 1.0 ± 0.11 | 1.18 ± 0.27 | 1.05 ± 0.15 |
| Casp3 | 1.0 ± 0.09 | 1.13 ± 0.18 | 1.04 ± 0.19 |
| Neurotransmission/ | |||
| P2xr4 | 1.0 ± 0.20 | 1.03 ± 0.06 | 1.28 ± 0.14 |
| P2xr7 | 1.0 ± 0.13 | 1.00 ± 0.09 | 0.99 ± 0.06 |
| P2yr1 | 1.0 ± 0.11 | 1.95 ± 0.34* | 1.73 ± 0.24* |
| P2yr6 | 1.0 ± 0.13 | 1.8 ± 0.29* | 1.27 ± 0.21* |
| Adora1 | 1.0 ± 0.09 | 1.28 ± 0.25 | 1.31 ± 0.11 |
| Adora2a | 1.0 ± 0.22 | 1.04 ± 0.15 | 1.16 ± 0.07 |
| Gabra1 | 1.0 ± 0.17 | 0.97 ± 0.15 | 1.02 ± 0.05 |
| Gabra4 | 1.0 ± 0.21 | 1.08 ± 0.23 | 1.11 ± 0.07 |
| Grin1 | 1.0 ± 0.13 | 1.72 ± 0.50 | 2.15 ± 0.29 |
| Grin2a | 1.0 ± 0.16 | 1.03 ± 0.17 | 1.01 ± .010 |
| Drd1 | 1.0 ± 0.02 | 5.05 ± 0.97* | 4.18 ± 1.39* |
| Drd2 | 1.0 ± 0.14 | 1.34 ± 0.06* | 1.27 ± 0.09* |
Data obtained using the Taqman custom gene expression plates
Gapdh was used as normalization control in the data analyses
- P < 0.05 compared to Control, n = 4/group for Control and Hybrid and n = 5 for Hybrid + A804598
We extended the study to test the protein levels of a few critical neuroinflammatory markers in hippocampus. Using Western blotting, we found that Hybrid exposure caused significant increases in the protein levels of pro-IL-1β and p65 NF-kB1, a key member of the NF-kB1 complex (Fig. 5a, b). Pro-IL-1β is the precursor for the mature form for IL-1β and is detected at 35 kDa on Western blots, while the cleaved, mature form of IL-1β (17 kDa) is readily secreted from the cells. Further, we demonstrated that P2X7R antagonist A804598 significantly reduced the increases in both, pro-IL-1β and p65 NF-kB1 (Fig. 5a, b). Changes in protein levels of pro-IL-1β and NF-kB1 correlated well with the changes in their respective genes (Figs. 3 and 4).
Fig. 5. P2X7R antagonist A804598 effects on Hybrid-induced increases in the protein levels of pro-IL-1β (a) and NF-kB1 in hippocampus (b).
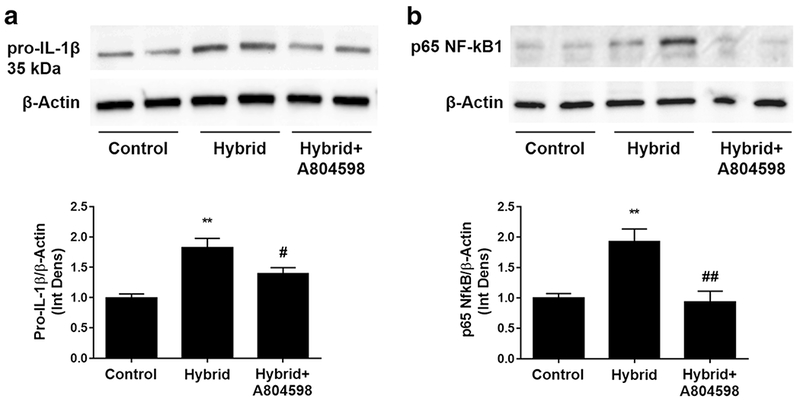
A804598 treatment reversed significant increases in the protein levels of pro-IL1β, the precursor of IL-1β, and p65 NF-kB1, a component of NF-kB1 complex. Upper panels - Western blots of representative bands for the proteins. Lower panels – densitometry analyses of the pro-IL-1β (a) and NF-kB1 (b) levels normalized to their corresponding actin levels. Data are presented as fold differences from the Control values, which were averaged and set as “1”. Data are reported as mean ± SEM. * P < 0.05, ** P < 0.01 compared to Control, # P < 0.05, # P < 0.01 compared to Hybrid, (n = 5/Control, n = 4 for Hybrid and Hybrid + A804598)
P2X7R Antagonist Did Not Alter Neuroprotective Response to Hybrid Exposure in Hippocampus
There were no significant changes in the gene markers for neurotoxicity/apoptosis during Hybrid exposure (Table 3). These data suggest that neurotoxicity is counter-balanced by activation of neuroprotective pathways. To further test if the antagonist affected the neuroprotective neuron-glia communication via fractalkine (CX3CL1) and its receptor CX3CR1, we ran a conventional RT-qPCR for mRNAs of these genes. Hybrid significantly increased mRNA levels of Cx3cl1 and Cx3cr1 and these increases were not attenuated by A804598 administration (Fig. 6a). In contrast, the antagonist abolished the effects of Hybrid on neuroinflammatory marker Cxcl5 and its receptor Cxcr2 (Fig. 6b).
Fig. 6. Effects of A804598 treatment on neuroprotective versus neuroinflammatory response caused by Hybrid exposure.

A804598 did not affect the neuroprotective response whereas it abolished neuroinflammatory response caused by Hybrid. Data were obtained using a conventional RT-qPCR with Gapdh used as a normalization control. Values reported as fold changes compared to Control; mean ± SEM. * P < 0.05, # P < 0.05 compared to Hybrid (n = 5/group)
P2X7R Antagonist Did Not Affect Gene Expression Changes in Neurotransmission Systems Caused by Hybrid Treatment in Hippocampus
Hybrid exposure affected the expression of a few genes in major neurotransmitter pathways (Table 3). Hybrid caused a significant increase in mRNA expression for metabotropic purinergic receptors P2yr1 and P2yr6 but not for P2xr7 or P2xr4. We also found significant increases in gene expression of dopamine receptors (Drd1 and Drd2). Furthermore, no changes were found for adenosine receptors (Adora1a and Adora2a) or glutamate receptors (Grin1 and Grin2a), or any of the tested genes encoding for GABA alpha 1 and alpha 4 receptors (respectively Gabra1 and Gabra4). A804598 administration did not affect any changes in neurotransmission systems caused by Hybrid (Table 3).
P2X7R Antagonist Reduced Inflammation but Not Steatosis in Liver Caused by Hybrid Exposure
H&E staining showed significant liver steatosis after Hybrid exposure (Fig. 7a). There was a large increase in fat deposits calculated as the ratio of white areas versus background color with the adjusted threshold. A804598 treatment did not affect the extent of the steatosis (Fig. 7a).
Fig. 7. Effects of P2X7R antagonist treatment on liver steatosis and inflammation.
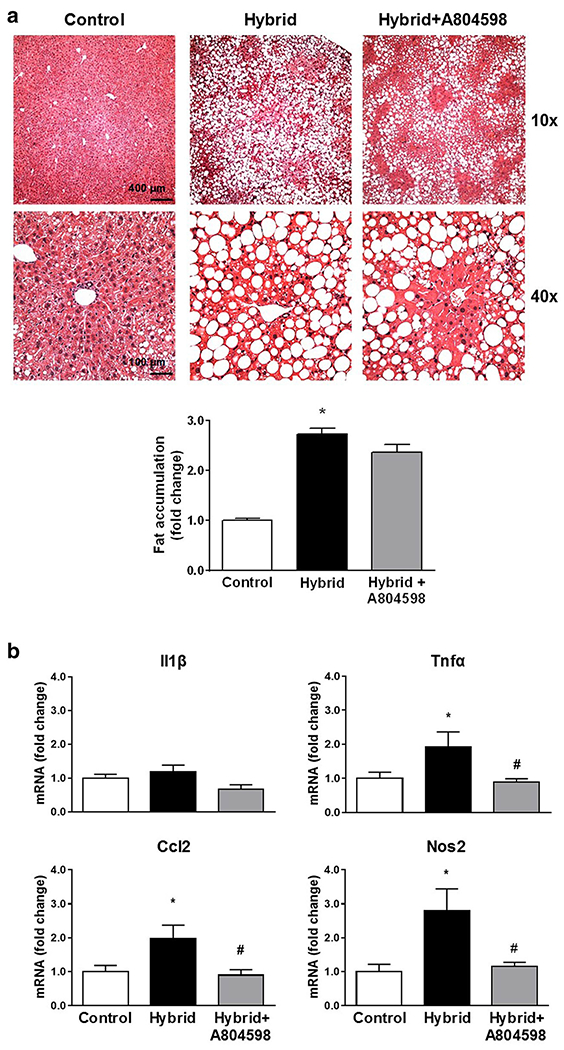
a Upper panel - Representative images of H&E staining of liver tissues; lower panel - bar graph of the analyzed data. Accumulation of fat after 4 weeks of the Hybrid exposure was not affected by A804598 treatment. b mRNA levels of inflammatory markers in liver. Hybrid induced inflammatory response which was abolished by A804598. Data were obtained using a conventional RT-qPCR. Gapdh was used as a normalization control in the data analyses. All values reported as a fold change compared to control, mean ± SEM. * P < 0.05 compared to Control; # P < 0.05 compared to Hybrid (4–7 images/each mouse, n = 6/group)
Hybrid exposure caused increases in pro-inflammatory markers in liver. There were significant increases in mRNA for Tnfα, Ccl2 and Nos2. These increases were abolished by administration of A804598 (Fig. 7b).
Discussion
In our previous study, we reported that exposure to 8 weeks of iG chronic alcohol and high fat diet (Hybrid) resulted in increased neuroinflammatory responses that paralleled increased expression of P2X7Rs in hippocampus of C57BL/6J mice (Asatryan et al. 2015). Current studies expanded on this work by testing the potential of pharmacologically reducing P2X7R activity to attenuate the inflammatory responses in the brain induced by 4 weeks of Hybrid exposure in C57BL/6J mice. Using the P2X7R antagonist A804598 resulted in the elimination of Hybrid-induced neuroinflammation. There was no effect by the antagonist on the neuroprotective response in hippocampus. In addition, antagonizing P2X7R also protected from liver inflammation but not liver steatosis caused by Hybrid exposure.
In this study, we modified the Hybrid paradigm to shorten the exposure duration to 4 weeks. We used this approach to reduce the time of the antagonist treatment, due to the lack of information on potential adverse effects of chronic treatment with this particular antagonist. Despite the shorter duration of exposure to Hybrid, we found neuroinflammatory responses largely similar to the findings of our previous study (Asatryan et al. 2015). That was evident from changes in glial cells in the hippocampus, which is one of the ethanol-sensitive brain regions in regard to alcohol-related neurodegeneration (Kane et al. 2014). Glial cells respond to insults with changes in morphology and production of pro-inflammatory factors (Graeber 2010). Morphological changes in microglia and astrocytes were present following 4 weeks of Hybrid exposure. These changes were evidenced by rounding of cell bodies and less prominent processes for microglia. Microglial marker Iba1 protein expression was significantly decreased suggesting that these cells were rather dysfunctional after 4 weeks of Hybrid exposure. These results are in good agreement with our earlier findings with 8 weeks of Hybrid exposure (Asatryan et al. 2015) and studies using other models of ethanol exposure in rodents (Marshall et al. 2013; McClain et al. 2011). Notably, treatment of P2X7R antagonist reversed both the morphological changes and the decrease in Iba1 expression in microglia.
Hybrid also caused morphological changes in astrocytes in that there were larger cell bodies and shorter, denser processes suggesting the presence of activated cells. Despite the fact there was no increase in GFAP-fluorescence intensity, we found an increase in GFAP-positive cell number with Hybrid. These findings suggested that Hybrid caused proliferation of astrocytes in hippocampus. These effects are similar to our earlier findings on the presence of activated astrocytes in hippocampus after 8 weeks of Hybrid (Asatryan et al. 2015). In the current study, A804598 application reversed the observed phenotypic changes in astrocytes suggesting that P2X7Rs participate in astrocyte activation caused by Hybrid exposure.
As part of the innate neuroimmune response to alcohol exposure, activation of glial cells results in the production of pro-inflammatory mediators. MCP-1, TNFα, iNOS and other major neuroinflammatory factors were shown to be upregulated in the brain tissues from post-mortem alcoholic human brains (He and Crews 2008) and rodents exposed to a variety of alcohol exposure paradigms (Alfonso-Loeches et al. 2010; Crews and Vetreno 2014). In addition, in our previous study using the 8-week Hybrid paradigm we found increases in mRNA for IL-1β, CCL2 and iNOS (Asatryan et al. 2015). Similarly, in the current study using shorter time Hybrid exposure, Taqman gene expression findings demonstrated increases in mRNA for IL-1β, TNFα, CCL2, iNOS, and CXCR2 in hippocampus. We did not find changes in other mediators such as IL-6, IL-18 or CCL5. We also found increases in the protein levels of pro-IL-1β with Hybrid exposure that correlated well with changes in mRNA levels. Together, these findings suggest that activated microglia and astrocytes both contribute to Hybrid-induced increases in inflammatory markers.
In the present study, antagonizing P2X7R activity with A804598 resulted in inhibition of the neuroinflammatory response to the Hybrid exposure as evidenced by significant reduction in the increases in IL-1β, iNOS and CXCR2 mRNAs. P2X7R activation in glial cells can lead to increases in the production of major pro-inflammatory mediators. For example, P2X7R is an established co-activator of the signaling pathway that leads to the production of IL-1β, a known player in ischemia and neurodegeneration (Bartlett et al. 2014; Ferrari et al. 2006; Idzko et al. 2014; Takenouchi et al. 2009). Furthermore, P2X7R was shown to be upstream of pathways regulating TNFα, CCL2 secretion, and NO release (Barbera-Cremades et al. 2017; Chen et al. 2016; Panenka et al. 2001; Suzuki et al. 2004). A804598 administration also reversed the increases in mRNAs levels of several components of inflammatory signaling pathways such as endotoxin receptor TLR2 and transcriptional activators NF-kB1 and CREB1 that are known to increase production of pro-IL-1β and other inflammatory factors, or Casp 1, which converts the pro-IL-1β to its mature form IL-1β (Alfonso-Loeches and Guerri 2011). Our findings of changes in the protein levels of pro-IL-1β and NF-kB1, which correlated well with the mRNA levels of these markers, further supported the involvement of the NF-kB pathway in Hybrid-induced processes. As such, A804598 treatment resulted in significant reduction of the increases in the protein levels of pro-IL-1β and p65 NF-kB1, an important component of the NF-kB1 complex. Further, we did not detect changes in mRNA for TLR4 or MyD88 with Hybrid. TLRs are found on glial cells and have been attributed to regulating NFκB signaling to induce inflammatory responses to ethanol with emphasis on TLR4 involvement in this mechanism (Crews et al. 2015; Crews and Vetreno 2016; Szabo and Lippai 2014). However, TLR2 were also found to play a similar role in regulating ethanol induced cytokine release and behavioral changes (Pascual et al. 2015). Involvement in Hybrid-induced processes of P2X7R-related molecule PANX1, NLRP3 inflammasome or HMGB1 transcriptional regulator, which have been implicated in alcohol effects (Crews and Vetreno 2016), was not obvious as we did not find significant changes in the mRNA of these markers. Taken together, the findings with P2X7R antagonist treatment support the functional involvement of P2X7Rs in Hybrid-induced neuroinflammatory response in hippocampus.
P2X7R antagonist administration did not affect the neuroprotective response caused by Hybrid paradigm. We did not find changes in mRNA levels for neurotoxicity markers Rbfox3/NeuN, Bax, Bcl-2, Trp53, Casp3, which suggested the Hybrid-induced activation of adaptive processes leading to neuroprotection. To test this possibility, we assessed the expression of fractalkine (CX3CL1) and its receptor CX3CR1. Fraktalkine is a chemokine expressed at high levels in hippocampal neurons and is considered largely neuroprotective with a role in neurogenesis (reviewed in (Sheridan and Murphy 2013)). On the other hand, neurons and microglia but not astrocytes express the receptor for fraktalkine, CX3CR1. Fraktalkine secretion and binding to its receptor on microglia initiates a migratory response and neuroprotective action. Interaction with CX3CR1 on neurons can also promote neuroprotective action against toxic insults (Sheridan and Murphy 2013). We found increases in mRNA levels for CX3CL1 and CX3CR1 in hippocampus after Hybrid exposure. However, these increases were not attenuated with the administration of P2X7R antagonist. This was in contrast to the reducing effect of the antagonist on mRNA expression for another inflammatory chemokine CXCL5 (the murine ortholog of IL-8) and its respective receptor CXCR2. Besides their role in inflammatory signaling, recent studies suggest a new role for IL-8 and CXCR2 in the development of alcoholic steatohepatitis (Wieser et al. 2017). However, very little information is available on the changes in brain IL-8 and CXCR2 in the studied experimental models. Overall, our findings suggest that there is a functional link between P2X7Rs and CXCR2 but not CX3CR1 pathways.
P2X7R antagonist did not affect changes in neurotransmitter systems caused by Hybrid exposure. Interestingly, we did not find changes in mRNA levels for P2X7R or P2X4R. Rather, increases were observed for the metabotropic purinergic receptors, P2Y1R and P2Y6R. Thus far, based on the lack of previous information, these findings are the first to point to the potential role of these receptors in alcohol-induced effects on the brain. Further, increases were found in mRNA levels of dopamine receptors (Drd1, Drd2) but no changes in adenosine (Adora1, Adora2a), glutamate (Grin1, Grin2) or GABA receptors (Gabra1, Gabra4). All these neurotransmitter systems are established mediators of alcohol effects on brain (Lovinger 2008; Vengeliene et al. 2008). The findings with P2X7R antagonist suggested that P2X7R-mediated effects during Hybrid exposure are primarily originating from the glial and not the neuronal cells.
Hybrid paradigm was developed as a model to study alcoholic liver disease and is characterized by significant changes in the liver in terms of fat accumulation (steatosis) and inflammation suggestive of liver damage (Lazaro et al. 2015; Xu et al. 2011). We evaluated P2X7R antagonist for its protective effects on liver characteristics. A804598 treatment resulted in the attenuation of liver inflammation caused by Hybrid. This was evident from the reversal of the mRNA levels for inflammatory TNFα, Ccl2 and iNOS. However, the antagonist treatment did not affect the reduction in body weight towards the end of Hybrid exposure, increases in liver weight, liver/body weight, or liver steatosis. These effects may be explained by the fact that P2X7R antagonist did not alter the alcohol metabolism as seen from the BECs. Antagonizing P2X7Rs and/or deleting the P2X7R gene protected the liver from steatosis in obese mice in carbon tetrachloride liver injury model (Chatterjee et al. 2012). Targeting P2X7R may have beneficial protective effects in non-alcohol liver disease as opposed to alcoholic steatohepatitis. P2X7Rs are expressed in hepatic Kupffer cells as well as blood mononuclear cells (Chatterjee et al. 2012; Karmakar et al. 2016) that are shown to infiltrate into the liver tissue during the Hybrid exposure (Lazaro et al. 2015). It is likely that P2X7Rs in these cells play a role in Hybrid-induced inflammatory response in liver.
Conclusion
Taken together, findings from the present study suggest a functional role for P2X7Rs in inflammatory response in brain and liver to the exposure of combined chronic alcohol and high-fat diet. Antagonizing P2X7Rs in brain resulted in reduction/abolishment in the increase of inflammatory markers, signaling components in hippocampus. Additionally, P2X7R antagonist was beneficial in that it did not affect the neuroprotective response as well as did not alter many neurotransmitter receptors suggesting that it can be used to specifically target the neuroinflammatory component during chronic alcohol exposure. Moreover, the potential to reduce hepatic inflammation but not steatosis makes it an attractive tool to differentiate between the two processes during Hybrid exposure.
Lastly, the present study was limited with the numbers of experimental animals per each group which was due to labor-intensive makeup of the Hybrid model. This did not allow detection of larger and significant differences in several markers between the groups upon different treatment conditions. Future studies with increased sample size, increased dose of the antagonist or utilization of P2X7-knockout mouse model will be required to provide more support for the role of P2X7 in ethanol-induced inflammatory responses and for validation of this molecule as a therapeutic target to confer alcohol-related organ damage.
Acknowledgements
We would like to thank the Animal Core of the Southern California Research Center for ALPD and Cirrhosis (Director Dr. H. Tsukamoto) for providing us with the experimental animals, Elliott Cheung for Western blots and Lisa Walter for proof reading the manuscript and valuable recommendations. This work was conducted as partial fulfilment of the requirements for the MS. degree in Pharmaceutical Sciences, University of Southern California (D.F.). Sources of support: NIH/NIAAA AA017243 and Zumberge Individual Research Fund (L.A.), NIH/NIAAA A022448 (D.L.D.), NIH P50AA011999 (H.T.).
Abbreviations
- P2X7R
purinergic P2X7 receptor
- BEC
blood ethanol concentration
- iG
intragastric
- HFD
high fat diet
- HCFD
high cholesterol high fat diet
- RT-qPCR
reverse transcriptase – quantitative polymerase chain reaction
- Gapdh
glyceraldehyde 3-phosphate dehydrogenase
- GFAP
glial fibrillary acidic protein
- Iba1
ionized calcium binding adaptor molecule 1
Footnotes
Conflict of Interest The authors declare that they have no conflict of interest.
References
- Able SL, Fish RL, Bye H, Booth L, Logan YR, Nathaniel C, Hayter P, Katugampola SD (2011) Receptor localization, native tissue binding and ex vivo occupancy for centrally penetrant P2X7 antagonists in the rat. Br J Pharmacol 162:405–414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alfonso-Loeches S, Guerri C (2011) Molecular and behavioral aspects of the actions of alcohol on the adult and developing brain. Crit Rev Clin Lab Sci 48:19–47 [DOI] [PubMed] [Google Scholar]
- Alfonso-Loeches S, Pascual-Lucas M, Blanco AM, Sanchez-Vera I, Guerri C (2010) Pivotal role of TLR4 receptors in alcohol-induced neuroinflammation and brain damage. J Neurosci 30:8285–8295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asatryan L, Khoja S, Rodgers KE, Alkana RL, Tsukamoto H, Davies DL (2015) Chronic ethanol exposure combined with high fat diet up-regulates P2X7 receptors that parallels neuroinflammation and neuronal loss in C57BL/6J mice. J Neuroimmunol 285:169–179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asatryan L, Ostrovskaya O, Lieu D, Davies DL (2018) Ethanol differentially modulates P2X4 and P2X7 receptor activity and function in BV2 microglial cells. Neuropharmacol 128:11–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbera-Cremades M, Gomez AI, Baroja-Mazo A, Martinez-Alarcon L, Martinez CM, de Torre-Minguela C, Pelegrin P (2017) P2X7 receptor induces tumor necrosis factor-alpha converting enzyme activation and release to boost TNF-alpha production. Front Immunol 8: 862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartlett R, Stokes L, Sluyter R (2014) The P2X7 receptor channel: recent developments and the use of P2X7 antagonists in models of disease. Pharmacol Rev 66:638–675 [DOI] [PubMed] [Google Scholar]
- Catanzaro JM, Hueston CM, Deak MM, Deak T (2014) The impact of the P2X7 receptor antagonist A-804598 on neuroimmune and behavioral consequences of stress. Behav Pharmacol 25:582–598 [DOI] [PubMed] [Google Scholar]
- Chatterjee S, Rana R, Corbett J, Kadiiska MB, Goldstein J, Mason RP (2012) P2X7 receptor-NADPH oxidase axis mediates protein radical formation and Kupffer cell activation in carbon tetrachloride-mediated steatohepatitis in obese mice. Free Radic Biol Med 52: 1666–1679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chavda S, Luthert PJ, Salt TE (2016) P2X7R modulation of visually evoked synaptic responses in the retina. Purinergic Signal 12:611–625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Q, Wu H, Qin S, Liu C, Chen Y, Yang Y, Xu C (2016) The P2X7 receptor involved in gp120-induced cell injury in BV2 microglia. Inflammation 39:1814–1826 [DOI] [PubMed] [Google Scholar]
- Chessell IP, Hatcher JP, Bountra C, Michel AD, Hughes JP, Green P, Egerton J, Murfin M, Richardson J, Peck WL (2005) Disruption of the P2X7 purinoceptor gene abolishes chronic inflammatory and neuropathic pain. Pain 114:386–396 [DOI] [PubMed] [Google Scholar]
- Crews FT, Sarkar DK, Qin L, Zou J, Boyadjieva N, Vetreno RP (2015) Neuroimmune function and the consequences of alcohol exposure. Alcohol Res 37:344–351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crews FT, Vetreno RP (2014) Neuroimmune basis of alcoholic brain damage. Int Rev Neurobiol 118:315–357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crews FT, Vetreno RP (2016) Mechanisms of neuroimmune gene induction in alcoholism. Psychopharmacol (Berl) 233:1543–1557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz-Hernandez M, Diez-Zaera M, Sanchez-Nogueiro J, Gomez-Villafuertes R, Canals JM, Alberch J, Miras-Portugal MT, Lucas JJ (2009) Altered P2X7-receptor level and function in mouse models of Huntington’s disease and therapeutic efficacy of antagonist administration. FASEB J 23:1893–1906 [DOI] [PubMed] [Google Scholar]
- Donnelly-Roberts DL, Namovic MT, Surber B, Vaidyanathan SX, Perez-Medrano A, Wang Y, Carroll WA, Jarvis MF (2009) [3H]A-804598 ([3H]2-cyano-1-[(1S)-1-phenylethyl]-3-quinolin-5-ylguanidine) is a novel, potent, and selective antagonist radioligand for P2X7 receptors. Neuropharmacol 56:223–229 [DOI] [PubMed] [Google Scholar]
- Ferrari D, Pizzirani C, Adinolfi E, Lemoli RM, Curti A, Idzko M, Panther E, Di Virgilio F (2006) The P2X7 receptor: a key player in IL-1 processing and release. J Immunol 176:3877–3883 [DOI] [PubMed] [Google Scholar]
- Graeber MB (2010) Changing face of microglia. Science 330:783–788 [DOI] [PubMed] [Google Scholar]
- He J, Crews FT (2008) Increased MCP-1 and microglia in various regions of the human alcoholic brain. Exp Neurol 210:349–358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Idzko M, Ferrari D, Eltzschig HK (2014) Nucleotide signalling during inflammation. Nature 509:310–317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung YH, Kim YO, Han JH, Kim YC, Yoon MH (2017) Isobolographic analysis of drug combinations with intrathecal BRL52537 (kappa-opioid agonist), Pregabalin (Calcium Channel modulator), AF 353 (P2X3 receptor antagonist), and A804598 (P2X7 receptor antagonist) in neuropathic rats. Anesth Analg 125:670–677 [DOI] [PubMed] [Google Scholar]
- Kane CJ, Phelan KD, Douglas JC, Wagoner G, Johnson JW, Xu J, Phelan PS, Drew PD (2014) Effects of ethanol on immune response in the brain: region-specific changes in adolescent versus adult mice. Alcohol Clin Exp Res 38:384–391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karmakar M, Katsnelson MA, Dubyak GR, Pearlman E (2016) Neutrophil P2X7 receptors mediate NLRP3 inflammasome-dependent IL-1beta secretion in response to ATP. Nat Commun 7: 10555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khakh BS, North RA (2006) P2X receptors as cell-surface ATP sensors in health and disease. Nature 442:527–532 [DOI] [PubMed] [Google Scholar]
- Kneer K, Green MB, Meyer J, Rich CB, Minns MS, Trinkaus-Randall V (2018) High fat diet induces pre-type 2 diabetes with regional changes in corneal sensory nerves and altered P2X7 expression and localization. Exp Eye Res 175:44–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazaro R, Wu R, Lee S, Zhu NL, Chen CL, French SW, Xu J, Machida K, Tsukamoto H (2015) Osteopontin deficiency does not prevent but promotes alcoholic neutrophilic hepatitis in mice. Hepatol 61:129–140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Zhao Z, Ji R, Zhu J, Sui QQ, Knight GE, Burnstock G, He C, Yuan H, Xiang Z (2017) Inhibition of P2X7 receptors improves outcomes after traumatic brain injury in rats. Purinergic Signal 13:529–544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovinger DM (2008) Communication networks in the brain: neurons, receptors, neurotransmitters. and alcohol Alcohol Res Health 31: 196–214 [PMC free article] [PubMed] [Google Scholar]
- Marshall SA, McClain JA, Kelso ML, Hopkins DM, Pauly JR, Nixon K (2013) Microglia activation is not equivalent to neuroinflammation in alcohol-induced neurodegeneration: the importance of mciroglia phenotype. Neurobiol Dis 54:239–251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matute C, Torre I, Perez-Cerda F, Perez-Samartin A, Alberdi E, Etxebarria E, Arranz AM, Ravid R, Rodriguez-Antiguedad A, Sanchez-Gomez M, Domercq M (2007) P2X(7) receptor blockade prevents ATP excitotoxicity in oligodendrocytes and ameliorates experimental autoimmune encephalomyelitis. J Neurosci 27:9525–9533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClain JA, Morris SA, Deeny MA, Marshall SA, Hayes DM, Kiser ZM, Nixon K (2011) Adolescent binge alcohol exposure induces long-lasting partial activation of microglia. Brain Behav Immun 25(Suppl 1):S120–S128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLarnon JG, Ryu JK, Walker DG, Choi HB (2006) Upregulated expression of purinergic P2X7 receptor in Alzheimer disease and amyloid-[beta] peptide-treated microglia and in peptide-injected rat hippocampus. J Neuropathol Exp Neurol 65:1090–1097 [DOI] [PubMed] [Google Scholar]
- Monif M, Burnstock G, Williams DA (2010) Microglia: proliferation and activation driven by the P2X7 receptor. Int J Biochem Cell Biol 42: 1753–1756 [DOI] [PubMed] [Google Scholar]
- North RA, Jarvis MF (2013) P2X receptors as drug targets. Mol Pharmacol 83:759–769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandolfi JB, Ferraro AA, Sananez I, Gancedo MC, Baz P, Billordo LA, Fainboim L, Arruvito L (2016) ATP-induced inflammation drives tissue-resident Th17 cells in metabolically unhealthy obesity. J Immunol 196:3287–3296 [DOI] [PubMed] [Google Scholar]
- Panenka W, Jijon H, Herx LM, Armstrong JN, Feighan D, Wei T, Yong VW, Ransohoff RM, MacVicar BA (2001) P2X7-like receptor activation in astrocytes increases chemokine monocyte chemoattractant protein-1 expression via mitogen-activated protein kinase. J Neurosci 21:7135–7142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pascual M, Balino P, Aragon CM, Guerri C (2015) Cytokines and chemokines as biomarkers of ethanol-induced neuroinflammation and anxiety-related behavior: role of TLR4 and TLR2. Neuropharmacol 89:352–359 [DOI] [PubMed] [Google Scholar]
- Peng W, Cotrina ML, Han X, Yu H, Bekar L, Blum L, Takano T, Tian GF, Goldman SA, Nedergaard M (2009) Systemic administration of an antagonist of the ATP-sensitive receptor P2X7 improves recovery after spinal cord injury. PNAS USA 106:12489–12493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sacks JJ, Gonzales KR, Bouchery EE, Tomedi LE, Brewer RD (2015) 2010 national and state costs of excessive alcohol consumption. Am J Prev Med 49:e73–e79 [DOI] [PubMed] [Google Scholar]
- Schmittgen TD, Livak KJ (2008) Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc 3:1101–1108 [DOI] [PubMed] [Google Scholar]
- Sheridan GK, Murphy KJ (2013) Neuron-glia crosstalk in health and disease: fractalkine and CX3CR1 take Centre stage. Open Biol 3: 130181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skaper SD, Debetto P, Giusti P (2010) The P2X7 purinergic receptor: from physiology to neurological disorders. FASEB J 24:337–345 [DOI] [PubMed] [Google Scholar]
- Stahre M, Roeber J, Kanny D, Brewer RD, Zhang X (2014) Contribution of excessive alcohol consumption to deaths and years of potential life lost in the United States. Prev Chronic Dis 11:E109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki T, Hide I, Ido K, Kohsaka S, Inoue K, Nakata Y (2004) Production and release of neuroprotective tumor necrosis factor by P2X7 receptor-activated microglia. J Neurosci 24:1–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabo G, Lippai D (2014) Converging actions of alcohol on liver and brain immune signaling. Int Rev Neurobiol 118:359–380 [DOI] [PubMed] [Google Scholar]
- Takenouchi T, Sekiyama K, Sekigawa A, Fujita M, Waragai M, Sugama S, Iwamaru Y, Kitani H, Hashimoto M (2010) P2X7 receptor signaling pathway as a therapeutic target for neurodegenerative diseases. Arch Immunol Therap Exp 58:91–96 [DOI] [PubMed] [Google Scholar]
- Takenouchi T, Sugama S, Iwamaru Y, Hashimoto M, Kitani H (2009) Modulation of the ATP-induced release and processing of IL-1beta in microglial cells. Crit Rev Immunol 29:335–345 [DOI] [PubMed] [Google Scholar]
- Tewari M, Seth P (2015) Emerging role ofP2X7 receptors in CNS health and disease. Ageing Res Rev 24:328–342 [DOI] [PubMed] [Google Scholar]
- Ueno A, Lazaro R, Wang PY, Higashiyama R, Machida K, Tsukamoto H (2012) Mouse intragastric infusion (iG) model. Nat Protoc 7:771–781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vengeliene V, Bilbao A, Molander A, Spanagel R (2008) Neuropharmacology of alcohol addiction. Br J Pharmacol 154: 299–315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vetreno RP, Crews FT (2014) Current hypotheses on the mechanisms of alcoholism. Handb Clin Neurol 125:477–497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volonte C, Apolloni S, Carri MT, D’Ambrosi N (2011) ALS: focus on purinergic signalling. Pharmacol Ther 132:111–122 [DOI] [PubMed] [Google Scholar]
- Volonte C, Apolloni S, Skaper SD, Burnstock G (2012) P2X7 receptors: channels, pores and more. CNS & neurological disorders drug targets 11:705–721 [DOI] [PubMed] [Google Scholar]
- Wieser V, Adolph TE, Enrich B, Kuliopulos A, Kaser A, Tilg H, Kaneider NC (2017) Reversal of murine alcoholic steatohepatitis by pepducin-based functional blockade of interleukin-8 receptors. Gut 66:930–938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Lai KK, Verlinsky A, Lugea A, French SW, Cooper MP, Ji C, Tsukamoto H (2011) Synergistic steatohepatitis by moderate obesity and alcohol in mice despite increased adiponectin and p-AMPK. J Hepatol 55:673–682 [DOI] [PMC free article] [PubMed] [Google Scholar]