

Abstract
Friedreich ataxia (FRDA) is an autosomal recessive neurodegenerative disease caused by the deficiency of frataxin, a mitochondrial protein crucial for iron–sulfur cluster biogenesis and adenosine triphosphate (ATP) production. Currently, there is no therapy to slow down the progression of FRDA. Recent evidence indicates that posttranslational regulation of residual frataxin levels can rescue some of the functional deficit of FRDA, raising the possibility of enhancing levels of residual frataxin as a treatment for FRDA. Here, we present evidence that mitochondrial molecular chaperone GRP75, also known as mortalin/mthsp70/PBP74, directly interacts with frataxin both in vivo in mouse cortex and in vitro in cortical neurons. Overexpressing GRP75 increases the levels of both wild-type frataxin and clinically relevant missense frataxin variants in human embryonic kidney 293 cells, while clinical GRP75 variants such as R126W, A476T and P509S impair the binding of GRP75 with frataxin and the effect of GRP75 on frataxin levels. In addition, GRP75 overexpression rescues frataxin deficiency and abnormal cellular phenotypes such as the abnormal mitochondrial network and decreased ATP levels in FRDA patient-derived cells. The effect of GRP75 on frataxin might be in part mediated by the physical interaction between GRP75 and mitochondrial processing peptidase (MPP), which makes frataxin more accessible to MPP. As GRP75 levels are decreased in multiple cell types of FRDA patients, restoring GRP75 might be effective in treating both typical FRDA patients with two guanine–adenine–adenine repeat expansions and compound heterozygous patients with point mutations.
Introduction
Friedreich ataxia (FRDA) is an autosomal recessive neurodegenerative disease characterized by progressive gait and limb ataxia, cerebellar, pyramidal and dorsal column involvement, visual defects, scoliosis and cardiomyopathy (1). It is largely caused by homozygous, expanded guanine–adenine–adenine (GAA) repeats in the first intron of frataxin gene, with a small percentage (4%) being caused by compound heterozygosity for a point mutation in one allele and an expanded GAA repeat on the other (2,3). The expanded GAA repeats cause transcriptional silencing and consequent reduction in the expression of frataxin (2). Frataxin is involved in iron homeostasis (4,5), biosynthesis of iron–sulfur clusters (ISCs) (6–8) and energy production in the cell (9,10), but its precise cellular function is controversial. Frataxin deficiency leads to reduced activity of ISC containing enzymes (6,7,11), impairment in mitochondrial respiration and energy generation (9,11–13) and increased mitochondrial iron and oxidative stress (14,15), all of which ultimately result in pathological changes in affected tissues. Currently, there is no therapy to slow down the progression of FRDA.
GRP75 is a mitochondrial molecular chaperone of the heat shock protein family that functions in mitochondrial protein import and quality control, ISC protein biogenesis, mitochondrial homeostasis and regulation of p53 (16–21). Loss of GRP75 function leads to mitochondrial dysfunction including abnormal mitochondrial morphology and decreased adenosine triphosphate (ATP) production (22,23), while overexpression of GRP75 inhibits apoptosis induced by glucose deprivation and extends the lifespan of both human cells and nematode Caenorhabditis elegans (24–26). GRP75 also participates in the maturation of frataxin. Mutation of yeast GRP75 homologs SSC1 and SSQ1, which shares 66% and 49% identity to GRP75, respectively, impairs the import and processing of yeast frataxin homolog Yfh1p. In contrast, GRP75 complements the function of yeast homologs in the maturation of Yfh1p (27–29). GRP75 physically interacts with frataxin in human embryonic kidney 293 (HEK293) and COS7 cells, and knockdown of GRP75 decreases the level of frataxin in cancer cell lines (27). However, whether GRP75 overexpression rescues the functional defects of frataxin deficiency is unknown. In the present study, we studied the physical and functional interaction between GRP75 and frataxin using cellular and molecular approaches and suggest that GRP75 can regulate frataxin as overexpression of GRP75 increases frataxin protein levels and rescues mitochondrial fragmentation and decreased ATP levels in FRDA patient-derived cells.
Results
GRP75 physically interacts with frataxin both in mouse brain cortex and neuronal cells
GRP75 interacts with frataxin in cell lines (27). To examine whether this interaction occurs in brain, an affected tissue of FRDA, we immunoprecipitated GRP75 from mouse cortex homogenates using anti-GRP75 antibody. GRP75 but not control Immunoglobulin G (IgG) pulled down mature frataxin (Fig. 1A). As brain cortex homogenates contain both neurons and astrocytes, we next performed co-immunoprecipitation assay in cultured primary cortical neurons. Similarly, GRP75 but not control IgG immunoprecipitated mature frataxin (Fig. 1B). Consistent with the role of GRP75 in ISC biogenesis (27,30), the ISC biogenesis protein ISCU2, a mitochondrial form of ISCU, was also co-immunoprecipitated by GRP75 but not control IgG in both cortical homogenates and cortical neurons (Fig. 1A and B). These results indicate that GRP75 binds to frataxin both in vivo in cortex and in vitro in neurons where it forms a complex with the ISC biogenesis protein ISCU2.
Figure 1.
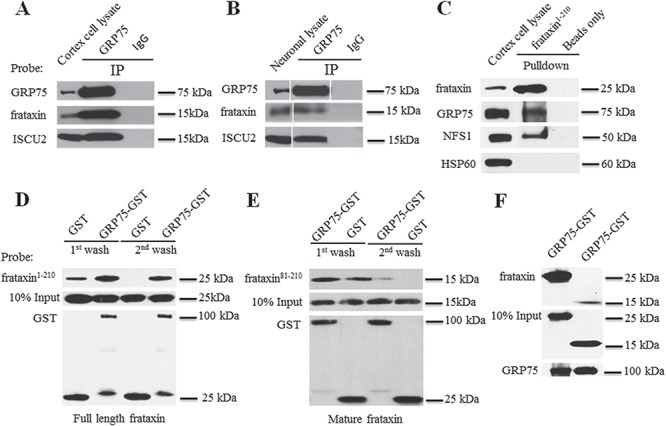
GRP75 interacts with frataxin both in vivo and in vitro. GRP75 but not control IgG immunoprecipitated frataxin and ISCU2 from both cortical homogenates (A) and neuronal lysates (B). Purified human full-length frataxin (frataxin1–210-6XHis) (precursor) also pulled down GRP75 from cortical homogenates while control experiments with frataxin1–210-6XHis omitted (beads only) showed no immunoreactivity. NFS1 and HSP60 served as a positive and negative controls, respectively (C). Similarly, purified GST-GRP75 but not GST pulled down both frataxin1–210-6XHis (D) and mature frataxin (frataxin81–210-6XHis) (E) in an in vitro binding assay. GST-GRP75 bound strongly to frataxin precursor when compared with mature frataxin in an in vitro binding assay (F).
The interaction between GRP75 and frataxin was further confirmed by pull-down assays from mouse cortex homogenates using full-length human frataxin (precursor) fused to a C-terminal 6XHis tag (frataxin1–210-6XHis). Purified frataxin1–210-6XHis protein led to the pull down of endogenous GRP75 and the ISC biogenesis protein cysteine desulfurase (NFS1) but not HSP60, another mitochondrial chaperone serving as a negative control. No GRP75, NFS1 or HSP60 proteins were detected when frataxin1–210-6XHis proteins were omitted in the assays (beads only) (Fig. 1C). To determine whether GRP75 directly interacts with frataxin, we performed in vitro binding assays using purified glutathione S-transferase (GST)-GRP75 fusion proteins and frataxin1-210–6XHis proteins. To monitor the binding specificity, frataxin1–210-6XHis bound to GST-GRP75 or GST-conjugated glutathione beads were eluted after both the first and second washes. Binding between frataxin1–210-6XHis and GST-GRP75 was detected after both the first and second washes. While low levels of binding of frataxin1–210-6XHis to GST were detected after the first wash, no frataxin1–210-6XHis bound to GST after the second wash (Fig. 1D), suggesting that frataxin1–210-6XHis specifically bind to GST-GRP75. The binding of mature frataxin to GRP75 was also detected in in vitro binding assays using the purified 15 kDa mature form of human frataxin (amino acids 81–210) fused to a C-terminal 6XHis tag (frataxin81–210-6XHis) (Fig. 1E and F) but at a much lesser degree compared with full-length frataxin. These results indicate that GRP75 directly binds to frataxin, preferentially to the frataxin precursor.
GRP75 potentiates the interaction of frataxin with MPP
The maturation of frataxin requires two cleavages by the mitochondrial processing peptidase (MPP) (31). As GRP75 participates in the maturation of frataxin (27–29), we examined whether GRP75 physically interacts with MPP to facilitate this process. Mouse cortex homogenates were immunoprecipitated with anti-GRP75 antibody or control IgG. MPP subunit beta was co-immunoprecipitated by anti-GRP75 but not control IgG (Fig. 2A), suggesting a physical interaction between GRP75 and MPP. The interaction between MPP and GRP75 was further confirmed by pull-down assays from mouse cortex homogenates using purified GST-GRP75. In agreement with the Co-immunoprecipitation (Co-IP) assay, purified GRP75 but not GST led to the pull down of endogenous MPP (Fig. 2B). To determine whether GRP75 directly interacts with MPP, we then performed an in vitro binding assay using purified GST-GRP75 proteins and MPP fused to a C-terminal MYC/DDK tag (MPP-MYC/DDK). Purified GST-GRP75 bound to MPP-MYC/DDK after both the first and second washes. While low levels of binding of MPP-MYC/DDK to GST were detected after the first wash, no MPP bound to GST after the second wash (Fig. 2C), indicating the binding specificity of GRP75 to MPP and a direct interaction between these proteins.
Figure 2.
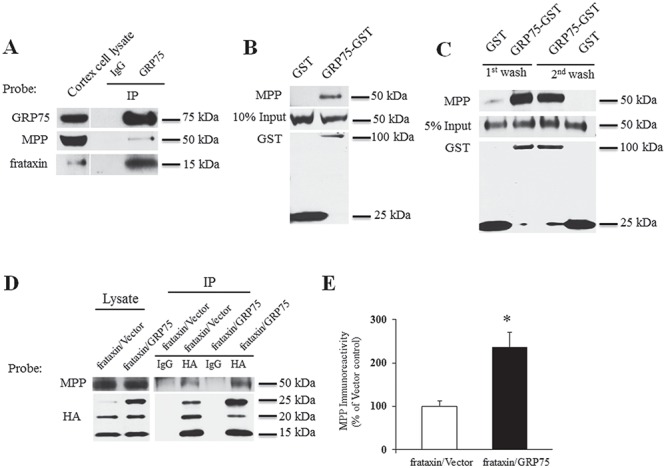
GRP75 potentiates the interaction of frataxin with MPP. Cortical homogenates were immunoprecipitated with GRP75 antibody and the precipitated proteins were probed with the indicated antibodies. GRP75 but not control IgG immunoprecipitated MPP from cortical homogenates (A). Purified GST-GRP75 but not GST pulled down MPP from cortical homogenates (B) and in an in vitro binding assay (C), indicating a direct interaction between GRP75 and MPP. In HEK293 cells transfected with frataxin-HA along with GRP75-c-Myc-6XHis or vector, HA antibody but not control IgG immunoprecipitated MPP in cells with or without additional GRP75. The presence of GRP75 significantly increased MPP associating with frataxin (D and E) (n = 3). *P < 0.05. Data were shown as mean ± SE (error bars).
We then investigated whether GRP75 potentiates the interaction between frataxin and MPP. HEK293 cells transiently transfected with frataxin fused to a C-terminal human influenza hemagglutinin (HA) tag (frataxin-HA) along with GRP75 containing a C-terminal c-Myc-6XHis tag (GRP75-c-Myc-6XHis) or vector control were lysed and subjected to co-immunoprecipitation followed by western blot analysis. Anti-HA antibody precipitated MPP in cells without additional GRP75; however, the presence of GRP75 significantly increased the levels of MPP associating with frataxin (136% increase over vector control; P < 0.05; n = 3) (Fig. 2D and E), suggesting that GRP75 potentiates the interaction of frataxin with MPP.
Frataxin does not regulate GRP75 in both HEK293 cells and human skin fibroblasts
As GRP75 directly interacts with frataxin, we sought to investigate whether frataxin regulates GRP75. We first examined GRP75 protein levels in FRDA patient-derived cells. Both skin fibroblasts and buccal cells from FRDA patients (Fig. 3) had significantly reduced frataxin (fibroblasts, Fig. 3A and B; 78% decrease from the control, P < 0.01, n = 3; buccal cells, Fig. 3C and D; 95% decrease from the control, P < 0.01, n = 20 for FRDA patients, n = 7 for control). Similarly, GRP75 was also significantly reduced in both fibroblasts (Fig. 3A and B; 60% decrease from the control, P < 0.05; n = 3) and buccal cells (Fig. 3C and D; 42% decrease from the control, P < 0.05, n = 20 for FRDA patients and n = 7 for control), indicating that GRP75 reduction correlates with frataxin deficiency.
Figure 3.
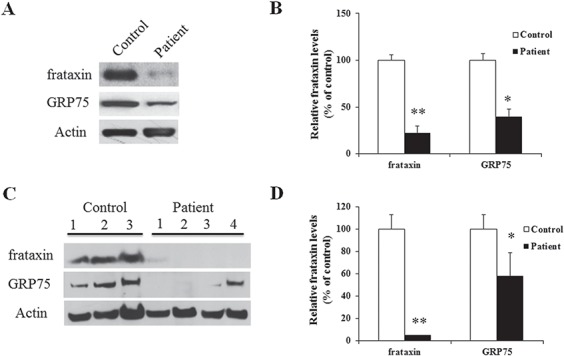
GRP75 protein levels are decreased in both FRDA patient skin fibroblasts and buccal cells. FRDA patient skin fibroblasts or buccal cells were lysed and subjected to western blotting with the indicated antibodies. The amount of immunoreactivity in the lysates was quantified as a percentage of the controls. Representative blots and bar graphs demonstrate reduced frataxin in both fibroblasts (A and B) (n = 3) and buccal cells (C and D) (n = 20 for patients and n = 7 for controls). Accordingly, GRP75 was also decreased in both fibroblasts (A, B) (n = 3) and buccal cells (C, D) (n = 20 for patients and n = 7 for controls). *P < 0.05; **P < 0.01. Data were shown as mean ± SE.
We next examined whether such a decrease in GRP75 is a direct downstream event of frataxin deficiency. Overexpressing frataxin in HEK293 cells had no effect on GRP75 (Fig. 4A and B; P > 0.05, n = 4) as detected by anti-GRP75 antibody. Similarly, frataxin depletion with siRNA had no effect on GRP75, as transfection of frataxin siRNA in HEK293 cells for 3 or 5 days resulted in 40% or 22% residual mature frataxin in comparison with control siRNA (Fig. 4C and D; P < 0.01, n = 8 for 3 days and P < 0.01, n = 4 for 5 days), but no reduction in GRP75 was observed. Similarly, no change in GRP75 levels was observed in human skin fibroblasts after frataxin depletion with siRNA for either 3 or 5 days (frataxin: 44% decrease from the control, P < 0.01, n = 4 for 3 days and 89% decrease from the control, P < 0.01, n = 5 for 5 days; Fig. 4E and F). These results indicate that both frataxin overexpression and knockdown have no effect on GRP75 and that GRP75 reduction in FRDA patient-derived cells is not a direct event but rather a chronic, secondary effect of frataxin deficiency.
Figure 4.
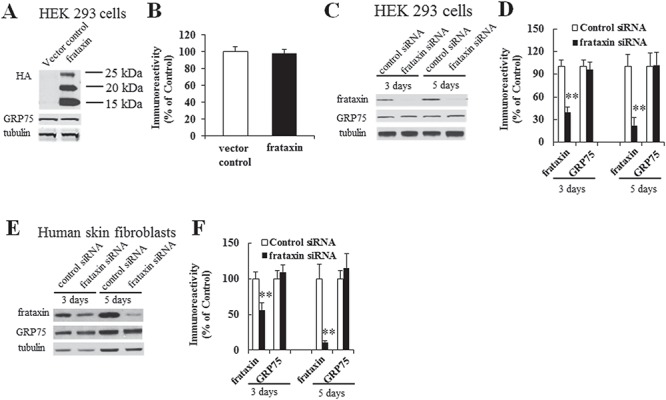
Frataxin overexpression or knockdown has no effect on GRP75 protein levels. HEK293 cells transfected with frataxin plasmid DNA or siRNA were lysed and subjected to western blotting with the indicated antibodies. The amount of immunoreactivity in the lysates was quantified as a percentage of vector or siRNA control. No change in GRP75 was found in cells transfected with frataxin-HA in comparison with vector control (A and B) (n = 6; P > 0.05). While frataxin knockdown resulted in 40% and 22% residual frataxin at 3 and 5 days, respectively, no decrease in GRP75 was observed at either condition (C and D) (n = 8, P > 0.05 for 3 days and n = 4, P > 0.05 for 5 days). Similar results were noted in human skin fibroblasts with frataxin siRNA leading to 56% and 11% residual frataxin at 3 and 5 days, respectively (E and F) (n = 4, P > 0.05 for both 3 and 5 days) **P < 0.01. Data were shown as mean ± SE.
GRP75 overexpression increases the protein levels of wild-type frataxin and frataxin variants in HEK 293 cells
We next examined whether GRP75 overexpression increases frataxin. Compared with vector control, GRP75 overexpression significantly increased precursor (10-fold increase over vector control, P < 0.01, n = 8), intermediate (0.63-fold increase over vector control, P < 0.05, n = 8) and mature frataxins (0.75-fold increase over vector control, P < 0.05, n = 8; Fig. 5A and B). We next asked whether GRP75 overexpression increases frataxin variants such as frataxinG130V, frataxinW155R, frataxinI154F, frataxinG137V, frataxinW168R, frataxinL106S and frataxinR165C, which are found in compound heterozygote patients and in general lead to lower frataxin levels compared with patients with two GAA repeat expansions (32,33). Similarly, when HEK293 cells were transfected with frataxinG130V, frataxinW155R, frataxinI154F, frataxinG137V, frataxinW168R, frataxinL106S or frataxinR165C along with GRP75-c-Myc-6XHis or vector control, GRP75 overexpression significantly increased frataxinG130V and other frataxin variant protein levels (Fig. 5, Table 1 and Supplementary Material, Fig. S1). While GRP75 overexpression increased the levels of all the tested frataxin variants, it has the largest effect on frataxinW168R precursor (Table 1).
Figure 5.
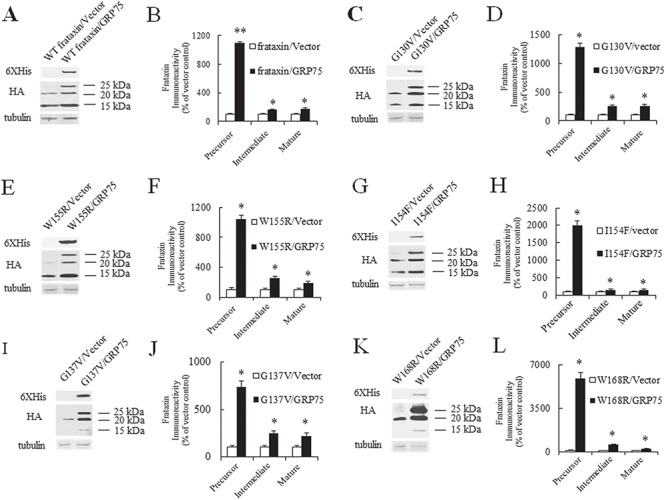
GRP75 overexpression increases both WT frataxin and frataxin variant protein levels. HEK293 cells co-transfected with WT frataxin-HA, frataxin variant-HA, GRP75-c-Myc-6XHis or vector control were lysed and subjected to western blotting with the indicated antibodies. Representative blots and bar graphs demonstrate increased precursor, intermediate and mature forms of WT frataxin (A and B) (n = 8) and frataxin variants (frataxinG130V: C and D, n = 5; frataxinW155R: E and F, n = 5; frataxinI154F: G and H, n = 5; frataxinG137V: I and J, n = 7; frataxinW168R: K and L, n = 5). *P < 0.05, **P < 0.01. Data were shown as mean ± SE.
Table 1.
GRP75 overexpression increases frataxin variants
| Fold increase | ||||
|---|---|---|---|---|
| Variants | Precursor | Intermediate | Mature | Number |
| G130V | 11.9* | 1.52* | 1.55* | 5 |
| W155R | 9.42* | 1.56* | 0.85* | 5 |
| I154F | 19* | 0.51* | 0.54* | 5 |
| G137V | 6.4* | 1.5* | 1.19* | 7 |
| W168R | 58* | 4.98* | 1.12* | 5 |
| L106S | 12.55* | 2.37* | Unchanged | 5 |
| R165C | 7.69* | Unchanged | Unchanged | 5 |
Precursor, intermediate and mature forms in HEK293 cells co-transfected with frataxin variants and GRP75 were quantified and listed as fold increase over vector control. *P < 0.05.
To investigate whether the mutations in frataxin variants affect binding of GRP75 to frataxin, we performed co-immunoprecipitation in HEK293 cells transfected with wild-type (WT) frataxin or frataxin variants using anti-GRP75 antibody. GRP75 bound preferentially to the precursor and intermediate forms (Supplementary Material, Fig. S2). Binding of GRP75 to mature frataxin was also detected but to a much lesser degree (data not shown). Compared with WT frataxin, the G130V, W168R, G137V and L106S mutations had increased the levels of frataxin interacting with GRP75 while the R165C, I154F and W155R mutations had no effect (Supplementary Material, Fig. S2), indicating that these mutations do not interfere with the binding of GRP75 to frataxin.
Mature frataxin levels in variants carrying W168R, G137V and L106S mutations were significantly reduced in transfected cells. The intermediate forms of frataxinW168R and frataxinG137V were comparable to other frataxin variants but was barely detectable for frataxinL106S (Supplementary Material, Fig. S2). The L106S mutation also led to aberrant mobility of both the precursor and intermediate forms by electrophoresis (Supplementary Material, Figs. S1A and F and S2). Although frataxin maturation involves two cleavages by MPP, frataxinL106S may have been cleaved at a third site as evidenced by the presence of two intermediate forms (Supplementary Material, Fig. S1A), suggesting that the L106S mutation also interferes with frataxin processing.
GRP75-controlled frataxin expression is reduced by the R126W, A476T and P509S mutations in GRP75 variants
GRP75 variants such as A476T, P509S and R126W, associated with Parkinson’s disease (PD), result in loss of function (23). We examined the effect of these mutations on GRP75-controlled frataxin expression. In HEK293 cells co-transfected with WT frataxin-HA along with WT GRP75 fused to a C-terminal V5-6XHis tag (GRP75-V5-6XHis), GRP75 variants (R126W, A476T and P509S)-V5-6XHis or vector control, GRP75-V5-6XHis overexpression significantly increased precursor (Fig. 6A and B; 292% increase over vector control, n = 5, P < 0.05) with less effect on intermediate and mature frataxins. In comparison with GRP75-c-Myc-6XHis, which induced a 10-fold increase in precursor (Fig. 5), GRP75-V5-6XHis has a relatively small effect, suggesting a possible conformational change on the binding of GRP75 to frataxin. No significant increase in intermediate or mature frataxin was observed for GRP75-V5-6XHis. Thus, we compared precursor levels among WT GRP75 and GRP75 variants. Compared with WT GRP75, mutations in GRP75 variants significantly reduced precursor (Fig. 6A and B; 29% decrease from WT GRP75, n = 5, P < 0.05 for A476T; 44% decrease from WT GRP75, n = 5, P < 0.05 for P509S; 34% decrease from WT GRP75, n = 5, P < 0.05 for R126W). These results indicate that mutations found in GRP75 variants reduce GRP75-controlled frataxin expression.
Figure 6.
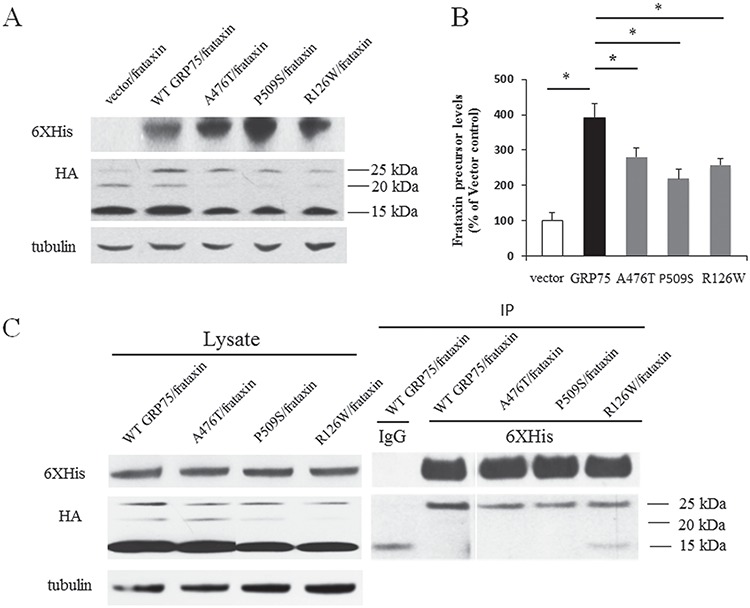
GRP75 variants (A476T, P509S and R126W) reduce GRP75-controlled frataxin expression and the binding of GRP75 to frataxin. HEK293 cells co-transfected with frataxin-HA along with WT GRP75-V5-6XHis, GRP75 variant-V5-6XHis or vector control were lysed and subjected to western blotting with the indicated antibodies. The amount of frataxin precursor immunoreactivity in the lysates was quantified as a percentage of vector control. Compared with vector control, WT GRP75 overexpression significantly increased frataxin precursor while A476T, P509S and R126W mutations reduced the effect of GRP75 on frataxin precursor (A and B) (n = 5). The comparison was carried out in the same blot. A476T, P509S and R126W mutations also reduced the amounts of frataxin precursor interacting with GRP75 (C). Results are representative of three separate experiments. *P < 0.05. Data were shown as mean ± SE.
We then examined whether these mutations alter the interaction of GRP75 with frataxin. HEK293 cells co-transfected with frataxin-HA along with WT GRP75-V5-6XHis or GRP75 variants (R126W, A476T and P509S)-V5-6XHis were immunoprecipitated with anti-6XHis antibody. 6XHis antibody immunoprecipitated frataxin precursor in cells transfected with either WT GRP75 or GRP75 variants, but precursor associating with GRP75 variants were significantly reduced in comparison with WT GRP75 (Fig. 6C). This indicates that these amino acids are critical for the binding of GRP75 to frataxin.
Both mitochondria-targeted GRP75 and cytosolic GRP75 overexpression increase frataxin in FRDA patient-derived cells
While GRP75 predominantly localizes in mitochondria, a substantial amount of GRP75 is found in the cytosol (34,35). To differentiate the role of GRP75 at different subcellular localizations and to investigate whether GRP75 overexpression increases frataxin in FRDA patient cells, we transduced FRDA patient-derived skin fibroblasts with lentivirus carrying pHAGE-GRP75 gene with or without an HA tag at the N-terminus. About 60–70% cells were transduced with GRP75 gene after 5 days transduction. This HA tag targets GRP75 into the cytosol rather than the mitochondria likely because of mitochondrial targeting sequence interference. GRP75 without an HA tag was targeted into the mitochondria and displayed punctate and globular staining as well as the colocalization with mitotracker, which specifically labels mitochondria. In contrast, the staining of GRP75 with an HA tag appeared diffusely in the cytosol (Fig. 7A). Interestingly, both mitochondrial (Fig. 7B and C) and cytosolic GRP75 transduction (Fig. 7D and E) increased mature frataxin in fibroblasts from both typical FRDA patients (3.43-fold increase from vector control, n = 4, P < 0.05 for mitochondrial GRP75; 1-fold increase from vector control, n = 4, P < 0.05 for cytosolic GRP75) and G130V patients (1.63-fold increase from vector control, n = 4, P < 0.05 for mitochondrial GRP75; 1.7-fold increase from vector control, n = 4, P < 0.05 for cytosolic GRP75), with concomitant increases in GRP75. Mitochondrial and cytosolic GRP75 transductions also increased ISC biogenesis protein ISCU2 and non-ISC protein HSP60 in fibroblasts from typical FRDA patients (Supplementary Material, Fig. S3A and B; ISCU2: 1.51-fold increase from vector control, n = 5, P < 0.05 for mitochondrial GRP75; 0.82-fold increase from vector control, n = 5, P < 0.05 for cytosolic GRP75; HSP60: 1.14-fold increase from vector control, n = 5, P < 0.05 for mitochondrial GRP75; 1.59-fold increase from vector control, n = 5, P < 0.05 for cytosolic GRP75). No change was found in the levels of beta tubulin, indicating that lentivirus transduction does not globally increase non-specific protein expression. These results indicate that both mitochondrial and cytosolic GRP75 promote the accumulation of mature frataxin in FRDA patient-derived cells.
Figure 7.
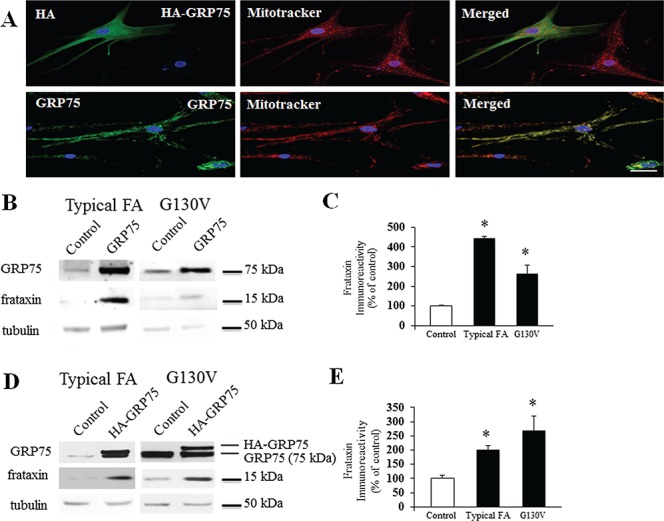
The subcellular location and effect of HA-GRP75 and GRP75 on frataxin levels in FRDA patient-derived cells. Skin fibroblasts from FRDA patient with G130V mutation were transduced with lentivirus carrying pHAGE-HA-GRP75 or pHAGE-GRP75 gene for 5 days and subjected to immunofluorescence or western blotting with the indicated antibodies. HA-GRP75 showed diffused staining in the cytosol while GRP75 showed punctate and globular staining and colocalized with mitotracker (A), suggesting the mitochondrial localization. Both HA-GRP75 and GRP75 increased mature frataxin in both typical FRDA patient fibroblasts (pHAGE-GRP75: B and C, n = 4; pHAGE-HA-GRP75: D and E, n = 4) and G130V fibroblasts (pHAGE-GRP75: B and C, n = 4; pHAGE-HA-GRP75: D and E, n = 4). *P < 0.05. Data were shown as mean ± SE. Scale bar = 50 μm.
Mitochondria-targeted GRP75 but not cytosolic GRP75 overexpression rescues mitochondria structure from fragmentation in FRDA patient-derived cells
Frataxin deficiency is associated with abnormal mitochondrial morphology (36,37). We next examined whether GRP75 overexpression rescues the mitochondrial phenotype in FRDA patient-derived skin fibroblasts. Fibroblasts transduced with vector control displayed fragmented mitochondria (Fig. 8A). However, mitochondrial GRP75 transduction recreated a tubular network and increased mitochondrial branching as indicated by the form factor (FF; Fig. 8B and C; 17-fold increase from vector control, n = 24 cells, P < 0.001) and mitochondrial length as indicated by the aspect ratio (AR; Fig. 8B and D; 0.57-fold increase from vector control, n = 24 cells, P < 0.001). No change was observed in fibroblasts transduced with cytosolic GRP75 (data not shown). These results indicate that mitochondrial GRP75 rescues the abnormalities in mitochondrial morphology in FRDA patient-derived cells.
Figure 8.
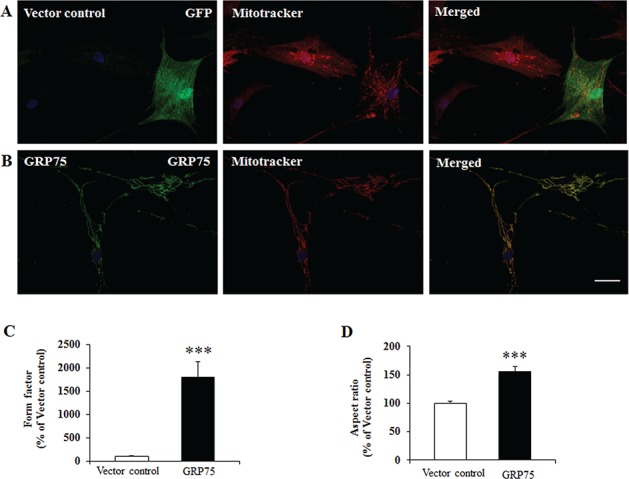
Mitochondrial GRP75 rescues mitochondria fragmentation in FRDA patient-derived cells. Skin fibroblasts from typical FRDA patient were transduced with lentivirus carrying pHAGE-GRP75 gene or vector control for 5 days and subjected to immunofluorescence by using mitotracker and GRP75 antibody. Vector-transduced fibroblasts displayed fragmented mitochondria (A) while GRP75-transduced fibroblasts revealed tubular mitochondrial network (B). Compared with vector control, GRP75 transduction exhibited significantly increased mitochondrial branching as indicated by the FF (C) and mitochondrial length as indicated by the AR (D) (n = 24 cells from three independent experiments). ***P < 0.001. Data were shown as mean ± SE. Scale bar = 50 μm.
Both mitochondria-targeted GRP75 and cytosolic GRP75 overexpression rescue the ATP deficit in FRDA patient-derived cells
Finally, we investigated the functional significance of GRP75 overexpression by examining ATP levels in FRDA patient-derived skin fibroblasts. Compared with control fibroblasts, both typical FRDA patient fibroblasts and G130V patient fibroblasts showed decreased ATP levels (Fig. 9A; 44% decrease from control, n = 4, P < 0.05 for typical FRDA patient; 37% decrease from control, n = 4, P < 0.05 for G130V). Both mitochondrial and cytosolic GRP75 transductions significantly increased ATP levels in fibroblasts from both typical FRDA patients (Fig. 9B and C; 22% increase from control, n = 4, P < 0.05 for mitochondrial GRP75; 26% increase from control, P < 0.05, n = 4 for cytosolic GRP75) and G130V patients (Fig. 9B and C; 25% increase from control, n = 4, P < 0.05 for mitochondrial GRP75; 33% increase from control, n = 4, P < 0.05 for cytosolic GRP75). These results indicate that both mitochondrial and cytosolic GRP75 overexpressions rescue ATP deficit in FRDA patient-derived cells.
Figure 9.
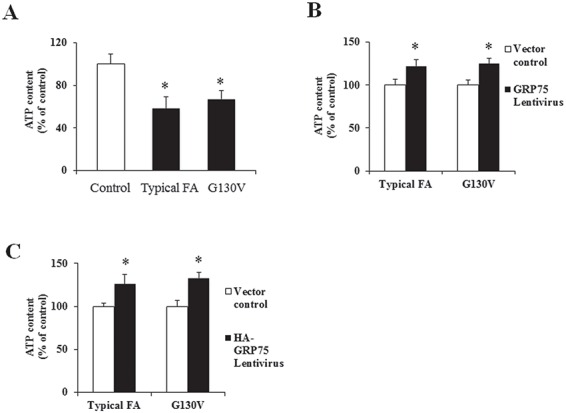
Both mitochondrial and cytosolic GRP75 transductions increase ATP levels in FRDA patient-derived cells. Compared with control, ATP levels were decreased in fibroblasts from both typical FRDA patient (n = 4) and patient with G130V mutation (n = 4) (A). Transduction with lentivirus carrying pHAGE-GRP75 or pHAGE–HA-GRP75 gene for 5 days resulted in increased ATP levels in both typical FRDA patient fibroblasts (B and C) (n = 4) and G130V fibroblasts (B, C) (n = 4). *P < 0.05. Data were shown as mean ± SE.
Discussion
In the present study, we have demonstrated that the interaction of GRP75 with frataxin controls frataxin and pathophysiological events downstream from frataxin deficiency. While WT GRP75 overexpression increases frataxin both in HEK293 cells and in FRDA patient-derived cells, GRP75 variants carrying R126W, A476T and P509S mutations impair the interaction of GRP75 with frataxin and reduce the effect of GR75 on frataxin. In addition, GRP75 overexpression partially rescues the cellular phenotypes of FRDA as demonstrated by the increased ATP levels and improved integrity of the mitochondrial network. These results define GRP75 as a major posttranslational regulator of frataxin level and function.
Although the mechanisms leading to frataxin deficiency in FRDA are transcriptional in typical FRDA, frataxin levels are posttranslationally regulated. Interfering with frataxin ubiquitination and degradation with ubiquitin-competing molecules promotes the accumulation of frataxin precursor and mature forms (38). In agreement with these findings, GRP75 overexpression also increases frataxin. Although GRP75 predominantly increases precursor, it also increases the mature form of frataxin, suggesting that increasing the pool of precursor by GRP75 aids in generating mature frataxin. GRP75 regulates frataxin both before and after mitochondrial import as evidenced by the increased levels of mature frataxin by both cytosolic and mitochondrial GRP75 overexpression in FRDA patient-derived cells. The effect of cytosolic GRP75 could be mediated by the chaperone activity of GRP75 which prevents the aggregation and degradation of frataxin precursor during its trafficking to mitochondria, while mitochondrial GRP75 imports frataxin and assists its folding. In addition, the physical interaction of GRP75 with MPP contributes to the maturation of frataxin as demonstrated by the increased amounts of MPP associating with frataxin by GRP75. GRP75 directly interacts with frataxin and MPP, suggesting that GRP75 may form a ternary complex with frataxin and MPP to make frataxin more accessible to MPP and the subsequent processing more efficient. Thus, in addition to its role in protein import, GRP75 could also function as a scaffold protein to promote the accumulation of frataxin. GRP75 also interacts with multiple proteins involved in the biogenesis of ISC such as NFS1, ISCU and Nfu in vivo (27,30), further supporting such a scaffolding role.
The predominant effect of GRP75 on precursor is supported by its preferential binding to frataxin precursor in both in vitro binding assays (Fig. 1) and Co-IP experiments in HEK cells (Fig. 6C and Supplementary Material, Fig. S2). This would be more beneficial for frataxin variants that have reduced efficiency of protein folding and higher tendency toward proteolytic degradation (39,40), leading to lower-than-normal levels of frataxin and more severe clinical phenotypes. Indeed, we noticed more prominent effects on frataxin variants by GRP75 (Fig. 5, Table 1 and Supplementary Material, Fig. S1). The largest effect was seen on the W168R mutant with 58-fold increase in precursor. The effects on intermediate and mature forms of frataxin were also observed to a lesser extent, indicating a major role of GRP75 in frataxin precursor stabilization.
Consistent with energy impairment in FRDA cells (9,10), we noted the ATP deficit in FRDA patient fibroblasts as measured by the generation of a luminescent signal from luciferase reaction, which is proportional to the amount of ATP present in the cells. GRP75 overexpression rescues the ATP deficit in FRDA cells. While increased frataxin in FRDA could lead to increased activity of ISC containing enzymes in Krebs cycle such as aconitase and electron transport chain enzymes, improved mitochondrial network integrity could also increase the activity of ATP synthase, Oxidative phosphorylation and decrease ATP hydrolysis (41,42). Interestingly, both mitochondrial and cytosolic GRP75 overexpressions increase ATP levels to a similar degree, but only mitochondrial GRP75 overexpression rescues the mitochondrial network, suggesting that mitochondrial network restoration involves mechanisms other than the chaperone activity of GRP75 in the cytosol. The mitochondrial fission proteins Drp1 and Opa1 participate in GRP75 depletion-mediated mitochondria fragmentation (43,44). How these proteins might interact with GRP75 to restore mitochondria network structure in FRDA remains to be investigated. The present findings also provide mechanistic support for GRP75-mediated facilitation of ATP production in other cellular and animal models of neurodegenerative diseases (45–47). As cellular ATP levels are a sensitive indicator of viability in multiple cells (48,49), alternatively, increased ATP levels by GRP75 may confer an increased ability to cope with cellular stress to FRDA cells.
GRP75 plays an essential role in mitochondrial biogenesis through its capacity to direct the import and folding of nuclear-encoded mitochondrial matrix proteins (16,17). In neurodegenerative diseases, GRP75 has been linked to neurodegeneration in PD and Alzheimer’s disease (AD) based on reduced levels of GRP75 in brain samples of patients (43,50). GRP75 reduction also correlates with the disease stage of PD (51). Consistent with findings from brain homogenates of FRDA animal models and patient blood cells (52,53), GRP75 levels are reduced in both FRDA patient fibroblasts and buccal cells, and effects of GRP75 overexpression suggest that GRP75 deficiency may contribute to the pathogenesis of FRDA. GRP75 reduction could lead to further decreases in frataxin and ISC containing enzymes and produce mitochondrial biogenesis deficits as observed in FRDA patient cells and animal models at presymptomatic ages (52,53). This could contribute to the progression of FRDA. Both frataxin overexpression and acute knockdown had no effect on GRP75 levels, suggesting that GRP75 reduction is a secondary effect of frataxin deficiency and may reflect decreased mitochondrial number seen in FRDA patient cells and animal models (54).
GRP75 variants (R126W, A476T and P509S) have been implicated in the pathogenesis of PD. All of these variants are substitutions found in highly conserved regions of GRP75, and accumulation of these mutations leads to mitochondrial dysfunction both in neuronal and non-neuronal cell lines (23). Analogous mutations in yeast recapitulate multiple in vivo phenotypes and biochemical defects associated with these variants (55). GRP75 variants and frataxin deficiency share similar mitochondrial abnormalities such as abnormal mitochondrial morphology, reduced mitochondrial membrane potential and increased production of oxidative species (23,36,37,56). GRP75 may represent one of a series of proteins involved in the pathophysiological overlap between FRDA and other neurodegenerative disorders like PD and AD.
Materials and Methods
Preparation of brain homogenates
Mice were deeply anesthetized and euthanized by decapitation. The cortex was rapidly removed and homogenized with Potter-Elvehjem homogenizer (Thermo Fisher Scientific Inc., Hampton, NH) in radioimmunoprecipitation assay buffer (RIPA buffer) (100 mm Tris–HCL; 150 mm NaCl; 1% Octylphenoxy poly(ethyleneoxy)ethanol (IGEPAL CA-630); 1 mm ethylenediaminetetraacetic acid (EDTA); pH 7.4) containing protease inhibitor cocktail (1:500 dilution; Calbiochem, Darmstadt, Germany). The homogenates were centrifuged at 13 000 rpm for 15 min. The supernatant was immediately frozen and stored at −80°C until use. Protein concentration was estimated by Pierce BCA Protein Assay Kit (Thermo Fisher Scientific Inc.).
Preparation of primary neuronal cultures
Primary rat cortical neurons were derived from embryonic day 17 Sprague Dawley rat embryos, as described previously (57). Cortical tissue was dissected and subsequently minced and trypsinized (0.027%, 37°C, 7% CO2 for 20 min) and then washed with 1XHBSS. Cells were plated in neurobasal medium supplemented with B27 and grown at a density of 6 × 105 viable cells per 35 mm culture dish. Cultures were maintained at 37°C with 5% CO2. Non-neuronal cell growth was inhibited with cytosine arabinoside at 7–10 days in vitro. Cells were used after at least 14 days in vitro.
RNA interference-mediated downregulation of frataxin and overexpression of plasmid DNAs in HEK293 cells
HEK293 cells were cultured in a 5% CO2 humidified atmosphere in Minimum Essential Medium containing 1% penicillin/streptomycin, 1% L-glutamine (200 mm), 2.5% horse serum and 7.5% fetal bovine serum (FBS). Frataxin knockdown was achieved by transfection of human frataxin siRNA (5′-rGrArArCrCrUrArUrGrUrGrArUrCrArArCrArArGrCrArGAC-3′; Dr Robert Wilson, University of Pennsylvania and Children’s Hospital of Philadelphia, Philadelphia, PA) into HEK293 cells using Lipofectamine RNAiMax reagent (Thermo Fisher Scientific Inc.). Scrambled siRNA was used as a negative control. Cells were collected after 3 or 5 days.
For overexpression of plasmid DNAs, HEK293 cells were transfected with plasmid DNAs containing WT frataxin fused to a C-terminal HA tag, frataxin mutants (33), GRP75 fused to a C-terminal c-Myc and 6XHis (Abgent, San Diego, CA), GRP75 or GRP75 mutants (A476T, P509S and R126W) fused to a C-terminal V5-6XHis tag (Dr Rejko Krüger, University of Tubingen, Germany) using Lipofectamine™ 2000 reagent (Thermo Fisher Scientific Inc.) for 24 h. Cells were then collected and subjected to co-immunoprecipitation or western blot.
Lentiviral transduction of human skin fibroblasts
FRDA patient-derived skin fibroblasts were cultured in 5% CO2 humidified atmosphere in Dulbecco’s Modified Eagle Medium: Nutrient Mixture F-12 containing 1% penicillin/streptomycin, 1% non-essential amino acids and 10% FBS. Lentivirus containing the GRP75 gene with or without an HA tag in the N-terminus (pHAGE-HA-GRP75 or pHAGE-GRP75; Dr Jong-In Park, Medical College of Wisconsin, Milwaukee, WI) or vector control (pHAGE-CMV-dsRed-UBC-GFP-W; Addgene, Cambridge, MA 02139), made at the research vector core at the Children’s Hospital of Philadelphia, was transduced into cultured fibroblasts in the presence of polybrene (8 μg/ml; Sigma, St Louis, MO). One day after transduction, the medium was replaced with normal culture medium. Five days after transduction, fibroblasts were collected and used for experiments.
Co-immunoprecipitation
Cultured rat cortical neurons or HEK293 cells were rinsed with phosphate-buffered saline (PBS) and lysed in RIPA buffer (100 mm Tris–HCL; 150 mm NaCl; 1% IGEPAL CA-630; 1 mm EDTA; pH 7.4) with a protease inhibitor cocktail (1:500 dilution; Calbiochem) at 4°C for 1 h. Cell lysates were then centrifuged at 13 000 rpm for 10 min. The supernatant was transferred to a new tube and incubated with 30 μl of protein G beads (Invitrogen, Carlsbad, CA) at 4°C for 1 h to remove non-specific binding to the beads. The samples were then centrifuged at 500 g for 5 min and the supernatant was incubated with antibodies against GRP75 (Abcam, Cambridge, MA at 1/300), HA (Cell Signaling, Danvers, MA at 1/100) or 6XHis (Thermo Fisher Scientific Inc. at 1/300) overnight at 4°C. On the second day, 30 μl of protein G beads were added to the samples and incubated at 4°C for 2 h. The samples were then centrifuged at 500 g for 5 min and the beads were washed four times with RIPA buffer. A total of 50 μl of sample buffer for sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) was then added, and the mixture was boiled for 5 min. Beads were pelleted by centrifugation, and a fixed volume of supernatant was applied to a NuPAGE 4–12% Bis-Tris gel for SDS-PAGE.
Western blot
Cultured human fibroblasts and HEK293 cells were collected in Laemmli sample buffer (50 mm Tris–HCl, pH 6.8, 2% SDS, 5 mm EDTA, 0.1% bromophenol blue, 10% glycerol, and 2% β-mercaptoethanol). Buccal cells from controls and FRDA patients were collected in extraction buffer (Abcam) as previously described (58) and diluted with Laemmli sample buffer. A sample (15–50 μg) of total protein was boiled for 5 min and then loaded onto NuPAGE 4–12% Bis-Tris gel. Following SDS-PAGE, proteins were transferred to nitrocellulose, blocked with 3% dry milk and incubated with antibodies against frataxin (Abcam at 1/500), ISCU2 (Proteintech, Rosemont, IL at 1/1000), MPP (Proteintech at 1/1000), GRP75 (Abcam at 1/1000), HSP60 (Cell Signaling at 1/1000), actin (Sigma at 1/1000), beta tubulin (Cell Signaling at 1/1000), HA (Cell Signaling at 1/1000), 6XHis (Thermo Fisher Scientific Inc. at 1/1000) and GST (Sigma at 1/1000). Blots were then incubated with appropriate horseradish peroxidase-conjugated secondary antibodies and developed using enhanced chemiluminescence (Pierce, Rockford, IL).
Pull-down assay and in vitro binding assay
Mouse cortex homogenates (500 μg) were precleared with protein G beads for 1 h then incubated with recombinant human full-length frataxin (precursor) fused to a C-terminal 6XHis tag (frataxin1–210-6XHis) (Proteintech) at 4°C for 2 h. Anti-frataxin antibody (Abcam at 1/300; catalog # ab113691) was then added. Samples were incubated at 4°C overnight. On the second day, 25 μl of protein G beads were added to the samples and incubated at 4°C for 2 h. Samples were centrifuged at 500 g for 5 min, and the beads were washed four times with RIPA buffer including 300 mm NaCl. A total of 50 μl of Laemmli sample buffer was added, and the samples were boiled for 5 min. Beads were pelleted by centrifugation, and a fixed volume of supernatant was applied to a NuPAGE 4–12% Bis-Tris gel for SDS-PAGE.
To determine whether exogenous GRP75 pulls down MPP, recombinant human GRP75 protein fused to a N-terminal GST tag (GST-GRP75; Rockland Immunochemicals, Pottstown, PA) or GST (Sigma) were bound to glutathione beads in RIPA buffer (100 mm Tris–HCL; 150 mm NaCl; 1% IGEPAL CA-630; 1 mm EDTA; pH 7.4) overnight at 4°C. After 4 washes, 500 μg mouse cortex homogenates precleared with protein G beads were added and incubated overnight at 4°C. Samples were centrifuged at 500 g for 5 min, and the beads were washed four times with RIPA buffer including 300 mm NaCl. A total of 50 μl of Laemmli sample buffer was added, and the samples were boiled for 5 min. Beads were pelleted by centrifugation, and a fixed volume of supernatant was applied to a NuPAGE 4–12% Bis-Tris gel for SDS-PAGE.
For in vitro binding assays, 2 μg GST-GRP75 or GST were bound to glutathione beads in RIPA buffer (100 mm Tris–HCL, 150 mm NaCl, 1% IGEPAL CA-630, 0.5% sodium deoxycholate; 1 mm EDTA, pH 7.4) overnight at 4°C. After four washes, 2 μg human frataxin1–210-6XHis (Proteintech), frataxin81–210-6XHis made from bacteria (Dr Andrew Dancis, University of Pennsylvania, Philadelphia, PA), or human MPP fused to a C-terminal MYC/DDK tag (MPP-MYC/DDK; Origene, Rockville, MD, catalog # TP315039) was added and incubated overnight at 4°C. Samples were centrifuged at 500 g for 5 min, and the glutathione beads were washed twice at 4°C for 10 min each and eluted with Laemmli sample buffer after each wash and subjected to SDS-PAGE.
Immunofluorescence
Coverslips were coated with poly-D-lysine (0.5 μg/ml) and seeded with FRDA patient skin fibroblasts transduced with lentivirus carrying pHAGE-GRP75, pHAGE-HA-GRP75 or vector control. Cells were loaded with MitoTracker® Red CMXRos (Thermo Fisher Scientific Inc.) for 30 min and washed three times with PBS followed by fixation in 4% paraformaldehyde at room temperature for 10 min. Cells were then permeabilized with 0.3% Triton X-100 and blocked with 10% bovine serum albumin for 1 h at 37°C. Anti-GRP75 (at 1/200), anti-HA (at 1/100) or anti-GFP (Neuromab, Davis, CA at 1/200) antibody was then added and incubated overnight at 4°C. After washing with PBS, cells were incubated with Alexa Fluor® 488 (Life Technology, Eugene, OR) in the dark for 1 h at 37°C. Coverslips were mounted with Vectashield mounting medium containing 4′,6-diamidino-2-phenylindole (DAPI) (Vector Laboratory, Burlingame, CA). Fluorescent images were captured using Leica TCS SP8 confocal laser scanning microscope.
Analysis of mitochondrial morphology
Mitochondrial morphology was analyzed as reported previously (23,59). Briefly, using Image J 1.48v software, fluorescence microscopy images were optimized by adjusting the contrast and subsequently binarized by conversion to eight bit images. A threshold was applied to the images to define mitochondrial structures. Every single mitochondrion labeled by MitoTracker® Red CMXRos of the investigated cells was marked to analyze morphological characteristics such as its area, perimeter and major and minor axes. On the basis of these parameters, the AR of a mitochondrion (ratio between the major and the minor axes of the ellipse equivalent to the object) representing the length of mitochondrion and its FF [perimeter2/(4π × area)], consistent with the degree of branching, were calculated.
Determination of ATP content
An equal number of skin fibroblasts transduced with lentivirus containing pHAGE-HA-GRP75, pHAGE-GRP75 or vector control were seeded in a 96-well plate. ATP content of the cells was quantified with the CellTiter-Glo Luminescent Cell Viability Kit (catalog # G7570, Promega, Madison, WI).
Statistical analysis
Calculations of statistical differences of the experiments were assessed by two-tailed Student’s t-test and a probability value of P < 0.05 was considered to be statistically significant.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
Acknowledgements
We are grateful to Amy J. Salovin for technical support and editing and Dr Hajime Takano for aid in confocal imaging.
Conflict of Interest statement. None declared.
Funding
National Institute of Health Grant (R21NS087343); Friedreich’s Ataxia Research Alliance (Center of Excellence Grant to D.R.L.).
References
- 1. Strawser C., Schadt K., Hauser L., McCormick A., Wells M., Larkindale J., Lin H. and Lynch D.R. (2017) Pharmacological therapeutics in Friedreich ataxia: the present state. Expert. Rev. Neurother., 17, 895–907. [DOI] [PubMed] [Google Scholar]
- 2. Campuzano V., Montermini L., Moltò M.D., Pianese L., Cossée M., Cavalcanti F., Monros E., Rodius F., Duclos F., Monticelli A. et al. (1996) Friedreich’s ataxia: autosomal recessive disease caused by an intronic GAA triplet repeat expansion. Science, 271, 1423–1427. [DOI] [PubMed] [Google Scholar]
- 3. Cossée M., Dürr A., Schmitt M., Dahl N., Trouillas P., Allinson P., Kostrzewa M., Nivelon-Chevallier A., Gustavson K.H., Kohlschütter A. et al. (1999) Friedreich’s ataxia: point mutations and clinical presentation of compound heterozygotes. Ann. Neurol., 45, 200–206. [DOI] [PubMed] [Google Scholar]
- 4. Babcock M., Silva D., Oaks R., Davis-Kaplan S., Jiralerspong S., Montermini L., Pandolfo M. and Kaplan J. (1997) Regulation of mitochondrial iron accumulation by Yfh1p, a putative homolog of frataxin. Science, 276, 1709–1712. [DOI] [PubMed] [Google Scholar]
- 5. Foury F. and Cazzalini O. (1997) Deletion of the yeast homologue of the human gene associated with Friedreich’s ataxia elicits iron accumulation in mitochondria. FEBS Lett., 411, 373–377. [DOI] [PubMed] [Google Scholar]
- 6. Rötig A., Lonlay P., Chretien D., Foury F., Koenig M., Sidi D., Munnich A. and Rustin P. (1997) Aconitase and mitochondrial iron–sulphur protein deficiency in Friedreich ataxia. Nat. Genet., 17, 215–217. [DOI] [PubMed] [Google Scholar]
- 7. Stehling O., Elsässer H.P., Brückel B., Mühlenhoff U. and Lill R. (2004) Iron–sulfur protein maturation in human cells: evidence for a function of frataxin. Hum. Mol. Genet., 13, 3007–3015. [DOI] [PubMed] [Google Scholar]
- 8. Gerber J., Mühlenhoff U. and Lill R. (2003) An interaction between frataxin and Isu1/Nfs1 that is crucial for Fe/S cluster synthesis on Isu1. EMBO Rep., 4, 906–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lodi R., Cooper J.M., Bradley J.L., Manners D., Styles P., Taylor D.J. and Schapira A.H. (1999) Deficit of in vivo mitochondrial ATP production in patients with Friedreich ataxia. Proc. Natl. Acad. Sci. U. S. A., 96, 11492–11495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Heidari M.M., Houshmand M., Hosseinkhani S., Nafissi S. and Khatami, M. (2009) Complex I and ATP content deficiency in lymphocytes from Friedreich’s ataxia. Can. J. Neurol. Sci., 36, 26–31. [DOI] [PubMed] [Google Scholar]
- 11. Seznec H., Simon D., Bouton C., Reutenauer L., Hertzog A., Golik P., Procaccio V., Patel M., Drapier J.C., Koenig M. and Puccio H. (2005) Friedreich ataxia: the oxidative stress paradox. Hum. Mol. Genet., 14, 463–474. [DOI] [PubMed] [Google Scholar]
- 12. Wilson R.B. and Roof D.M. (1997) Respiratory deficiency due to loss of mitochondrial DNA in yeast lacking the frataxin homologue. Nat. Genet., 16, 352–357. [DOI] [PubMed] [Google Scholar]
- 13. Zarse K., Schulz T.J., Birringer M. and Ristow M. (2007) Impaired respiration is positively correlated with decreased life span in Caenorhabditis elegans models of Friedreich ataxia. FASEB J., 21, 1271–1275. [DOI] [PubMed] [Google Scholar]
- 14. Puccio H., Simon D., Cossée M., Criqui-Filipe P., Tiziano F., Melki J., Hindelang C., Matyas R., Rustin P. and Koenig M. (2001) Mouse models for Friedreich ataxia exhibit cardiomyopathy, sensory nerve defect and Fe–S enzyme deficiency followed by intramitochondrial iron deposits. Nat. Genet., 27, 181–186. [DOI] [PubMed] [Google Scholar]
- 15. Al-Mahdawi S., Pinto R.M., Varshney D., Lawrence L., Lowrie M.B., Hughes S., Webster Z., Blake J., Cooper J.M., King R. and Pook M.A. (2006) GAA repeat expansion mutation mouse models of Friedreich ataxia exhibit oxidative stress leading to progressive neuronal and cardiac pathology. Genomics, 88, 580–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Geissler A., Rassow J., Pfanner N. and Voos W. (2001) Mitochondrial import driving forces: enhanced trapping by matrix Hsp70 stimulates translocation and reduces the membrane potential dependence of loosely folded preproteins. Mol. Cell. Biol., 21, 7097–7104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Horst M., Oppliger W., Rospert S., Schönfeld H.J., Schatz G. and Azem A. (1997) Sequential action of two hsp70 complexes during protein import into mitochondria. EMBO J., 16, 1842–1849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Voisine C., Cheng Y.C., Ohlson M., Schilke B., Hoff K., Beinert H., Marszalek J. and Craig E.A. (2001) Jac1, a mitochondrial J-type chaperone, is involved in the biogenesis of Fe/S clusters in Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. U. S. A., 98, 1483–1488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Cai K., Frederick R.O., Kim J.H., Reinen N.M., Tonelli M. and Markley J.L. (2013) Human mitochondrial chaperone (mtHSP70) and cysteine desulfurase (NFS1) bind preferentially to the disordered conformation, whereas co-chaperone (HSC20) binds to the structured conformation of the iron–sulfur cluster scaffold protein (ISCU). J. Biol. Chem., 288, 28755–28770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Burbulla L.F., Fitzgerald J.C., Stegen K., Westermeier J., Thost A.K., Kato H., Mokranjac D., Sauerwald J., Martins L.M., Woitalla D. et al. (2014) Mitochondrial proteolytic stress induced by loss of mortalin function is rescued by Parkin and PINK1. Cell Death Dis., 5, e1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ma Z., Izumi H., Kanai M., Kabuyama Y., Ahn N.G. and Fukasawa K. (2006) Mortalin controls centrosome duplication via modulating centrosomal localization of p53. Oncogene, 25, 5377–5390. [DOI] [PubMed] [Google Scholar]
- 22. Zhu J.Y., Vereshchagina N., Sreekumar V., Burbulla L.F., Costa A.C., Daub K.J., Woitalla D., Martins L.M., Krüger R. and Rasse T.M. (2013) Knockdown of Hsc70-5/mortalin induces loss of synaptic mitochondria in a Drosophila Parkinson’s disease model. PLoS One, 8, e83714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Burbulla L.F., Schelling C., Kato H., Rapaport D., Woitalla D., Schiesling C., Schulte C., Sharma M., Illig T., Bauer P. et al. (2010) Dissecting the role of the mitochondrial chaperone mortalin in Parkinson’s disease: functional impact of disease-related variants on mitochondrial homeostasis. Hum. Mol. Genet., 19, 4437–4452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Yang L., Liu X., Hao J., Yang Y., Zhao M., Zuo J. and Liu W. (2008) Glucose-regulated protein 75 suppresses apoptosis induced by glucose deprivation in PC12 cells through inhibition of Bax conformational change. Acta Biochim. Biophys. Sin. (Shanghai), 40, 339–348. [DOI] [PubMed] [Google Scholar]
- 25. Kaul S.C., Yaguchi T., Taira K., Reddel R.R. and Wadhwa R. (2003) Overexpressed mortalin (mot-2)/mthsp70/GRP75 and hTERT cooperate to extend the in vitro lifespan of human fibroblasts. Exp. Cell Res., 286, 96–101. [DOI] [PubMed] [Google Scholar]
- 26. Yokoyama K., Fukumoto K., Murakami T., Harada S., Hosono R., Wadhwa R., Mitsui Y. and Ohkuma S. (2002) Extended longevity of Caenorhabditis elegans by knocking in extra copies of hsp70F, a homolog of mot-2 (mortalin)/mthsp70/Grp75. FEBS Lett., 516, 53–57. [DOI] [PubMed] [Google Scholar]
- 27. Shan Y., Napoli E. and Cortopassi G. (2012) Mitochondrial frataxin interacts with ISD11 of the NFS1/ISCU complex and multiple mitochondrial chaperones. Hum. Mol. Genet., 21, 1457–1469. [DOI] [PubMed] [Google Scholar]
- 28. Knight S.A., Sepuri N.B., Pain D. and Dancis A. (1998) Mt-Hsp70 homolog, Ssc2p, required for maturation of yeast frataxin and mitochondrial iron homeostasis. J. Biol. Chem., 273, 18389–18393. [DOI] [PubMed] [Google Scholar]
- 29. Voisine C., Schilke B., Ohlson M., Beinert H., Marszalek J. and Craig E.A. (2000) Role of the mitochondrial Hsp70s, Ssc1 and Ssq1, in the maturation of Yfh1. Mol. Cell. Biol., 20, 3677–3684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Maio N., Singh A., Uhrigshardt H., Saxena N., Tong W.H. and Rouault T.A. (2014) Cochaperone binding to LYR motifs confers specificity of iron sulfur cluster delivery. Cell Metab., 19, 445–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Cavadini P., Adamec J., Taroni F., Gakh O. and Isaya G. (2000) Two-step processing of human frataxin by mitochondrial processing peptidase. Precursor and intermediate forms are cleaved at different rates. J. Biol. Chem., 275, 41469–41475. [DOI] [PubMed] [Google Scholar]
- 32. Lazaropoulos M., Dong Y., Clark E., Greeley N.R., Seyer L.A., Brigatti K.W., Christie C., Perlman S.L., Wilmot G.R., Gomez C.M. et al. (2015) Frataxin levels in peripheral tissue in Friedreich ataxia. Ann. Clin. Transl. Neurol., 2, 831–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Clark E., Butler J.S., Isaacs C.J., Napierala M. and Lynch D.R. (2017) Selected missense mutations impair frataxin processing in Friedreich ataxia. Ann. Clin. Transl. Neurol., 4, 575–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wadhwa R., Pereira-Smith O.M., Reddel R.R., Sugimoto Y., Mitsui Y. and Kaul S.C. (1995) Correlation between complementation group for immortality and the cellular distribution of mortalin. Exp. Cell Res., 216, 101–106. [DOI] [PubMed] [Google Scholar]
- 35. Ran Q., Wadhwa R., Kawai R., Kaul S.C., Sifers R.N., Bick R.J., Smith J.R. and Pereira-Smith O.M. (2000) Extramitochondrial localization of mortalin/mthsp70/PBP74/GRP75. Biochem. Biophys. Res. Commun., 275, 174–179. [DOI] [PubMed] [Google Scholar]
- 36. Hick A., Wattenhofer-Donzé M., Chintawar S., Tropel P., Simard J.P., Vaucamps N., Gall D., Lambot, L., André, C., Reutenauer, L.et al. (2013) Neurons and cardiomyocytes derived from induced pluripotent stem cells as a model for mitochondrial defects in Friedreich’s ataxia. Dis. Model Mech., 6, 608–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lefevre S., Sliwa D., Rustin P., Camadro J.M. and Santos R. (2012) Oxidative stress induces mitochondrial fragmentation in frataxin-deficient cells. Biochem. Biophys. Res. Commun., 418, 336–341. [DOI] [PubMed] [Google Scholar]
- 38. Rufini A., Fortuni S., Arcuri G., Condò I., Serio D., Incani O., Malisan F., Ventura N. and Testi R. (2011) Preventing the ubiquitin-proteasome-dependent degradation of frataxin, the protein defective in Friedreich’s ataxia. Hum. Mol. Genet., 20, 1253–1261. [DOI] [PubMed] [Google Scholar]
- 39. Correia A.R., Adinolfi S., Pastore A. and Gomes C.M. (2006) Conformational stability of human frataxin and effect of Friedreich’s ataxia-related mutations on protein folding. Biochem. J., 398, 605–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Correia A.R., Pastore C., Adinolfi S., Pastore A. and Gomes C.M. (2008) Dynamics, stability and iron-binding activity of frataxin clinical mutants. FEBS J., 275, 3680–3690. [DOI] [PubMed] [Google Scholar]
- 41. Gomes L.C., Di Benedetto G. and Scorrano L. (2011) During autophagy mitochondria elongate, are spared from degradation and sustain cell viability. Nat. Cell Biol., 13, 589–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Tondera D., Grandemange S., Jourdain A., Karbowski M., Mattenberger Y., Herzig S., Da Cruz S., Clerc P., Raschke I., Merkwirth C. et al. (2009) SLP-2 is required for stress-induced mitochondrial hyperfusion. EMBO J., 28, 1589–1600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Park S.J., Shin J.H., Jeong J.I., Song J.H., Jo Y.K., Kim E.S., Lee E.H., Hwang J.J., Lee E.K., Chung S.J. et al. (2014) Down-regulation of mortalin exacerbates A β-mediated mitochondrial fragmentation and dysfunction. J. Biol. Chem., 289, 2195–2204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Banerjee S. and Chinthapalli B. (2014) A proteomic screen with Drosophila Opa1-like identifies Hsc70–5/mortalin as a regulator of mitochondrial morphology and cellular homeostasis. Int. J. Biochem. Cell Biol., 54, 36–48. [DOI] [PubMed] [Google Scholar]
- 45. Voloboueva L.A., Duan M., Ouyang Y., Emery J.F., Stoy C. and Giffard R.G. (2008) Overexpression of mitochondrial Hsp70/Hsp75 protects astrocytes against ischemic injury in vitro. J. Cereb. Blood Flow Metab., 28, 1009–1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Xu L., Voloboueva L.A., Ouyang Y., Emery J.F. and Giffard R.G. (2009) Overexpression of mitochondrial Hsp70/Hsp75 in rat brain protects mitochondria, reduces oxidative stress, and protects from focal ischemia. J. Cereb. Blood Flow Metab., 29, 365–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Qu M., Zhou Z., Xu S., Chen C., Yu Z. and Wang D. (2011) Mortalin overexpression attenuates beta-amyloid-induced neurotoxicity in SH-SY5Y cells. Brain Res., 1368, 336–345. [DOI] [PubMed] [Google Scholar]
- 48. Zhou Y., Tozzi F., Chen J., Fan F., Xia L., Wang J., Gao G., Zhang A., Xia X., Brasher H. et al. (2012) Intracellular ATP levels are a pivotal determinant of chemoresistance in colon cancer cells. Cancer Res., 72, 304–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Zhou M., Ottenberg G., Sferrazza G.F., Hubbs C., Fallahi M., Rumbaugh G., Brantley A.F. and Lasmézas C.I. (2015) Neuronal death induced by misfolded prion protein is due to NAD+ depletion and can be relieved in vitro and in vivo by NAD+ replenishment. Brain, 138, 992–1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Jin J., Hulette C., Wang Y., Zhang T., Pan C., Wadhwa R. and Zhang J. (2006) Proteomic identification of a stress protein, mortalin/mthsp70/GRP75: relevance to Parkinson disease. Mol. Cell. Proteomics, 5, 1193–1204. [DOI] [PubMed] [Google Scholar]
- 51. Shi M., Jin J., Wang Y., Beyer R.P., Kitsou E., Albin R.L., Gearing M., Pan C. and Zhang J. (2008) Mortalin: a protein associated with progression of Parkinson disease? J. Neuropathol. Exp. Neurol., 67, 117–124. [DOI] [PubMed] [Google Scholar]
- 52. Selak M.A., Lyver E., Micklow E., Deutsch E.C., Onder O., Selamoglu N., Yager C., Knight S., Carroll M., Daldal F. et al. (2010) Blood cells from Friedreich ataxia patients harbor frataxin deficiency without a loss of mitochondrial function. Mitochondrion, 11, 342–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Lin H., Magrane J., Rattelle A., Stepanova A., Galkin A., Clark E.M., Dong Y.N., Halawani S.M. and Lynch D.R. (2017) Early cerebellar deficits in mitochondrial biogenesis and respiratory chain complexes in the KIKO mouse model of Friedreich ataxia. Dis. Model. Mech., 10, 1343–1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Jasoliya M.J., McMackin M.Z., Henderson C.K., Perlman S.L. and Cortopassi G.A. (2017) Frataxin deficiency impairs mitochondrial biogenesis in cells, mice and humans. Hum. Mol. Genet., 26, 2627–2633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Goswami A.V., Samaddar M., Sinha D., Purushotham J. and D’Silva P. (2012) Enhanced J-protein interaction and compromised protein stability of mtHsp70 variants lead to mitochondrial dysfunction in Parkinson’s disease. Hum. Mol. Genet., 21, 3317–3332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Bolinches-Amorós A., Mollá B., Pla-Martín D., Palau F. and González-Cabo P. (2014) Mitochondrial dysfunction induced by frataxin deficiency is associated with cellular senescence and abnormal calcium metabolism. Front. Cell. Neurosci., 8, 124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Estus S., Tucker H.M., Rooyen C., Wright S., Brigham E.F., Wogulis M. and Rydel R.E. (1997) Aggregated amyloid-beta protein induces cortical neuronal apoptosis and concomitant ‘apoptotic’ pattern of gene induction. J. Neurosci., 17, 7736–7745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Deutsch E.C., Santani A.B., Perlman S.L., Farmer J.M., Stolle C.A., Marusich M.F. and Lynch D.R. (2010) A rapid, noninvasive immunoassay for frataxin: utility in assessment of Friedreich ataxia. Mol. Genet. Metab., 101, 238–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Koopman W.J., Verkaart S., Visch H.J., Westhuizen F.H., Murphy M.P., Heuvel L.W., Smeitink J.A. and Willems P.H. (2005) Inhibition of complex I of the electron transport chain causes O2-. -mediated mitochondrial outgrowth. Am. J. Physiol. Cell Physiol., 288, C1440–C1450. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.