

Abstract
Abl family nonreceptor tyrosine kinases regulate cellular morphogenesis and motility through functional interactions with the actin cytoskeleton. Although Abl family kinases are known to contain filamentous (F)-actin-binding domains at their C termini, it is unclear how Abl family kinases regulate the structure and/or function of the actin cytoskeleton. We show here that the Abl-related kinase Arg binds with positive cooperativity to F-actin in vitro with binding saturating at a ratio of one Arg/two actin molecules. Measurements of the F-actin-binding properties of Arg deletion mutants led to the identification of a second, previously uncharacterized internal F-actin-binding domain in Arg. Purified Arg can bundle F-actin in vitro, and this bundling activity requires both F-actin-binding domains. An Arg-yellow fluorescent protein fusion protein can induce the formation of actin-rich structures at the lamellipodia of Swiss 3T3 fibroblasts. Both of Arg's F-actin-binding domains are necessary and sufficient for the formation of these actin-rich structures. Together, our data suggest that Arg can use its F-actin-bundling activity to directly regulate actin cytoskeletal structure in vivo.
Abl family nonreceptor tyrosine kinases, which include Drosophila (D)-Abl and the mammalian Abl and Abl-related gene (Arg) proteins, are important regulators of cellular morphogenesis in developing metazoan nervous systems. For example, D-Abl is localized to the growth cones of developing neurons, where it is required for normal fasciculation and axon pathfinding (1–4). Activated Abl mutants also promote neurite outgrowth when introduced into cultured mouse cortical neurons (5). In developing mice, the mammalian Abl family kinases Abl and Arg are most abundant in the neural tube, where they colocalize with bundles of apical actin filaments in neuroepithelial cells (6). abl−/−arg−/− mouse embryos suffer from a severe “buckling” of the neural tube that may result from the disorder of filamentous (F)-actin bundles observed in these embryos (6).
Abl family kinases regulate cellular morphology and motility through functional interactions with the actin cytoskeleton. Dosage-sensitive modifiers of D-abl mutant phenotypes include several regulators of cytoskeletal dynamics (3, 4, 7–9). These proteins may act in one or more D-Abl-regulated pathways to exert regulatory control over the actin cytoskeleton during neuronal morphogenesis. Vertebrate Abl family kinases can also direct functional rearrangements of the actin-based cytoskeleton. For example, the activated Bcr-Abl fusion oncoprotein can evoke multiple changes in the actin cytoskeleton, leading to increases in membrane ruffling and filopodial extension (10). In addition, although treatment of mouse fibroblasts with platelet-derived growth factor leads to an F-actin-mediated membrane ruffling, Abl-deficient fibroblasts exhibit a greatly reduced membrane ruffling response in this assay. This deficiency can be rescued by reexpression of Abl (11), demonstrating a direct role for Abl in the control of ruffling.
Despite the identification of multiple Abl family kinase pathway components, the molecular mechanisms by which Abl family kinases regulate cytoskeletal structure remain to be elucidated. Abl contains binding domains for globular (G)- and F-actin at its C terminus (9, 12, 13). Elimination of the C-terminal F-actin-binding domain severely compromises the oncogenic potential of Bcr-Abl (14, 15), suggesting that interactions of Bcr-Abl with F-actin may contribute to substrate selection. F-actin-binding domains likely tether Abl family kinases near important regulators or substrates, which may be associated themselves with the actin cytoskeleton. We examined the F-actin-binding properties of Arg as a first step toward understanding how interactions between this Abl family kinase and F-actin might influence kinase activity, substrate targeting, and F-actin organization within the cell.
We show here that Arg binds cooperatively to F-actin in vitro. Moreover, Arg can assemble F-actin into tight bundles in vitro, an activity that requires both the C-terminal F-actin-binding domain and a previously uncharacterized internal F-actin-binding domain. We also show that both of these F-actin-binding domains are required for Arg to induce the formation of actin-rich structures at the lamellipodia of Swiss 3T3 fibroblasts. Together, our studies suggest that Abl family kinases may regulate cytoskeletal structure by bundling F-actin in vivo.
Materials and Methods
Molecular Cloning and Production of Recombinant Proteins.
Full-length murine Arg and Arg mutant cDNAs were cloned into pFastBac HTa plasmids. All Arg constructs begin with the first common exon (E74 in myristoylated Arg). Amino acids are numbered as for the myristoylated form of murine Arg. Deletion mutants lack the amino acids indicated in parentheses: ArgΔGF (1034–1182), ArgΔC (558–1182), ArgΔInt (688–1034), and ArgΔSH3/2 (1). Arg protein fragments include the indicated amino acids: Arg688–1182, Arg688–1034, and Arg1034–1182. Arg proteins were produced in Hi5 insect cells and purified on nickel-nitrilotriacetic acid (Qiagen, Chatsworth, CA) and UnoS (Bio-Rad) columns. Gel filtration of full-length Arg was performed on a Sephacryl S-200 (Amersham Pharmacia) column. All glutathione S-transferase (GST) fusion proteins were expressed in Escherichia coli and purified as described (16). Full-length or mutant versions of Arg were cloned into the pEYFPN1 plasmid (CLONTECH) for localization experiments.
Cosedimentation and Bundling Assays.
F-actin was purified from chicken skeletal muscle (17) and stabilized by addition of phalloidin (Sigma). For binding assays, increasing amounts of full-length Arg or Arg mutant were incubated with 1 μM F-actin for 15 min at room temperature in a buffer containing 20 mM Pipes, pH 7.0, 10 mM imidazole, pH 7.0, 100 mM KCl, 2 mM MgCl2, and 0.01% Nonidet P-40. The mixtures were centrifuged at 120,000 × g for 30 min at 4°C. Equal amounts of supernatant and pellet were separated on an SDS/PAGE gel. After Coomassie blue staining, protein bands were quantified by densitometry. For Scatchard analysis, P/S was plotted versus P, where P = the concentration of Arg bound to F-actin and S = the concentration of Arg in the supernatant. Bundling assays were performed similarly except Arg/F-actin mixtures were centrifuged at 10,000 × g for 10 min (18).
Electron Microscopy.
Phalloidin-stabilized F-actin (0.5 μM) was incubated with 0.5 μM Arg or Arg mutant for 15 min at room temperature. The mixture was applied to nitrocellulose and carbon-coated copper grids and negatively stained with 1% uranyl acetate. Specimens were viewed on a Zeiss 1°C electron microscope at an accelerating voltage of 80 kV.
Confocal Microscopy.
Arg-yellow fluorescent protein (YFP) or Arg mutant-YFP DNA constructs were transfected into Swiss 3T3 cells using PolyFect reagent (Qiagen). Immunoblotting with anti-YFP antibodies revealed that each of the YFP fusion proteins were of the expected size and expressed at similar levels in transfected cells. After 24 h, cells were fixed with 2% paraformaldehyde and stained with rhodamine phalloidin (Molecular Probes). Coverslips were viewed on a Zeiss confocal microscope at a magnification of ×63.
Results
Arg Binds F-Actin Cooperatively.
We measured the ability of purified recombinant Arg (Fig. 1 A and B) to bind F-actin by using a cosedimentation assay. Increasing amounts of Arg (0.125–1 μM) were mixed with a fixed amount of F-actin (1 μM). After high-speed centrifugation at 120,000 × g, the amount of Arg cosedimenting with F-actin in the pellet fraction was measured. Arg could be recovered in the pellet fraction, demonstrating an association of Arg with F-actin (Fig. 1B). However, when incubated alone, Arg remained in the supernatant after centrifugation (Fig. 1B). Scatchard analysis revealed that Arg binding to F-actin saturated at 0.5 μM of bound Arg when the F-actin concentration was kept constant at 1 μM (Fig. 1G), a binding ratio of one Arg/two actin molecules. The convex shape of the Scatchard plot indicated that Arg binds to F-actin with positive cooperativity (Fig. 1G; ref. 19). A similar convex Scatchard plot has been observed for the actin-bundling protein adducin (20). Hill plot analysis confirmed that Arg binding to F-actin was cooperative, exhibiting a Hill coefficient of ≈1.9 (data not shown).
Figure 1.

Arg binds F-actin. (A) Diagram of purified recombinant Arg proteins. (B–F) Cosedimentation of Arg or Arg mutants with F-actin. A fixed concentration of F-actin (1 μM) was mixed with increasing concentrations of Arg from 0.125 to 1 μM (lanes 1–8). For each panel, Arg or the Arg mutants (1 μM) also were centrifuged in the absence of F-actin as a control (lanes 9–10). After ultracentrifugation at 120,000 × g for 30 min, equivalent amounts of pellet (P) and supernatant (S) fractions were subjected to SDS/PAGE followed by Coomassie blue staining. (B) Arg; (C) ArgΔGF; (D) ArgΔInt; (E) ArgΔC; (F) Arg688–1182. A plot of concentration (x axis) vs. amount bound (y axis) for each protein is shown to the right of the binding data. (G) Scatchard analysis of Arg, ArgΔGF, ArgΔInt, and Arg688–1182 binding to F-actin.
To better define which Arg residues were required for F-actin binding, we measured the F-actin-binding activity of Arg deletion mutants (Fig. 1A). Van Etten et al. (13) have identified distinct G- and F-actin-binding domains at the C-terminal end of Abl. These G- and F-actin-binding domains are 26% identical (32% similar) and 57% identical (67% similar) in Arg, respectively (Fig. 1A). The ArgΔGF mutant lacking these domains retained F-actin-binding activity, although in comparison to full-length Arg, less ArgΔGF bound to F-actin at each concentration tested (Fig. 1 B and C). Removal of the Src homology 3 and 2 domains of Arg had no effect on the F-actin binding properties of Arg (data not shown), but an Arg mutant lacking all residues C-terminal to the tyrosine kinase domain (ArgΔC) failed to bind F-actin (Fig. 1E). Together, these observations suggested that Arg contained a second F-actin-binding domain located between the tyrosine kinase domain and the putative G-actin-binding domain. Similar to ArgΔGF, the internal deletion mutant ArgΔInt bound comparatively less well to F-actin than full-length Arg (Fig. 1 B and D). These observations suggested that the residues missing from ArgΔInt (amino acids 688-1034) contributed to the second Arg F-actin-binding domain.
Although the cooperativity of Arg binding to F-actin precluded an accurate determination of the dissociation constant of Arg for F-actin (19), the apparent Kd was ≈0.3 μM. Scatchard analysis revealed ArgΔGF and ArgΔInt did not bind cooperatively to F-actin, allowing us to determine that ArgΔGF bound to F-actin with a Kd of 4.1 μM, whereas ArgΔInt bound with a Kd of 3.1 μM (Fig. 1G). These observations suggest that the F-actin-binding domains retained in each mutant bound F-actin with similar affinity.
We also measured the F-actin-binding properties of Arg688–1182, a C-terminal Arg fragment containing both putative F-actin-binding domains (Fig. 1A and F). A Scatchard plot indicated that Arg688–1182 did not exhibit cooperativity in its binding to F-actin (Fig. 1G). However, Arg688–1182 bound F-actin with a Kd of 0.7 μM, ≈5-fold higher affinity for F-actin than either the ArgΔGF or ArgΔInt deletion mutants. These data illustrated that an Arg fragment containing both the putative F-actin-binding domains bound F-actin more tightly than Arg mutants that retained only one of the F-actin-binding domains. We also find that Arg688–1182 failed to bind to microtubules in a cosedimentation assay (data not shown), suggesting that Arg688–1182 does not associate with polymers via a nonspecific electrostatic effect.
Arg Bundles F-Actin.
Proteins that contain two F-actin-binding domains sometimes can bundle F-actin. Because the evidence presented above suggested that Arg might contain two distinct F-actin-binding domains, we investigated whether Arg could bundle F-actin. We used a low-speed centrifugation assay to measure F-actin bundling by Arg (18). F-actin remains in the supernatant after low-speed centrifugation at 10,000 × g (Fig. 2A). However, when included in the incubations, Arg assembled F-actin into high molecular mass aggregates that were recovered in the pellet fraction (Fig. 2A). We confirmed that these aggregates were F-actin bundles using electron microscopy (Fig. 2 E and F). When incubated under conditions identical to those used in the bundling experiments, Arg eluted from a Sephacryl S-200 gel filtration column at a position corresponding to a monomer of 130 kDa (Fig. 5, which is published as supporting information on the PNAS web site, www.pnas.org). This observation suggested that Arg might bundle F-actin by binding two distinct actin filaments via each of its F-actin-binding domains. In support of this theory, both ArgΔGF and ArgΔInt failed to bundle F-actin (Fig. 2 B, C, and G), indicating that both of the putative actin-binding domains are required for F-actin-bundling activity.
Figure 2.

Arg bundles F-actin. (A–D) F-actin-bundling activity of Arg and Arg mutants. F-actin (1 μM) was mixed with 0.125–1 μM Arg or Arg mutant proteins (lanes 3–10), and the mixture was centrifuged at 10,000 × g for 10 min to pellet F-actin bundles. Equivalent amounts of the pellet (P) and supernatant (S) fractions were fractionated by SDS/PAGE followed by Coomassie blue staining. As a control in each experiment, F-actin was centrifuged without Arg or Arg mutant (lanes 1 and 2 in each panel). (A) Arg; (B) ArgΔGF; (C) ArgΔInt; (D) Arg688–1182. (E–I) Electron microscopy of F-actin in the presence of Arg (E, F), ArgΔGF (G), and Arg688–1182 (H). F-actin alone is shown also as a control (I). Mixtures of 0.5 μM phalloidin-stabilized F-actin in the absence or presence of 0.5 μM Arg or Arg mutants were incubated for 15 minutes at room temperature before negative staining with 1% uranyl acetate. Bar in E, 5 μm; bar in F, 0.1 μm. G–I were photographed at a magnification similar to F.
Arg688–1182 assembled F-actin into bundles that were recovered in the low-speed pellet fraction (Fig. 2D). These data indicate that Arg's two F-actin-binding domains are sufficient for F-actin-bundling activity. We consistently noted that less Arg688–1182 was brought down in the F-actin bundles than full-length Arg, which we attributed to Arg688–1182's lower affinity for F-actin than full-length Arg. However, Arg688–1182 assembled F-actin into bundles that were indistinguishable from those assembled with full-length Arg (compare Fig. 2F with 2H). We conclude that Arg688–1182 can bundle F-actin effectively even when bound to F-actin at lower amounts than those achieved by full-length Arg.
Two Distinct Classes of F-Actin-Binding Domains in Arg.
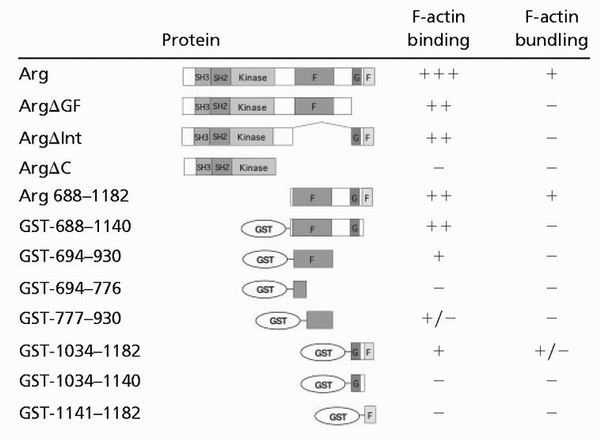
Although ArgΔGF and ArgΔInt could each bind F-actin, both mutants failed to bundle actin filaments (Fig. 2 B, C, and G; summarized in Table 1). These data suggested that the residues missing in ArgΔGF (amino acids 1034–1182) and ArgΔInt (amino acids 688-1034) harbored distinct F-actin-binding domains.
Table 1.
Summary of binding and bundling data
 |
We aligned the amino acid sequences of the two putative Arg F-actin-binding domains to sequences of known actin-binding domains. The internal F-actin-binding domain contains sequences (amino acids 695–930) that align with members of the I/LWEQ class of F-actin-binding domains (named for the conserved initial residue in each of four blocks of homology; ref. 21). The Arg I/LWEQ domain is most similar to the I/LWEQ domains of the talin proteins from mouse and Caenorhabditis elegans (Fig. 6A, which is published as supporting information on the PNAS web site). The Arg I/LWEQ domain is identical to one or both talin I/LWEQ domains at 28% (52/188) of the positions examined. Although the Arg I/LWEQ domain contains amino acid insertions into blocks two and four of the canonical I/LWEQ domain sequences, the identity between the Arg and talin I/LWEQ domains extends to areas between the I/LWEQ homology blocks.
We determined that the C-terminal G- and F-actin-binding domains of Arg aligned with members of the CH class of F-actin-binding domains (22). The Arg CH domain is most similar to the human β-spectrin CH domain and the N-terminal CH domain from human fimbrin (Fig. 6B). The Arg CH domain is identical to one or both of these CH domains at 27% (26/98) of the positions examined. The Arg CH domain contains a large amino acid insertion in the middle separating it into N- and C-terminal portions of homology with the fimbrin and β-spectrin CH domains. In conjunction with peptide-binding and genetic studies, atomic modeling of fimbrin binding to F-actin strongly suggests that the F-actin-binding residues in the CH domain are concentrated in its C-terminal portion (23). The Arg CH domain is 38% (16/42) similar to the fimbrin CH domain in this putative actin-interaction region. Single CH domains may function differently than actin-binding domains, which contain two CH domains in tandem (22). Because both fimbrin and spectrin have tandem CH domains, whereas Arg contains a single CH domain, we also compared Arg to another single CH domain-containing protein, mouse H2-calponin (ref. 24; Fig. 6C). Overall, the Arg CH domain is identical to the H2-calponin CH domain at 20% (29/148) of the positions examined.
We observed little F-actin binding by Arg fragments identified as distinct putative F-actin-binding domains (amino acids 688–1034 and amino acids 1034–1182). We examined whether dimerization of the domains by fusion to GST might allow for detectable interactions with F-actin (Fig. 3A; refs. 24 and 25). We detected no binding of GST alone to F-actin, as has been reported previously (ref. 13; Fig. 3B). The GST-694–930 and GST-1034–1182 fusion proteins each bound to F-actin as revealed by high-speed cosedimentation experiments (Fig. 3 C and D). Gel filtration analysis indicate that these GST-694–930 and GST-1034–1182 fusion proteins formed dimers of approximately twice their expected monomeric size (data not shown). We conclude that F-actin binding by the individual F-actin-binding domains is detectable only when these domains are dimerized by fusion to GST. When fused to GST, smaller pieces of the F-actin-binding domains (amino acids 694–776, 1034–1140, and 1141–1182) all failed to bind F-actin, and GST-777–930 bound F-actin weakly (Table 1).
Figure 3.

Arg contains two independent F-actin binding domains. (A) Diagram of recombinant purified GST-Arg protein fragments. (B–D) Cosedimentation of GST or GST-Arg fragments with F-actin. A fixed concentration of F-actin (1 μM) was mixed with 2.5–10 μM GST (lanes 1–6) or 1.25–10 μM GST-Arg fragment fusion protein (lanes 1–8). For C and D, GST-fusion proteins (10 μM) were centrifuged also in the absence of F-actin as a control (lanes 9–10). After ultracentrifugation at 120,000 × g for 30 min, all the pellet fractions (P) and one third of the supernatant (S) fractions were subjected to SDS/PAGE followed by Coomassie blue staining. (B) GST; (C) GST-694–930; (D) GST-1034–1182. (E) Competitive binding of F-actin-binding domains to F-actin. For each reaction, F-actin (1 μM) was mixed with GST-694–930 (10 μM) and 0–10 μM GST-1034–1182. Samples were incubated, centrifuged, and analyzed as described for C and D.
We also determined whether the distinct F-actin-binding domains could compete for binding sites on F-actin using a high-speed cosedimentation assay. Increasing the concentration of GST-1034–1182 in incubations containing a constant amount (10 μM) of GST-694–930 resulted in decreased amounts of GST-694–930 bound to F-actin (Fig. 3E). By gel filtration, we found no evidence that GST-694–930 or GST-1034–1182 dimers could exchange subunits when incubated together in the absence of F-actin. Our observations suggest that the two Arg F-actin-binding domains compete for binding to F-actin.
Arg-YFP Fusion Directs the Formation of F-Actin-Rich Structures in Fibroblasts.
We expressed full-length Arg and Arg fragments fused to YFP in Swiss 3T3 fibroblasts to examine whether Arg's F-actin-bundling function could influence cytoskeletal structure in vivo. Arg-YFP was associated with F-actin-rich structures that appeared in the lamellipodia of transfected cells (Fig. 4A, a–l). These F-actin-rich structures were not observed in control YFP-transfected cells where the YFP signal remained diffusely distributed (Fig. 4A, v–x). Formation of the F-actin-rich structures by Arg-YFP required the presence of both F-actin-binding domains, because ArgΔC-YFP, ArgΔInt-YFP, and ArgΔGF-YFP each failed to induce actin-rich structures or colocalize with F-actin (Fig. 4A, m–u). However, Arg688–1182-YFP could direct the formation of the F-actin-rich structures (Fig. 4B, a–f). Neither of Arg's F-actin-binding domains alone (Arg688–1034-YFP or Arg1034–1182-YFP) colocalized with F-actin or directed the formation of the actin-rich structures (Fig. 4B, g–l). We conclude that both of Arg's F-actin-binding domains are necessary and sufficient for the formation of the F-actin-rich structures in Swiss 3T3 cells.
Figure 4.

Arg directs the formation of actin-rich structures at the lamellipodia of Swiss 3T3 cells. Swiss 3T3 cells were transfected with Arg, Arg deletion, or Arg fragment-YFP fusions, plated on coverslips, and fixed 24 h posttransfection. F-actin was visualized with rhodamine phalloidin. (A) Colocalization of Arg-YFP and Arg deletion-YFP fusions with F-actin. (a–l) Arg-YFP; (m–o) ArgΔC-YFP; (p and r) ArgΔGF-YFP; (s–u) ArgΔInt-YFP; (v–x) YFP alone. (d–f) Enlargements of the boxed areas indicated in a–c. (j–l) Enlargements of the boxed areas indicated in g–i. (B) Colocalization of Arg fragment-YFP fusions with F-actin. (a–f) Arg688–1182-YFP; (g–i) Arg688-1034-YFP; (j–l) Arg1034–1182-YFP. (d–f) Enlargements of the boxed areas indicated in a–c.
Discussion
In this paper, we identified the presence of two distinct classes of F-actin-binding domains in Arg and demonstrated that both F-actin-binding domains are required for Arg to bundle F-actin in vitro and direct the formation of actin-rich structures in vivo.
Our results suggest that by bundling F-actin, Abl family kinases can directly regulate cytoskeletal structure in vivo. We showed previously that both Abl and Arg colocalize with the contractile F-actin latticework at the apical surface of neuroepithelial cells in the developing mouse neural tube (6). A loss of this F-actin-bundling activity may lead to the disordered F-actin observed in abl−/−arg−/− neuroepithelium (6). We show here that Arg and Arg fragments that retain F-actin-bundling activity promote the formation of actin-rich structures in Swiss 3T3 cells, strongly suggesting that Arg's bundling activity is necessary and sufficient to direct rearrangements of the actin cytoskeleton in cells. However, we cannot rule out the possibility that Abl family kinases influence actin cytoskeletal structure via additional mechanisms in vivo.
Arg represents ≈0.004% of protein in whole brain extract (6). Considering that the in vivo concentration of proteins probably is ≈70–100 mg/ml, we estimate the concentration of Arg (130 kDa) to be ≈20–30 nM if Arg were distributed diffusely throughout cells. However, our recent localization studies reveal that within neurons, Arg is localized to dendritic spines (A. J. Scheetz and A.J.K., unpublished data). Because dendritic spines represent 5% or less of total brain volume, we estimate the concentration of Arg in spines to be 400–600 nM, which is within the proper range for Arg to play a direct role in organizing F-actin in spines. We observe a similar concentration of the Arg-YFP and Arg688–1182-YFP fusion proteins with F-actin-rich structures at the lamellipodia of Swiss 3T3 fibroblasts. In addition to binding and bundling F-actin, residues 688-1182 may help to tether Arg to cell surface or cytoskeletal proteins that help to concentrate Arg within the cell.
Our studies have identified and characterized two distinct classes of F-actin-binding domains in Arg. Although the Arg C-terminal CH-like F-actin-binding domain had been suggested by sequence comparison to Abl, it had not been shown previously to bind F-actin. In fimbrin, each actin-binding domain is comprised of two tandem CH domains that seem to play nonequivalent roles in F-actin binding (23). Single CH domains bind only weakly to F-actin unless assisted by a second F-actin-binding domain (22). We indeed have identified a second F-actin-binding domain in Arg of the I/LWEQ class.
The ability of the isolated Arg I/LWEQ and CH domains to compete for F-actin binding strongly suggests that these domains have overlapping binding sites on the actin filament. This observation would explain why Arg binding to F-actin saturates at a molar ratio of one molecule of Arg/two molecules of F-actin. One actin unit would be required to accommodate each of the two Arg F-actin-binding domains when binding is saturated. An atomic modeling study indicates that the fimbrin CH domain docks into a cleft between subdomain 2 of one actin subunit (at position n along the filament) and subdomain 1 of the actin monomer above it (at position n + 2 along the filament; ref. 25). Because the Arg I/LWEQ domain competes with the Arg CH domain for F-actin binding, we predict that the I/LWEQ binding site on F-actin may include all or a portion of the CH-binding cleft.
Although Arg binding to F-actin saturates at a ratio of one Arg/two actin molecules, Arg688–1182 seems to bind to F-actin at a much lower density of approximately one Arg688–1182/five actin molecules. While it is possible that this fragment of Arg has low specific activity for F-actin binding, a plot of Arg688–1182 binding to F-actin (Fig. 1F) reveals that binding appeared to be reaching saturation in our experiments. One possible explanation is that the removal of N-terminal residues from Arg creates steric changes in the C-terminal half that prevent Arg688–1182 from binding F-actin at a maximal density.
Our finding that Arg binds cooperatively to F-actin suggests that either Arg molecules self-associate after initial contact with an actin filament or Arg binding to F-actin induces a conformational change in adjacent actin subunits that facilitates binding of the next Arg. In addition to its binding with lower affinity to F-actin than full-length Arg, Arg688–1182 fails to bind cooperatively to F-actin. Arg688–1182 may fail to bind cooperatively because it lacks sequences in the N-terminal two-thirds of Arg, which are required for Arg to self-associate or induce a conformational change in F-actin. Curiously, Arg and Arg688–1182 appear to assemble F-actin into bundles with similar spacing between filaments. These data suggest that the N-terminal half of Arg is accommodated readily in the space between bundled filaments and does not limit how tightly filaments can pack into these bundles.
F-actin has been reported recently to inhibit Abl kinase activity (26). The interactions between Arg and F-actin might similarly influence Arg kinase activity or substrate selection. It is possible that the two F-actin-binding domains might act as “sensors” of the local actin cytoskeleton conformation. Changes in cytoskeletal conformation could regulate Abl family kinases in one of two ways. One or both of the F-actin-binding domains of Arg might regulate the Arg kinase domain in a manner that depends on F-actin binding. Alternatively, rearrangement of actin microfilaments might bring actin-bound substrates in close proximity to the actin-bound kinases.
Supplementary Material
Acknowledgments
We are grateful to L. Crawford, A. J. Scheetz, K. Tanis, J. Wolf, and X. Ye for technical advice and assistance and L. Cooley, M. Koelle, S. Strobel, members of the Koleske lab, and the three anonymous reviewers for helpful advice and comments on the manuscript. This work was supported by U.S. Public Health Service Research Grants DK25387 and DK55389 (to M.S.M.) and NS39475 (to A.J.K.).
Abbreviations
- D
Drosophila
- Arg
Abl-related gene
- GST
glutathione S-transferase
- YFP
yellow fluorescent protein
- CH
calponin homology
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
See commentary on page 14745.
References
- 1.Gertler F B, Bennett R L, Clark M J, Hoffmann F M. Cell. 1989;58:103–113. doi: 10.1016/0092-8674(89)90407-8. [DOI] [PubMed] [Google Scholar]
- 2.Giniger E. Neuron. 1998;20:667–681. doi: 10.1016/s0896-6273(00)81007-7. [DOI] [PubMed] [Google Scholar]
- 3.Wills Z, Bateman J, Korey C A, Comer A, Van Vactor D. Neuron. 1999;22:301–312. doi: 10.1016/s0896-6273(00)81091-0. [DOI] [PubMed] [Google Scholar]
- 4.Wills Z, Marr L, Zinn K, Goodman C S, Van Vactor D. Neuron. 1999;22:291–299. doi: 10.1016/s0896-6273(00)81090-9. [DOI] [PubMed] [Google Scholar]
- 5.Zukerberg L R, Patrick G N, Nikolic M, Humbert S, Wu C L, Lanier L M, Gertler F B, Vidal M, Van Etten R A, Tsai L H. Neuron. 2000;26:633–646. doi: 10.1016/s0896-6273(00)81200-3. [DOI] [PubMed] [Google Scholar]
- 6.Koleske A J, Gifford A M, Scott M L, Nee M, Bronson R T, Miczek K A, Baltimore D. Neuron. 1998;21:1259–1272. doi: 10.1016/s0896-6273(00)80646-7. [DOI] [PubMed] [Google Scholar]
- 7.Gertler F B, Comer A R, Juang J L, Ahern S M, Clark M J, Liebl E C, Hoffmann F M. Genes Dev. 1995;9:521–533. doi: 10.1101/gad.9.5.521. [DOI] [PubMed] [Google Scholar]
- 8.Liebl E C, Forsthoefel D J, Franco L S, Sample S H, Hess J E, Cowger J A, Chandler M P, Shupert A M, Seeger M A. Neuron. 2000;26:107–118. doi: 10.1016/s0896-6273(00)81142-3. [DOI] [PubMed] [Google Scholar]
- 9.Lanier L M, Gertler F B. Curr Opin Neurobiol. 2000;10:80–87. doi: 10.1016/s0959-4388(99)00058-6. [DOI] [PubMed] [Google Scholar]
- 10.Salgia R, Li J L, Ewaniuk D S, Pear W, Pisick E, Burky S A, Ernst T, Sattler M, Chen L B, Griffin J D. J Clin Invest. 1997;100:46–57. doi: 10.1172/JCI119520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Plattner R, Kadlec L, DeMali K A, Kazlauskas A, Pendergast A M. Genes Dev. 1999;13:2400–2411. doi: 10.1101/gad.13.18.2400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McWhirter J R, Wang J Y. EMBO J. 1993;12:1533–1546. doi: 10.1002/j.1460-2075.1993.tb05797.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Van Etten R A, Jackson P K, Baltimore D, Sanders M C, Matsudaira P T, Janmey P A. J Cell Biol. 1994;124:325–340. doi: 10.1083/jcb.124.3.325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Renshaw M W, McWhirter J R, Wang J Y. Mol Cell Biol. 1995;15:1286–1293. doi: 10.1128/mcb.15.3.1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Heisterkamp N, Voncken J W, Senadheera D, Gonzalez-Gomez I, Reichert A, Haataja L, Reinikainen A, Pattengale P K, Groffen J. Blood. 2000;96:2226–2232. [PubMed] [Google Scholar]
- 16.Smith D B, Johnson K S. Gene. 1988;67:31–40. doi: 10.1016/0378-1119(88)90005-4. [DOI] [PubMed] [Google Scholar]
- 17.Spudich J A, Watt S. J Biol Chem. 1971;246:4866–4871. [PubMed] [Google Scholar]
- 18.Meyer R K, Aebi U. J Cell Biol. 1990;110:2013–2024. doi: 10.1083/jcb.110.6.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cornish-Bowden A. Fundamentals of Enzyme Kinetics. London: Portland Press; 1995. pp. 216–219. [Google Scholar]
- 20.Mische S M, Mooseker M S, Morrow J S. J Cell Biol. 1987;105:2837–2845. doi: 10.1083/jcb.105.6.2837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McCann R O, Craig S W. Biochem Biophys Res Commun USA. 1999;266:135–140. doi: 10.1006/bbrc.1999.1776. [DOI] [PubMed] [Google Scholar]
- 22.Stradal T, Kranewitter W, Winder S J, Gimona M. FEBS Lett. 1998;431:134–137. doi: 10.1016/s0014-5793(98)00751-0. [DOI] [PubMed] [Google Scholar]
- 23.Hanein D, Volkmann N, Goldsmith S, Michon A M, Lehman W, Craig R, DeRosier D, Almo S, Matsudaira P. Nat Struct Biol. 1998;5:787–792. doi: 10.1038/1828. [DOI] [PubMed] [Google Scholar]
- 24.McTigue M A, Bernstein S L, Williams D R, Tainer J A. Proteins. 1995;22:55–57. doi: 10.1002/prot.340220108. [DOI] [PubMed] [Google Scholar]
- 25.Walker J, Crowley P, Moreman A D, Barrett J. Mol Biochem Parasitol. 1993;61:255–264. doi: 10.1016/0166-6851(93)90071-5. [DOI] [PubMed] [Google Scholar]
- 26.Woodring P J, Hunter T, Wang J Y. J Biol Chem. 2001;276:27104–27110. doi: 10.1074/jbc.M100559200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}