

Summary
Background
Previous study demonstrated that overstimulation of TRPM7 substantially contributes to zinc‐mediated neuronal toxicity. Inhibition of TRPM7 activity and TRPM7‐mediated intracellular Zn2+ accumulation may represent a promising strategy in the treatment of stroke.
Aims
To investigate whether local anesthetics lidocaine could inhibit TRPM7 channel and TRPM7‐mediated zinc toxicity.
Methods
Whole‐cell patch‐clamp technique was used to investigate the effect of local anesthetics on TRPM7 currents in cultured mouse cortical neurons and TRPM7‐overexpressed HEK293 cells. Fluorescent Zn2+ imaging technique was used to study the effect of lidocaine on TRPM7‐mediated intracellular Zn2+ accumulation. TRPM7‐mediated zinc toxicity in neurons was used to evaluate the neuroprotective effect of lidocaine.
Results
(1) Lidocaine dose dependently inhibits TRPM7‐like currents, with an IC50 of 11.55 and 11.06 mM in cultured mouse cortical neurons and TRPM7‐overexpressed HEK293 cells, respectively; (2) Lidocaine inhibits TRPM7 currents in a use/frequency‐dependent manner; (3) Lidocaine inhibits TRPM7‐mediated intracellular Zn2+ accumulation in both cortical neurons and TRPM7‐overexpressed HEK293 cells; (4) TRPM7‐mediated Zn2+ toxicity is ameliorated by lidocaine in cortical neurons; (5) QX‐314 has a similar inhibitory effect as lidocaine on TRPM7 currents when applied extracellularly; (6) Procaine also shows potent inhibitory effect on the TRPM7 currents in cortical neurons.
Conclusion
Our data provide the first evidence that local anesthetic lidocaine inhibits TRPM7 channel and TRPM7‐mediated zinc toxicity.
Keywords: Anesthetics, Lidocaine, Stroke, TRPM7, Zinc
Introduction
The transient receptor potential (TRP) channels, cation‐permeable channels, form a large superfamily of versatile channels that are widely expressed in mammalian tissues 1. There are seven subfamilies, including TRPC, TRPV, TRPM, TRPA, TRPN, TRPP, and TRPML, functioning as either homo‐ or heteromultimers composed of four TRP subunits 1. TRPM7 is one of the eight members of the transient receptor potential melastatin (TRPM) subfamily of ion channels and is ubiquitously expressed throughout mammalian tissues. TRPM7 has been demonstrated to be implicated in several important cellular and biological processes such as cellular Mg2+ homeostasis 1, 2, neurotransmitter release 3, and in some pathological conditions including cancer cell growth/proliferation, hyperglycemia‐induced endothelial cell injury and cerebral ischemia 4, 5, 6, 7. The role of TRPM7 in stroke has been well demonstrated. When primary cultured cortical neurons were subjected to oxygen–glucose deprivation (OGD) for a prolonged period, there was an increase in reactive oxygen species (ROS) production, which mediated TRPM7 activation and subsequent cell deaths 6. Knockdown of the expression of TRPM7 by TRPM7‐siRNA largely diminished neuronal death under anoxia conditions 6.
Growing evidence shows that, in addition to Ca2+, the ubiquitous metal ion Zn2+ contributes substantially to the neuronal injury during stroke 8, 9, 10. The accumulation of Zn2+ in neurons following cerebral ischemia is now well recognized, and a striking correlation between zinc accumulation and cell viability was revealed 11, 12, 13. For example, neuronal toxicity could be largely reduced by Zn2+ chelation 8, 9. Considering the high permeability of TRPM7 to Zn2+, TRPM7 channels might mediate Zn2+ toxicity in stroke. Recently, we demonstrated that Zn2+‐induced neuronal toxicity was largely reduced by TRPM7 channel inhibition or TRPM7 knockdown 14. In this regard, the agents that inhibit TRPM7 activation would provide neuroprotection in stroke.
In the current study, we, for the first time, demonstrate that local anesthetic lidocaine inhibits TRPM7 currents, TRPM7‐mediated zinc accumulation and neuronal death. This study not only enhances our understanding of local anesthetics but also provides clues for the design and development of TRPM7 inhibitors for stroke intervention.
Materials and methods
Cell Culture
Mouse cortical neurons were cultured as described previously and modified 14. The use of mice for neuronal cultures was approved by the Institutional Animal Care and Use Committee of Morehouse School of Medicine. Briefly, pregnant Swiss mice were anesthetized with halothane followed by cervical dislocation. Brains of fetuses (embryonic day 16) were removed rapidly and placed in cold Ca2+/Mg2+‐free phosphate‐buffered saline. Cerebral cortices were dissected under a dissection microscope and incubated with 0.05% trypsin‐EDTA for 10 min at 37°C, followed by trituration with fire‐polished glass pipettes. Cells were counted and plated in poly‐L‐ornithine‐coated culture dishes at a density of 1 × 106 cells/dish or 24‐well plates at a density of 2 × 105 cells/well, respectively. Cells were initially cultured in minimal essential medium (MEM) with 10% fetal bovine serum (FBS), 10% horse serum and 25 mM glucose, and maintained at 37°C in a humidified 5% CO2 atmosphere incubator for 24 h. After 24 h, culture medium was replaced by Neurobasal medium supplemented with B‐27 (Invitrogen, Carlsbad, CA, USA). Cultures were fed twice a week with half change of the culture medium. Neurons were used for the experiments between days 10 and 14 in vitro. Human embryonic kidney (HEK293) cells, with inducible expression of human TRPM7 channels, were cultured in MEM supplemented with 10% FBS and antibiotics. For the induction of TRPM7, the cells were treated with 1 μg/mL of tetracycline, as described in our previous studies 14.
Cytotoxicity Assay
Cytotoxicity was measured by lactate dehydrogenase (LDH) assay, which was performed as described previously 14. At the end of the experiments, 50 μL medium was taken from each well and placed into 96‐well plate for LDH release measurement. Cells were then incubated with Triton X‐100 (final concentration 0.5%) for 30 min at 37°C. Fifty microlitre supernatant was withdrawn from each well for maximal LDH measurement. Fifty microlitre assay reagent from cytotoxicity detection kit (Roche Diagnostics) was added to each sample and mixed. After 30 min, the absorbance at 492 and 620 nm was examined by spectrometer (SpectraMax Plus; Molecular devices, Sunnyvale, CA, USA), and the values of the absorbance at 492 nm were subtracted by those at 620 nm to yield the values of LDH release.
Fluorescein Diacetate Staining and Propidium Iodide Uptake
Cortical neurons were incubated in standard extracellular solution with addition of 5 μM fluorescein diacetate (FDA) and 2 μM propidium iodide (PI) for 15 min followed by wash with standard extracellular solution. Alive (FDA‐positive) and dead (PI‐positive) cells were viewed on a microscope (Zeiss, Thornwood, NY, USA) equipped with epifluorescence at 580/630 nm excitation/emission for PI and 500/550 nm for FDA.
Electrophysiology
Patch‐clamp recordings were performed as described previously 7. Patch electrodes were constructed from thin‐walled borosilicate glass and had resistances of 3‐5 MΩ. Currents were recorded using Axopatch 200B amplifier with pCLAMP software (Axon Instruments, Union City, CA, USA) and were filtered at 2 kHz and digitized at 5 kHz using Digidata 1322A. Data were eliminated from statistical analysis when access resistance was >10 MΩ or leak current was >100 pA at −60 mV. A multibarrel perfusion system was used to achieve a rapid exchange of external solutions. Standard extracellular solution contained (in mM) 140 NaCl, 5.4 KCl, 2 CaCl2, 1 MgCl2, 20 HEPES, and 10 glucose (pH 7.4 adjusted with NaOH; 320–330 mOsm). For activation of TRPM7, extracellular CaCl2 and MgCl2 were removed. Patch electrodes contained (in mM) 140 CsF, 10 HEPES, 1 CaCl2, 11 EGTA, and 2 TEA (pH 7.3 adjusted with CsOH, 290–300 mOsm). All experiments were performed at room temperature.
Zinc Imaging
Intracellular zinc level in mouse cortical neurons or HEK‐293 cells were imaged using a zinc‐sensitive fluorescent dye, FluoZin‐3 (Invitrogen). As described previously 14, cells were incubated with 5 μM FluoZin‐3‐AM in standard extracellular fluid (ECF) for 30 min at 37°C, followed by de‐esterification of the dye for another 30 min at room temperature (22–25°C). MK‐801 (10 μM; Sigma, St. Louis, MO, USA), CNQX (10 μM; Sigma), and nimodipine (5 μM; Sigma) were included in the extracellular solutions to block potential zinc entry through NMDA and AMPA/kainate receptors, and voltage‐gated calcium channels. The coverslips containing dye‐loaded cells were held in a recording chamber placed on the stage of an inverted microscope (EclipseTE2000‐U; Nikon, Melville, NY, USA), and perfused with standard ECF at room temperature for at least 10 min prior to experiments. FluoZin‐3 was excited at a wavelength of 490 nm, and emitted light was filtered with a 500–550‐nm band pass filter. Zinc fluorescence was detected with ×40 objective lens (Super Fluor ×40, numerical aperture = 0.90; Nikon) and a CCD camera (CoolSNAP ES2; Photometrics, Tucson, AZ, USA), using Imaging Workbench software (INDEC BioSystems, Santa Clara, CA, USA). Fluorescence intensity (ΔF) was normalized to the values of basal fluorescence intensity (F) before the change of solutions.
Statistics
All data were expressed as mean ± SE. GraphPad Prism 4 (San Diego, CA, USA) and Sigma Plot (San Jose, CA, USA) were used for statistical analysis. Student's t test or ANOVA followed by Bonferroni posttests were used to examine the statistical significance where appropriate. The criterion for significance was set at P < 0.05. The dose–response curve was fitted with three parameters logistic nonlinear regression model.
Results
Lidocaine Inhibits TRPM7‐Like Currents in Primary Cultured Mouse Cortical Neurons
As described previously 4, 6, 15, decreasing the extracellular Ca2+/Mg2+‐induced TRPM7 currents in cultured cortical neurons (Figure 1A). After recording of at least three stable current traces, we started to examine the effect of lidocaine, Gd3+ and 2‐APB on TRPM7 currents. Gd3+ and 2‐APB are nonspecific TRPM7 channel blockers. As expected, TRPM7‐like currents were significantly inhibited by 30 μM Gd3+ (76% inhibition, n = 4) and 100 μM 2‐APB (42% inhibition, n = 5) (Figure 1A–D). We further examined the effect of lidocaine on TRPM7‐like currents. As shown in Figure 1E,F, lidocaine inhibited TRPM7 currents in a dose‐dependent manner. The dose–response analysis yielded a half‐maximal inhibitory concentration (IC50) of 11.55 ± 0.95 mM (Figure 1F, n = 4). These electrophysiological studies provide evidence, for the first time, supporting the inhibition of TRPM7 current by lidocaine.
Figure 1.
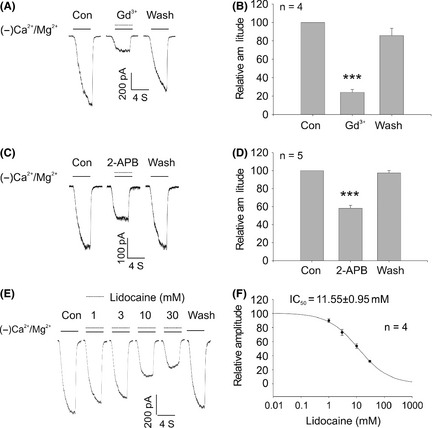
Inhibition of the TRPM7 current by lidocaine in primary cultured cortical neurons. (A and B) Representative current traces and summary data showing that 30 μM Gd3+ inhibits TRPM7 current in cortical neurons (***P < 0.001). (C and D) Representative current traces and summary data showing that 100 μM 2‐APB inhibits TRPM7 current in cortical neurons (***P < 0.001). (E) Representative current traces showing that lidocaine inhibits TRPM7 current in a concentration‐dependent manner. TRPM7 current was induced by deprivation of the extracellular Ca2+/Mg2+. (F) Dose–response curves were inferred from A. For each concentration, the 5th traces in the presence of lidocaine were used for dose–response analysis. The IC50 was 11.55 ± 0.95 mM (n = 4). Data were expressed as mean ± SE. MK‐801 (10 μM) and TTX (0.3 μM) were included in the extracellular solutions to block potential activation of NMDA and voltage‐gated Na+ currents.
Lidocaine Dose Dependently and Voltage Independently Inhibits TRPM7 Currents in HEK‐293 Cells Overexpressing TRPM7 Channels
We further examined the effect of lidocaine on heterogeneously expressed TRPM7 channel in HEK‐293 cells. Lidocaine was applied after at least three stable currents were obtained (Figure 2A). Lidocaine inhibits ~10% of TRPM7 current at a threshold concentration of 0.3 mM, and the half‐maximal inhibition concentration (IC50) is 11.06 ± 0.62 mM (Figure 2B). We next characterized the current–voltage relationships in the absence or presence of 10 mM lidocaine using a ramp depolarization protocol (from –80 mV to +60 mV) (Figure 2C). Consistent with the previous findings, the rectification was largely diminished and became approximately linear when the extracellular divalent cations were removed (Figure 2D). In the presence of lidocaine, the linear current–voltage relationship did not change, suggesting that lidocaine inhibits TRPM7 currents in a voltage‐independent manner.
Figure 2.
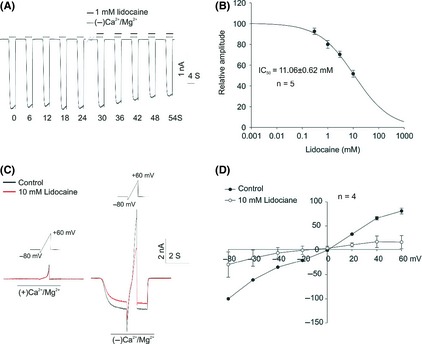
Inhibition of the TRPM7 current by lidocaine in HEK‐293 cells overexpressing TRPM7 channels. (A) Representative traces show the inhibition of TRPM7 current by 1 mM lidocaine in HEK‐293 cells that overexpress TRPM7 channels. (B) Dose–response curve was inferred from A. For each concentration, the 10th traces in the presence of lidocaine were used for dose–response analysis. The IC50 was 11.06 ± 0.62 mM (n = 5). (C) Voltage ramp (−80 to +60 mV) was applied for 4 seconds at a holding potential of −60 mV in HEK293 cells overexpressing TRPM7 channels. TRPM7 current was induced by deprivation of Ca2+ and Mg2+ (−Ca2+/Mg2+) in the absence or presence of 10 mM lidocaine. (D) Current–voltage relationship (I‐V curve) was inferred from C. Current amplitude recorded in (−)Ca2+/Mg2+ minus that recorded in (+)Ca2+/Mg2+ was used for data analysis; n = 4.
Lidocaine Inhibits TRPM7 Current in a Frequency‐Dependent Manner
The time‐dependent decrease of TRPM7 by lidocaine in Figure 2A is reminiscent of a use‐ or frequency‐dependent inhibition. To test this hypothesis, we compared the effects of lidocaine on TRPM7 currents in HEK293 cells with two different stimulation frequencies at 6 seconds (Figure 3A,B) and 16 seconds intervals (Figure 3C). No significant current rundown was observed after three stable currents were obtained (Figure 3A). However, in the presence of 10 mM lidocaine, a significant time‐dependent decrease in current was observed under both stimulation frequencies of 6 seconds (Figure 3B) and 16 seconds intervals (Figure 3C). Interestingly, during a same time period, the more activation (6 seconds interval) of TRPM7 channel produced more inhibition (Figure 3D). For example, at the end of 72 seconds (as indicated by the dashed blue line), higher‐frequency stimulation (6 seconds interval) causes ~50% TRPM7 current inhibition in the presence of 10 mM lidocaine, whereas, lower‐frequency stimulation (16 seconds interval) produces <20% current inhibition (Figure 3D). Interestingly, the inhibition of TRPM7 currents by lidocaine under both stimulating protocols (6 seconds and 16 seconds intervals) was almost the same after 10 times of stimulation (as shown by the dashed purple line), both of which were ~50% (Figure 3D). In addition, TRPM7 current was not inhibited (Figure 3E,F) when lidocaine was applied only when the channels are closed. Together, these results imply that lidocaine preferentially binds to the activated channel or functions as an open‐channel blocker, which property supports the use/frequency‐dependent inhibition.
Figure 3.
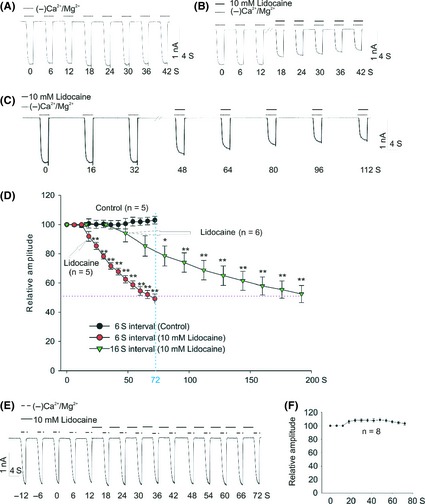
Frequency‐dependent inhibition of the TRPM7 current by lidocaine in HEK‐293 cells overexpressing TRPM7 channels. (A and B) TRPM7 current was recorded, with an interval of 6 seconds, in the absence or presence of 10 mM lidocaine, respectively. Three stable currents were recorded before the treatment with lidocaine. (C) TRPM7 current was recorded in the presence of 10 mM lidocaine with an interval of 16 seconds. (D) Summary data showing time‐dependent decrease of TRPM7 current in the absence (black circle, stimulating interval of 6 seconds, n = 5) or presence of 10 mM lidocaine (red circle, stimulating interval of 6 seconds, n = 5; green triangle, stimulating interval of 16 seconds, n = 6). (Two‐way ANOVA followed by Bonferroni posttests, *P < 0.5, **P < 0.01). Arrows represent the initial administration of lidocaine. (E and F) Representative current traces and summary data showing the lack of inhibition on TRPM7 current by lidocaine. Lidocaine was applied only when the channel was inactivated (n = 8).
Lidocaine Inhibits TRPM7‐Mediated Intracellular Zinc Accumulation
TRPM7 is highly permeable to zinc. Activation of TRPM7 increases zinc entry and resultant intracellular zinc accumulation. Inhibition of TRPM7 activity, on the other hand, decreases TRPM7‐mediated zinc accumulation. As lidocaine inhibits TRPM7 currents, we speculate that lidocaine could inhibit TRPM7‐mediated intracellular zinc accumulation. Using a zinc indicator FluoZin‐3, we examined the effect of lidocaine on TRPM7‐mediated intracellular zinc accumulation in primary cultured cortical neurons. As shown in Figure 4A, in the absence of extracellular zinc, activation of TRPM7 channels by deprivation of extracellular calcium and magnesium did not alter the basal zinc fluorescence intensity. However, a dramatic increase of FluoZin‐3 fluorescence intensity was observed upon the activation of TRPM7 in the presence of 30 μM extracellular zinc (Figure 4A), which is consistent with our previous observations 14. Our previous study also showed that zinc alone, without the activation of TRPM7 channel, caused no intracellular zinc accumulation, implying that TRPM7 contributes greatly to zinc entry. As expected, lidocaine (10 mM) dramatically inhibited TRPM7‐mediated FluoZin‐3 fluorescence increase. More than 50% of zinc increase, evaluated at 1000 seconds time point, was inhibited by lidocaine (Figure 4B,C). Addition of 10 mM lidocaine did not affect the basal FluoZin‐3 fluorescence intensity (Figure 4D), implying that lidocaine specifically inhibits TRPM7‐mediated zinc accumulation. We further validated the effect of lidocaine on TRPM7‐mediated intracellular zinc accumulation in HEK293 cells overexpressing TRPM7. Consistently, lidocaine significantly inhibited TRPM7‐mediated intracellular zinc accumulation in HEK293 cells (Figure 4E,F), but had no effect on the basal zinc fluorescence (data not shown).
Figure 4.
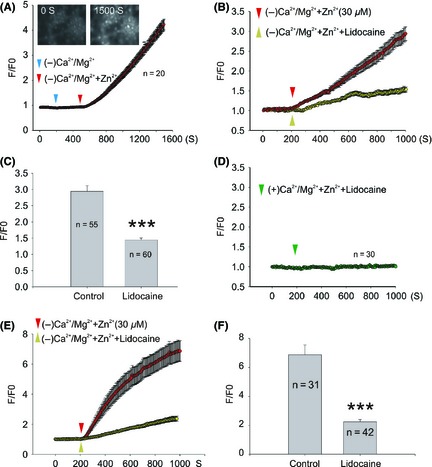
Lidocaine inhibits TRPM7‐mediated [Zn2+]i accumulation in cortical neurons and HEK‐293 cells overexpressing TRPM7 channels. (A) Representative images (inset images) and traces showing FluoZin‐3 fluorescence change in normal ECF (0–200S), Ca2+/Mg2+ deprivation ECF (200–500S), and Ca2+/Mg2+ deprivation with zinc addition ECF (500–1500S). (B) Time‐dependent change of FluoZin‐3 fluorescence with (yellow triangle) or without (red triangle) 10 mM lidocaine. Neurons were treated with normal ECF before the activation of TRPM7 by Ca2+/Mg2+ deprivation. Each trace represents an average fluorescent intensity from randomly selected cells from three to four independent experiments. (C) Summary bar graph inferred from B showing the normalized fluorescence intensity at the 1000 S time point (***P < 0.001). (D) The effect of 10 mM lidocaine on the basal FluoZin‐3 fluorescence. (E) Time‐dependent changes of FluoZin‐3 fluorescence with (yellow triangle) or without (red triangle) 10 mM lidocaine in HEK‐293 cells. HEK‐293 cells were treated with normal ECF before TRPM7 activation by Ca2+/Mg2+ deprivation. Each trace represents an average fluorescent intensity from randomly selected 31–42 cells from three to four independent experiments. (F) Summary bar graph represents the normalized fluorescence intensity at the 1000 S time point (***P < 0.001).
Lidocaine Inhibits TRPM7‐Mediated Zinc Toxicity in Primary Cultured Cortical Neurons
Our previous study clearly demonstrated that TRPM7 contributes to zinc‐induced neuronal toxicity and that suppression of TRPM7 is neuroprotective 14. As lidocaine can inhibit TRPM7 current and TRPM7‐mediated intracellular zinc accumulation, it is expected to ameliorate TRPM7‐mediated zinc toxicity. Consistent with the previous findings 14, deprivation of the extracellular Ca2+/Mg2+ or incubation of 30 μM zinc alone for 1 h, did not cause significant neuronal injury. However, the combination of zinc addition and Ca2+/Mg2+ deprivation dramatically increases neuronal injury (Figure 5A). These results confirm our previous findings that TRPM7 channels mediate zinc toxicity in native neurons. In the presence of lidocaine (1 and 10 mM), the morphological changes of injury including shrinked cell bodies and broken dendrites (Figure 5B) were largely ameliorated. Consistent with the morphological change, alive staining with fluorescein diacetate (FDA, green) and dead staining with propidium iodide (PI, red) (Figure 5C) or LDH release (Figure 5D) showed similar increases in cell death and injury by 1 h zinc treatment with TRPM7 activation. Incubation of lidocaine significantly decreased the cell death and injury (Figure 5C,D), and lidocaine alone did not change the basal LDH release (Figure 5E).
Figure 5.
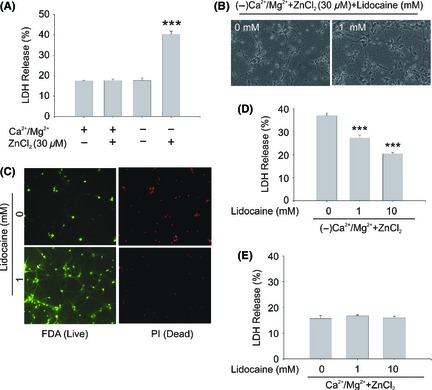
The effect of lidocaine on TRPM7‐mediated zinc toxicity in mouse cortical neurons. (A) Relative lactate dehydrogenase (LDH) release from various treatments as indicated. Neurons were incubated in different ECF for 1 h and then incubated with normal culture medium for 23 h. In all experiments, the blockers for NMDA receptors, AMPA/kainate receptors, and VDCCs were included in the extracellular solutions to eliminate the potential zinc entry through these pathways (n = 4). (B) Representative phase‐contrast images showing cultured mouse cortical neurons taken at 23 h following 1 h treatment with or without lidocaine. (C) Fluorescein diacetate (FDA) staining of cell bodies of alive neurons and propidium iodide (PI) staining of nuclei of dead neurons. (D) LDH release in the presence or absence of lidocaine. The cells were incubated in zinc containing and Ca2+/Mg2+ deprivation ECF, with or without lidocaine as indicated for 1 h. Then, cells were incubated with normal culture medium for 23 h, followed by LDH release assay (n = 5, ***P < 0.001). (E) Relative LDH release in the absence or presence of different concentrations of lidocaine. Neurons were incubated in normal ECF for 1 h and then incubated with normal culture medium for 23 h (n = 7).
QX‐314 and Procaine Inhibit TRPM7 Current in Cultured Cortical Neurons
Local anesthetics bind to voltage‐gated Na+ channels within the transmembrane domains. QX‐314, features an additional N‐ethyl group attached to the amine moiety, is a permanent charged form of lidocaine (Figure 6A), which makes it membrane impermeable. Thus, QX‐314, unlike the membrane permeable local anesthetics, has no inhibitory effect on Na+ currents when applied extracellularly 16. In this study, we used QX‐314 to probe whether the binding location of lidocaine in TRPM7 channel is similar to that in voltage‐gated Na+ channels. Our results show that QX‐314 has a similar efficacy as lidocaine in inhibiting TRPM7 current, which causes ~50% TRPM7 currents inhibition at 10 mM concentration in cortical neurons (Figure 6B,C). Based on their structures, local anesthetics are grouped into amino esters and amino amides. QX‐314 and lidocaine belong to amino amides, and procaine belongs to amino esters (Figure 6A). Interestingly, amino ester procaine inhibits ~70% of the TRPM7 current in cortical neurons, the potency of which is higher than that of the amino amides lidocaine and QX‐314 (Figure 6D,E), suggesting that the amino ester structure helps to increase the potency of local anesthetics in inhibiting TRPM7 currents. A further systematic structure–activity analysis based on lidocaine structure may help to indentify a potent TRPM7 inhibitor.
Figure 6.
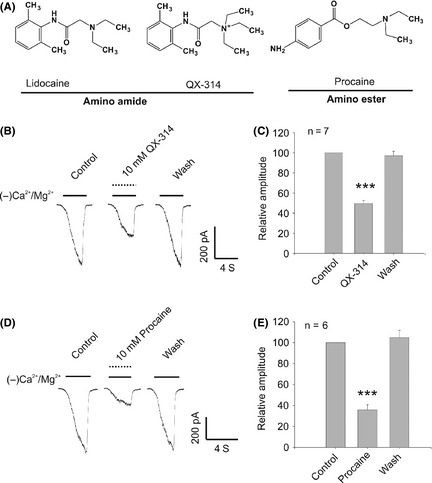
The effect of QX‐314 and procaine on TRPM7 current in cortical neurons. (A) The structure of lidocaine, QX‐314, and procaine. Based on their structure, local anesthetics are classified into two series of compounds including amino amide and amino ester. (B) and (C) The representative traces and summary data showing the effect of 10 mM QX‐314 on TRPM7 current in cortical neurons (n = 7, ***P < 0.001). (D) and (E) The representative traces and summary data showing the effect of 10 mM procaine on TRPM7 current in cortical neurons (n = 6, ***P < 0.001). MK‐801 (10 μM) and TTX (0.3 μM) were included in the extracellular solutions to block potential activation of NMDA and voltage‐gated Na+ currents.
Discussion
During cerebral ischemia, the excessive activation of voltage‐gated calcium channel and NMDA receptor results in overwhelming influx of Ca2+ into the neurons which makes a decrease of extracellular Ca2+ 17. In addition to Ca2+, a large reduction of the extracellular Mg2+ was observed in the ischemic brain 18. TRPM7 currents could be activated by decreasing extracellular divalent cations including Ca2+ and Mg2+ 6, 19. The decrease of extracellular Mg2+/Ca2+, during stroke, contributes to the activation of TRPM7 to some extent. In addition to the activation by decreased extracellular Mg2+/Ca2+, TRPM7 current is inhibited by intracellular Mg2+ 20, 21. In the current study, we induce TRPM7 current by deprivation of both extracellular Ca2+/Mg2+ and intracellular Mg2+ and, for the first time, demonstrate that local anesthetic lidocaine could inhibit TRPM7 currents.
The accumulation of Zn2+ in neurons following cerebral ischemia is now well recognized, and a striking correlation between zinc accumulation and cell viability is revealed 11, 12, 13. Zn2+‐induced neuronal toxicity, for example, could be reduced by Zn2+ chelation 8, 9. TRPM7 is highly permeable to divalent cations, with an order of Zn2+≈Ni2+≥Ba2+>Co2+>Mg2+≥Mn2+≥Sr2+≥Cd2+≥Ca2+ 22. The high permeability to zinc implies that TRPM7 may contribute to zinc‐mediated neuronal injury during stoke. Our previous study has clearly demonstrated the activation of TRPM7 channels enhances zinc toxicity in mouse cortical neurons. In the current study, we show that local anesthetic lidocaine decreases TRPM7‐mediated intracellular zinc increase and subsequent neuronal injury.
Lidocaine blocks voltage‐gated Na+ currents with an IC50 of 204 μM 23. The concentrations used in the current study cannot be used in clinical practice owing to CNS side effects such as coma and respiratory arrest when systemic administration of lidocaine reaches a plasma concentration of >200 μM 24. A systematic structure–activity analysis and further structure modification of lidocaine may help to obtain a compound with increased inhibitory effect on TRPM7 currents and decreased inhibitory effect on voltage‐gated Na+ current, which might be acceptable for clinical use in stroke intervention.
Local anesthetics are lipophilic in their uncharged form and can gain access to the intracellular sodium channel pore binding site located at the cytoplasmic (inner) portion by diffusion across the cell membranes 16. QX‐314, a permanently charged quaternary derivative of lidocaine, is membrane impermeable, which makes it unable to reach the binding site on Na+ channel. Thus, QX‐314 has no effect on sodium channels when applied extracellularly 16. Interestingly, in the current study, QX‐314 has the same efficacy as lidocaine in inhibiting TRPM7 current, suggesting that the binding sites for lidocaine might be located at the extracellular domain in TRPM7 channel. Two basic categories of local anesthetics exist including the amino amides and the amino esters. Amino amides have an amide link between the intermediate chain and the aromatic end, whereas amino esters have an ester link between the intermediate chain and the aromatic end. Besides amino amides lidocaine and its derivative QX‐314, we tested the effect of amino ester procaine on TRPM7 current. In comparison with QX‐314 and lidocaine, procaine is more potent in inhibiting TRPM7 current, which suggests that the replacement of amino amide by amino ester would increase the inhibitory potency of local anesthetics on TRPM7 current.
To date, there is lacking of specific TRPM7 inhibitor. Some compounds, including 2‐aminoethyl‐diphenylborinate (2‐APB), gadolinium (Gd3+), lanthanum (La3+), SKF‐96365, spermine, and carvacrol could inhibit TRPM7, however, the selectivity is poor 6, 25, 26, 27. Future identification of highly selective TRPM7 inhibitors would enhance our understanding of TRPM7 physiological and pathological function. Of interesting, QX‐314, unlike lidocaine and procaine, has no direct effect on voltage‐gated sodium channel when applied extracellularly, which might be a useful tool for the investigation of TRPM7 channel functions.
Conflict of Interest
The authors declare no conflict of interest.
Acknowledgments
This work was supported by R01NS066027, NIMHD S21MD000101, U54 NS083932, and AHA 0840132N.
The first two authors contributed equally to this work.
References
- 1. Venkatachalam K, Montell C. TRP channels. Annu Rev Biochem 2007;76:387–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Schmitz C, Perraud AL, Johnson CO, et al. Regulation of vertebrate cellular Mg2+ homeostasis by TRPM7. Cell 2003;114:191–200. [DOI] [PubMed] [Google Scholar]
- 3. Krapivinsky G, Mochida S, Krapivinsky L, Cibulsky SM, Clapham DE. The TRPM7 ion channel functions in cholinergic synaptic vesicles and affects transmitter release. Neuron 2006;52:485–496. [DOI] [PubMed] [Google Scholar]
- 4. Sun HS, Jackson MF, Martin LJ, et al. Suppression of hippocampal TRPM7 protein prevents delayed neuronal death in brain ischemia. Nat Neurosci 2009;12:1300–1307. [DOI] [PubMed] [Google Scholar]
- 5. Jiang J, Li MH, Inoue K, Chu XP, Seeds J, Xiong ZG. Transient receptor potential melastatin 7‐like current in human head and neck carcinoma cells: Role in cell proliferation. Cancer Res 2007;67:10929–10938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Aarts M, Iihara K, Wei WL, et al. A key role for TRPM7 channels in anoxic neuronal death. Cell 2003;115:863–877. [DOI] [PubMed] [Google Scholar]
- 7. Sun H, Leng T, Zeng Z, Gao X, Inoue K, Xiong ZG. Role of TRPM7 channels in hyperglycemia‐mediated injury of vascular endothelial cells. PLoS One 2013;8:e79540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Koh JY, Suh SW, Gwag BJ, He YY, Hsu CY, Choi DW. The role of zinc in selective neuronal death after transient global cerebral ischemia. Science 1996;272:1013–1016. [DOI] [PubMed] [Google Scholar]
- 9. Frederickson CJ, Koh JY, Bush AI. The neurobiology of zinc in health and disease. Nat Rev Neurosci 2005;6:449–462. [DOI] [PubMed] [Google Scholar]
- 10. Chen M, Chen Q, Cheng XW, et al. Zn2+ mediates ischemia‐induced impairment of the ubiquitin‐proteasome system in the rat hippocampus. J Neurochem 2009;111:1094–1103. [DOI] [PubMed] [Google Scholar]
- 11. Frederickson CJ, Cuajungco MP, Frederickson CJ. Is zinc the link between compromises of brain perfusion (excitotoxicity) and Alzheimer's disease? J Alzheimers Dis 2005;8:155–160. [DOI] [PubMed] [Google Scholar]
- 12. Sensi SL, Paoletti P, Bush AI, Sekler I. Zinc in the physiology and pathology of the CNS. Nat Rev Neurosci 2009;10:780–791. [DOI] [PubMed] [Google Scholar]
- 13. Shuttleworth CW, Weiss JH. Zinc: New clues to diverse roles in brain ischemia. Trends Pharmacol Sci 2011;32:480–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Inoue K, Branigan D, Xiong ZG. Zinc‐induced neurotoxicity mediated by transient receptor potential melastatin 7 channels. J Biol Chem 2010;285:7430–7439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wei WL, Sun HS, Olah ME, et al. TRPM7 channels in hippocampal neurons detect levels of extracellular divalent cations. Proc Natl Acad Sci USA 2007;104:16323–16328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Theile JW, Cummins TR. Recent developments regarding voltage‐gated sodium channel blockers for the treatment of inherited and acquired neuropathic pain syndromes. Front Pharmacol 2011;2:54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Macdonald JF, Xiong ZG, Jackson MF. Paradox of Ca2+ signaling, cell death and stroke. Trends Neurosci 2006;29:75–81. [DOI] [PubMed] [Google Scholar]
- 18. Lin MC, Huang YL, Liu HW, Yang DY, Lee JB, Cheng FC. Microdialysis analyzer and flame atomic absorption spectrometry in the determination of blood glucose, lactate and magnesium in gerbils subjected to cerebral ischemia/reperfusion. J Am Coll Nutr 2004;23:556S–560S. [DOI] [PubMed] [Google Scholar]
- 19. Nadler MJ, Hermosura MC, Inabe K, et al. LTRPC7 is a Mg.ATP‐regulated divalent cation channel required for cell viability. Nature 2001;411:590–595. [DOI] [PubMed] [Google Scholar]
- 20. Langeslag M, Clark K, Moolenaar WH, van Leeuwen FN, Jalink K. Activation of TRPM7 channels by phospholipase C‐coupled receptor agonists. J Biol Chem 2007;282:232–239. [DOI] [PubMed] [Google Scholar]
- 21. Kozak JA, Cahalan MD. MIC channels are inhibited by internal divalent cations but not ATP. Biophys J 2003;84:922–927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Monteilh‐Zoller MK, Hermosura MC, Nadler MJ, Scharenberg AM, Penner R, Fleig A. TRPM7 provides an ion channel mechanism for cellular entry of trace metal ions. J Gen Physiol 2003;121:49–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Brau ME, Vogel W, Hempelmann G. Fundamental properties of local anesthetics: Half‐maximal blocking concentrations for tonic block of Na+ and K+ channels in peripheral nerve. Anesth Analg 1998;87:885–889. [DOI] [PubMed] [Google Scholar]
- 24. Putrenko I, Schwarz SK. Lidocaine blocks the hyperpolarization‐activated mixed cation current, I(h), in rat thalamocortical neurons. Anesthesiology 2011;115:822–835. [DOI] [PubMed] [Google Scholar]
- 25. Kerschbaum HH, Kozak JA, Cahalan MD. Polyvalent cations as permeant probes of MIC and TRPM7 pores. Biophys J 2003;84:2293–2305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Li M, Jiang J, Yue L. Functional characterization of homo‐ and heteromeric channel kinases TRPM6 and TRPM7. J Gen Physiol 2006;127:525–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Parnas M, Peters M, Dadon D, et al. Carvacrol is a novel inhibitor of Drosophila TRPL and mammalian TRPM7 channels. Cell Calcium 2009;45:300–309. [DOI] [PMC free article] [PubMed] [Google Scholar]