

Abstract
Peripheral nerve injury results in persistent motor deficits, even after the nerve regenerates and muscles are reinnervated. This lack of functional recovery is partly explained by brain and spinal cord circuit alterations triggered by the injury, but the mechanisms are generally unknown. One example of this plasticity is the die-back in the spinal cord ventral horn of the projections of proprioceptive axons mediating the stretch reflex (Ia afferents). Consequently, Ia information about muscle length and dynamics is lost from ventral spinal circuits, degrading motor performance after nerve regeneration. Simultaneously, there is activation of microglia around the central projections of peripherally injured Ia afferents, suggesting a possible causal relationship between neuroinflammation and Ia axon removal. Therefore, we used mice (both sexes) that allow visualization of microglia (CX3CR1-GFP) and infiltrating peripheral myeloid cells (CCR2-RFP) and related changes in these cells to Ia synaptic losses (identified by VGLUT1 content) on retrogradely labeled motoneurons. Microgliosis around axotomized motoneurons starts and peaks within 2 weeks after nerve transection. Thereafter, this region becomes infiltrated by CCR2 cells, and VGLUT1 synapses are lost in parallel. Immunohistochemistry, flow cytometry, and genetic lineage tracing showed that infiltrating CCR2 cells include T cells, dendritic cells, and monocytes, the latter differentiating into tissue macrophages. VGLUT1 synapses were rescued after attenuating the ventral microglial reaction by removal of colony stimulating factor 1 from motoneurons or in CCR2 global KOs. Thus, both activation of ventral microglia and a CCR2-dependent mechanism are necessary for removal of VGLUT1 synapses and alterations in Ia-circuit function following nerve injuries.
SIGNIFICANCE STATEMENT Synaptic plasticity and reorganization of essential motor circuits after a peripheral nerve injury can result in permanent motor deficits due to the removal of sensory Ia afferent synapses from the spinal cord ventral horn. Our data link this major circuit change with the neuroinflammatory reaction that occurs inside the spinal cord following injury to peripheral nerves. We describe that both activation of microglia and recruitment into the spinal cord of blood-derived myeloid cells are necessary for motor circuit synaptic plasticity. This study sheds new light into mechanisms that trigger major network plasticity in CNS regions removed from injury sites and that might prevent full recovery of function, even after successful regeneration.
Keywords: CCR2, CX3CR1, microglia, nerve injury, stretch reflex, VGLUT1
Introduction
Injury or dysfunction in remote regions of the body can cause long-term modifications of synaptic network structure and function in undamaged brain and spinal cord regions connected to them. A well-studied case that causes life-long deficits in patients is the modification of brain and spinal cord circuitry following peripheral nerve injury (PNI) (Lundborg, 2003; Navarro et al., 2007; Davis et al., 2011; Mohanty et al., 2015). One such change is the permanent loss of muscle stretch reflexes (Cope et al., 1994; Haftel et al., 2005; Bullinger et al., 2011). Stretch-sensitive Ia afferents innervate muscle spindles peripherally and after entering the spinal cord, they direct axon collateral branches ventrally where they establish monosynaptic connections with motoneurons (MNs) forming the stretch reflex circuit (Burke et al., 1979; Brown and Fyffe, 1981). Ventral horn Ia axons and synapses are withdrawn after PNI and never return, even after motor and Ia axons reinnervate muscles in the periphery (Alvarez et al., 2010, 2011; Rotterman et al., 2014). Consequently, the stretch reflex becomes functionally disconnected in regenerated MNs (Bullinger et al., 2011). Absence of Ia input implies that ventral horn motor circuitries operate after regeneration without feedback about muscles lengths and dynamics, and force signals from Ib Golgi afferents become unopposed, both affecting many spinal control mechanisms. Thus, motor tasks involving high forces and/or rapid and large muscle lengthening (steep slopes) show deficits (Abelew et al., 2000; Maas et al., 2007; Sabatier et al., 2011; Lyle et al., 2017; Chang et al., 2018). The mechanisms implicated in this die-back of ventral horn Ia axons are unknown.
After PNI, several coincident phenomena occur around cell bodies of axotomized MNs and sensory neurons: (1) activated microglia surround MNs; (2) blood-borne immune cells surround sensory neurons; and (3) synapses become displaced from axotomized MN cell bodies (Blinzinger and Kreutzberg, 1968; Svensson and Aldskogius, 1993; Aldskogius et al., 1999; Cullheim and Thams, 2007; Aldskogius, 2011; Niemi et al., 2013). The significance of these phenomena and their relation to the disappearance of Ia synapses are not clear. After PNI, microglia proliferate and migrate toward axotomized MNs interposing themselves between MN cell bodies and synapses undergoing detachment (Blinzinger and Kreutzberg, 1968). These observations led to the suggestion that microglia “lift” these synapses, a phenomenon known as “synaptic stripping.” However, follow-up studies failed to confirm the necessity of microglia activation for “synaptic stripping” (Svensson and Aldskogius, 1993; Kalla et al., 2001; Berg et al., 2013), and the role of microglia in this process continues to be debated (Perry and O'Connor, 2010). Ia synapses were proposed to follow similar “stripping” fates (Mendell, 1984), but most Ia synapses target and are lost from dendrites (Brown and Fyffe, 1981; Rotterman et al., 2014), not the cell body where “synaptic stripping” occurs. Moreover, their disappearance is permanent and associated with die-back of ventral Ia axons (Alvarez et al., 2011), suggesting a degradative process.
Neuroinflammation influences synapse stability and loss (Hong et al., 2016; Nisticò et al., 2017) and after PNI, there is intense microgliosis in the spinal cord side ipsilateral to the injury and sometimes infiltration of myeloid cells, including chemokine receptor 2 (CCR2)-expressing macrophages. CCR2 cells surrounding the cell bodies of axotomized sensory neurons promote the regeneration of their axons in the peripheral nerve (Niemi et al., 2013), but CCR2 cells are also responsible for removing injured sensory axons in the spinal cord (Evans et al., 2014) and peripheral nerves (Siebert et al., 2000). This study was therefore designed to investigate whether microglia and/or blood-derived CCR2 macrophages participate in removing Ia axons/synapses from the ventral horn. To this end, we used transgenic mouse models to distinguish resident microglia from infiltrating myeloid cells and to manipulate each cell type independently. The results suggest that microglia and CCR2 activation are both necessary for Ia synapse removal, demonstrating a link between the alteration of the stretch reflex after PNI and the neuroinflammatory response in the ventral horn.
Materials and Methods
All experiments were performed at Emory University and approved by the Institutional Animal Care and Use Committee.
Transgenic models.
All procedures were performed in adult mice (∼3 months of age; 20–25 g) with comparable representation of males and females. When sex differences were found, these are noted in Results. We used several transgenic lines obtained from The Jackson Laboratory and maintained on a mixed C57BLk/6J and 6N background (Table 1). Cx3cr1EGFP animals (RRID:IMSR_JAX:005582) carry an EGFP gene “knocked in” to replace the first 390 bp of the second exon region of the fractalkine receptor gene (Jung et al., 2000). In this model, EGFP is expressed in CNS resident microglia and at various levels in subsets of peripheral myeloid cells. We also used Ccr2RFP animals (RRID:IMSR_JAX:017586) to genetically label infiltrating peripheral myeloid cells after PNI. In these animals, red fluorescent protein (RFP) replaces the first 279 bp of the CCR2 open reading frame (Saederup et al., 2010). This model labels peripheral monocytes, T cells, dendritic cells, and other small populations of myeloid-derived cells. Experimental mice were produced by crossing Cx3cr1EGFP/EGFP with Ccr2RFP/RFP mice to generate dual-heterozygous Cx3cr1EGFP/+::Ccr2 RFP/+ mice. In both knock-in models, the allele carrying the fluorescent protein gene is disrupted; therefore, homozygous mice are functional “KOs” for each respective receptor. Thus, we performed sciatic nerve injuries in Ccr2RFP/RFP mice to determine whether interfering with CCR2 mechanisms could prevent the loss of Ia afferent synapses on MNs. In addition, we crossed Cx3cr1CreER mice (RRID:IMSR_JAX:020940) with the Ai9 R26-tdTomato reporter line to obtain a genetic permanent label of all resident microglia before nerve injury. This allowed us to search after injury for possible microglia derived from peripheral immune cells that escaped the tamoxifen-induced recombination event (see below). Finally, to test the hypothesis that microglia activation is mediated via a signal from the MNs to the microglia, we deleted colony stimulating factor 1 (CSF1) from MNs by crossing ChatIRES-Cre mice (RRID:IMSR_JAX:006410) with csf1flox/flox mice (Harris et al., 2012) generously donated by Dr. Jean X. Jiang (University of Texas, San Antonio, TX). These animals showed a lack of CSF1 upregulation in injured MNs and a blunted microglia reaction after PNI.
Table 1.
Transgenic mice
| Mouse | Jax # | Background | Donating laboratory | Reference |
|---|---|---|---|---|
| CX3CR1EGFP | 005582 | C57BL/6 | Dr. Dan R. Littman, New York University | Jung et al., 2000 |
| CCR2RFP | 017586 | C57BL/6J | Dr. Israel F. Charo, Gladstone Institute of Cardiovascular Disease, University of California at San Francisco | Saederup et al., 2010 |
| CSF1-flox | NA | C57BL/6J | Dr. Jean X. Jiang, University of Texas Health Science Center | Harris et al. 2012 |
| Chat-IRES-Cre | 006410 | C57BL/6 | Dr. Bradford B. Lowell, Beth Israel Deaconess Medical Center Harvard | Rossi et al., 2011 |
| CX3CR1CreER | 020940 | C57BL/6J | Dr. Steffen Jung, Weizmann Institute of Science | Yona et al., 2013 |
| Ai9 R26-tdTomato | 007909 | C57BL/6J | Dr. Hongkui Zeng, Allen Institute for Brain Science | Madisen et al., 2010 |
MN labeling and PNI procedures.
All animals were anesthetized with isoflurane until a surgical plane of anesthesia was achieved (induction: 4%; maintenance: 2%, both in 100% O2) and given a subcutaneous injection of 0.05–0.10 mg/kg buprenorphine to reduce postsurgical pain. The left hindlimb was secured and the lateral gastrocnemius (LG) muscle exposed by making a posterior mid-line incision at the popliteal fossa level. The overlaying biceps femoris was retracted to access the muscle. The muscle was then injected with 4–5 μl of 1.5% Fast Blue (Polysciences) using a 10 μl Hamilton syringe. The skin was aligned and sutured back together. Animals recovered for ∼1 week to allow sufficient retrograde transport of the tracer to MN cell bodies and dendrites before they underwent a second surgery in which either the tibial nerve (TN) or the sciatic nerve (SN) was exposed, isolated, and transected. A small piece of sterile silk was tied around the proximal nerve stump to prevent regeneration from occurring. In the case of tibial nerve injuries, the nerve was transected immediately after the trifurcation of the sciatic branch into the common peroneal, sural, and tibial nerves. The sciatic nerve was transected just distal to the top head of the femur. Once the appropriate nerve was transected and ligated, muscle and skin were closed using absorbable sutures.
In global and conditional KO animals, we performed sciatic nerve cut-repair surgeries, and the same procedure was done in parallel in heterozygous mice or control mice lacking Cre recombinase expression. In this case, the sciatic nerve was exposed as above; and after a complete transection, the proximal and distal nerve stumps were immediately aligned and joined using a fibrin glue mixture containing thrombin, fibronectin, and fibrinogen to repair the nerve and facilitate regeneration (Guest et al., 1997). This procedure results in >80% reinnervation of neuromuscular junctions by 8 weeks after injury (data not shown). All animals were monitored daily for the first week after surgery; none of the animals exhibited signs of pain or distress.
In animals carrying Cx3cr1CreER alleles and crossed with the Ai9 tdTomato line, we induced Cre recombination 5–6 weeks before nerve injuries with two subcutaneous injections of 2 mg tamoxifen (dissolved in peanut oil) at P12 and P14. At the time of injection, all CX3CR1-expressing cells (resident microglia and also some peripheral immune cells) underwent Cre recombination. Given that microglia are long-lived with little turnover (Tay et al., 2017), and peripheral immune cells are fully replaced in a few weeks by progenitors lacking CX3CR1 expression, the only genetically Cre-recombined cells remaining 5–6 weeks after tamoxifen injections correspond to resident microglia. This strategy to specifically target microglia has been validated in previous studies (Goldmann et al., 2013; Tay et al., 2017).
Harvesting tissue for histological analysis.
Animals were allowed to survive for variable times ranging from 3 d to 8 weeks after nerve injury, at which times they were deeply anesthetized with Euthasol (100 mg/kg) and transcardially perfused, first with vascular rinse containing heparin followed by 4% PFA in 0.1 m PB, pH 7.4. The spinal cords were dissected out and postfixed overnight in the same fixative solution at 4°C. The following day, the spinal cords were rinsed with PB and cryoprotected in 30% sucrose solution at 4°C. Spinal segments L4-L5 were blocked out, and 50-μm-thick transverse sections cut on a freezing sliding microtome. Sections were collected and processed for immunohistochemistry as free-floating sections.
Histological processing and immunohistochemistry.
In dual-heterozygous Cx3cr1EGFP/+::Ccr2 RFP/+ mice, we usually performed triple immunocytochemical labeling using specific primary antibodies against GFP, RFP, and VGLUT1, each raised in a different species. The sections were washed in 0.01 m PBS with 0.3% Triton X-100 (PBS-T) and then blocked for 1 h with 10% normal donkey serum diluted in PBS-T. Sections were then incubated at room temperature with gentle agitation for ∼24 h in a primary antibody mixture containing chicken anti-GFP (1:1000, RRID:AB_620519), rabbit anti-DsRed (1:1000, RRID:AB_10013483), and guinea pig anti-VGLUT1 (1:1000, RRID:AB_887878), all mixed in PBS-T. Immunoreactive sites were revealed with species-specific secondary antibodies raised in donkey (Jackson ImmunoResearch Laboratories). Secondary antibodies (diluted 1:100 in PBS-T) were conjugated to either FITC (anti-chicken IgY, RRID:AB_2340356) for EGFP amplification, Cy3 (anti-rabbit IgG, RRID:AB_2307443) for enhancing RFP, or DyLight 647 (anti-guinea pig IgG, RRID:AB_2340476) for immunodetection of VGLUT1. In Ccr2 global and conditional KO mice, we used rabbit anti-Iba1 (1:500, RRID:AB_839504) revealed with donkey anti-rabbit IgG antibodies conjugated to FITC (RRID:AB_2315776) to detect microglia/macrophages in the CNS. In some experiments, infiltrating immune cells were tested for CD3ε immunoreactivity (T-cell marker) using Syrian Hamster anti-CD3ε antibodies (1:100, RRID:AB_394727). T-cell antibodies were mixed with rabbit anti-DsRed (1:1000, RRID:AB_10013483) to amplify detection of CCR2-RFP cells. Immunoreactive sites were revealed with Cy3 (anti-rabbit IgG, RRID:AB_2307443) for enhancing RFP, and AlexaFluor-647 (anti-Syrian Hamster IgG, RRID:AB_2337482) for detection of CD3ε immunoreactivity.
To study synaptic stripping in the ChatIRES-Cre::csf1flox/flox background, we prepared animals in which LG MNs were labeled unilaterally or bilaterally by Fast Blue injection in one or both LG muscles. One week after the animals underwent a sciatic nerve injury unilaterally (as above), they were perfusion-fixed 14 d after injury (as above) to compare injured versus control MNs 14 d after injury. Sections were equally divided into three vials. Each group was immunolabeled with Iba1 antibodies (as above) and antibodies against one of three synaptic markers: VGLUT1, VGLUT2, and VGAT. In this case, we used rabbit polyclonal antibodies for VGLUT1 and VGLUT2 (1:500 RRID:AB_887875 and RRID:AB_887883, respectively) and a monoclonal antibody against (VGAT 1:200 RRID:AB_887872), each revealed with DyLight 647-conjugated antibodies against their respective primary antibody species. In addition, we confirmed deletion of CSF1 from MNs with dual immunofluorescence for CSF1 and NeuN. In this case, a polyclonal antibody against CSF1 (1:2000 RRID:AB_355351) was mixed with a monoclonal antibody against NeuN (1:500 RRID:AB_2298772). Immunoreactivities were revealed with species-specific secondary antibodies conjugated to Cy3 (CSF1) or DyLight 647 (NeuN). A summary of antibody sources and catalog numbers appears in Table 2.
Table 2.
Antibodies used for immunohistochemistry
| Antigen | Immunogen | Host/type | Manufacturer | RRID # | Dilution |
|---|---|---|---|---|---|
| EGFP | Recombinant GFP 6-his tag | Chicken/polyclonal | Serotec catalog #obt1644 | AB_620519 | 1:1000 |
| DsRed | Variant of Discosoma sp. | Rabbit/polyclonal | Clontech catalog #632496 | AB_10013483 | 1:1000 |
| VGLUT1 | Recombinant protein (aa 456 to 560 from rat VGLUT1) | Guinea pig/polyclonal | Synaptic Systems catalog #135304 | AB_887878 | 1:1000 |
| VGLUT1 | Recombinant protein (aa 456 to 560 from rat VGLUT1) | Rabbit/polyclonal | Synaptic Systems catalog #135303 | AB_887875 | 1:500 |
| VGLUT2 | Recombinant protein (aa 510 to 582 from rat VGLUT2) | Rabbit/polyclonal | Synaptic Systems catalog #135402 | AB_887883 | 1:500 |
| VGAT | Synthetic peptide (aa 75 to 87 from rat VGAT) | Mouse/monoclonal | Synaptic Systems catalog #131011 | AB_887872 | 1:200 |
| Iba1 | C-terminus of Iba1 | Goat/polyclonal | Wako catalog #019-19741 | AB_839504 | 1:500 |
| CD3ϵ | Clone 500A2 25-kDA ϵ chain | Syrian hamster/monoclonal | BD Biosciences catalog #553238 | AB_394727 | 1:100 |
| CSF1 | Escherichia coli-derived recombinant mouse M-CSF Lys33-Glu262 | Goat/polyclonal | R&D Systems catalog #AF416 | AB_355351 | 1:2000 |
| NeuN | Purified cell nuclei from mouse brain | Mouse/monoclonal A60 clone | EMD Millipore catalog #MAB377 | AB_2298772 | 1:500 |
Densitometric analysis of VGLUT1.
Six spinal cord sections per animal, all containing labeled LG MNs, were randomly selected and imaged on an Olympus FV1000 confocal microscope at low magnification (10×1, z step: 1.5 μm, NA 0.4). The sections were imaged on both sides with identical parameters. Confocal images were uploaded to ImageJ, and a circular ROI with a diameter of 270 μm was placed around the labeled motor pool on the injured side. A symmetrical ROI was placed on the noninjured side to measure VGLUT1 immunofluorescence-integrated pixel densities (see Figs. 2A,B, 3G,H). All measurements were done in a single optical plane per section and presented as a ratio of the immunofluorescence density over retrogradely labeled MNs normalized to the contralateral side. Differences in VGLUT1 fluorescence were estimated in each animal by averaging ratios obtained from six sections per animal.
Figure 2.

VGLUT1 synaptic loss after nerve injury. A, VGLUT1 immunoreactivity (IR) shows a unilateral decrease in the injured side around the region containing Fast Blue-axotomized LG MNs. ROIs (white circles) are placed on LG motor pools on both sides of the spinal cord, and the integrated pixel density of VGLUT1 immunofluorescence was measured. B, VGLUT1 immunofluorescence density changes with time. Data are presented as a ratio of estimates ipsilateral versus contralateral to the injury. VGLUT1 fluorescence decreases ipsilateral to the injury after sciatic (blue line) or tibial (red line) nerve cut-ligation injuries. y axis represents integrated pixel density ipsilateral/contralateral. Each dot represents estimates from 1 animal (n = 4 mice per time point per injury type). Lines indicate mean ± SE at each time point. Black asterisks are comparisons with control: ***p < 0.001. Red asterisk at 8 weeks is a comparison between injuries: *p < 0.05. C, High-magnification confocal image of a retrogradely labeled LG MN (blue, Fast Blue) from a sham control animal receiving VGLUT1 synaptic inputs (white). D, Neurolucida 3D reconstruction of the same MN with VGLUT1 contacts mapped along soma and dendrites. E1–E3, Imaris 3D surface renderings of dendritic segments shown in box highlighted in C, D. Rotations demonstrate VGLUT1 synapses attachment to the dendrite. F, D, VGLUT1 density in cell bodies and dendrites of dual-heterozygous sham control animals is not different from control MNs in WT animals. In dual-heterozygous animals, VGLUT1 densities are decreased in cell bodies both after either tibial or sciatic nerve transection compared with sham controls. Black asterisks are comparisons with sham: ***p < 0.001. Red asterisk compares the two injuries: *p < 0.05. Dendrite linear densities of VGLUT1 contacts are significantly decreased compared with sham controls only after sciatic nerve injury (black asterisk; *p < 0.05), but not after tibial nerve injury. H, Sholl analysis (bin size: 25 μm distance increments from cell body center): VGLUT1 densities compared with sham controls show significant decreases in the first 25 μm bin after both injuries, but at further distances depletions were significant only after sciatic nerve injury: ***p < 0.001; **p < 0.01. I, VGLUT1 bouton surface area is similarly reduced after tibial or sciatic nerve injury: **p < 0.01 to control. F–H, Error bars indicate mean ± SE (n = 4–6 mice); each mouse estimate is from 6 MNs per mouse. Scale bars: A, 100 μm; C, D, 50 μm.
Figure 3.

Microglia activation around injured MNs. A–C, CX3CR1-GFP microglia (green) 3 d (A), 14 d (B), and 8 weeks (C) after tibial (A1, B1, C1) or sciatic (A2, B2, C2) cut ligation. LG MNs are labeled with Fast Blue (blue). D, Quantification of GFP+ microglia using unbiased automatic Imaris spot function. D1, GFP fluorescence. D2, Cell body identification. A line traced above the dorsal tip of the central canal separates dorsal from ventral regions. E, Number of CX3CR1-GFP microglia per ventral horn in 50-μm-thick sections at different times after injury after a sciatic (blue line) or a tibial (red line) nerve injury. There is a significant increase in the number of GFP+ microglia after both tibial and sciatic ligation from 3 to 21 d after injury: ***p < 0.001, all versus control after either injury. Microglia numbers return to baseline by 8 weeks after injury. No statistical differences were detected between sciatic and tibial nerve injury in GFP+ microglial cell numbers. Each dot represents 1 animal. Lines indicate mean ± SE at each time point (n = 4 mice; 6 ventral horns were counted per mouse). F, High-magnification 2D projections of single CX3CR1-GFP microglia in control and 14 d after a tibial or sciatic nerve injury. Bottom, Imaris filament tracker reconstructions used for quantification. G, Microglia cell body volume significantly increased compared with control after both nerve injuries: **p < 0.01; ***p < 0.001. H, Total microglia process length shows significant decreases compared with controls: ***p < 0.001. G, H, Each data point represents an individual microglia cell (n = 12 in each group). Error bars indicate SE. I, Sholl analysis compares the total number of microglia filaments in increasing 10 μm distance bins from the cell body center. Control microglia have significantly fewer processes in the first 10 μm than microglia in the injured conditions (blue asterisks); but past 20 μm, there are significantly fewer microglia processes in injured animals. Asterisks represent significant differences compared with control (blue) after sciatic (red) or tibial (green) nerve injury: ***p < 0.001. Inset, Sholl analysis bins with color-coded distances. Scale bars: A, D1, 100 μm; F, 10 μm.
Analysis of VGLUT1 densities on 3D reconstructed FB-labeled MNs.
Retrogradely labeled LG MNs were imaged at high magnification (60×1, z step: 0.5 μm, NA 1.35, oil-immersion) for VGLUT1 bouton quantification on MN surfaces. MN images were uploaded to the neural reconstruction software Neurolucida (version 12.0, MicroBrightField). Cell bodies and proximal dendritic arbors contained within single sections were manually traced through all optical planes. VGLUT1-immunoreactive boutons in direct contact with the soma or dendrites were identified and a “marker” placed and attached to the corresponding position on the 3D tracing. The number of VGLUT1 terminals per cell body was obtained and their density calculated by dividing the number of VGLUT1 contacts by the total cell body surface (per 100 μm2). VGLUT1 contacts on dendrites were estimated as linear densities by dividing the number of VGLUT1 terminals by total dendritic length of the traced dendrites (per 100 μm) (Alvarez et al., 2011; Schultz et al., 2017). Sholl analysis was used to examine VGLUT1 distributions in dendrite segments at 25 μm bins of incremental radial distance from the center of the cell body. Average VGLUT1 densities in soma, dendrite, or Sholl bins were obtained for each animal from 6 reconstructed MNs.
Analysis of VGLUT1, VGLUT2, and VGAT densities on FB-labeled MN cell bodies.
The cell bodies of MNs retrogradely labeled with Fast Blue were imaged at high magnification (60 × 2, z step: 0.5 μm). A mid-cell slab of the cell body surface with a 5–7 μm depth (10–15 optical sections) was reconstructed and all VGLUT1, VGLUT2, and VGAT clusters in contact with the cell body surface counted. The surface reconstructed always corresponded to optical planes of the cell body containing the nucleus and nucleolus and therefore correspond to mid-cross-sections of the cells analyzed with the membrane surface orthogonal to the imaging plane. The surface area of the reconstructed region was estimated and the area corresponding to dendrites stemming from this region subtracted. We then obtained a surface density (per 100 μm2) for each synaptic marker. Four or five animals were analyzed per genotype. From each animal and each side (ipsilateral or contralateral to the injury), we reconstructed from 3 to 10 MN cell body surface slabs per animal.
Quantification of microglia cells.
Spinal cord hemi-sections were imaged with confocal microscopy at 20× magnification (z step: 1.0 μm, NA 0.75) with automatic tiling, each containing six stitched tiles. Stitched images were uploaded to Imaris 3D neural analysis software (Bitplane), and we used the “spot function” to automatically detect and count the number of GFP+ cell bodies in 50-μm-thick spinal cord sections (see Fig. 3D,E). We counted all GFP+ microglia cells in dorsal (data not shown) and ventral horns. The ventral horn was defined as the region below a horizontal line traced over the dorsal tip of the central canal. Each animal estimate was derived from the average of 6 hemi-sections.
Microglia morphological analysis.
Twelve microglia cells (3 cells per animal, n = 4 animals per condition) were randomly sampled from confocal images containing retrogradely labeled MNs acquired at 60× (see above) from control animals or animals 14 d after tibial or sciatic nerve injury and uploaded into the Imaris program. All microglia analyzed were near and/or touching labeled MN cell bodies in lamina IX. We used the Imaris filament tracker module to reconstruct in 3D all the cellular processes associated with individual cells and quantified overall filament lengths as well as total number of filaments per cell. We also performed a Sholl analysis for the number of filaments at 10 μm bins of incremental radial distance from the center of the cell body. Finally, we digitally cropped all microglia processes and surface rendered the isolated cell body to calculate cell body volumes.
RFP cell quantification.
CCR2-RFP cells were counted manually using an Olympus epifluorescence microscope (BX52) at 20× magnification (NA 0.75). All 50-μm-thick sections containing labeled LG MNs were included in the analysis and averaged to obtain an estimate in each animal (average of 23.2 sections per animal; n = 4 animals). To quantify the percentage of CCR2-RFP cells CD3ε, six sections from each mouse (dual-heterozygous and CCR2 KO) were randomly selected from sections containing CCR2-RFP cells 21 d after sciatic nerve transection and ligation.
Quantification of Iba1 cells after injury that are not derived from resident microglia.
We crossed Cx3cr1CreER homozygous mice with Ai9 R26 tdTomato reporter mice and injected the offspring with tamoxifen (see above) to induce Cre recombination and tdTomato expression. When mice were ∼3 months of age, the LG muscle was injected with Fast Blue. One week later, the sciatic nerve was exposed, transected, and then ligated. Mice were allowed to survive for 21 d or 8 weeks before being perfusion-fixed. Control (sham) mice were injected with tamoxifen but had no surgical procedures performed on them. Spinal cords were then sectioned (50-μm-thick) and immunolabeled for Iba1 revealed with FITC-conjugated secondary antibodies (as above). All Iba1 cells derived from resident microglia are therefore expected to express tdTomato, whereas Iba1 cells derived from other sources should lack tdTomato. Tiled confocal microscope images of the entire ventral horn were obtained at 20× magnification (n = 4 mice, 6 sections per mouse). Image files were uploaded to Neurolucida, and all tdTomato+:Iba1+ dual-expressing cells, and Iba1+ only cells were manually counted within the ventral horn on both the ipsilateral and contralateral sides of the spinal cord. Because microglia numbers were quite different between sides due to microgliosis after the nerve injury, we also obtained percentage estimates of the number of Iba1+ cells lacking tdTomato labeling.
Experimental design and statistical analysis for histological analyses.
For each injury type, genotype, and time point, we averaged data from 3 to 6 mice. The exact details of the sampling strategy and numbers used can be found in preceding sections detailing each of the analyses. Further explanations are reported in the appropriate results sections. We used either a one- or two-way ANOVA to reveal significant differences according to injury type (sciatic, tibial, or sham) and interactions with time after injury (3 d, 7 d, 14 d, 21 d, and 8 weeks). Significant differences were followed by Bonferroni post hoc t tests for pairwise comparisons. In cases where normality failed, we used nonparametric Kruskal–Wallis one-way ANOVA on ranks followed by Dunn's method post hoc tests. In comparisons between different combination pairs of dual-heterozygous mice, controls, and global or conditional KOs, we used two-tailed t tests. Specific tests used in each experiment are indicated in the text. We pooled together data from males and females because we did not detect statistically significant differences in our measurements according to sex. All α values were set at 0.05. Sample sizes were set to power = 0.80 and varied according to sample variance and the size of the effect. If effect sizes were too small (<10% difference), we did not seek incrementing sample sizes to increase power but interpreted any change too small to be of relevance. “n” usually refers to number of animals (except when indicated in Results) and interanimal variability was usually kept low by performing repetitive measurements in each animal before obtaining one average per animal (for example, 6 MNs per animal estimating VGLUT1 densities; >20 sections per animal for counts of CCR2 cells; 6 sections per animal for microglia cell counts). Some genotypes occur at very low frequencies; and in some cases, “n” is necessarily low because it is unfeasible to obtain larger samples (situations in which we did not achieve the desired statistical power are indicated in the text).
For analyses of different synaptic marker (VGLUT1, VGLUT2, VGAT) densities, we performed paired t tests comparing MNs ipsilateral and contralateral to the injury. This approach avoided biases introduced by differences in immunoreactivity intensity from animal to animal, in particular with the more variable VGLUT2 and VGAT immunoreactions (VGLUT1 immunofluorescence is very consistent from animal to animal).
Peripheral blood mononuclear cell (PBMC) isolation and spinal cord tissue dissociation for myeloid cell analysis:
A total of 12 Ccr2RFP/+ heterozygous and 12 Ccr2RFP/RFP KO mice were used for these experiments. All mice were subjected to a bilateral sciatic transection injury and were allowed to survive for 21 d. On the day of the experiment, each individual mouse was first subjected to a cheek bleed by using a blood lancet to puncture the submandibular vein. Approximately 200 μl of blood was collected in a sterile tube and placed on ice. Red blood cells were lysed using red blood cell lysis buffer (Biolegend), and PBMCs were pelleted by centrifugation. Cells were suspended in 1× HBSS (Hank's Balance Salt Solution without calcium, Invitrogen), and 1 million cells were stained for flow cytometry. Methods are as previously described in more detail (MacPherson et al., 2017).
To harvest the spinal cords, all mice were anesthetized with isoflurane and decapitated. The entire lumbar enlargement was rapidly extracted and placed in a sterile Petri dish containing 1× HBSS where it was finely minced using a scalpel. The tissue solution was transferred to an enzymatic DDP solution (DMEM/F12 media containing 1 mg/ml papain, Roche Diagnostics) and 220 U/ml DNase I (Invitrogen) for dissolution of tissue. The mixture was then incubated at 37°C for 20 min before being neutralized with 10% FBS (Atlanta Biologicals). The solution was centrifuged to create a tissue pellet, and the supernatant was aspirated off. The pellet was homogenized in ice-cold 1× HBSS using a fine-tip glass pipette, filtered through a 70 μm cell strainer, and then resuspended in 37% Percoll, pH 8.5–9.5 (Sigma-Aldrich), with a 70% Percoll layer below and with 30% Percoll and 1× HBSS layers on top. The Percoll gradient was centrifuged, and the myeloid cell cloud was identified and collected between the 70% and 37% layer. Collected cells were washed twice with 1× HBSS.
Experimental design and statistics for flow cytometry.
Each individual blood sample was used for a single “n” in the analysis (CCR2 heterozygotes: n = 12 and CCR2 KO: n = 12). However, preliminary experiments showed a low yield of CCR2 cells in the spinal cord after injury; therefore, 4 mice from each group were pooled together and analyzed as a single set: CCR2 heterozygotes: n = 3 (4 mice in each) and CCR2 KO: n = 3 (4 mice in each). Isolated immune cells from the spinal cord and PBMCs were stained with Live/Dead Fixable Aqua (1:2000, Invitrogen, L34957) and incubated with anti-mouse CD3 PE eFluor 610 (1:100, eBioscience, 61-0031), anti-mouse CD45 PerCP-CY5.5 (1:100, eBioscience, 45-0451), anti-mouse CD11b FITC (1:100, eBioscience, 11-0112), anti-mouse CD11c AlexaFluor-700 (1:50, Biolegend, 117320), anti-mouse Ly6G PacBlue (1:100, Biolegend, 127611), and anti-mouse CD19 PE (1:100, BD Bioscience, 561736), all diluted in FACS buffer. Samples were run on an LSRII Flow Cytometer (BD Biosciences). The antibody panel was extensively validated by antibody titrations using blood and spleen (MacPherson et al., 2017). Background levels were set using antibody isotypes and FMO (fluorescence minus one) controls. The data were analyzed using FlowJo software (version 10.0.6). Isolated cells were gated for size (forward scatter area) and granularity (side scatter area) followed by forward scatter height versus forward scatter area to select single cells. Live cells were detected with Fixable Aqua Live/Dead and gated for CD45. CD45+ immune cells were gated for CCR2-RFP. The following gating was applied to both total cells (CD45+) and CCR2+ cells (CD45+CCR2+). Cells were gated on CD19 (B cells) versus CD3 (T cells). The CD19− CD3− population was gated on CD11b versus Ly6G to identify neutrophils (CD11b+Ly6G+) and microglia/macrophage populations (CD11b+ Ly6G−). Within the microglia/macrophage populations, gates were applied to identify “resting” microglia (CD45low) and activated microglia/macrophage (CD45high). The CD45low and CD45high populations were assessed for CD11c expression. We obtained both raw cell counts and percentages for each cell type with respect to all CCR2 cells. Differences in cell numbers and percentages for each cell type between Ccr2RFP/+ heterozygous and 12 Ccr2RFP/+RFP KO mice were statistically compared using t tests independently in blood and spinal cord samples.
Results
Animal model and experimental design
To investigate the extent of neuroinflammation inside the spinal cord after PNI, we used dual-heterozygous mice in which one Cx3cr1 allele encodes for GFP (Jung et al., 2000) and one Ccr2 allele encodes for RFP (Saederup et al., 2010). CX3CR1 is the receptor for fractalkine and is known to be specifically expressed by microglia within the CNS (Mizutani et al., 2012) and at varying levels in peripheral immune cells, such as circulating Ly6C− noninflammatory monocytes, dendritic cells, and natural killer cells (Harrison et al., 1998; Papadopoulos et al., 1999; Huang et al., 2006). CCR2 is a chemokine receptor known to be expressed by T cells and Ly6C+ CD45high CD11b+ inflammatory monocytes, and it is involved in monocyte tissue trafficking in a variety of neurodegenerative diseases (Bonecchi et al., 1998; Ajami et al., 2011). It is also known to promote hyperalgesia in the spinal cord dorsal horn after PNI (Zhang et al., 2007; Gu et al., 2016). Cx3cr1-GFP and Ccr2-RFP alleles have their respective fluorescent protein-gene constructs knocked into the coding regions of the target cytokine receptor gene resulting in their inactivation. In Cx3cr1GFP/+::Ccr2RFP/+ dual-heterozygotes, one allele expresses the reporter protein, whereas the other maintains expression of the receptors at functional levels. This reporter mouse has been extensively used to differentiate resident microglia from blood-derived monocytes/macrophages in various neurodegenerative models (Ajami et al., 2011; Prinz et al., 2011; Shi and Pamer, 2011), after spinal cord injury (Donnelly et al., 2011; Evans et al., 2014) and after traumatic brain injury (Morganti et al., 2015; Katsumoto et al., 2018), and to study dorsal horn pain mechanisms after PNI (Gu et al., 2016). This is the first study to use this dual-heterozygote model to study neuroinflammation in the ventral horn after PNI. All mice were maintained on a C57BL/6J and N mix genetic background to avoid variability in inflammatory responses and/or differences in synaptic plasticity after PNI, which are known to occur among different strains of mice (Sabha et al., 2008; Jinno and Yamada, 2011).
Our analyses focused on MNs projecting to the LG muscle that were retrogradely labeled with Fast Blue 1 week before any nerve injury, or sham operation (Fig. 1A,B). In some cases, animals did not receive an operation to have a noninjured cohort. CX3CR1-GFP microglia and CCR2-RFP cell infiltration were studied specifically in the locations of retrogradely labeled LG MNs (Fig. 1C,D). Most sections were analyzed in the transverse plane, but we also obtained longitudinal sections in 2 animals at each of 7 and 14 d after injury, to examine the rostrocaudal extent of microgliosis, CCR2-RFP infiltration, and VGLUT1 synapse depletion. Microglia activation was found in dorsal and ventral horns corresponding with areas occupied by the sensory afferent projections and motor pools of the injured sciatic nerve (Fig. 1B,C). Infiltration of CCR2-RFP cells occurs primarily in the ventral horn after a period of strong microgliosis; very few CCR2-RFP cells were seen in the dorsal horn (Fig. 1D). Clusters of infiltrated CCR2-RFP cells were discontinuous in the longitudinal axis, with regions of high and low infiltration through the rostrocaudal extent of the sciatic motor pool (not shown). The same sections were immunostained against VGLUT1 to study changes in the density of muscle spindle afferent synapses (Fig. 1E). Regions with microgliosis, and particularly regions with high numbers of infiltrated CCR2-RFP cells, showed large depletions in VGLUT1 immunoreactivity. Ia afferents are the predominant muscle spindle afferent monosynaptic to MNs; however, Group II afferents also contribute many synapses in the ventral horn and are also VGLUT1-positive (Vincent et al., 2017).
Figure 1.
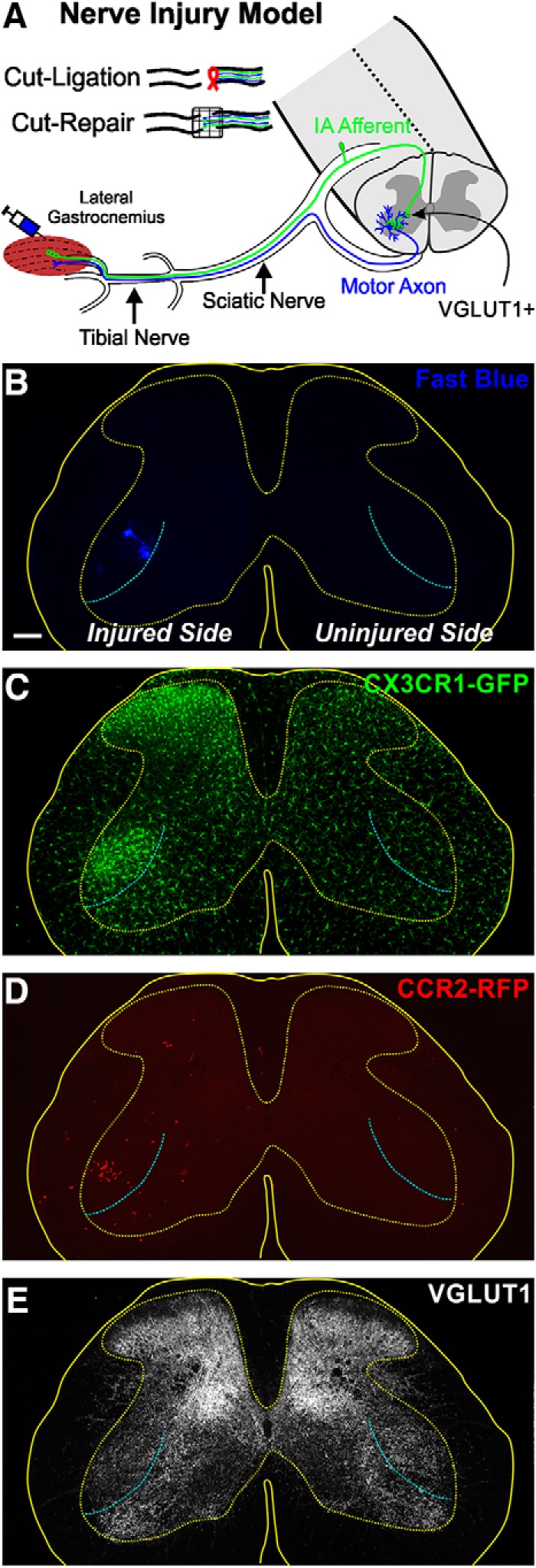
Nerve injury and mouse models. A, The LG muscle of Cx3cr1GFP/+::Ccr2RFP/+ dual-heterozygous mice was injected with Fast Blue to retrogradely label one pool of axotomized MNs. Then, the tibial or sciatic nerve was transected and the proximal stump either ligated to prevent regeneration or aligned to the distal stump with fibrin glue to allow regeneration. Spinal cords were analyzed 3 d to 8 weeks after injury. B–E, 2D projections of confocal image stacks through the 50 μm section thickness showing the four fluorescent signals analyzed: Fast Blue MNs (B), CX3CR1-GFP+ microglia (C), CCR2-RFP+ infiltrating myeloid cells (D), and VGLUT1-immunofluorescence (E). This spinal cord was processed 14 d after sciatic nerve transection. Solid yellow line indicates the spinal cord edge. Dotted line indicates the gray matter. Cyan line indicates the approximate boundary of the sciatic and nonsciatic motor pools. GFP+ microgliosis is visible in the dorsal and ventral horns ipsilateral to the nerve injury. Ventrally microglia accumulate around the axotomized sciatic motor pools. Infiltrating RFP+ myeloid cells accumulate preferentially within the area of ventral microgliosis with little evidence of CCR2-RFP cells in the dorsal horn or the contralateral side. The sciatic motor pool region also shows depleted VGLUT1-immunoreactive punctae ipsilateral to the nerve injury side. Scale bars: B, 100 μm. C–E, Same magnification.
Because LG MNs and afferents might be misguided to incorrect targets, peripheral regeneration is increasingly impaired the more proximal and larger the injured nerve (Sunderland, 1968). To avoid confounding factors related to differences in efficiency and specificity of regeneration in the periphery after SN or TN injuries, we initially prevented regeneration in both nerves by placing a small ligature in the proximal end of the cut nerve. These two injury models are thus referred to as tibial or sciatic transection and ligation (TTL and STL). In these models, we noted shrinkage of LG MNs disconnected from their targets after long-term survival (see below). To analyze resulting VGLUT1 synapse organization after regeneration (8 weeks survival) in KO models and avoid the confounding influence of cellular shrinkage, we also performed sciatic nerve cut and repair injuries (STR). This procedure is permissive of motor axon regeneration and prevents cell shrinkage.
VGLUT1 synapse depletions on MNs following tibial or sciatic nerve injury
We first compared changes in VGLUT1 immunofluorescence densities in the spinal cord ipsilateral and contralateral to the nerve injury. To obtain these measurements, we placed an ROI around the labeled LG MN pool on the side ipsilateral to the injury and a corresponding symmetrical ROI on the contralateral side (Fig. 2A). ROI placement and measurements were performed in single optical planes of low-magnification confocal images obtained with a 10× objective (NA = 0.4) and with a theoretical optical thickness of 4.2 μm. We calculated the integrated pixel density ratio for each pair of images. In sham control mice, the average VGLUT1-immunofluorescence ratio between the two sides was 1.05 ± 0.02 (± SE, n = 4 animals), indicating no differences between sides (Fig. 2B). After TTL or STL, there was a decrease in VGLUT1 fluorescence in the ipsilateral side that was significant by 7 d after injury, continued to decline up to 21 d, and remained depleted 8 weeks after injury. At 8 weeks, VGLUT1 immunofluorescence depletion was significantly larger after STL than TTL (Fig. 2B; two-way ANOVA, time: F(5,45) = 35.663, p < 0.001; type of injury: F(1,45) = 6.143, p = 0.018; STL vs TTL at 8 weeks post hoc Bonferroni STL vs TTL comparison, t test, t = 2.479, p = 0.018).
Changes in VGLUT1 immunofluorescence integrated pixel density can occur because synapses are lost and/or VGLUT1 content decreases (Hughes et al., 2004; Alvarez et al., 2011). To specifically analyze synaptic loss, we compared densities of VGLUT1 synaptic contacts on the surface of reconstructed LG MNs. We selected Fast Blue-labeled MNs with long and well-stained dendrites for high magnification confocal imaging (60×, NA = 1.35) and analysis (Fig. 2C). Cell bodies and proximal dendrites were reconstructed in Neurolucida to obtain estimates of available surface and dendritic lengths (Fig. 2D; Table 2). VGLUT1 boutons in contact with the MN surface were identified and plotted along the soma and dendritic arbor (Fig. 2C,D). If necessary, bouton contacts on MN surfaces were confirmed with 3D visualization using Imaris (Fig. 2E). We found no significant differences in VGLUT1 densities over the cell body or dendrites between sham control mice with a dual-heterozygous genotype and uninjured MNs in WT animals (n = 4 WT animals and 6 dual-heterozygous sham-operated animals, 6 MNs are included in each individual animal estimate; two-tailed t tests, cell bodies: t = −1.150 with 8 df, p = 0.884; dendrites: t = −1.143 with 8 df, p = 0.286). These data show that sham control dual-heterozygous animals have VGLUT1 densities identical to uninjured MNs in WT animals (Fig. 2F,G), and thus they are an appropriate control to compare with dual-heterozygous injured animals. MNs in sham control mice had, on average, a somatic density of 0.75 ± 0.03 (± SE) VGLUT1 contacts per 100 μm2 and overall dendritic linear density of 17.17 ± 0.72 VGLUT1 contacts per 100 μm of dendrite length (n = 6 animals) (Fig. 2F,G). Eight weeks after injury (n = 4 animals in each of TTL and STL injuries), VGLUT1 contacts were significantly depleted on the soma of LG MNs following both TTL and STL (one-way ANOVA, F(2,13) = 62.696, p < 0.001; post hoc Bonferroni t tests vs control, TTL: t = 7.504, p < 0.001, STL: t = 10.619, p < 0.001). VGLUT1 somatic density depletions were significantly greater after STL compared with TTL (post hoc Bonferroni TTL vs STL t test, t = 2.844, p = 0.048). We also noticed that the cell bodies of LG MNs prevented from reinnervating muscle for 8 weeks tended to be smaller and had less surface area available for synaptic connections (Table 2; one-way ANOVA, F(2,13) = 3.563, p = 0.064). Changes in surface area might underestimate synapse losses when comparing densities per unit surface. For this reason, we also compared the absolute number of VGLUT1 contacts per cell body and found a significant 46% reduction with respect to control after TTL and 70% loss after STL (one-way ANOVA, F(2,13) = 55.619, p < 0.001; post hoc Bonferroni t tests, TTL: t = 6.654, p < 0.001; STL: t = 10.161, p < 0.001). Thus, VGLUT1 bouton contacts are lost over MN cell bodies in both types of injuries, and this is partially offset by cell body shrinkage 8 weeks after nerve injury when regeneration is prevented.
While losses in somatic VGLUT1 synapses are significant, most VGLUT1 synapses innervate the proximal dendritic arbor (Rotterman et al., 2014), and this region therefore best represents changes in total input. Overall dendritic VGLUT1 densities were significantly different when comparing sham controls, TTL and STL animals (one-way ANOVA, F(2,13) = 5.410, p = 0.026). While VGLUT1 dendrite densities after STL were significantly depleted (post hoc Bonferroni t test STL vs control, t = 3.259, p = 0.023), VGLUT1 densities after TTL were intermediate between STL and controls and did not reach statistical significance against either (post hoc Bonferroni t test, TTL vs control, t = 1.713, p = 0.344; TTL vs STL, t = 1.411, p = 0.557). To avoid interpretation problems due to differences in the lengths of the dendrites analyzed in different samples (Table 2) and to test whether VGLUT1 bouton contacts were preferentially lost along particular regions of the dendrite, we performed Sholl analysis with bin sizes increasing by 25 μm distance from the cell body center (Table 3; Fig. 2H). In the first 25 μm of dendrite, there was a significant depletion in VGLUT1 linear density for both TTL and STL compared with sham controls (one-way ANOVA, F(2,13) = 23.829, p < 0.001; post hoc Bonferroni t tests vs control, TTL: t = 2.332, p = 0.004; STL: t = 6.659, p < 0.001). In the 25–50 and 50–75 μm bins, we detected significant depletions of VGLUT1 contacts, compared with sham control, following STL but not after TTL (Fig. 2H) (one-way ANOVA, 25–50 μm bin: F(2,13) = 7.996, p = 0.007, post hoc Bonferroni t test vs control, TTL: t = 2.493, p = 0.090; STL: t = 3.863, p = 0.008; 50–75 μm bin: F(2,13) = 9.581, p = 0.004; post hoc Bonferroni t test vs control, TTL: t = 0.783, p = 1.000; STL: t = 4.260, p = 0.004). In conclusion, VGLUT1 densities in different dendritic regions after TTL were always intermediate between controls and STL, and there were no significant differences detected between TTL and STL or TTL and sham controls at distances further than 25 μm from the cell body center.
Table 3.
Morphological characteristics of reconstructed motoneurons and VGLUT1 numbersa
| Control | Tibial ligation | Sciatic ligation | |
|---|---|---|---|
| Soma surface area (μm2) | 3118 ± 142 | 2989 ± 268 | 2377 ± 239 |
| Dendritic length (μm) | 431 ± 28 | 324 ± 55 | 249 ± 45 |
| Dendritic surface area (μm2) | 5014 ± 292 | 3553 ± 920 | 2044 ± 364b |
| # of VGLUT1 contacts on soma | 23.2 ± 1.0 | 12.6 ± 1.6b | 7.0 ± 1.3b |
| # of VGLUT1 contacts on dendrites | 71.6 ± 4.8 | 45.0 ± 6.7b | 28.0 ± 4.8b |
aData are reported as mean ± SE; n = 4 mice, 6 motoneurons per mouse.
bSignificant difference from controls (p < 0.05).
The pattern of VGLUT1 synapse loss is consistent with results from densitometry, suggesting a greater VGLUT1 reduction occurred after STL compared with TTL. However, another variable that could influence densitometry is a change in the size of VGLUT1 puncta. VGLUT1 sizes were compared by surface-rendering VGLUT1 puncta contacting FB-labeled MNs 8 weeks after injury. Average VGLUT1 puncta surface areas were similarly reduced by 52%–55% after TTL and STL injuries from an average value of 12.89 ± 1.33 μm2 (± SE) in sham controls. The reductions in synaptic size were significant for both injuries (Fig. 2I) (one-way ANOVA, F(2,11) = 16.876, p < 0.001; post hoc Bonferroni t test vs control, TTL: t = 5.347, p = 0.001, STL: t = 4.640, p = 0.004), but there was no difference between TTL or STL in VGLUT1 punctae size (post hoc Bonferroni t test TTL vs STL, t = 0.707, p = 1.000). In conclusion, VGLUT1 loss on LG MN cell bodies and proximal dendrites is greater after SN injury compared with TN injury but both injuries result in similar reductions in VGLUT1 puncta size. The latter might be the result of chronic long-term inactivity after preventing reconnection to muscle spindles.
Microglia responses after tibial and sciatic nerve injuries
Starting 3 d after injury, both TTL and STL trigger a microglia response around injured motor pools and in the dorsal horn (Fig. 3A–C). We used the “spot function” algorithm in Imaris to automatically detect and quantify the number of CX3CR1-GFP microglia present in the ventral horn (Fig. 3D) (n = 3 or 4 animals per condition, each animal estimated from 6 ventral horns). Sham control mice had an average of 200.8 ± 16.9 (±SE) CX3CR1-GFP microglia per ventral horn in 50-μm-thick sections. Their distribution was identical to that of Iba1-immunoreactive microglia in WT animals and dual labeling between Iba1 and CX3CR1-GFP in dual-heterozygous animals demonstrated labeling of identical cells with both markers (data not shown). In both tibial and sciatic models, there was a significant increase in the number of CX3CR1-GFP microglia cells with time after injury (Fig. 3E), but there were no significant differences between the two injury types (two-way ANOVA, time: F(5,44) = 25.250, p < 0.001, injury: F(1,44) = 1.701, p = 0.201). Microglia numbers were significantly increased already by 3 d after both injuries (post hoc Bonferroni t tests vs control, t = 6.136, p < 0.001), and the number of microglia remained significantly elevated 21 d after injury before returning to control levels by 8 weeks. These data suggest that, regardless of the site of the nerve injury or the degree of VGLUT1 synaptic loss, microglia proliferation in the spinal cord ventral horn is similar.
Next, we examined whether there were differences in the morphology of activated microglia between TTL and STL (Fig. 3F). Following PNI, microglia processes retract and become thicker, whereas cell bodies increase in volume. These morphological changes are associated with “activation” (Kreutzberg, 1996; Nimmerjahn et al., 2005) and cytokine production (Hanisch, 2002; Hu et al., 2015). We compared the morphologies of randomly selected microglia close to LG MNs in controls or 14 d after either TTL or STL (n = 12 cells per condition, 3 microglia cells per animal). First, we calculated cell body volumes after rendering them using the Imaris “surface module” (Fig. 3G). Control microglia had an average cell body volume of 142.6 ± 11.58 μm3 (± SE, n = 12); and after either TTL or STL, cell body volumes significantly increased by 83% and 107%, on average, with respect to control (one-way ANOVA, F(2,35) = 12.574, p < 0.001; post hoc Bonferroni t test vs control, TTL: t = 3.719, p = 0.001; STL: t = 4.773, p < 0.001). There were no significant differences between TTL and STL injuries (Fig. 3G). Microglia were then reconstructed using Imaris “filament tracker” to estimate process lengths and numbers. For filament skeletonization, we thresholded microglia branches and extensions, not filopodia. Control microglia had an average combined process length of 861.4 ± 96.9 μm (Fig. 3H) and an average of 110.0 ± 17.4 individual filament branches per microglial cell. After both TTL and STL, overall processes lengths were 51.6%–52.6% significantly shorter (one-way ANOVA, F(2,35) = 16.059, p < 0.001, post hoc Bonferroni t test vs control, TTL: t = 4.859, p < 0.001; STL: t = 4.956, p < 0.001), and the number of filament branches was also significantly reduced by 59.0%–57.2% (one-way ANOVA, F(2,35) = 11.378, p < 0.001, post hoc Bonferroni t test vs control, TTL: t = 4.194, p < 0.001; STL: t = 4.07, p < 0.001). Again, we found no differences between injuries (Fig. 3H). Finally, we performed Sholl analysis on the number of processes in regions at increasing 10 μm distances from the cell body center (Fig. 3I; Table 4). In the first 10 μm bin, we found more branches in microglia after TTL and STL compared with control (one-way ANOVA, F(2,35) = 11.420, p < 0.001, post hoc Bonferroni t test vs control, TTL: t = 3.992, p < 0.001, STL: t = 4.271, p < 0.001). In the 20 μm distance bin, there was no difference between groups, but thereafter there were significantly fewer branches after TTL or STL compared with controls (one-way ANOVA, F(2,35) = 17.871, p < 0.001, post hoc Bonferroni t test vs control, TTL: t = 4.873, p < 0.001; STL: t = 5.436 p < 0.001). This indicates that TTL and STL lesions caused similar morphological changes in ventral horn microglia located close to injured LG MNs.
Table 4.
Sholl analysisa
| Control |
Tibial ligation |
Sciatic ligation |
|||||||
|---|---|---|---|---|---|---|---|---|---|
| 25 μm | 50 μm | 75 μm | 25 μm | 50 μm | 75 μm | 25 μm | 50 μm | 75 μm | |
| Dendritic length (μm) | 51.0 ± 3.7 | 177.9 ± 10.1 | 124.7 ± 11.8 | 52.1 ± 4.3 | 139.1 ± 14.6 | 54.7 ± 16.2b | 60.3 ± 4.8 | 140.2 ± 21.2 | 42.3 ± 15.9b |
| # of VGLUT1 | 10.6 ± 0.9 | 33.2 ± 1.9 | 17.6 ± 1.9 | 7.6 ± 0.6 | 20.9 ± 2.8b | 10.3 ± 2.9 | 7.6 ± 1.2 | 16.9 ± 2.4b | 3.8 ± 1.4b |
| Dendritic density (per 100 μm) | 21.7 ± 0.8 | 19.2 ± 1.0 | 14.5 ± 0.5 | 14.6 ± 0.4b | 15.0 ± 0.8 | 13.1 ± 1.9 | 10.8 ± 2.0b | 12.7 ± 1.9b | 7.2 ± 1.5b |
aMotoneuron morphological details by increasing bins 25 μm from cell body. Data are reported as mean ± SE; n = 4 mice, 6 motoneurons per mouse.
bSignificant difference from controls (p < 0.05).
Microglia activation is necessary for VGLUT1 synapse loss, but not for “synapse stripping”
To test whether microglia activation is necessary for inducing the loss of VGLUT1 synapses in the ventral horn, we used animals carrying floxed csf1 alleles to remove this gene from MNs. The csf1 gene encodes for CSF1, a secreted cytokine that acts on microglia through its receptor (CSF1R) to maintain microglia in the basal state or induce microglia proliferation and activation after injury or disease (Elmore et al., 2014). Removal of csf1 from all spinal neurons using a nestin-Cre driver prevented spinal microglia activation after PNI in both the dorsal and ventral horn without changes in basal microglia numbers before the injury (Guan et al., 2016). Thus, CSF1 might be a key signal secreted by axotomized MNs to induce activation of specifically ventral horn microglia after PNI. We removed csf1 from MNs using Chat-IRES-Cre mice and compared microglia proliferation 14 d after injury and VGLUT1 synaptic densities on FB LG MN 8 weeks after injury. In this case, we prevented MN shrinkage by repairing the nerve with fibrin-glue immediately after injury. This nerve injury and repair model results in extensive motor axon regeneration and reinnervation of neuromuscular junctions by 8 weeks after injury (data not shown).
Microglia were detected in these animals by Iba1-IR. We compared Chat+/+::csf1flox/flox with ChatIRES-Cre/+::csf1flox/flox animals. Removal of CSF1 from MNs prevented microglia proliferation in the ventral horn after PNI, whereas dorsal horn microgliosis remained intact (Fig. 4A,B). Numbers of Iba1-IR microglia in Chat+/+::csf1flox/flox increased to 304.9 ± 7.3 cells per ventral horn after injury (similar to dual-heterozygote Cx3cr1GFP/+::Ccr2RFP/+ animals after a comparable injury) but did not increase after injury in ChatIRES-Cre/+::csf1flox/flox (Fig. 4G) (177 ± 6.3 cells per ventral horn). These differences in microglia numbers after nerve injury were significant between genotypes (two-tailed t test, t(6) = 13.188, p < 0.001, n = 4 animals per genotype; each animal was analyzed in 6 ventral horns containing FB MNs). We did note that Iba1-IR was nevertheless more robust in ventral horn microglia on the injured side, suggesting that CSF1 removal did not completely abolish all microglia activation, but fully prevented microglia proliferation and migration toward injured motor pools. We confirmed deletion of CSF1 using antibodies against CSF1 combined with NeuN (Fig. 4C,D). Axotomized MNs in Chat+/+::csf1flox/flox animals downregulated NeuN and upregulated CSF1 (Fig. 4C; n = 3 of 3 animals), but CSF1 expression was not visible in MNs in the injured side of ChatIRES-Cre/+::csf1flox/flox animals (Fig. 4D; n = 4 of 4 animals).
Figure 4.

VGLUT1 synapses are rescued by genetic attenuation of microglia responses in the ventral horn after deleting CSF1 from MNs. A, B, Microglia activation in the dorsal and ventral horn 14 d after sciatic nerve transection and repair in animals in which csf1 is genetically deleted from MNs (B) or not (A). Ventral horn microglia response is significantly attenuated after removing csf1 from MNs. C, D, Confirmation of CSF1 deletion. CSF1 is upregulated in axotomized MNs after nerve injury, but CSF1 is not present following csf1 deletion in Chat-expressing neurons. Section counterstained with NeuN. NeuN is downregulated in axotomized MNs (Alvarez et al., 2011). E, F, VGLUT1 synapses on Fast Blue LG MNs with csf1 preserved (E) or removed (F). Images represent a single confocal plane (left) and the 3D Neurolucida reconstructions (right). Reconstructions correspond to MNs with an asterisk. G, Quantification of microglia numbers 14 days post injury in the ventral horn of animals with csf1 removed from MNs (ChatCre/+::csf1 flox/flox) and genetic controls (Chat+/+::csf1flox/flox). Dashed lines indicate the average number of ventral horn microglia per section in dual-heterozygote Cx3cr1GFP/+::Ccr2RFP/+ animals in sham controls (dark dashed line) or after sciatic nerve transection and ligation (dashed dotted line). Removal of csf1 from MNs prevents the increase in microglia numbers after nerve injury. ***p < 0.001, between animals with csf1 removed or preserved (t tests). H, I, VGLUT1 densities on the cell body (surface densities, H) and on the dendrites (linear density, I) eight weeks after injury in injured animals (white bars) with csf1 removed or not compared with uninjured animals of the same genotypes. MNs with csf1 preserved display the excepted reductions in VGLUT1 densities. After removal of csf1, MNs display densities similar to uninjured, suggesting rescue. Moreover, removal of csf1 from Chat-expressing neurons did not affect VGLUT1 densities (these are similar to uninjured WTs and sham controls in Fig. 2). Error bars in all histograms indicate SE. Each dot represents 1 animal n = 4 animals per bar. For microglia numbers, each animal value is the average of 6 ventral horns. For estimating VGLUT1 densities, each animal's estimate results from averaging 6 reconstructed MNs. H, ***p < 0.001, compared with all other groups (post hoc Bonferroni t tests). I, **p < 0.01, compared with all other groups (post hoc Bonferroni t tests). Scale bars: A–D, 200 μm; C, Inset, 50 μm; E1, F1, 20 μm.
Downregulation of the microglia reaction specifically in the ventral horn rescued VGLUT1 synapses on LG MNs, both on their dendrites and cell bodies when analyzed 8 weeks after the injury (Fig. 4E,F,H,I). In Chat+/+::csf1flox/flox animals (n = 4), VGLUT1 densities were reduced to similar levels as those observed in dual-heterozygote Cx3cr1GFP/+::Ccr2RFP/+ animals after SN transection. VGLUT1 densities in ChatIRES-Cre/+::csf1flox/flox animals (n = 4) were similar to uninjured MNs in ChatIRES-Cre/+::csf1flox/flox (n = 4) or Chat+/+::csf1flox/flox animals (n = 4) (Fig. 4H,I) These were also similar to dual-heterozygote sham control animals and uninjured WT MNs (compare with Fig. 2F,G). Thus, soma and dendritic VGLUT1 synaptic densities were significantly different between MNs in ChatIRES-Cre/+::csf1flox/flox and Chat+/+::csf1flox/flox animals 8 weeks after injury, but there were no significant differences between injured MNs in ChatIRES-Cre/+::csf1flox/flox and uninjured MNs in ChatIRES-Cre/+::csf1flox/flox and Chat+/+::csf1flox/flox (somatic analyses: one-way ANOVA, between the four groups of animals, F(3,15) = 24.057 p < 0.001; post hoc Bonferroni t tests, p < 0.001 between MNs in injured Chat+/+::csf1flox/flox and all three other groups; p > 0.05 in all comparisons of injured Chat+/+::csf1flox/flox and the other groups: injured ChatIRES-Cre/+::csf1flox/flox animals and uninjured ChatIRES-Cre/+::csf1flox/flox and Chat+/+::csf1flox/flox; dendritic analyses: one-way ANOVA, between the four groups of animals, F(3,15) = 12.462 p < 0.001; post hoc Bonferroni t tests, p < 0.01 between MNs in injured ChatIRES-Cre/+::csf1flox/flox and all three other groups; p = 1.000 in all comparisons between the other groups).
We compared these results with the “synaptic stripping” of all synapses on cell bodies after nerve injury by analyzing synaptic coverage of their cell bodies by all excitatory (VGLUT1 and VGLUT2) and inhibitory synapses (marked with vesicular GABA/glycine amino acid transporter [VGAT]). We elected to analyze 14 d after injury because there is significant “synaptic stripping” of different types of synapses at this time, whereas “stripped” VGLUT2 and VGAT synapses generally come back after MNs reinnervate muscle (Alvarez et al., 2011). Because there is higher interanimal variability in VGLUT2 and VGAT immunostaining, we compared synaptic coverage on FB-retrogradely labeled LG MNs ipsilateral and contralateral with the injury side within animals. We analyzed 3–10 MNs per side and per animal (mean ± SD, 7 ± 3 MNs; n = 5 animals). Each MN was imaged at high magnification (60×2) with confocal microscopy (Fig. 5A), and the cell body membrane surface was reconstructed for a 5–7 μm depth at a mid-plane region (a level containing nucleus and nucleolus) (Fig. 5B). The area corresponding to emerging dendrites in the reconstructed surface was estimated and subtracted (Fig. 5C). This method allowed us to increase sample sizes by permitting inclusion of FB-labeled MN cell bodies, even when not fully contained within the 50 μm thickness of the tissue sections. It also prevented synapse density overestimations by counting synapses only in planes orthogonal to the imaging. Paired comparisons removed any biases due to immunolabeling differences among animals (Fig. 5E). We counted all appositions of VGLUT and VGAT clusters. Many excitatory synapses establish only one active zone over MN cell bodies, corresponding to one presynaptic cluster. However, inhibitory synapses frequently establish multiple well-separated active zones (Alvarez et al., 1997); thus, VGAT immunofluorescent puncta frequently occur in groups of close-spaced clusters possibly belonging to the same synaptic bouton (Fig. 5D). Because we did not label the totality of the presynaptic bouton, we counted all immunoreactive puncta appositions for each of the three vesicular markers. Given the differences in active zone number and structure, VGAT densities were higher than expected when compared with VGLUT1 and VGLUT2 densities, considering known relative abundances of inhibitory and excitatory synapses on MNs (Conradi et al., 1979; Kellerth et al., 1979, 1983).
Figure 5.

“Synaptic stripping” is independent of microglia activation. A–C, Method for assessing synaptic densities on MN cell bodies. A, Three representative confocal optical sections separated by 2 μm z distance through the midplane of a Fast Blue MN. White represents VGLUT2-immunoreactive puncta. B, A slab of mid-cell body region reconstructed from 15 such optical sections (7 μm wide; z step: 0.5 μm) and VGLUT2 contacts marked on its surface (red circles). C, A similar reconstruction on a different MN marking exits of dendrites on the reconstructed surface. These areas were subtracted from the total surface of the neuronal slab to estimate the density only on the membrane surface available for synapses on the cell body. D1, VGAT contacts (white) on the cell surface; single optical plane confocal image. D2, D3, High-magnification and rotation showing a cluster of closely spaced VGAT contacts. Each was counted separately but likely represents independent vesicular accumulations in a single terminal and related to the multiplicity of active zones that inhibitory synapses form over cell bodies of MNs. E, Quantification of vesicular marker contact densities comparing MNs in the side ipsilateral (injured MNs) or contralateral (control MNs) to the injury in animals with normal microglia activation (Chat+/+::csf1flox/flox) or blunted microglia activation (ChatCre/+::csf1 flox/flox) (n = 5 animals per genotype; 3–10 MNs analyzed per genotype and side; all MNs were pooled together in a single average per animal, genotype, and side). VGLUT1 and VGLUT2 synapses were both significantly depleted after injury independent of genotype (p < 0.001). VGAT synapses were also depleted with microglia activation preserved or blunted, but depletions were lower compared with VGLUTs, but still significant (p < 0.05). There was more interanimal variability in VGAT depletions, and this was identified as originating in sex differences (in all graphs, blue represents female data points). All replace for comparisons, plural were paired t tests between control and injured side. Scale bars, 20 μm. *p < 0.05; **p < 0.01; ***p < 0.001.
Comparison of synaptic vesicle puncta densities in axotomized MNs and control MNs revealed significant depletions for all three synaptic markers 14 d after injury in Chat+/+::csf1flox/flox and ChatIRES-Cre/+::csf1flox/flox animals, even when, in the latter, the ventral microglia reaction after PNI was significantly reduced (for both genotypes: VGLUT1, p < 0.001; VGLUT2, p < 0.001; VGAT, p < 0.05; paired t tests) (Fig. 5E). Reductions in synaptic density were always higher for excitatory synaptic vesicle clusters (VGLUT1: −46.4 ± 5.4% [SEM] reduction; VGLUT2: −45.4 ± 14.0% [SEM] reduction) than for inhibitory synapses (VGAT: −25.3 ± 17.7% [SEM] reduction) in agreement with previous studies reporting the better preservation of inhibitory synapses in axotomized spinal MNs (Lindå et al., 2000; Alvarez et al., 2011). The higher variation in synaptic reductions observed for VGAT synapses resulted from a better preservation in females than in males (1 female analyzed in the Chat+/+::csf1flox/flox sample and 2 in ChatIRES-Cre/+::csf1flox/flox animals, all with no reductions in VGAT synapse densities). This is the only sex difference noted in all datasets in this manuscript, and its significance will need to be investigated in future studies. Comparison of percentage reductions in synaptic density between Chat+/+::csf1flox/flox and ChatIRES-Cre/+::csf1flox/flox animals showed no differences for VGLUT2 and VGAT synapses; the synapses were reduced to similar levels after PNI with or without ventral microglia activation (one-tailed t tests: VGLUT2, t(8) = −1.321, p = 0.117; VGAT, t(8) = −1.091, p = 0.154; in both, 5 animals for each genotype). However, reductions in VGLUT1 synapses were significantly different between genotypes, suggesting partial preservation when microglia are not fully activated (−34.1 ± 2.8% [SE] reduction in ChatIRES-Cre/+::csf1flox/flox animals, compared with −46.4 ± 5.4 [SE] reduction in Chat+/+::csf1flox/flox animals; t(7) = −12.225, p = 0.00487).
These results suggest that “synaptic stripping” is not prevented in the absence of microglia activation. The fact that VGLUT1 synapses are not significantly depleted in MNs 8 weeks after injury after MNs reinnervate muscle indicates that, in the absence of microglia activation, “stripped” VGLUT1 synapses are re-formed after regeneration, similar to the transient plasticity of VGLUT2 and VGAT synapses (Alvarez et al., 2011). These data thus suggest that “synaptic stripping” mechanisms are microglia-independent, but the permanent disappearance of VGLUT1 synapses (and their axons) from the ventral horn is microglia-dependent. The results also demonstrate that: (1) dorsal and ventral horn microglia are activated after PNI by signaling from different cell types (dorsal horn microglia are activated by CSF1 released from sensory afferents [Guan et al., 2016], ventral microglia by MN-derived CSF1); (2) CSF1 receptor activation is necessary for proliferation and migration of ventral horn microglia after PNI; (3) 8 weeks after transection of the sciatic nerve, VGLUT1 synapse loss is identical in MNs that are either prevented from (ligation) or allowed to (repair) regenerate; and (4) blunting microglia activation, specifically in the ventral horn, prevents the permanent removal of VGLUT1 synapses from axotomized and then regenerated MNs.
The number of CCR2-RFP cells infiltrating the ventral horn depends on injury type
Although microglia activation appears necessary for permanent removal of VGLUT1 synapses, further mechanisms may account for the difference in synaptic losses between tibial and sciatic nerve injuries. To address this issue, we analyzed the possibility that infiltration of peripheral immune cells labeled by expression of CCR2 plays a role. In the spinal cord of sham control mice, there is an average of 1.3 ± 0.2 CCR2-RFP cells (±SE, n = 4 animals) per 50-μm-thick section (all sections analyzed contained FB-labeled MNs). Most CCR2-RFP cells in the spinal cord of naive animals are elongated perivascular macrophages that also express weak CX3CR1-GFP (see below). After nerve injury, many CCR2-RFP cells infiltrate the spinal cord (Fig. 6A–D), more prominently the ventral horn, where they associate with injured motor pools and are sometimes opposed to FB-labeled MN cell bodies. However, CCR2-RFP cell entry is delayed with respect to microglia proliferation, peaking 21 d after injury. There was significant variability in the number of CCR2-RFP cells from section to section because entry is not homogeneous throughout the rostrocaudal extent of the injured sciatic motor pools. To best estimate CCR2-RFP cell entry, we counted cells in the ventral horns of every transverse section containing FB-labeled MNs (15–26 sections per animal; n = 3 or 4 animals per time point and injury type).
Figure 6.

CCR2+ cells infiltrate the ventral horn of the spinal cord after nerve injury, and CCR2 activation is necessary for VGLUT1 losses from dendrites. A–C, 2D projections of confocal image stacks (50-μm-thick section) showing CCR2+ cells (white cells) infiltrated in the spinal cord 3 or 21 d after either tibial or sciatic nerve transections. These cells are located predominantly in the ventral horn, close to retrogradely labeled LG MNs (blue). The area highlighted in the rectangle is shown at higher magnification below. D, Time course of CCR2+ cell infiltration after tibial (red line) or sciatic (blue line) nerve injury. Data are represented as average number of total RFP+ cells in the ventral horn of 50-μm-thick spinal cord sections. Each data point represents the average from all L4-L5 sections containing Fast Blue MNs in each mouse (average of 23.2 sections per animal). Lines indicate the average of 3 or 4 mice per time point. Error bars indicate SE. Significant increases in CCR2+ cells compared with control are found from 3 d to 8 weeks after injury for both injuries (black asterisks), whereas significant differences between STL and TTL are found at 14 d, 21 d, and 8 weeks after injury (gray asterisks): *p < 0.05; ***p < 0.001. E, Time course of CX3CR1-GFP+ cell activation (green lines), infiltration of CCR2-RFP+ cells (red lines), and overall VGLUT1 loss after sciatic or tibial nerve injury (blue lines) normalized to maximum change for each condition and measurement. The time course of VGLUT1 loss overlaps with CCR2+ cell entry; both occur at a delay after the onset of microglia activation. F, VGLUT1 synapses on LG reconstructed MNs after injury in dual-heterozygote Cx3cr1GFP/+::Ccr2RFP/+ animals compared with CCR2 KOs. G, Quantification of VGLUT1 synapses on LG MN dendrites. Dual-heterozygote animals, but not CCR2 KOs, show a significant decrease in dendritic synapses (***p < 0.001), each compared with their respective sham controls (n = 6 animals per group; each animal estimate is the average of 6 MNs per animal; error bars indicate SE). H, VGLUT1 densities on cell bodies. There was only partial preservation in CCR2 KO animals: *p < 0.05; ***p < 0.001. Scale bars: A1, A2, 100 μm; B1, C1, B2, C2, respectively, at the same, F, 50 μm.
CCR2-RFP cells in the ventral horn significantly increased with time after injury, and there was also a significant difference between injuries (two-way ANOVA: time, F(5,43) = 74.099, p < 0.001; injury type, F(1,43) = 27.930, p < 0.001). The number of intraparenchymal CCR2-RFP cells started to increase the first week after injury. Although this infiltration was small 3 and 7 d after injury (Fig. 6D), it was nevertheless significant given the practical nonexistence of these cells before the injury (post hoc Bonferroni t tests vs control, 3 d: t = 3.728. p = 0.011; 7 d: t = 6.643, p < 0.001). These cells greatly increased in number by 14 d after injury, peaked at 21 d, and remained elevated 8 weeks after injury (post hoc Bonferroni t tests vs control, 14 d: t = 14.179, p < 0.001; 21 d: t = 16.254, p < 0.001; 8 weeks: t = 6.522, p < 0.001). At 14 and 21 d after injury, there were significantly more CCR2-RFP cells following STL compared with TTL, and a small difference was maintained at 8 weeks (post hoc Bonferroni t tests STL vs TTL, 14 d: t = 5.183, p < 0.001; 21 d: t = 4.739, p < 0.001; 8 weeks: t = 2.156, p = 0.039).
CCR2 activation is necessary for elimination of VGLUT1 synapses from axotomized MN dendrites
Comparing the time courses of the CX3CR1-GFP microglia reaction, CCR2-RFP cell infiltration, and VGLUT1 immunofluorescence decrease (Fig. 6E), we found that in both STL and TTL nerve injuries, microglia activation precedes infiltration of CCR2-RFP cells, and the latter tightly associates with the time course of VGLUT1 loss. The temporal coincidence between CCR2-RFP cell entry and VGLUT1 decrease suggests a possible role for these cells in the permanent degradation of Ia/II VGLUT1 afferent synapses. Because CCR2 activation is necessary for monocyte kinesis and infiltration into peripheral tissues (Deshmane et al., 2009), we took advantage of Ccr2RFP/RFP homozygotes in which both Ccr2 alleles are inactivated (CCR2 KOs) while preserving CCR2-RFP labeling. We tested whether VGLUT1 synapses are preserved in CCR2 KOs by analyzing VGLUT1 densities on reconstructed LG FB-labeled MNs 8 weeks after sciatic nerve injury and repair (STR) (Fig. 6F–H). We first compared baseline somatic and dendritic VGLUT1 densities in sham-operated CCR2 KOs (n = 6 animals) with their dual-heterozygote counterparts (n = 6) and found no significant differences on cell bodies (post hoc Bonferroni t test dual-heterozygote control vs CCR2 KO, t = 0.760, p = 1.000) or dendrites (post hoc Bonferroni t test dual-heterozygote control vs CCR2 KO, t = 0.913, p = 1.000). Therefore, lack of CCR2 has no impact on VGLUT1 synapse density on MNs in noninjured animals. STR injury significantly reduced somatic (post hoc Bonferroni t test dual-heterozygote sham vs dual-heterozygote cut, t = 6.170, p < 0.001) and dendritic densities (post hoc Bonferroni t test dual-heterozygote sham vs dual-heterozygote cut, t = 4.708, p < 0.001) in dual-heterozygote mice compared with sham controls, but in CCR2 KOs VGLUT1 synapses on dendrites were preserved, with densities similar to age- and genotype-matched sham controls (post hoc Bonferroni t tests, vs sham CCR2 KO, t = 0.0167, p = 1.000; vs sham dual-heterozygote, t = 0.930, p = 1.000). Consequently, VGLUT1 densities on dendrites after STR injury were significantly larger in CCR2 KOs compared with dual-heterozygotes after STL injuries (post hoc Bonferroni t test, t = 3.778, p = 0.007). On the cell bodies, we found intermediate densities of VGLUT1 synapses in CCR2 KOs compared with sham control animals or dual-heterozygotes after STR. Compared with their respective sham control animals, there was a 54.1% loss in VGLUT1 density in dual-heterozygotes but only a 30.5% loss in CCR2 KO animals. Differences reached significance compared with sham control animals (post hoc Bonferroni t tests, vs sham CCR2 KO, t = 3.251, p = 0.024), but not with injured dual-heterozygote animals (post hoc Bonferroni t tests, control vs injured CCR2 KOs, t = 3.251, p = 0.024; injured dual-heterozygote vs injured CCR2 KO, t = 2.159, p = 0.259). Thus, CCR2 genetic deletion rescues many VGLUT1 synapses, particularly on MN dendrites. Examination of the sections nevertheless revealed many CCR2-RFP cells still present inside the spinal cord of CCR2 KOs after injury. Next, we investigated the types of CCR2-RFP cells inside the spinal cord of Ccr2RFP/+ heterozygotes and Ccr2RFP/RFP KOs after sciatic nerve injury.
Heterogeneity of CCR2-RFP cells infiltrating the spinal cord after nerve injury
To better understand the types of CCR2-RFP cells infiltrating the spinal cord, we performed histological analyses 21 d after STL injuries (peak infiltration: Fig. 6D). These analyses revealed heterogeneity in regard to morphology, CX3CR1-GFP expression, and relations with CX3CR1-GFP microglia. The majority of CCR2-RFP cells had no CX3CR1-GFP expression and displayed a ruffled round or ellipsoid morphology (Fig. 7A). When tested with CD3ε, a marker of T cells, 65.4 ± 5.2% (± SE) of ventral horn CCR2-RFP cells were positive (n = 6 animals, 6 sections with retrogradely labeled LG MNs analyzed per animal), and almost all CD3ε+ cells express CCR2-RFP (87.1 ± 1.7%) (Fig. 7B). CCR2-RFP T cells were frequently located close to microglia and sometimes formed large cell clusters with activated microglia.
Figure 7.

CCR2-RFP cells infiltrating the spinal cord ventral horn. A1–A3, Round and elongated CCR2-RFP cells lacking CX3CR1-GFP. Some of these cells are isolated (top arrow), but more frequently interact with microglia (bottom arrow). B1–B3, Many CCR2-RFP cells are positive for CD3ε, a T-cell marker. C1, D1, E1, F1, Confocal images (2D projections) of different types of dual-labeled CCR2-RFP/CX3CR1-GFP cells. C2, D2, E2, F2, CX3CR1-GFP expression. C3, D3, E3, F3, CCR2-RFP and CX3CR1-GFP merge images. C4, C5, D4, D5, E4, E5, F4, F5, CCR2-RFP content inside these cells, confirmed by surface rendering CX3CR1-GFP cells in Imaris and rotating the 3D volume. F4, F5, The surface of a blood vessel (stained by background VGLUT1 labeling) is also surface rendered. C, Rounded cell with low CX3CR1 fluorescence. D, Cell with few processes. E, Cell with multiple process and high CX3CR1 fluorescence and microglia-like morphology. F, Perivascular cell. G, Time course of CCR2-RFP/CX3CR1-GFP dual-expressing cells after tibial (red line and dots) or sciatic (blue line and dots) injuries. Data points represent individual animals (average of n = 23.2 sections per animal/data point). Lines indicate averages at each time point. Scale bars indicate SE (n = 4 animals per time point). Black and gray asterisks, respectively, represent significant differences with controls or between injury types at individual time points: **p < 0.01; ***p < 0.001. A significant infiltration of dual-labeled cells occurs after both injuries starting at 3 d and lasting for 8 weeks, but a peak increase in entry at 14 d occurs only after sciatic injuries. H, Genetic lineage labeling demonstrating cells derived from resident microglia (Iba1 and tdTomato colabeled) or sources other than resident microglia (labeled only with Iba1) 21 d after sciatic injuries. These include cells in close apposition to MN cell bodies (arrow). I–L, Change in microglia and CCR2-RFP cell numbers in CCR2 heterozygotes compared with CCR2 KOs 21 d after sciatic nerve injury and repair (STR). CCR2 heterozygotes are CX3CR1-EGFP:CCR2-RFP dual-heterozygotes. In all cases, n = 6 animals (each represented by a different dot). Errors bars indicate SE. *p < 0.05 (t tests). ***p < 0.001 (t tests). Deltas indicate effect sizes. Scale bars: A–F, 10 μm; H, 20 μm.
Non-T cells represent ∼35% of CCR2-RFP cells and could contain a number of different cell types, including monocytes/macrophages. A minority of these cells coexpress CX3CR1-GFP. Dual-labeled cells displayed variable morphologies, from round/ellipsoid (Fig. 7C1–C5) to microglia-like with many processes (Fig. 7E1–E5) and some intermediate (Fig. 7D1–D5). RFP and GFP fluorescence were inversely correlated and varied according to morphology. Rounded cells were CCR2-RFPhigh, CX3CR1-GFPlow, whereas microglia-like cells were CCR2-RFPlow, CX3CR1-GFPhigh. RFP intensity was reduced with increased size and number of processes, suggesting downregulation of CCR2 while establishing a CX3CR1high microglia-like phenotype. A less abundant type of dual RFP/GFP dual-labeled cell corresponds to elongated perivascular macrophages (Fig. 7F1–F5). These cells were always apposing blood vessel surfaces (the VGLUT1 polyclonal antibody renders weak nonspecific background immunolabeling of blood vessels; for description of this nonspecific immunoreactivity in VGLUT1 KOs, see Siembab et al., 2016). A few perivascular macrophages were present before the injury, and their number increased after the injury.
Dual-labeled cell numbers varied according to injury type (Fig. 7G). They significantly increased after both injuries (two-way ANOVA time: F(5,43) = 52.265, p < 0.001, injury: F(5,43) = 14.631, p < 0.001; post hoc Bonferroni t test vs control, TTL: t = 4.42–7.15, all time points, p < 0.001; STL: t = 4.94–13.73, all time points, p < 0.001), and peaked at 14 d after STL. This peak more than doubles the number of dual-labeled cells present at 3 and 7 d after injury. In contrast, dual-labeled cell numbers were similar from 3 d to 8 weeks after TTL. The interaction between injury type and time after injury was significant for the number of dual-labeled cells (two-way ANOVA time × injury: F(5,43) = 7.740, p < 0.001). Correspondingly, pairwise comparisons showed significantly larger numbers of dual-labeled cells at 14 and 21 d after STL (post hoc Bonferroni t tests: TTL vs STL, 14 d: t = 6.627, p < 0.001; 21 d, t = 2.789, p = 0.009). Because many of these cells seem to actively downregulate CCR2-RFP after entering the spinal cord, their numbers are best interpreted as a reflection of steady-state infiltration rates and not total cell accumulations. Thus, maximum presence of dual-labeled cells occurs 14 d after injury, and CCR2-RFP downregulation may account in part for their rapid decrease. In contrast, the total number of CCR2-RFP cells further increases from 14 to 21 d after injury in both STL and TTL injuries (Fig. 5D). This might represent increases in infiltration of “RFP only” T cells or upregulation of CCR2 in infiltrated T cells.
The results suggest the possible presence of a dynamic population of CCR2-RFP cells converting into CX3CR1-GFP microglia-like cells, specifically after STL injuries. To test whether a proportion of microglia after STL injuries are blood-derived, we genetically labeled all resident microglia in juveniles before any injury using Cx3cr1CreER mice crossed with Ai9 R26 tdTomato reporter mice. In these mice, tamoxifen drives tdTomato expression in microglia and several other CX3CR1-expressing peripheral immune cells. However, due to the fast replacement of peripheral immune cells compared with the low turnover of microglia (Tay et al., 2017), only resident microglia remain labeled 4–5 weeks after tamoxifen injections (Goldman et al., 2013). Tamoxifen was injected at postnatal days 12 and 14, and sham (n = 3 animals) or nerve injury surgeries (STL) (n = 8 animals) were performed at 3 months of age 1 week after retrogradely labeling LG MNs with Fast Blue. The spinal cord sections were collected at 21 d after surgery for sham and half of the nerve injury animals (n = 4), the rest of the animals were analyzed 8 weeks after injury. Iba1 immunolabeling enabled identification of microglia-like cells. In sham control animals, 99.8% of Iba1+ cells were tdTomato+ and only an average of 0.3 ± 0.2 (± SE) cells per ventral horn section lacked tdTomato. A similar result was found at 8 weeks after injury (99.3% of Iab1+ cells were tdTomato+ representing 1.4 ± 1.8 cells per ventral horn). In contrast, Iba1+ cells lacking tdTomato were frequently observed at 21 d after injury, sometimes in direct contact with axotomized FB-labeled LG MNs (Fig. 7H). Their numbers were low, however, representing 2.6% of the total Iba1+ cell population in the ventral horn or, on average, 8.2 ± 5.2 cells per ventral horn section. They were variable from animal to animal, ranging from 1.1% to 4.8% of the total ventral horn Iba1+ microglia population at 21 d after injury. It is quite possible that these are underestimates given that Cx3cr1CreER mice reportedly exhibit some level of tamoxifen-independent recombination (Fonseca et al., 2017). The data nevertheless demonstrate the presence of a transient population of blood-derived Iba1+ microglia at 21 d after injury, whose exact number and fate will need to be confirmed with alternative lineage-labeling strategies.
KO of CCR2 depletes dual CCR2-RFP/CX3CR1-GFP cells but not CCR2-RFP T cells
To examine which populations of CCR2-RFP cells are affected in CCR2 KOs, we analyzed microglia, T cells, and dual-labeled RFP/GFP cell numbers in the ventral horn after sciatic nerve transection with repair (STR). The number of Iba1+ microglia in the ventral horn of CCR2 KO mice 21 d after STR was significantly decreased by 15% compared with the number of CX3CR1-GFP microglia cells in dual-heterozygote mice (n = 6 animals per condition; two-tailed t test, t = 2.563, p = 0.03) (Fig. 7I). There was also a significant 29% decrease in the overall number of CCR2-RFP cells in CCR2 KOs (one-tailed t test, t = 2.195, p = 0.026) (Fig. 7J). The biggest reduction was found comparing RFP+/GFP+ cells in dual-heterozygote mice to the number of RFP+/Iba1+ cells in CCR2 KOs; this constituted a 79% decrease (Mann–Whitney Rank Sum Test, t = 57.000, p = 0.002) (Fig. 7K). Indeed, remaining dual-labeled cells were mostly perivascular macrophages and were so few in number that the data distribution did not reach normality. This can be interpreted as evidence that a population of CCR2-RFP cells converts into CX3CR1 microglia explaining the parallel reductions in dual CCR2/CX3CR1 cells, CCR2-RFP cells overall, and also the total number of Iba1+ microglia. In contrast, T cells were not prevented from entry into the spinal cord in CCR2 KOs (Fig. 7L). Indeed, the proportion of CCR2-RFP cells expressing CD3ε was significantly augmented by 24% (two-tailed t test, t = −2.567, p = 0.028), as expected if non-T cells are partially depleted. This might suggest that T-cell entry is unrelated to VGLUT1 synapse losses. In contrast, dual-labeled CCR2/CX3CR1 cells are prevented from entry in parallel with preservation of VGLUT1 synapses. The most likely origin of these cells are CCR2+ CD45high CD11bhigh Ly6C+ inflammatory monocytes known to acquire the phenotype of resident macrophages in the tissues they infiltrate (i.e., microglia in the spinal cord). To further identify these cells, we performed flow cytometry analyses (Fig. 7).
Flow cytometry characterization of CCR2 cells
We isolated CCR2-RFP cells from heterozygous and KO CCR2-RFP animals 21 d after bilateral sciatic transections to maximize CCR2 cell recovery within the spinal cord. Experiments were run in parallel for cells isolated from spinal cord or blood of the same animals. Lymphocytes were selected according to size and granularity and single and live cells gated for CD45 (Fig. 8A–D). CD45+ cells were then gated on CCR2 histogram to identify CD45+CCR2+ immune cells (Fig. 8E). The following gates were applied to both the CD45+ total immune cell population and the CD45+ CCR2+ immune cell population. Populations were gated on CD3 and CD19 to identify CD3+ T cells and CD19+ B cells (Fig. 8F). The CD3− CD19− population was then gated on Ly6G versus CD11b to identify CD11b+ Ly6G+ neutrophils (Fig. 8G). CD11b+ Ly6G− cells were gated CD11b versus CD11c to identify CD11b+ CD11c+ dendritic cells (Fig. 8H). CD11b+ CD11c− cells were gated for CD45hi and CD45lo populations to identify resting microglia (CD45low) and activated microglia and monocyte/macrophages (CD45high) (Fig. 8I). We observed three peaks according to CD45 expression; cells in the highest CD45 expression peak expressed CCR2-RFP.
Figure 8.

Flow cytometry analysis of CCR2+ cells inside the spinal cord and in the blood in CCR2 hets and CCR2 KOs 21 d after bilateral sciatic nerve injuries. A–C, Gating for cells (A), single cells (B), and live cells (C) from spinal cord Percoll isolates. D, Resulting cells were gated for CD45 and (E) CCR2. F, All CD45 or CD45+ CCR2+ cells were gated on CD19 (B cells) versus CD3 (T cells). G, The CD3− CD19− population was then gated on Ly6G versus CD11b to identify neutrophiles (Ly6G+ CD11b+). H, CD11b+ Ly6G− cells were on CD11b versus CD11c to identify dendritic cells. I, CD45 cells were analyzed for low or high expression to identify populations of microglia (CD45lo) and activated microglia and macrophages (CD45hi). Only CD45hi CD11b cells express CCR2, suggesting that they have trafficked from the periphery. The results show all cell types represented in the spinal cord, but some lack CCR2. CCR2+ subtypes include CD3 T cells, CD11b monocytes/macrophages, and CD11c+ CD11b+ dendritic cells. J, Different cell types in the blood of injured CCR2 heterozygous animal (gray bars) and CCR2 KOs (black bars). Each data point represents a different animal (n = 12 animals). ***p < 0.001 (t tests). The data show the expected reduced numbers of monocyte/macrophages and increased numbers of neutrophils in the blood of CCR2 KOs. K, Percentages of CCR2+ cell populations in spinal cord 21 d after sciatic nerve injuries in CCR2 heterozygous (gray bars) and CCR2 KO animals (black bars). Each data point represents pooling together 4 animals to increase CCR2 cell yield inside the spinal cord (n = 12 animals total, n = 3). *p < 0.05 (t tests). Error bars indicate SE. The results show a depletion of monocyte/macrophages with p = 0.055.
For quantitative comparisons, a total of 12 Ccr2RFP/+ heterozygous and 12 Ccr2RFP/RFP KO mice were used. We maintained each animal as an individual data point for analyses in the blood; but for spinal cord analyses, we increased the yield of CCR2-RFP cells by pooling together 4 mice from each group, each analyzed as a single set (n = 3, 4 mice in each, for both CCR2 heterozygotes and CCR2 KOs) (Fig. 8J,K). The numbers and proportions of each cell population in blood samples from Ccr2RFP/+ heterozygous animals were similar to previously published materials, suggesting effective detection and discrimination (MacPherson et al., 2017). CCR2-RFP fluorescence was somewhat stronger in Ccr2RFP/RFP KO mice (Fig. 8E) likely due to expression of RFP from both Ccr2 alleles. Interestingly, there was a significant increase in blood neutrophils in CCR2 KOs. This compensatory increase in neutrophils has been reported before in CCR2 KOs (Saederup et al., 2010; Lindborg et al., 2017) and further confirms the KO phenotype and our detection methods.
The results indicate that many different cell types are detectable inside the spinal cord after nerve injuries (this include cells in the spinal cord parenchyma and also in capillaries and meninges). CCR2+ cells consist of T cells, monocyte/macrophages, and CD11b+ CD11c+ cells. We defined the latter as possible dendritic cells, but it is also possible they represent a population of highly activated monocytes/macrophages with increased CD11c expression. The relative proportions of CCR2+ T cells to monocytes inside the spinal cord with flow cytometry are lower than estimated with immunocytochemistry. This difference likely arises because, by difference to histological preparations, the spinal cord samples collected for flow cytometry originate from animals not exsanguinated by perfusion and therefore counts include CCR2+ cells that infiltrated the spinal cord parenchyma, and also cells in spinal vascular beds, capillaries, and meninges.
In CCR2 KOs, we detected a significant depletion of CCR2+ monocyte/macrophages in the blood (heterozygote vs KO t test: t(22) = 8.346, p < 0.001), whereas the proportion of T cells and neutrophils increased as expected. In the spinal cord, there was a strong trend toward a decrease in the proportion of CD11b+ CCR2+ monocyte/macrophages inside the spinal cord (heterozygote vs KO t test: t(4) = 2.042, p = 0.0553) (Fig. 8K). This decrease was less than expected from results in the blood and could have been offset by populations of meningeal and perivascular macrophages that, over time, form stable populations in brain and spinal cord. With histology, we found an increased number of perivascular macrophages expressing CCR2-RFP after nerve injury. Nevertheless, CCR2+ monocytes/macrophages seem to be the most affect cell types in both spinal cord and blood. Changes in their number can occur because these cells are known to require CCR2 activation for infiltrating peripheral tissues (see above) as well as exiting the bone marrow and traveling into the blood (Serbina and Pamer, 2006). Blockade of either or both mechanisms reduces infiltration into the spinal cord.
In conclusion, flow cytometry confirmed that most infiltrating CCR2+ cells in the spinal cord after nerve injury were T cells and monocyte/macrophages, and that the monocyte/macrophage population was preferentially affected in CCR2 KOs. Thus, CCR2-independent mechanisms likely regulate T-cell traffic to the spinal cord following PNI and may also influence some populations of monocyte/macrophages, likely including perivascular and meningeal macrophages.
Discussion
The results suggest an association between Ia/II-VGLUT1 synapse removal and neuroinflammation, both modulated by nerve injury severity. They also indicate that microglia activation is necessary, but not sufficient, for VGLUT1 synapse removal; a CCR2-dependent mechanism is also required. Spinal cord infiltration of CCR2 monocytes/macrophages diminishes in CCR2 KOs, correlating with preservation of Ia/II-VGLUT1 synapses. Moreover, injuries causing similar microgliosis, but different CCR2 cell entry, differ in Ia/II-VGLUT1 loss. Thus, microglia activation and CCR2 cell infiltration are both related to the loss of Ia/II afferent synapses in the ventral horn, resulting in loss of stretch reflexes and spindle-derived information. The physiological significance of this Ia afferent circuit plasticity in the spinal cord ventral horn is not fully clear. Loss of Ia inputs manifests in joint discoordination and diminished performance after muscles are reinnervated, in particular for motor tasks requiring fast and accurate proprioceptive feedback (Cope et al., 1994; Abelew et al., 2000; Haftel et al., 2005; Maas et al., 2007; Bullinger et al., 2011; Sabatier et al., 2011; Lyle et al., 2017; Chang et al., 2018). On the other hand, deletion of Ia/II inputs might reduce connectivity incongruences between proprioceptors and MNs inside the spinal cord after both sensory and motor axons regenerate in the periphery. Different types of nerve injuries result in axon regeneration with variable success on targeting the original muscles, which correlates with more or less loss of Ia afferent synapses (Schultz et al., 2017). CCR2 cell entry similarly fluctuates with injury severity. Thus, microglia activation is a necessary step for the loss of Ia synapses, but modulation of the inflammatory state might offer a mechanism to variably adjust central circuits to possible reorganizations in peripheral innervation according to differences in the specificity of regeneration after different injuries.
It should also be noted that the synaptic losses detected in mice were of smaller intensity that those we previously reported in Wistar rats (Alvarez et al., 2011; Rotterman et al., 2014). The difference may be related to species differences in neuroimmune responses because upregulation of proinflammatory mediators is known to be larger in Wistar rats compared with C57BLk/6 mice after brain injury (Schroeter et al., 2003).
Microglia role in deletion of Ia/II-VGLUT1 synapses
Microglia become activated around MNs after PNI, but their exact roles in MNs survival, motor axon regeneration, synapse removal, and remodeling the central projections of peripherally injured sensory afferents have been remarkably difficult to elucidate with precision (Aldskogius, 2011). In their pioneering study, Blinzinger and Kreutzberg (1968) found that facial MNs lose many synapses on their cell bodies and become covered by microglia after PNI. Microglia were suggested to “lift” synapses from the MN surface, a phenomenon thereafter named “synaptic stripping” and extended to many other MNs (Kerns and Hinsman, 1973; Sumner and Sutherland, 1973; Chen, 1978; Graeber et al., 1993; Brännström and Kellerth, 1998). However, blocking microglia proliferation and/or activation failed to prevent “synaptic stripping” on MNs (Svensson and Aldskogius, 1993; Kalla et al., 2001), casting doubts on the proposed mechanism (Perry and O'Connor, 2010). We confirmed microglia-independent decreases in cell body synaptic coverage of VGLUT1, VGLUT2, and VGAT synapses on axotomized MNs within the “synaptic stripping” time window. Alternatively, “synaptic stripping” might depend on cell-autonomous mechanisms intrinsic to axotomized MNs, such as nitric oxide destabilization of presynaptic boutons (Sunico et al., 2005, 2010; Sunico and Moreno-López, 2010) as well as downregulation of postsynaptic receptors and synaptic adhesion proteins (Piehl et al., 1995; Alvarez et al., 2000; Zelano et al., 2007; Berg et al., 2010). This microglia-independent transient synaptic plasticity on MN cell bodies has been associated with development of a “regenerative state” (Carlstedt and Cullheim, 2000). The deletion of ventral Ia/II axon projections represent more permanent circuit adjustments that might compensate for scrambled connectivity in the periphery after regeneration and require microglia activation.
Decreases in Ia EPSP amplitude on axotomized MNs were first attributed to “synaptic stripping” (Mendell, 1984), but now we know that downregulation of Ia/II-VGLUT1 synapses and stretch-evoked EPSPs differ from “synaptic stripping” in time course, fate of afferent axons, and lack of recovery (Alvarez et al., 2011; Bullinger et al., 2011). Importantly, while “stripping” involves detachment from axotomized MNs of presynaptic inputs unaffected by the injury, Ia/II-VGLUT1 synapse removal depends on injury to Ia/II afferents in the periphery and die-back of ventral Ia/II axon projections. This affects synapses on cell bodies and dendrites of axotomized MNs (Alvarez et al., 2011; Rotterman et al., 2014), as well as on uninjured MNs and ventral interneurons (Horstman, 2012). Removal of Ia/II axons from the ventral horn likely prevents reestablishment of these synapses after peripheral nerve regeneration is completed. Microglia activation is thus necessary for transforming transient plasticity of Ia/II-VGLUT1 synapses on MN cell bodies (similar to the stripping of other synapses) into permanent losses on MN dendrites and throughout the ventral horn. Ia/II-VGLUT1 synapse elimination is therefore more similar to the “transganglionic anterograde degeneration” previously described after PNI for the central synapses and axons of cutaneous mechanoreceptors (Grant and Arvidsson, 1975; Aldskogius et al., 1985; Arvidsson, 1986).
Microglia proliferation and activation occur in all brain and spinal cord areas receiving projections from axotomized primary afferents (Eriksson et al., 1993), including dorsal horn projection territories of cutaneous mechanoreceptors and muscle proprioceptors (Figs. 1, 3, 4) and Clarke's column in thoracic segments (no shown); however, Ia afferents are lost preferentially in the ventral horn. The role of activated dorsal horn microglia enhancing responses of spinal neurons to cutaneous sensory inputs after PNI has been extensively studied (Ferrini and De Koninck, 2013), but there is no defined role for activated microglia on the central synapses of the sensory afferent themselves. Like Ia/II afferents, cutaneous mechanoreceptor synapses express VGLUT1 (Todd et al., 2003; Alvarez et al., 2004), and virally infected VGLUT1 synapses are removed by microglia using complement-dependent mechanisms (Vasek et al., 2016). There is also electron microscopy evidence of microglia engulfment of vibrissae afferent synapses and axons after PNI (Arvidsson, 1986). These observations agree with our conclusion that microglia are related to the removal of Ia/II-VGLUT1 synapses in the ventral horn. However, Ia/II-VGLUT1 synapses are preserved around dorsal horn activated microglia, and reducing ventral horn microglia activation is enough for rescuing Ia/II-VGLUT1 synapses on MNs, suggesting limited influence from dorsal horn microglia. Moreover, the large anatomical remodeling of the ventral projections of neurobiotin-filled Ia axons that have regenerated in the periphery (Alvarez et al., 2011) is not paralleled by similar large structural plasticity in the dorsal horn projections of neurobiotin-filled cutaneous mechanoreceptors (Koerber et al., 2006). Thus, dorsal and ventral horn-activated microglia differentially affect the stability of synapses from sensory afferents axotomized in the peripheral nerve. Moreover, ventral microgliosis does not always result in large deletions of Ia/II-VGLUT1 synapses, suggesting that microglia activation alone is not sufficient. One important difference, first revealed in this study, is the recruitment of hematogenous immune cells identified by CCR2 expression to ventral horn regions where permanent removal of Ia/II-VGLUT1 synapses occurs.
CCR2 activation and monocyte entry are critical for VGLUT1 synapse plasticity
Infiltration of CCR2 blood-derived immune cells follows microglia activation and temporally correlates with Ia/II-VGLUT1 synapse removal. Moreover, global deletion of CCR2 prevents Ia/II-VGLUT1 losses on dendrites of regenerated MNs and partially protects those on the soma. CCR2 is well known for its role in trafficking specific leukocytes into the CNS (Ransohoff, 2009). Specifically, entry of CD45high Ly6Chigh CD11bhigh proinflammatory monocytes is CCR2-dependent, and these cells differentiate into macrophages within the CNS parenchyma (Ma et al., 2002; Mahad and Ransohoff, 2003; Serbina and Pamer, 2006; El Khoury et al., 2007; Serbina et al., 2008; Ajami et al., 2011; Shi and Pamer, 2011; Shi et al., 2011; Goldmann et al., 2016). CCR2 macrophages actively remove damaged axons in injured nerves; and after spinal cord injuries, they become the principal phagocytic cell responsible for secondary die-back of sensory myelinated axons in the dorsal columns (Ma et al., 2002; McPhail et al., 2004; Evans et al., 2014). Flow cytometry indicated that a subgroup of CD45high CD11bhigh monocytes/macrophages is most affected in CCR2 KOs in parallel with the rescue of Ia/II-VGLUT1 synapses, suggesting a possible relation between the two. One possibility is that CCR2 monocytes differentiate into phagocytic CX3CR1/Iba1 microglia-like cells, and we revealed with lineage labeling microglia-like cells after PNI derived from sources other than resident microglia. Alternatively, CCR2 activation might directly modulate resident microglia phagocytic activity. Recently, CCR2 activation was found necessary to promote microglia synapse phagocytosis in a murine model of encephalitis (Di Liberto et al., 2018). Both mechanisms are not exclusive, and future studies using novel CCR2-Cre lines (Croxford et al., 2015) and time-lapse two photon microscopy may help resolve the functions of CCR2-derived cells and/or CX3CR1 microglia after PNI and their respective roles during removal of Ia/II-VGLUT1 synapses and axons.
In conclusion, we revealed that the permanent loss of VGLUT1 Ia/II synapses differs from synaptic stripping mechanistically: it requires microglia activation and CCR2 activation. The latter seems to be modulated according to nerve injury type and might be instrumental in adjusting central synaptic circuit plasticity to different types of nerve injuries.
Footnotes
This work was supported by National Institutes of Health, National Institute of Neurological Disorders and Stroke Grants P01NS057228 and R56NS099092 to F.J.A., and Ruth L. Kirschstein National Research Service Award F31NS095528 fellowship to T.M.R. We thank Dr. Jean X. Jiang (University of Texas, San Antonio, TX) for the donation of the csf1-flox mouse line; and Myriam Alvarez for quantification of microglia in CSF1 motoneuron KOs.
The authors declare no competing financial interests.
References
- Abelew TA, Miller MD, Cope TC, Nichols TR (2000) Local loss of proprioception results in disruption of interjoint coordination during locomotion in the cat. J Neurophysiol 84:2709–2714. 10.1152/jn.2000.84.5.2709 [DOI] [PubMed] [Google Scholar]
- Ajami B, Bennett JL, Krieger C, McNagny KM, Rossi FM (2011) Infiltrating monocytes trigger EAE progression, but do not contribute to the resident microglia pool. Nat Neurosci 14:1142–1149. 10.1038/nn.2887 [DOI] [PubMed] [Google Scholar]
- Aldskogius H. (2011) Mechanisms and consequences of microglial responses to peripheral axotomy. Front Biosci (School Ed) 3:857–868. 10.2741/192 [DOI] [PubMed] [Google Scholar]
- Aldskogius H, Arvidsson J, Grant G (1985) The reaction of primary sensory neurons to peripheral nerve injury with particular emphasis on transganglionic changes. Brain Res 357:27–46. [DOI] [PubMed] [Google Scholar]
- Aldskogius H, Liu L, Svensson M (1999) Glial responses to synaptic damage and plasticity. J Neurosci Res 58:33–41. 10.1002/(SICI)1097-4547(19991001)58:1<33::AID-JNR5>3.0.CO;2-M [DOI] [PubMed] [Google Scholar]
- Alvarez FJ, Dewey DE, Harrington DA, Fyffe RE (1997) Cell-type specific organization of glycine receptor clusters in the mammalian spinal cord. J Comp Neurol 379:150–170. 10.1002/(SICI)1096-9861(19970303)379:1<150::AID-CNE10>3.0.CO;2-T [DOI] [PubMed] [Google Scholar]
- Alvarez FJ, Fyffe RE, Dewey DE, Haftel VK, Cope TC (2000) Factors regulating AMPA-type glutamate receptor subunit changes induced by sciatic nerve injury in rats. J Comp Neurol 426:229–242. 10.1002/1096-9861(20001016)426:2<229::AID-CNE5>3.0.CO;2-W [DOI] [PubMed] [Google Scholar]
- Alvarez FJ, Villalba RM, Zerda R, Schneider SP (2004) Vesicular glutamate transporters in the spinal cord, with special reference to sensory primary afferent synapses. J Comp Neurol 472:257–280. 10.1002/cne.20012 [DOI] [PubMed] [Google Scholar]
- Alvarez FJ, Bullinger KL, Titus HE, Nardelli P, Cope TC (2010) Permanent reorganization of Ia afferent synapses on motoneurons after peripheral nerve injuries. Ann N Y Acad Sci 1198:231–241. 10.1111/j.1749-6632.2010.05459.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez FJ, Titus-Mitchell HE, Bullinger KL, Kraszpulski M, Nardelli P, Cope TC (2011) Permanent central synaptic disconnection of proprioceptors after nerve injury and regeneration: I. Loss of VGLUT1/IA synapses on motoneurons. J Neurophysiol 106:2450–2470. 10.1152/jn.01095.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arvidsson J. (1986) Transganglionic degeneration in vibrissae innervating primary sensory neurons of the rat: a light and electron microscopic study. J Comp Neurol 249:392–403. 10.1002/cne.902490306 [DOI] [PubMed] [Google Scholar]
- Berg A, Zelano J, Cullheim S (2010) Netrin G-2 ligand mRNA is downregulated in spinal motoneurons after sciatic nerve lesion. Neuroreport 21:782–785. 10.1097/WNR.0b013e32833cadd8 [DOI] [PubMed] [Google Scholar]
- Berg A, Zelano J, Thams S, Cullheim S (2013) The extent of synaptic stripping of motoneurons after axotomy is not correlated to activation of surrounding glia or downregulation of postsynaptic adhesion molecules. PLoS One 8:e59647. 10.1371/journal.pone.0059647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blinzinger K, Kreutzberg G (1968) Displacement of synaptic terminals from regenerating motoneurons by microglial cells. Z Zellforsch Mikrosk Anat 85:145–157. 10.1007/BF00325030 [DOI] [PubMed] [Google Scholar]
- Bonecchi R, Bianchi G, Bordignon PP, D'Ambrosio D, Lang R, Borsatti A, Sozzani S, Allavena P, Gray PA, Mantovani A, Sinigaglia F (1998) Differential expression of chemokine receptors and chemotactic responsiveness of type 1 T helper cells (Th1s) and Th2s. J Exp Med 187:129–134. 10.1084/jem.187.1.129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brännström T, Kellerth JO (1998) Changes in synaptology of adult cat spinal alpha-motoneurons after axotomy. Exp Brain Res 118:1–13. 10.1007/s002210050249 [DOI] [PubMed] [Google Scholar]
- Brown AG, Fyffe RE (1981) Direct observations on the contacts made between Ia afferent fibres and alpha-motoneurones in the cat's lumbosacral spinal cord. J Physiol 313:121–140. 10.1113/jphysiol.1981.sp013654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bullinger KL, Nardelli P, Pinter MJ, Alvarez FJ, Cope TC (2011) Permanent central synaptic disconnection of proprioceptors after nerve injury and regeneration: II. Loss of functional connectivity with motoneurons. J Neurophysiol 106:2471–2485. 10.1152/jn.01097.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burke RE, Walmsley B, Hodgson JA (1979) HRP anatomy of group Ia afferent contacts on alpha motoneurones. Brain Res 160:347–352. 10.1016/0006-8993(79)90430-X [DOI] [PubMed] [Google Scholar]
- Carlstedt T, Cullheim S (2000) Spinal cord motoneuron maintenance, injury and repair. Prog Brain Res 127:501–514. 10.1016/S0079-6123(00)27025-8 [DOI] [PubMed] [Google Scholar]
- Chang YH, Housley SN, Hart KS, Nardelli P, Nichols RT, Maas H, Cope TC (2018) Progressive adaptation of whole-limb kinematics after peripheral nerve injury. Biol Open 7:bio028852. 10.1242/bio.028852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen DH. (1978) Qualitative and quantitative study of synaptic displacement in chromatolyzed spinal motoneurons of the cat. J Comp Neurol 177:635–664. 10.1002/cne.901770407 [DOI] [PubMed] [Google Scholar]
- Conradi S, Kellerth JO, Berthold CH, Hammarberg C (1979) Electron microscopic studies of serially sectioned cat spinal alpha-motoneurons: IV. Motoneurons innervating slow-twitch (type S) units of the soleus muscle. J Comp Neurol 184:769–782. 10.1002/cne.901840409 [DOI] [PubMed] [Google Scholar]
- Cope TC, Bonasera SJ, Nichols TR (1994) Reinnervated muscles fail to produce stretch reflexes. J Neurophysiol 71:817–820. 10.1152/jn.1994.71.2.817 [DOI] [PubMed] [Google Scholar]
- Croxford AL, Lanzinger M, Hartmann FJ, Schreiner B, Mair F, Pelczar P, Clausen BE, Jung S, Greter M, Becher B (2015) The cytokine GM-CSF drives the inflammatory signature of CCR2+ monocytes and licenses autoimmunity. Immunity 43:502–514. 10.1016/j.immuni.2015.08.010 [DOI] [PubMed] [Google Scholar]
- Cullheim S, Thams S (2007) The microglial networks of the brain and their role in neuronal network plasticity after lesion. Brain Res Rev 55:89–96. 10.1016/j.brainresrev.2007.03.012 [DOI] [PubMed] [Google Scholar]
- Davis KD, Taylor KS, Anastakis DJ (2011) Nerve injury triggers changes in the brain. Neuroscientist 17:407–422. 10.1177/1073858410389185 [DOI] [PubMed] [Google Scholar]
- Deshmane SL, Kremlev S, Amini S, Sawaya BE (2009) Monocyte chemoattractant protein-1 (MCP-1): an overview. J Interferon Cytokine Res. 29:313–326. 10.1089/jir.2008.0027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Liberto G, Pantelyushin S, Kreutzfeldt M, Page N, Musardo S, Coras R, Steinbach K, Vincenti I, Klimek B, Lingner T, Salinas G, Lin-Marq N, Staszewski O, Costa Jordão MJ, Wagner I, Egervari K, Mack M, Bellone C, Blümcke I, Prinz M, et al. (2018) Neurons under T cell attack coordinate phagocyte-mediated synaptic stripping. Cell 175:458–471.e19 10.1016/j.cell.2018.07.049 [DOI] [PubMed] [Google Scholar]
- Donnelly DJ, Longbrake EE, Shawler TM, Kigerl KA, Lai W, Tovar CA, Ransohoff RM, Popovich PG (2011) Deficient CX3CR1 signaling promotes recovery after mouse spinal cord injury by limiting the recruitment and activation of Ly6Clo/iNOS+ macrophages. J Neurosci 31:9910–9922. 10.1523/JNEUROSCI.2114-11.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Khoury J, Toft M, Hickman SE, Means TK, Terada K, Geula C, Luster AD (2007) Ccr2 deficiency impairs microglial accumulation and accelerates progression of Alzheimer-like disease. Nat Med 13:432–438. 10.1038/nm1555 [DOI] [PubMed] [Google Scholar]
- Elmore MR, Najafi AR, Koike MA, Dagher NN, Spangenberg EE, Rice RA, Kitazawa M, Matusow B, Nguyen H, West BL, Green KN (2014) Colony-stimulating factor 1 receptor signaling is necessary for microglia viability, unmasking a microglia progenitor cell in the adult brain. Neuron 82:380–397. 10.1016/j.neuron.2014.02.040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eriksson NP, Persson JK, Svensson M, Arvidsson J, Molander C, Aldskogius H (1993) A quantitative analysis of the microglial cell reaction in central primary sensory projection territories following peripheral nerve injury in the adult rat. Exp Brain Res 96:19–27. 10.1007/BF00230435 [DOI] [PubMed] [Google Scholar]
- Evans TA, Barkauskas DS, Myers JT, Hare EG, You JQ, Ransohoff RM, Huang AY, Silver J (2014) High-resolution intravital imaging reveals that blood-derived macrophages but not resident microglia facilitate secondary axonal dieback in traumatic spinal cord injury. Exp Neurol 254:109–120. 10.1016/j.expneurol.2014.01.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrini F, De Koninck Y (2013) Microglia control neuronal network excitability via BDNF signalling. Neural Plast 2013:429815. 10.1155/2013/429815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fonseca MI, Chu SH, Hernandez MX, Fang MJ, Modarresi L, Selvan P, MacGregor GR, Tenner AJ (2017) Cell-specific deletion of C1qa identifies microglia as the dominant source of C1q in mouse brain. J Neuroinflammation 14:48. 10.1186/s12974-017-0814-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldmann T, Wieghofer P, Müller PF, Wolf Y, Varol D, Yona S, Brendecke SM, Kierdorf K, Staszewski O, Datta M, Luedde T, Heikenwalder M, Jung S, Prinz M (2013) A new type of microglia gene targeting shows TAK1 to be pivotal in CNS autoimmune inflammation. Nat Neurosci 16:1618–1626. 10.1038/nn.3531 [DOI] [PubMed] [Google Scholar]
- Goldmann T, Wieghofer P, Jordão MJ, Prutek F, Hagemeyer N, Frenzel K, Amann L, Staszewski O, Kierdorf K, Krueger M, Locatelli G, Hochgerner H, Zeiser R, Epelman S, Geissmann F, Priller J, Rossi FM, Bechmann I, Kerschensteiner M, Linnarsson S, et al. (2016) Origin, fate and dynamics of macrophages at central nervous system interfaces. Nat Immunol 17:797–805. 10.1038/ni.3423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graeber MB, Bise K, Mehraein P (1993) Synaptic stripping in the human facial nucleus. Acta Neuropathol 86:179–181. 10.1007/BF00334886 [DOI] [PubMed] [Google Scholar]
- Grant G, Arvidsson J (1975) Transganglionic degeneration in trigeminal primary sensory neurons. Brain Res 95:265–279. 10.1016/0006-8993(75)90106-7 [DOI] [PubMed] [Google Scholar]
- Guan Z, Kuhn JA, Wang X, Colquitt B, Solorzano C, Vaman S, Guan AK, Evans-Reinsch Z, Braz J, Devor M, Abboud-Werner SL, Lanier LL, Lomvardas S, Basbaum AI (2016) Injured sensory neuron-derived CSF1 induces microglial proliferation and DAP12-dependent pain. Nat Neurosci 19:94–101. 10.1038/nn.4189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu N, Peng J, Murugan M, Wang X, Eyo UB, Sun D, Ren Y, DiCicco-Bloom E, Young W, Dong H, Wu LJ (2016) Spinal microgliosis due to resident microglial proliferation is required for pain hypersensitivity after peripheral nerve injury. Cell Rep 16:605–614. 10.1016/j.celrep.2016.06.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guest JD, Hesse D, Schnell L, Schwab ME, Bunge MB, Bunge RP (1997) Influence of IN-1 antibody and acidic FGF-fibrin glue on the response of injured corticospinal tract axons to human Schwann cell grafts. J Neurosci Res 50:888–905. 10.1002/(SICI)1097-4547(19971201)50:5<888::AID-JNR24>3.0.CO;2-W [DOI] [PubMed] [Google Scholar]
- Haftel VK, Bichler EK, Wang QB, Prather JF, Pinter MJ, Cope TC (2005) Central suppression of regenerated proprioceptive afferents. J Neurosci 25:4733–4742. 10.1523/JNEUROSCI.4895-04.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanisch UK. (2002) Microglia as a source and target of cytokines. Glia 40:140–155. 10.1002/glia.10161 [DOI] [PubMed] [Google Scholar]
- Harris SE, MacDougall M, Horn D, Woodruff K, Zimmer SN, Rebel VI, Fajardo R, Feng JQ, Gluhak-Heinrich J, Harris MA, Abboud Werner S (2012) Meox2Cre-mediated disruption of CSF-1 leads to osteopetrosis and osteocyte defects. Bone 50:42–53. 10.1016/j.bone.2011.09.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison JK, Jiang Y, Chen S, Xia Y, Maciejewski D, McNamara RK, Streit WJ, Salafranca MN, Adhikari S, Thompson DA, Botti P, Bacon KB, Feng L (1998) Role for neuronally derived fractalkine in mediating interactions between neurons and CX3CR1-expressing microglia. Proc Natl Acad Sci U S A 95:10896–10901. 10.1073/pnas.95.18.10896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong S, Beja-Glasser VF, Nfonoyim BM, Frouin A, Li S, Ramakrishnan S, Merry KM, Shi Q, Rosenthal A, Barres BA, Lemere CA, Selkoe DJ, Stevens B (2016) Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science 352:712–716. 10.1126/science.aad8373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horstman GM. (2012) Limitations of functional recovery of stretch reflex circuitry after peripheral nerve regeneration. Dayton, Ohio: Wright State University; Available at https://corescholar.libraries.wright.edu/etd_all/629/. [Google Scholar]
- Huang D, Shi FD, Jung S, Pien GC, Wang J, Salazar-Mather TP, He TT, Weaver JT, Ljunggren HG, Biron CA, Littman DR, Ransohoff RM (2006) The neuronal chemokine CX3CL1/fractalkine selectively recruits NK cells that modify experimental autoimmune encephalomyelitis within the central nervous system. FASEB J 20:896–905. 10.1096/fj.05-5465com [DOI] [PubMed] [Google Scholar]
- Hu X, Leak RK, Shi Y, Suenaga J, Gao Y, Zheng P, Chen J (2015) Microglial and macrophage polarization-new prospects for brain repair. Nat Rev Neurol 11:56–64. 10.1038/nrneurol.2014.207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes DI, Polgár E, Shehab SA, Todd AJ (2004) Peripheral axotomy induces depletion of the vesicular glutamate transporter VGLUT1 in central terminals of myelinated afferent fibres in the rat spinal cord. Brain Res 1017:69–76. 10.1016/j.brainres.2004.05.054 [DOI] [PubMed] [Google Scholar]
- Jinno S, Yamada J (2011) Using comparative anatomy in the axotomy model to identify distinct roles for microglia and astrocytes in synaptic stripping. Neuron Glia Biol 7:55–66. 10.1017/S1740925X11000135 [DOI] [PubMed] [Google Scholar]
- Jung S, Aliberti J, Graemmel P, Sunshine MJ, Kreutzberg GW, Sher A, Littman DR (2000) Analysis of fractalkine receptor CX(3)CR1 function by targeted deletion and green fluorescent protein reporter gene insertion. Mol Cell Biol 20:4106–4114. 10.1128/MCB.20.11.4106-4114.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalla R, Liu Z, Xu S, Koppius A, Imai Y, Kloss CU, Kohsaka S, Gschwendtner A, Möller JC, Werner A, Raivich G (2001) Microglia and the early phase of immune surveillance in the axotomized facial motor nucleus: impaired microglial activation and lymphocyte recruitment but no effect on neuronal survival or axonal regeneration in macrophage-colony stimulating factor-deficient mice. J Comp Neurol 436:182–201. 10.1002/cne.1060 [DOI] [PubMed] [Google Scholar]
- Katsumoto A, Miranda AS, Butovsky O, Teixeira AL, Ransohoff RM, Lamb BT (2018) Laquinimod attenuates inflammation by modulating macrophage functions in traumatic brain injury mouse model. J Neuroinflammation 15:26. 10.1186/s12974-018-1075-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kellerth JO, Berthold CH, Conradi S (1979) Electron microscopic studies of serially sectioned cat spinal alpha-motoneurons: III. Motoneurons innervating fast-twitch (type FR) units of the gastrocnemius muscle. J Comp Neurol 184:755–767. 10.1002/cne.901840408 [DOI] [PubMed] [Google Scholar]
- Kellerth JO, Conradi S, Berthold CH (1983) Electron microscopic studies of serially sectioned cat spinal alpha-motoneurons: V. Motoneurons innervating fast-twitch (type FF) units of the gastrocnemius muscle. J Comp Neurol 214:451–458. 10.1002/cne.902140409 [DOI] [PubMed] [Google Scholar]
- Kerns JM, Hinsman EJ (1973) Neuroglial response to sciatic neurectomy: I. Light microscopy and autoradiography. J Comp Neurol 151:237–254. 10.1002/cne.901510303 [DOI] [PubMed] [Google Scholar]
- Koerber HR, Mirnics K, Lawson JJ (2006) Synaptic plasticity in the adult spinal dorsal horn: the appearance of new functional connections following peripheral nerve regeneration. Exp Neurol 200:468–479. 10.1016/j.expneurol.2006.03.003 [DOI] [PubMed] [Google Scholar]
- Kreutzberg GW. (1996) Microglia: a sensor for pathological events in the CNS. Trends Neurosci 19:312–318. 10.1016/0166-2236(96)10049-7 [DOI] [PubMed] [Google Scholar]
- Lindå H, Shupliakov O, Ornung G, Ottersen OP, Storm-Mathisen J, Risling M, Cullheim S (2000) Ultrastructural evidence for a preferential elimination of glutamate-immunoreactive synaptic terminals from spinal motoneurons after intramedullary axotomy. J Comp Neurol 425:10–23. 10.1002/1096-9861(20000911)425:1<10::AID-CNE2>3.0.CO;2-# [DOI] [PubMed] [Google Scholar]
- Lindborg JA, Mack M, Zigmond RE (2017) Neutrophils are critical for myelin removal in a peripheral nerve injury model of Wallerian degeneration. J Neurosci 37:10258–10277. 10.1523/JNEUROSCI.2085-17.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundborg G. (2003) Richard P. Bunge memorial lecture: nerve injury and repair—a challenge to the plastic brain. J Peripheral Nerv Syst 8:209–226. 10.1111/j.1085-9489.2003.03027.x [DOI] [PubMed] [Google Scholar]
- Lyle MA, Nichols TR, Kajtaz E, Maas H (2017) Musculotendon adaptations and preservation of spinal reflex pathways following agonist-to-antagonist tendon transfer. Physiol Rep 5:e13201. 10.14814/phy2.13201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma M, Wei T, Boring L, Charo IF, Ransohoff RM, Jakeman LB (2002) Monocyte recruitment and myelin removal are delayed following spinal cord injury in mice with CCR2 chemokine receptor deletion. J Neurosci Res 68:691–702. 10.1002/jnr.10269 [DOI] [PubMed] [Google Scholar]
- Maas H, Prilutsky BI, Nichols TR, Gregor RJ (2007) The effects of self-reinnervation of cat medial and lateral gastrocnemius muscles on hindlimb kinematics in slope walking. Exp Brain Res 181:377–393. 10.1007/s00221-007-0938-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacPherson KP, Sompol P, Kannarkat GT, Chang J, Sniffen L, Wildner ME, Norris CM, Tansey MG (2017) Peripheral administration of the soluble TNF inhibitor XPro1595 modifies brain immune cell profiles, decreases beta-amyloid plaque load, and rescues impaired long-term potentiation in 5xFAD mice. Neurobiol Dis 102:81–95. 10.1016/j.nbd.2017.02.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madisen L, Zwingman TA, Sunkin SM, Oh SW, Zariwala HA, Gu H, Ng LL, Palmiter RD, Hawrylycz MJ, Jones AR, Lein ES, Zeng H (2010) A robust and high-throughput cre reporting and characterization system for the whole mouse brain. Nat Neurosci 13:133–140. 10.1038/nn.2467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahad DJ, Ransohoff RM (2003) The role of MCP-1 (CCL2) and CCR2 in multiple sclerosis and experimental autoimmune encephalomyelitis (EAE). Semin Immunol 15:23–32. 10.1016/S1044-5323(02)00125-2 [DOI] [PubMed] [Google Scholar]
- McPhail LT, Stirling DP, Tetzlaff W, Kwiecien JM, Ramer MS (2004) The contribution of activated phagocytes and myelin degeneration to axonal retraction/dieback following spinal cord injury. Eur J Neurosci 20:1984–1994. 10.1111/j.1460-9568.2004.03662.x [DOI] [PubMed] [Google Scholar]
- Mendell LM. (1984) Modifiability of spinal synapses. Physiol Rev 64:260–324. 10.1152/physrev.1984.64.1.260 [DOI] [PubMed] [Google Scholar]
- Mizutani M, Pino PA, Saederup N, Charo IF, Ransohoff RM, Cardona AE (2012) The fractalkine receptor but not CCR2 is present on microglia from embryonic development throughout adulthood. J Immunol 188:29–36. 10.4049/jimmunol.1100421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohanty CB, Bhat D, Devi BI (2015) Role of central plasticity in the outcome of peripheral nerve regeneration. Neurosurgery 77:418–423. 10.1227/NEU.0000000000000851 [DOI] [PubMed] [Google Scholar]
- Morganti JM, Jopson TD, Liu S, Riparip LK, Guandique CK, Gupta N, Ferguson AR, Rosi S (2015) CCR2 antagonism alters brain macrophage polarization and ameliorates cognitive dysfunction induced by traumatic brain injury. J Neurosci 35:748–760. 10.1523/JNEUROSCI.2405-14.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navarro X, Vivó M, Valero-Cabré A (2007) Neural plasticity after peripheral nerve injury and regeneration. Prog Neurobiol 82:163–201. 10.1016/j.pneurobio.2007.06.005 [DOI] [PubMed] [Google Scholar]
- Niemi JP, DeFrancesco-Lisowitz A, Roldán-Hernandez L, Lindborg JA, Mandell D, Zigmond RE (2013) A critical role for macrophages near axotomized neuronal cell bodies in stimulating nerve regeneration. J Neurosci 33:16236–16248. 10.1523/JNEUROSCI.3319-12.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nimmerjahn A, Kirchhoff F, Helmchen F (2005) Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 308:1314–1318. 10.1126/science.1110647 [DOI] [PubMed] [Google Scholar]
- Nisticò R, Salter E, Nicolas C, Feligioni M, Mango D, Bortolotto ZA, Gressens P, Collingridge GL, Peineau S (2017) Synaptoimmunology: roles in health and disease. Mol Brain 10:26. 10.1186/s13041-017-0308-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papadopoulos EJ, Sassetti C, Saeki H, Yamada N, Kawamura T, Fitzhugh DJ, Saraf MA, Schall T, Blauvelt A, Rosen SD, Hwang ST (1999) Fractalkine, a CX3C chemokine, is expressed by dendritic cells and is up-regulated upon dendritic cell maturation. Eur J Immunol 29:2551–2559. 10.1002/(SICI)1521-4141(199908)29:08<2551::AID-IMMU2551>3.0.CO;2-T [DOI] [PubMed] [Google Scholar]
- Perry VH, O'Connor V (2010) The role of microglia in synaptic stripping and synaptic degeneration: a revised perspective. ASN Neuro 2:e00047. 10.1042/AN20100024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piehl F, Tabar G, Cullheim S (1995) Expression of NMDA receptor mRNAs in rat motoneurons is down-regulated after axotomy. Eur J Neurosci 7:2101–2110. 10.1111/j.1460-9568.1995.tb00632.x [DOI] [PubMed] [Google Scholar]
- Prinz M, Priller J, Sisodia SS, Ransohoff RM (2011) Heterogeneity of CNS myeloid cells and their roles in neurodegeneration. Nat Neurosci 14:1227–1235. 10.1038/nn.2923 [DOI] [PubMed] [Google Scholar]
- Ransohoff RM. (2009) Chemokines and chemokine receptors: standing at the crossroads of immunobiology and neurobiology. Immunity 31:711–721. 10.1016/j.immuni.2009.09.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi J, Balthasar N, Olson D, Scott M, Berglund E, Lee CE, Choi MJ, Lauzon D, Lowell BB, Elmquist JK (2011) Melanocortin-4 receptors expressed by cholinergic neurons regulate energy balance and glucose homeostasis. Cell Metab 13:195–204. 10.1016/j.cmet.2011.01.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rotterman TM, Nardelli P, Cope TC, Alvarez FJ (2014) Normal distribution of VGLUT1 synapses on spinal motoneuron dendrites and their reorganization after nerve injury. J Neurosci 34:3475–3492. 10.1523/JNEUROSCI.4768-13.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabatier MJ, To BN, Nicolini J, English AW (2011) Effect of slope and sciatic nerve injury on ankle muscle recruitment and hindlimb kinematics during walking in the rat. J Exp Biol 214:1007–1016. 10.1242/jeb.051508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabha M Jr, Emirandetti A, Cullheim S, De Oliveira AL (2008) MHC I expression and synaptic plasticity in different mice strains after axotomy. Synapse 62:137–148. 10.1002/syn.20475 [DOI] [PubMed] [Google Scholar]
- Saederup N, Cardona AE, Croft K, Mizutani M, Cotleur AC, Tsou CL, Ransohoff RM, Charo IF (2010) Selective chemokine receptor usage by central nervous system myeloid cells in CCR2-red fluorescent protein knock-in mice. PLoS One 5:e13693. 10.1371/journal.pone.0013693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroeter M, Küry P, Jander S (2003) Inflammatory gene expression in focal cortical brain ischemia: differences between rats and mice. Brain Res Mol Brain Res 117:1–7. 10.1016/S0169-328X(03)00255-9 [DOI] [PubMed] [Google Scholar]
- Schultz AJ, Rotterman TM, Dwarakanath A, Alvarez FJ (2017) VGLUT1 synapses and P-boutons on regenerating motoneurons after nerve crush. J Comp Neurol 525:2876–2889. 10.1002/cne.24244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serbina NV, Pamer EG (2006) Monocyte emigration from bone marrow during bacterial infection requires signals mediated by chemokine receptor CCR2. Nat Immunol 7:311–317. 10.1038/ni1309 [DOI] [PubMed] [Google Scholar]
- Serbina NV, Jia T, Hohl TM, Pamer EG (2008) Monocyte-mediated defense against microbial pathogens. Annual Rev Immunol 26:421–452. 10.1146/annurev.immunol.26.021607.090326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi C, Pamer EG (2011) Monocyte recruitment during infection and inflammation. Nat Rev Immunol 11:762–774. 10.1038/nri3070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi YY, Wang YS, Zhang ZX, Cai Y, Zhou J, Hou HY, van Rooijen N (2011) Monocyte/macrophages promote vasculogenesis in choroidal neovascularization in mice by stimulating SDF-1 expression in RPE cells. Graefes Arch Clin Exp Ophthalmol 249:1667–1679. 10.1007/s00417-011-1699-4 [DOI] [PubMed] [Google Scholar]
- Siebert H, Sachse A, Kuziel WA, Maeda N, Brück W (2000) The chemokine receptor CCR2 is involved in macrophage recruitment to the injured peripheral nervous system J Neuroimmunol 110:177–185. 10.1016/S0165-5728(00)00343-X [DOI] [PubMed] [Google Scholar]
- Siembab VC, Gomez-Perez L, Rotterman TM, Shneider NA, Alvarez FJ (2016) Role of primary afferents in the developmental regulation of motor axon synapse numbers on Renshaw cells. J Comp Neurol 524:1892–1919. 10.1002/cne.23946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sumner BE, Sutherland FI (1973) Quantitative electron microscopy on the injured hypoglossal nucleus in the rat. J Neurocytol 2:315–328. 10.1007/BF01104033 [DOI] [PubMed] [Google Scholar]
- Sunderland S. (1968) Nerves and nerve injuries. Baltimore: Williams and Wilkins. [Google Scholar]
- Sunico CR, Moreno-López B (2010) Evidence for endothelial nitric oxide as a negative regulator of Schwann cell dedifferentiation after peripheral nerve injury. Neurosci Lett 471:119–124. 10.1016/j.neulet.2010.01.024 [DOI] [PubMed] [Google Scholar]
- Sunico CR, Portillo F, González-Forero D, Moreno-López B (2005) Nitric-oxide-directed synaptic remodeling in the adult mammal CNS. J Neurosci 25:1448–1458. 10.1523/JNEUROSCI.4600-04.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sunico CR, González-Forero D, Domínguez G, García-Verdugo JM, Moreno-López B (2010) Nitric oxide induces pathological synapse loss by a protein kinase G-, Rho kinase-dependent mechanism preceded by myosin light chain phosphorylation. J Neurosci 30:973–984. 10.1523/JNEUROSCI.3911-09.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svensson M, Aldskogius H (1993) Synaptic density of axotomized hypoglossal motorneurons following pharmacological blockade of the microglial cell proliferation. Exp Neurol 120:123–131. 10.1006/exnr.1993.1046 [DOI] [PubMed] [Google Scholar]
- Tay TL, Mai D, Dautzenberg J, Fernández-Klett F, Lin G, Sagar, Datta M, Drougard A, Stempfl T, Ardura-Fabregat A, Staszewski O, Margineanu A, Sporbert A, Steinmetz LM, Pospisilik JA, Jung S, Priller J, Grün D, Ronneberger O, Prinz M (2017) A new fate mapping system reveals context-dependent random or clonal expansion of microglia. Nat Neurosci 20:793–803. 10.1038/nn.4547 [DOI] [PubMed] [Google Scholar]
- Todd AJ, Hughes DI, Polgár E, Nagy GG, Mackie M, Ottersen OP, Maxwell DJ (2003) The expression of vesicular glutamate transporters VGLUT1 and VGLUT2 in neurochemically defined axonal populations in the rat spinal cord with emphasis on the dorsal horn. Eur J Neurosci 17:13–27. 10.1046/j.1460-9568.2003.02406.x [DOI] [PubMed] [Google Scholar]
- Vasek MJ, Garber C, Dorsey D, Durrant DM, Bollman B, Soung A, Yu J, Perez-Torres C, Frouin A, Wilton DK, Funk K, DeMasters BK, Jiang X, Bowen JR, Mennerick S, Robinson JK, Garbow JR, Tyler KL, Suthar MS, Schmidt RE, et al. (2016) A complement-microglial axis drives synapse loss during virus-induced memory impairment. Nature 534:538–543. 10.1038/nature18283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincent JA, Gabriel HM, Deardorff AS, Nardelli P, Fyffe RE, Burkholder T, Cope TC (2017) Muscle proprioceptors in adult rat: mechanosensory signaling and synapse distribution in spinal cord. J Neurophysiol 118:2687–2701. 10.1152/jn.00497.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yona S, Kim KW, Wolf Y, Mildner A, Varol D, Breker M, Strauss-Ayali D, Viukov S, Guilliams M, Misharin A, Hume DA, Perlman H, Malissen B, Zelzer E, Jung S (2013) Fate mapping reveals origins and dynamics of monocytes and tissue macrophages under homeostasis. Immunity 38:79–91. 10.1016/j.immuni.2012.12.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zelano J, Wallquist W, Hailer NP, Cullheim S (2007) Down-regulation of mRNAs for synaptic adhesion molecules neuroligin-2 and -3 and synCAM1 in spinal motoneurons after axotomy. J Comp Neurol 503:308–318. 10.1002/cne.21382 [DOI] [PubMed] [Google Scholar]
- Zhang J, Shi XQ, Echeverry S, Mogil JS, De Koninck Y, Rivest S (2007) Expression of CCR2 in both resident and bone marrow-derived microglia plays a critical role in neuropathic pain. J Neurosci 27:12396–12406. 10.1523/JNEUROSCI.3016-07.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]