

Summary
Aims
Oxidative stress is a direct cause of injury in various neural diseases. Manganese porphyrins (MnPs), a large category of superoxide dismutase (SOD) mimics, shown universally to have effects in numerous neural disease models in vivo. Given their complex intracellular redox activities, detailed mechanisms underlying the biomedical efficacies are not fully elucidated. This study sought to investigate the regulation of endogenous antioxidant systems by a MnP (MnTM‐4‐PyP) and its role in the protection against neural oxidative stress.
Methods
Primary cortical neurons were treated with MnTM‐4‐PyP prior to hydrogen peroxide–induced oxidative stress.
Results
MnTM‐4‐PyP increased cell viability, reduced intracellular level of reactive oxygen species, inhibited mitochondrial apoptotic pathway, and ameliorated endoplasmic reticulum function. The protein levels and activities of endogenous SODs were elevated, but not those of catalase. SOD2 transcription was promoted in a transcription factor–specific manner. Additionally, we found FOXO3A and Sirt3 levels also increased. These effects were not observed with MnTM‐4‐PyP alone.
Conclusion
Induction of various levels of endogenous antioxidant responses by MnTM‐4‐PyP has indispensable functions in its protection for cortical neurons against hydrogen peroxide‐induced oxidative stress.
Keywords: Endogenous antioxidant response, Manganese porphyrin, MnTM‐4‐PyP, Sirt3, Superoxide dismutase
Introduction
Oxidative stress is known as the unbalance between cellular oxidative and antioxidant systems that involves cellular injury and disruption of redox signaling. The role of oxidative stress has been well established in many disorders of the CNS, including Alzheimer's disease (AD) 1, Parkinson's disease (PD) 2, amyotrophic lateral sclerosis (ALS) 3, and ischemic stroke 4. While hyperphysiological level of reactive oxygen species (ROS) generation is triggered via diverse mechanisms, compromised antioxidant responses are also pertinent 5, 6, 7, 8. Excessive ROS that outweighs the capacity of cellular antioxidant systems can result in the dysfunction of major cellular systems, notably mitochondria and endoplasmic reticulum (ER) 9, 10. Among the damages caused by ROS, altered mitochondrial function and ER stress were found to play important roles in those neural disorders 10, 11.
Antioxidant strategies have been extensively investigated in therapeutics of the various neural disorders 12, 13. Superoxide dismutase (SOD) mimics are synthesized compounds designed to mimic endogenous SODs with sufficient activity of superoxide anion () dismutation and are usually complexed with metal ions as the redox‐active site 14. Manganese (Mn) complexes, especially manganese porphyrins (MnPs) are among the most studied SOD mimics with great potential for clinical applications 14. MnPs are characterized by the diversity of their structures and feasibility of optimization for activity, toxicity, and bioavailability 14. The efficacies of various MnPs have been confirmed in models of AD, PD, ALS, cerebral stroke, and other CNS disorders 15. However, detailed investigations of the mechanism of action in each case are limited. Explicit elucidation of the mechanisms can be the bottleneck of efforts pursuing the clinical use of MnPs 15.
Although initially designed as SOD mimics, it is recognized that no current MnP is fully comparable to endogenous SODs in specificity and activity in an intracellular context 14, 16. This fact highlights the importance of endogenous antioxidant systems in counteracting oxidative stress in pathological conditions. MnPs have complex redox chemistries that allow them to react with reactive species other than , cellular reductants such as glutathione (GSH) and other redox‐sensitive cellular components including transcription factors (TFs) 14; both antioxidant and pro‐oxidant activities can be involved. There are also cases in which direct reactive species‐scavenging cannot account for their biomedical efficacies 17, 18. Therefore, it is necessary to further investigate into MnPs' interaction with cellular redox systems and their potential impact on cellular antioxidant responses.
Manganese (III) meso‐tetrakis (N‐methylpyridinium‐4‐yl) porphyrin (MnTM‐4‐PyP5+ or MnTM‐4‐PyP, Figure 1A) is among the earliest‐discovered MnPs with a log kcat() of 6.58 19, 20, 21, 22. Other reported reactivities of MnTM‐4‐PyP involve peroxynitrite (ONOO−) reduction 23, hypochlorous acid (HOCl) reduction 24, and catalase‐like activity 25. Glutathione peroxidase (GPx)‐like activity for peroxide scavenging was also observed in isolated mitochondria 26, and similarly, reductase activity coupled with GSH redox cycling can be implied 14, 27. However, reactivity detected in solution such as SOD‐like and catalase‐like activity may not be competent in a cellular context 27, 28 and not much is known about its intracellular behavior. Until recently, MnTM‐4‐PyP was continually investigated in various CNS disease models in vivo, including ischemic stroke 29, global cerebral ischemia 30, PD 31, and epilepsy 32, and positive effects were observed. We propose that MnTM‐4‐PyP may be a suitable candidate for research on the interaction of MnPs with endogenous antioxidant systems in neurons. We also hypothesize that the potential to regulate endogenous antioxidant responses is important for the antioxidant effects of MnPs.
Figure 1.
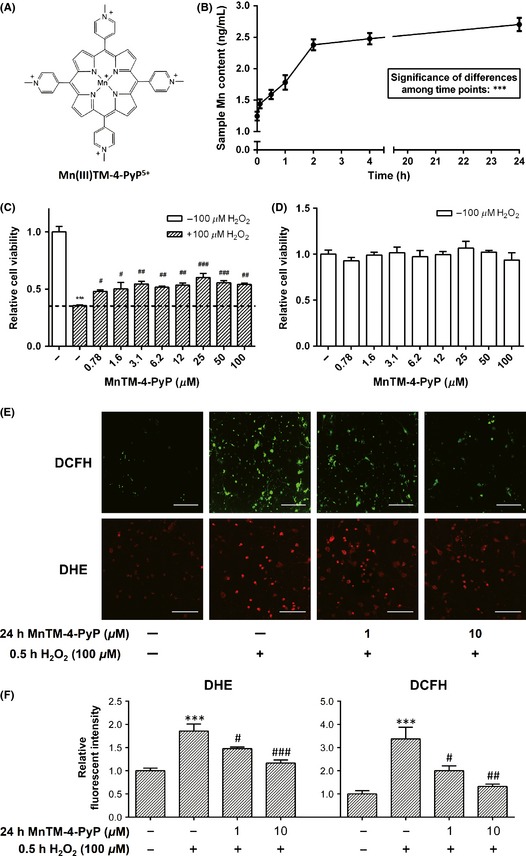
MnTM‐4‐PyP increases the viability of primary rat cortical neurons and reduces intracellular ROS levels. (A) Chemical structure of MnTM‐4‐PyP. (B) Intracellular manganese (Mn) content in the neurons at different time points after MnTM‐4‐PyP treatment assayed with inductively coupled plasma mass spectrometry (ICP‐MS). The data represent Mn concentrations in the test samples, which are in direct proportion to the respective intracellular Mn concentrations at the time points. ***P < 0.001. (C) Cell viability of primary rat cortical neurons with or without MnTM‐4‐PyP treatment for 24 h prior to H2O2 (30 min) ‐induced oxidative stress assayed by the AlamarBlue method. (D) Cell viability of the neurons treated with MnTM‐4‐PyP alone assayed by the AlamarBlue method. (E) ROS levels in primary rat cortical neurons with or without MnTM‐4‐PyP treatment prior to H2O2‐induced oxidative stress were detected by the fluorescent probes 2′, 7′‐dichlorodihydrofluorescein diacetate (DCFH‐DA) and dihydroethidium (DHE). Raw images acquired by a confocal laser scanning microscope (CLSM) are shown. Scale bars represent 100 μm. (F) Quantification of fluorescent intensity. In (C, D, and F), ***P < 0.001 compared to control groups; # P < 0.05, ## P < 0.01, and ### P < 0.001 compared to H2O2 groups. n = 3 for each group or time point in (B–D), n = 5 in (E, F).
In this study, we established the protection of MnTM‐4‐PyP against neural oxidative injury using an in vitro model of hydrogen peroxide (H2O2)–induced oxidative stress in primary rat cortical neurons. Cell viability, cellular oxidation and functions of mitochondrial and ER were inspected. We further investigated the underlying mechanisms by inspecting its impacts on the various components of cellular antioxidant systems, including antioxidant enzymes, redox‐responsive TFs, and other regulator proteins of redox responses.
Methods
Primary Rat Cortical Neuron Culture, MnTM‐4‐PyP Treatment, and Oxidative Injury by H2O2
The brains were isolated from postnatal day 1 Sprague‐Dawley rats, and whole cerebral neocortices were dissected. Tissues were incubated in 0.25% trypsin/EDTA and 30 U/mL DNAse for 15 min at 37°C and then triturated with a Pasteur pipette. The clumps were removed by filtering. The dissociated cortical neurons were cultured in 5 μg/mL poly‐L‐lysine (Sigma, St. Louis, MO, USA)‐coated wells in Neurobasal‐A with B‐27 (Life Technologies, Carlsbad, CA, USA), 0.5 mm L‐glutamine (Corning, Corning, NY, USA), and 1× penicillin–streptomycin. At 7 days in vitro, cells were treated with various concentrations of MnTM‐4‐PyP (Frontier Scientific, Logan, UT, USA) for 24 h, rinsed with PBS for 3 times, and treated with 100 μm H2O2 for 30 min. In other experiments, cells were treated with MnTM‐4‐PyP for 24 h without subsequent H2O2 treatment. Control experiments were performed with no MnTM‐4‐PyP or H2O2 treatment, and the H2O2‐injury group was treated with 100 μm H2O2 for 30 min only.
AlamarBlue Cell Viability Assay
The cells treated as described above were assayed for cell viability with AlamarBlue cell viability assay reagent (Life Technologies). Ten microlitre resazurin solution was added to the medium, and cells were incubated at 37°C for 12 h. OD570–OD600 was measured by a microtiter plate reader, and cell viability was calculated as (OD of test samples)/(OD of control samples) × 100%.
ICP‐MS Assay for Intracellular Manganese Content
Primary rat cortical neurons were treated with 10 μm MnTM‐4‐PyP for 0, 0.1, 0.5, 1, 2, 4, and 24 hours and rinsed with PBS for 3 times. The treated neurons were trypsinized and collected in centrifuge tubes. After centrifugation, the supernatant was abandoned and the pellet was resuspended in 1 mL ultrapure water, then the manganese content of each sample was assayed by ICP‐MS.
Fluorescent Probes
2′, 7′‐dichlorodihydrofluorescein diacetate (DCFH‐DA) and dihydroethidium (DHE; Life Technologies) were used for determination of the intracellular ROS levels, JC‐1 (Beyotime, Jiangsu, China) for mitochondrial membrane potential (ΔΨm), MitoTracker Green, and ER‐Tracker Red (Life Technologies) for mitochondrial and ER labeling, respectively. The treated cells were rinsed with PBS, incubated with the probes and observed with a confocal laser scanning microscope (CLSM; Leica, Wetzlar, Germany) according to the manufacturers' instructions. At least 5 fields of views were randomly selected for observation. Data were analyzed with Leica TCS‐NT or ImageJ Software.
Western Blot
Total protein was extracted from the treated cells with RIPA lysis buffer (with protease inhibitor cocktail), and the protein concentration was quantified by BCA method. 20 μg total protein from each sample was used for SDS‐PAGE and Western blot according to standard procedures. The bands were visualized by western chemiluminescent substrate (Millipore, Billerica, MA, USA). The optical densities of the bands were quantified using Quantity One 4.6 software (Bio‐Rad, Hercules, CA, USA). Antibodies for Western blot are as follows: GAPDH, Sirt3, pan Ac‐K, p53, Grp78, Calnexin (Cell Signaling Technology, Danvers, MA, USA); SOD2, SOD2 Ac‐K68 (Epitomics, Burlingame, CA, USA); SOD1, Catalase, FOXO3A, p‐IRE1, p‐eIF2α (Abcam, Cambridge, UK); p‐Akt, Bcl‐xL (Beyotime); goat anti‐rabbit or mouse IgG (H&L)‐HRP (GenScript, Piscataway, NJ, USA).
mRNA Quantitation by qRT‐PCR
Total RNA was extracted from treated neurons by the Trizol (Life Technologies) method according to standard procedure and quantified by spectrophotometry. 2.5 μg total RNA for each group was used for reverse transcription into cDNA with oligo dT primer, and q‐PCR was performed with GoTaq q‐PCR Master Mix (Promega, Madison, WI, USA) and Stratagene Mx3005P qPCR System (Agilent Technologies, Santa Clara, CA, USA) according to the manufacturer's instructions. All transcripts were normalized to β‐actin as internal control. Data were analyzed by MxPro‐Mx3005P 4.1 software. The following primers were used: β‐actin, forward 5′‐CCGCATCCTCTTCCTCCCT‐3′, reverse 5′‐GCCACAGGATTCCATACCCAG‐3′; sod1, forward 5′‐CGAGCATGGGTTCCATGTC‐3′, reverse 5′‐CTGGACCGCCATGTTTCTTAG‐3′; sod2, forward 5′‐ATTAACGCGCAGATCATGCA‐3′, reverse 5′‐CCTCGGTGACGTTCAGATTGT‐3′; catalase, forward 5′‐ACAACTCCCAGAAGCCTAAGAATG‐3′, reverse 5′‐GCTTTTCCCTTGGCAGCTATG‐3′.
Preparation of SOD2 Promoter‐Luciferase Reporter Construct and Assay of Transcription Activity
Sequence correlating to the promoter region of sod2 gene was amplified by PCR from the genome of HEK293T cell line using the following primers: forward 5′‐GCTGGCTCTACCCTCAGCTCATAG‐3′, reverse 5′‐TGCCGAAGCCACCACAGCCACGAGT‐3′. The sequence was cloned into pGL3 firefly luciferase reporter vector (Promega). The sod2 promoter‐luciferase reporter plasmid and pGL4.75 luciferase plasmid (Promega) as internal control were transfected with MegaTran 1.0 transfection reagent (Origene, Rockville, MD, USA) to C6 glioma cells at moderate confluency in culture. For assay of transactivational activity of NF‐κB or AP‐1, pNFκB‐TA‐luc or pAP1‐TA‐luc luciferase reporter plasmid (Beyotime) was transfected, respectively. The cells were treated with MnTM‐4‐PyP and/or H2O2 as described above 24 h after transfection, and luminescence was detected with dual‐luciferase assay system (Promega) as the manufacturer's instructions. Values were normalized to internal control for each group and reported as fold change to the untreated control group.
SOD and Catalase Activity Assay
Total protein was extracted from the treated cells as described in the Western blot section and quantified by BCA method. Twenty microgram total protein from each sample was used for SOD activity assay based on the WST method, and 10 μg total protein each was used for catalase activity assay with kits (Beyotime) according to the manufacturer's instructions.
Statistical Analysis
Data are presented as mean ± SEM for at least three replicates. Two‐tailed unpaired t‐test was performed for comparison of two groups. One‐way ANOVA was for comparison of multiple groups, including for the ICP‐MS data. Two‐way ANOVA was for the time‐dependent data of JC‐1 fluorescent intensity. Dunnett's post hoc test was for multiple comparisons. GraphPad Prism 6.0 Software (La Jolla, CA, USA) was used. P < 0.05 was considered statistically significant.
Results
MnTM‐4‐PyP Reduces Intracellular ROS Levels and Increases the Viability of Rat Cortical Neurons Subjected to Oxidative Injury
First, we established that MnTM‐4‐PyP protected cortical neurons against oxidative injury. Primary rat cortical neurons were treated as described in Methods and then assayed for cell viability. H2O2 treatment decreased the cell viability by approximately 70% compared with the untreated control, and the decrease in cell viability was significantly rescued by MnTM‐4‐PyP (Figure 1C). This effect did not show a strong concentration dependence in the range of 0.78–100 μm MnTM‐4‐PyP. Treatment with MnTM‐4‐PyP alone did not change the viability significantly compared with the untreated control (Figure 1D), which indicates a minimal influence by MnTM‐4‐PyP on the viability of normal cells.
Using inductively coupled plasma mass spectrometry (ICP‐MS), we found an increase in cellular Mn content upon MnTM‐4‐PyP treatment. Intracellular Mn content peaked at approximately 4 hours after treatment and remained nearly constant throughout 24 h (Figure 1B). This suggests that MnTM‐4‐PyP was able to enter the cells and function intracellularly.
To determine the antioxidant activity of MnTM‐4‐PyP, the intracellular ROS levels after H2O2 treatment were detected with fluorescent probes. General ROS level, as well as level in MnTM‐4‐PyP–pretreated cells was significantly lower than that in the cells without pretreatment (Figure 1E, F). Increasing the concentration of MnTM‐4‐PyP had a better effect of reducing intracellular ROS, measured by DCFA‐DA and DHE (Figure 1E, F). Additionally, treatment with MnTM‐4‐PyP alone at a higher concentration did not alter the basal level of intracellular superoxide (Figure 5D).
Figure 5.
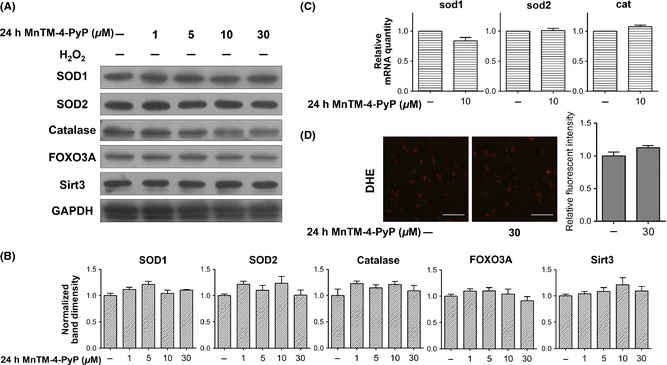
MnTM‐4‐PyP alone does not induce endogenous antioxidant responses. Primary rat cortical neurons were treated with MnTM‐4‐PyP alone (without subsequent H2O2 treatment). (A) Western blot assay of catalase, SOD1, SOD2, FOXO3A, and Sirt3 levels and (B) quantification of band densities relative to the control groups after normalization to GAPDH. (C) qRT‐PCR assay of mRNA levels of catalase, sod1, and sod2. The values represent fold changes to the control groups. (D) DHE staining for O2 − level and quantification of fluorescent intensity. Raw images acquired by a CLSM are shown. Scale bars represent 100 μm. n = 3 per group for Western blot or qRT‐PCR, and n = 5 per group for DHE staining.
MnTM‐4‐PyP Inhibits the Mitochondrial Apoptotic Pathway Triggered by H2O2 Treatment
As MnTM‐4‐PyP reduced ROS and counteracted the H2O2‐induced cellular injury, several major cellular events related to oxidative stress were investigated to further characterize the neuroprotection of MnTM‐4‐PyP. Oxidative stress can damage mitochondrial structure, disrupt normal mitochondrial functions, and trigger the mitochondrial pathway of apoptosis 10. Mitochondrial morphology was inspected with the MitoTracker fluorescent dye. The tubule network of mitochondria in normal neurons was disrupted by H2O2, and the disruption was partly rescued by MnTM‐4‐PyP pretreatment (Figure 2A).
Figure 2.
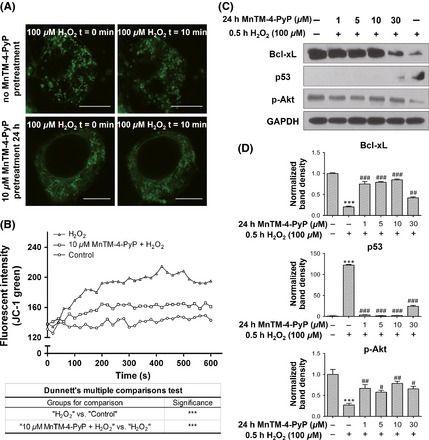
MnTM‐4‐PyP inhibits the mitochondrial pathway of apoptosis. Primary rat cortical neurons were treated with or without MnTM‐4‐PyP prior to H2O2 treatment. (A) The MitoTracker dye staining for mitochondrial morphology. Scale bars represent 7.5 μm. (B) Dynamic change of the fluorescent intensity of JC‐1 monomers after H2O2 treatment, which indicates the degree of mitochondrial membrane potential (ΔΨm) loss. JC‐1 form aggregates emitting red fluorescence at higher ΔΨm, while lowered ΔΨm leads to deaggregation into JC‐1 monomers which emit green fluorescence. ***P < 0.001. (C) Western blot assay for Bcl‐xL, p53 and p‐Akt levels and (D) quantification of band densities relative to the control groups after normalization to GAPDH. In (D), ***P < 0.001 compared to control groups; # P < 0.05, ## P < 0.01 and ### P < 0.001 compared to H2O2 groups. n = 3 for each group.
H2O2 induced mitochondrial depolarization, which typically occurs at the early stages of apoptosis 10. We examined mitochondrial membrane potential (ΔΨm) using the fluorescent probe JC‐1. MnTM‐4‐PyP pretreatment attenuated the loss of ΔΨm induced by H2O2 (Figure 2B). We next examined Bcl‐xL, an antiapoptotic member of the Bcl protein family 33, and p53, the major mediator of cell fate known to promote apoptosis 34. MnTM‐4‐PyP elevated the level of Bcl‐xL, which was suppressed by H2O2 (Figure 2C, D), whereas the change of p53 level was the opposite. p‐Akt, which is known to negatively regulate p53 and promote cell survival, showed a similar change in its level as Bcl‐xL. The above results indicate that MnTM‐4‐PyP inhibited the mitochondrial apoptotic pathway initiated by H2O2 treatment.
MnTM‐4‐PyP Effectively Orchestrated Unfolded Protein Response (UPR) Upon H2O2 Treatment
ER is the major organelle for oxidative folding of proteins. Oxidative stress can disrupt ER function, resulting in accumulation of misfolded proteins (known as ER stress) 9. The ER of cortical neurons was labeled with the ER‐Tracker dye, and ER morphology was inspected. H2O2 led to a compromised ER structure while MnTM‐4‐PyP pretreatment partly restored the normal morphology of ER (Figure 3A).
Figure 3.
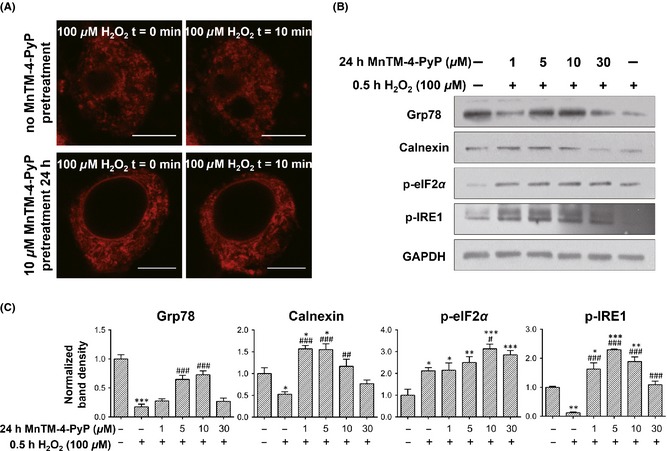
MnTM‐4‐PyP maintains intracellular calcium homeostasis and effectively orchestrates unfolded protein response (UPR). Primary rat cortical neurons were treated with or without MnTM‐4‐PyP prior to H2O2 treatment. (A) The ER‐Tracker dye staining for endoplasmic reticulum (ER) morphology. Scale bars represent 7.5 μm. (B) Western blot assay for UPR‐related proteins and (C) quantification of band densities relative to the control groups after normalization to GAPDH. *P < 0.05, **P < 0.01, and ***P < 0.001 compared to control groups; # P < 0.05, ## P < 0.01, and ### P < 0.001 compared to H2O2 groups. n = 3 for each group.
ER stress can trigger a collection of adaptive responses known as UPR 9, 11. A series of UPR‐related proteins were assayed by Western blot (Figure 3B, C). Upon H2O2 treatment, levels of the UPR transducer p‐IRE1 and ER chaperone Grp78, and Calnexin were decreased. By contrast, MnTM‐4‐PyP pretreatment inhibited or even reversed the decrease in protein levels, which indicates effective UPR induction. However, the level of p‐eIF2α, another component of UPR, increased after H2O2 treatment and was further elevated by MnTM‐4‐PyP.
MnTM‐4‐PyP Exhibits Differential Effects on Levels and Activities of Endogenous Antioxidant Enzymes
As shown above, the various aspects of the neuroprotection of MnTM‐4‐PyP against oxidative stress were established. Next, we explored the mechanisms underlying the antioxidation of MnTM‐4‐PyP by focusing on the endogenous antioxidant systems. Levels of several antioxidant enzymes including catalase, SOD1, and SOD2 in cells with or without MnTM‐4‐PyP treatment prior to H2O2‐induced injury were assayed. Unexpectedly, MnTM‐4‐PyP pretreatment exhibited differential effects on the various enzymes (Figure 4A, B). Only catalase was induced by H2O2, although its enzymatic activity was not significantly changed (Figure 4D). SOD2 level decreased upon H2O2 treatment. In the MnTM‐4‐PyP‐pretreated group, SOD2 level was efficiently rescued from decreasing, SOD1 level was increased, whereas catalase level was not significantly affected. In addition, level of FOXO3A, a TF intensively involved in antioxidant response 35, exhibited a similar although less remarkable change as the SODs. Changes in SOD activities were generally consistent with the respective changes of enzyme levels (Figure 4D).
Figure 4.
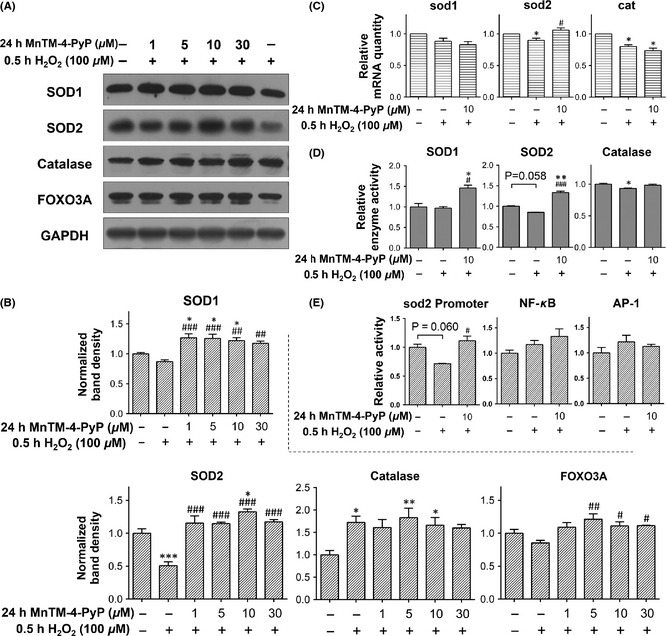
MnTM‐4‐PyP exhibits differential effects on endogenous antioxidant proteins. Primary rat cortical neurons were treated with or without MnTM‐4‐PyP prior to H2O2 treatment. (A) Western blot assay of catalase, SOD1, SOD2, and FOXO3A levels and (B) quantification of band densities relative to the control groups after normalization to GAPDH. (C) qRT‐PCR assay of mRNA levels of catalase, sod1, and sod2. The values represent fold changes to the control groups. (D) Assay of SOD and catalase activities. SOD activities are calculated as inhibition rate and catalase activity is calculated as activity units per milligram of total protein, then the data are normalized to those of the control group. (E) C6 glioma cells were treated with or without MnTM‐4‐PyP prior to H2O2 treatment, and the activities of sod2 promoter, NF‐κB responsive promoter, and AP‐1 responsive promoter were determined with luciferase reporter assays. *P < 0.05, **P < 0.01 and ***P < 0.001 compared to control groups; # P < 0.05, ## P < 0.01 and ### P < 0.001 compared to H2O2 groups. n = 3 for each group.
mRNA levels of the antioxidant enzymes were quantified by qRT‐PCR (Figure 4C). The result was not in perfect correlation with that of the protein levels, which is common 36. We observed the sod2 mRNA level was elevated by MnTM‐4‐PyP pretreatment with statistical significance, similar to the change of its protein level. By contrast, the mRNA levels of catalase were suppressed by H2O2, but not elevated by MnTM‐4‐PyP pretreatment.
As the sod2 mRNA level was increased by MnTM‐4‐PyP pretreatment, the transcription of sod2 gene was further investigated by luciferase reporter assays. The assays were performed in C6 rat glioma cells because of the difficulty in neuronal transfection. H2O2 repressed the sod2 promoter activity, whereas MnTM‐4‐PyP pretreatment inhibited the repression (Figure 4E). We further examined the TF‐specific effect on sod2 transcription. NF‐κB and AP‐1 are two well‐established classical TFs involved in cellular redox response, and their binding elements have been identified in the sequences of sod2 promoter and/or enhancer 37, 38. However, elevation of NF‐κB and AP‐1‐specific transcription by MnTM‐4‐PyP was not observed (Figure 4E), suggesting a minimal involvement of these TFs in SOD2 induction by MnTM‐4‐PyP.
MnTM‐4‐PyP Treatment Alone Does Not Significantly Induce Endogenous Antioxidant Responses
MnTM‐4‐PyP with subsequent H2O2 treatment induced endogenous antioxidant responses as indicated above. In addition, we also inspected the effect of MnTM‐4‐PyP alone (without H2O2). It was discovered that neither protein nor mRNA levels of the aforementioned proteins were significantly elevated by MnTM‐4‐PyP treatment alone, compared with the untreated control group (Figure 5). This suggests that noticeable onset of antioxidant response was absent.
MnTM‐4‐PyP Induces Sirt3 in Parallel with an Increase in the Deacetylated form of SOD2
SOD2 activity can be regulated on the level of posttranslational modification (PTM). Several lysine residues of SOD2, including K68, have been shown to undergo acetylation/deacetylation. The deacetylation, mediated by Sirt3, corresponds to increase in SOD2 activity 39, 40, 41. It was found that Sirt3 level was significantly elevated by MnTM‐4‐PyP (Figure 6A, B). Concomitantly, acetylation level of SOD2 detected by a site‐specific antibody of Ac‐K68 and a pan Ac‐K antibody did not increase comparably to that of total SOD2 (Figure 4A, B); thus, the deacetylated form of SOD2 was increased. This result can be better visualized by the ratio of acetylated SOD2 level to total SOD2 level (measured as band density) in each group (Figure 6C). Additionally, MnTM‐4‐PyP alone did not significantly increase Sirt3 level (Figure 5A, B).
Figure 6.
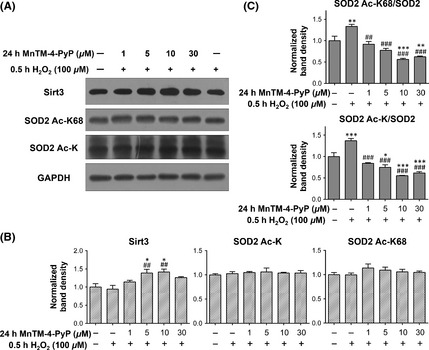
MnTM‐4‐PyP induces Sirt3 in parallel with SOD2 deacetylation. Primary rat cortical neurons were treated with or without MnTM‐4‐PyP prior to H2O2 treatment. (A) Western blot assay of Sirt3 and acetylated SOD2 levels. A SOD2 Ac‐K68 antibody and a pan Ac‐K antibody were used. (B) Quantification of band densities relative to the control groups after normalization to GAPDH. (C) The ratio of acetylated SOD2 level to total SOD2 level represented as the ratio of their respective band densities. *P < 0.05, **P < 0.01 and ***P < 0.001 compared to control groups; ## P < 0.01 and ### P < 0.001 compared to H2O2 groups. n = 3 for each group.
Discussion
We hypothesized that modulation or induction of endogenous antioxidant systems may comprise a major proportion of MnPs' pharmacology in CNS diseases. This study investigated this issue using MnTM‐4‐PyP, a MnP with moderate reactivity toward 22, and discovered that it regulated various levels of antioxidant response during H2O2‐induced oxidative stress in primary rat cortical neurons.
Consistent with previously established in vivo efficacies, we confirmed the protective effect of MnTM‐4‐PyP for primary rat cortical neurons against H2O2‐induced oxidative injury, indicated by the increased cell viability in the MnTM‐4‐PyP‐pretreated group. The neuroprotection of MnTM‐4‐PyP was further characterized by the inhibition of mitochondrial apoptotic pathway and the amelioration of the disrupted ER function. Mitochondria and ER are major targets of ROS‐induced injury. Excessive ROS damages the membranes of the organelles and inactivates different pore or channel proteins. For mitochondria, this leads to the transition of membrane permeability, the depletion of ΔΨm, and compromised mitochondrial respiration and initiation of apoptosis 10; for ER, ER stress can be induced 9. These events are relevant in the aforementioned neural diseases 10, 11. MnTM‐4‐PyP inhibited those events caused by H2O2. Notably, H2O2 treatment disrupted ER function, however; the cells did not seem to effectively mount the UPR as a response. Similar results were reported in rat cortical neurons that 100 μm H2O2 induced severe cell injury and downregulated ER stress response 42. By contrast, MnTM‐4‐PyP pretreatment elevated the levels of several UPR‐transducers and ER chaperones, thus contributing to the counteraction of ER stress.
DCFH‐DA and DHE were used as ROS probes in this study. DCFH‐DA is among the most widely used fluorescent probes for H2O2 and oxidative stress. Unfortunately, its reliability is largely compromised by its promiscuous reactivity 43. In our case, the fluorescent intensity of DCF was suppressed by MnTM‐4‐PyP pretreatment. Based on this result, we reason that MnTM‐4‐PyP probably exerted antioxidation or other protective effects. DHE, on the other hand, is a relatively acceptable probe for 43. MnTM‐4‐PyP treatment reduced oxidative stress, which can account for the aforementioned protective effects. However, it is not known to what degree the effect is credited to the direct ROS‐scavenging activity of MnTM‐4‐PyP. MnTM‐4‐PyP is among an earlier generation of MnPs as SOD mimics with a suboptimal structure and kcat for superoxide dismutation 19, 20, 21, and current evidence has not justified any direct ROS‐scavenging activity of MnTM‐4‐PyP to be significant in cells 16, 28.
Although we were unable to directly investigate the issue, our results highlighted the indirect effects of MnTM‐4‐PyP by showing that endogenous antioxidant response was significantly induced by MnTM‐4‐PyP pretreatment (compared with H2O2 treatment alone). Regulation on different levels of redox responses by MnTM‐4‐PyP was observed.
Protein levels of SODs were not increased, or even decreased by 100 μm H2O2 for 30 min, which correlated to several in vivo studies reporting repressed antioxidant status in acute neural injuries 5. MnTM‐4‐PyP pretreatment remarkably elevated the protein levels, and enzymatic activities correspondingly increased. Thus, a possibly incompetent SOD mimic was able to effectively scavenge (indicated by the DHE probe) at least partly by inducing the endogenous SOD proteins. The different response of catalase will be discussed later in this article.
On the transcriptional level, the mRNA quantity of sod2 correlated well with the protein level. The transcriptional activity of sod2 gene was specifically elevated by MnTM‐4‐PyP, although this was observed in C6 glioma cells. The mRNA levels of sod1 and catalase did not correlate well with their respective protein levels; mRNA levels can be much lower as in our case, which could be due to the short half‐life of mRNA or negative feedback of transcription 44. MnTM‐4‐PyP pretreatment also increased the level of FOXO3A, a TF of forkhead homeobox type O (FOXO) family, whose targets include the genes of antioxidant proteins 45, 46, 47, and is important in antioxidant responses 35.
As a regulation on the PTM level, MnTM‐4‐PyP pretreatment significantly elevated the protein level of Sirt3, a NAD+‐dependent protein lysine deacetylase of the sirtuin family that mainly localizes to mitochondria 48. By deacetylating and activating a large array of proteins involved in mitochondrial energy metabolism and antioxidation, Sirt3 has a pivotal role in maintaining normal mitochondrial physiology and reducing oxidative stress 48. Specially, Sirt3 deacetylates several lysine residues of SOD2 including K53, K89, K68, and K122, resulting in increased SOD2 activity 40, 41, 49. In our study, the elevated SOD2 activity correlated with the increase in deacetylated form of SOD2 and the concomitant induction of Sirt3 by MnTM‐4‐PyP. This suggests that SOD2 activity was regulated by Sirt3 in this case. Although induction of Sirt3 by MnTM‐4‐PyP points to the possibility that the MnP could exert a more extensive regulatory effect on endogenous redox systems via the PTM activity of Sirt3, it remains to be confirmed that the deacetylation activities of Sirt3 for various substrates were elevated.
As elaborated above, various aspects of endogenous antioxidant response were induced by MnTM‐4‐PyP. We speculate that the contribution of endogenous antioxidant systems was indispensable to the antioxidant and protective effect of MnTM‐4‐PyP in the neurons. Besides, MnTM‐4‐PyP appeared to regulate the endogenous antioxidant systems in a selective manner, possibly due to the specificity of redox signaling. Contrary to SODs, the protein level of catalase was increased by H2O2, whereas paradoxically the enzymatic activity slightly decreased. The discrepancy between protein level and activity may be explained as an inhibition of catalase activity by ROS 50. sod2 transcription seemed to be activated with TF specificity, in that the transcription activation activities of the well‐known redox‐responsive TFs, NF‐κB, and AP‐1 were not remarkably altered. Previous studies reported that MnPs were able to inhibit the activation of NF‐κB and AP‐1, and the inhibition of NF‐κB was mediated by the pro‐oxidant activity of MnPs 51, 52, 53. Thus, the redox behavior of MnTM‐4‐PyP in our study was possibly different. Indeed MnTM‐4‐PyP treatment alone did not exhibit an apparent pro‐oxidant effect (Figure 5). Other TFs may be responsible for the elevated sod2 transcription, such as SP‐1 and AP‐2, whose binding elements have been identified in the sod2 gene 38. FOXO3A activation also upregulates sod2 expression 45, the correlation between the levels of FOXO3A and SOD2 suggests the role of FOXO3A in SOD2 induction. Regarding how specific TFs were activated and antioxidants were differentially induced, further study on the intracellular redox activity of MnP is needed. Also note that MnTM‐4‐PyP appeared to induce the antiapoptotic and UPR‐related proteins at an optimal concentration in the range of 5–10 μm. Interestingly, the changes in SOD2 and Sirt3 levels showed a similar pattern of concentration dependence, which suggests again that endogenous antioxidant responses are associated with MnTM‐4‐PyP's efficacy. It is possible that a higher concentration (30 μm) of MnTM‐4‐PyP was not as efficient in reducing ROS, although it is sufficient to rescue the decrease in cell viability (Figure 1C). Investigation of the details of redox signaling is needed to exactly understand these aspects of MnTM‐4‐PyP's activity.
Conclusion
This study investigated the regulation of endogenous antioxidant systems by MnTM‐4‐PyP and sought to emphasize the contribution of this effect to the protection of MnTM‐4‐PyP against neural oxidative stress (Figure 7). In the in vitro model of H2O2‐induced oxidative stress in cortical neurons, MnTM‐4‐PyP exhibited an extensive yet differential impact on various components of endogenous redox systems, which was indispensable for the neuroprotection of the MnP. However, more detailed mechanisms underlying the regulatory effect await further investigations. This study highlights the significance of potential redox regulatory activities of MnPs and may promote further mechanistic studies into their biomedical efficacies, especially in CNS diseases. Elucidation of mechanisms will definitely lay the foundation for successful clinical applications of MnPs in therapeutics for CNS diseases.
Figure 7.
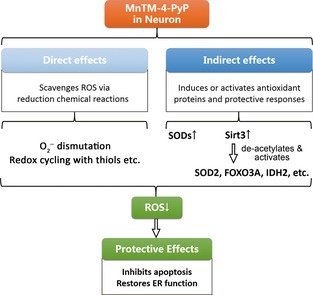
Scheme of the antioxidant functions of MnTM‐4‐PyP in neurons. In the cellular context, MnTM‐4‐PyP and possibly other manganese porphyrins may function both directly as reactive species scavengers (including via coupling reactions with cellular reductants) and indirectly as redox modulators that induce or activate diverse layers of antioxidant responses. This study focused on the indirect functions and discovered that SODs and Sirt3 were induced by MnTM‐4‐PyP in neurons, which contributes to the reduction of oxidative stress and the positive effects achieved. Details of the redox signaling involved await further investigation.
Conflict of interest
The authors declare no conflict of interest.
Acknowledgment
This study was supported by the National Natural Science Foundation of China (No. 21371013), Beijing Nova Program (No. 2010010), and the National Basic Research Program of China (No. 2010CB12300).
The first two authors contributed equally to this work.
References
- 1. Butterfield DA, Swomley AM, Sultana R. Amyloid beta‐peptide (1‐42)‐induced oxidative stress in Alzheimer disease: importance in disease pathogenesis and progression. Antioxid Redox Signal 2013;19:823–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Dias V, Junn E, Mouradian MM. The role of oxidative stress in Parkinson's disease. J Parkinsons Dis 2013;3:461–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Barber SC, Shaw PJ. Oxidative stress in ALS: key role in motor neuron injury and therapeutic target. Free Radic Biol Med 2010;48:629–641. [DOI] [PubMed] [Google Scholar]
- 4. Allen CL, Bayraktutan U. Oxidative stress and its role in the pathogenesis of ischaemic stroke. Int J Stroke 2009;4:461–470. [DOI] [PubMed] [Google Scholar]
- 5. Cojocaru IM, Cojocaru M, Sapira V, Ionescu A. Evaluation of oxidative stress in patients with acute ischemic stroke. Rom J Intern Med 2013;51:97–106. [PubMed] [Google Scholar]
- 6. Hitchler MJ, Domann FE. Regulation of CuZnSOD and Its Redox Signaling Potential: implications for Amyotrophic Lateral Sclerosis. Antioxid Redox Signal 2014;20:1590–1598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Venkateshappa C, Harish G, Mahadevan A, Srinivas BM, Shankar SK. Elevated oxidative stress and decreased antioxidant function in the human hippocampus and frontal cortex with increasing age: implications for neurodegeneration in Alzheimer's disease. Neurochem Res 2012;37:1601–1614. [DOI] [PubMed] [Google Scholar]
- 8. Venkateshappa C, Harish G, Mythri RB, Mahadevan A, Bharath MM, Shankar SK. Increased oxidative damage and decreased antioxidant function in aging human substantia nigra compared to striatum: implications for Parkinson's disease. Neurochem Res 2012;37:358–369. [DOI] [PubMed] [Google Scholar]
- 9. Csala M, Margittai E, Banhegyi G. Redox control of endoplasmic reticulum function. Antioxid Redox Signal 2010;13:77–108. [DOI] [PubMed] [Google Scholar]
- 10. Figueira TR, Barros MH, Camargo AA, et al. Mitochondria as a source of reactive oxygen and nitrogen species: from molecular mechanisms to human health. Antioxid Redox Signal 2013;18:2029–2074. [DOI] [PubMed] [Google Scholar]
- 11. Roussel BD, Kruppa AJ, Miranda E, Crowther DC, Lomas DA, Marciniak SJ. Endoplasmic reticulum dysfunction in neurological disease. Lancet Neurol 2013;12:105–118. [DOI] [PubMed] [Google Scholar]
- 12. Guerra‐Araiza C, Alvarez‐Mejia AL, Sanchez‐Torres S, et al. Effect of natural exogenous antioxidants on aging and on neurodegenerative diseases. Free Radic Res 2013;47:451–462. [DOI] [PubMed] [Google Scholar]
- 13. Uttara B, Singh AV, Zamboni P, Mahajan RT. Oxidative stress and neurodegenerative diseases: a review of upstream and downstream antioxidant therapeutic options. Curr Neuropharmacol 2009;7:65–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Batinic‐Haberle I, Tovmasyan A, Roberts ER, Vujaskovic Z, Leong KW, Spasojevic I. SOD therapeutics: latest insights into their structure‐activity relationships and impact on the cellular redox‐based signaling pathways. Antioxid Redox Signal 2014;20:2372–2415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sheng H, Chaparro RE, Sasaki T, et al. Metalloporphyrins as therapeutic catalytic oxidoreductants in central nervous system disorders. Antioxid Redox Signal 2014;20:2437–2464. [DOI] [PubMed] [Google Scholar]
- 16. Liochev SI. Superoxide dismutase mimics, other mimics, antioxidants, prooxidants, and related matters. Chem Res Toxicol 2013;26:1312–1319. [DOI] [PubMed] [Google Scholar]
- 17. Tauskela JS, Brunette E. Neuroprotection against staurosporine by metalloporphyrins independent of antioxidant capability. Neurosci Lett 2009;466:41–46. [DOI] [PubMed] [Google Scholar]
- 18. Tauskela JS, Brunette E, O'Reilly N, et al. An alternative Ca2 + ‐dependent mechanism of neuroprotection by the metalloporphyrin class of superoxide dismutase mimetics. FASEB J 2005;19:1734–1736. [DOI] [PubMed] [Google Scholar]
- 19. Pasternack RF, Banth A, Pasternack JM, Johnson CS. Catalysis of the disproportionation of superoxide by metalloporphyrins. III. J Inorg Biochem 1981;15:261–267. [DOI] [PubMed] [Google Scholar]
- 20. Peretz P, Solomon D, Weinraub D, Faraggi M. Chemical properties of water‐soluble porphyrins 3. The reaction of superoxide radicals with some metalloporphyrins. Int J Radiat Biol Relat Stud Phys Chem Med 1982;42:449–456. [DOI] [PubMed] [Google Scholar]
- 21. Weinraub D, Levy P, Faraggi M. Chemical properties of water‐soluble porphyrins. 5. Reactions of some manganese (III) porphyrins with the superoxide and other reducing radicals. Int J Radiat Biol Relat Stud Phys Chem Med 1986;50:649–658. [DOI] [PubMed] [Google Scholar]
- 22. Batinić‐Haberle I, Spasojević I, Hambright P, Benov L, Crumbliss AL, Fridovich I. Relationship among redox potentials, proton dissociation constants of pyrrolic nitrogens, and in vivo and in vitro superoxide dismutating activities of Manganese(III) and Iron(III) water‐soluble porphyrins. Inorg Chem 1999;38:4011–4022. [Google Scholar]
- 23. Ferrer‐Sueta G, Batinic‐Haberle I, Spasojevic I, Fridovich I, Radi R. Catalytic scavenging of peroxynitrite by isomeric Mn(III) N‐methylpyridylporphyrins in the presence of reductants. Chem Res Toxicol 1999;12:442–449. [DOI] [PubMed] [Google Scholar]
- 24. Carnieri N, Harriman A, Porter G. Photochemistry of manganese porphyrins. Part 6. Oxidation–reduction equilibria of manganese (III) porphyrins in aqueous solution. J Chem Soc, Dalton Trans 1982;931–938. [Google Scholar]
- 25. Day BJ, Fridovich I, Crapo JD. Manganic porphyrins possess catalase activity and protect endothelial cells against hydrogen peroxide‐mediated injury. Arch Biochem Biophys 1997;347:256–262. [DOI] [PubMed] [Google Scholar]
- 26. Araujo‐Chaves JC, Yokomizo CH, Kawai C, et al. Towards the mechanisms involved in the antioxidant action of MnIII [meso‐tetrakis(4‐N‐methyl pyridinium) porphyrin] in mitochondria. J Bioenerg Biomembr 2011;43:663–671. [DOI] [PubMed] [Google Scholar]
- 27. Faulkner KM, Liochev SI, Fridovich I. Stable Mn(III) porphyrins mimic superoxide dismutase in vitro and substitute for it in vivo . J Biol Chem 1994;269:23471. [PubMed] [Google Scholar]
- 28. Pessoto FS, Inada NM, Nepomuceno MF, et al. Biological effects of anionic meso‐tetrakis (para‐sulfonatophenyl) porphyrins modulated by the metal center. Studies in rat liver mitochondria. Chem Biol Interact 2009;181:400–408. [DOI] [PubMed] [Google Scholar]
- 29. Huang HF, Guo F, Cao YZ, Shi W, Xia Q. Neuroprotection by manganese superoxide dismutase (MnSOD) mimics: antioxidant effect and oxidative stress regulation in acute experimental stroke. CNS Neurosci Ther 2012;18:811–818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Sharma SS, Gupta S. Neuroprotective effect of MnTMPyP, a superoxide dismutase/catalase mimetic in global cerebral ischemia is mediated through reduction of oxidative stress and DNA fragmentation. Eur J Pharmacol 2007;561:72–79. [DOI] [PubMed] [Google Scholar]
- 31. Dixit A, Srivastava G, Verma D, et al. Minocycline, levodopa and MnTMPyP induced changes in the mitochondrial proteome profile of MPTP and maneb and paraquat mice models of Parkinson's disease. Biochim Biophys Acta 2013;1832:1227–1240. [DOI] [PubMed] [Google Scholar]
- 32. Folbergrova J, Otahal J, Druga R. Brain superoxide anion formation in immature rats during seizures: protection by selected compounds. Exp Neurol 2012;233:421–429. [DOI] [PubMed] [Google Scholar]
- 33. Czabotar PE, Lessene G, Strasser A, Adams JM. Control of apoptosis by the BCL‐2 protein family: implications for physiology and therapy. Nat Rev Mol Cell Biol 2013;15:49–63. [DOI] [PubMed] [Google Scholar]
- 34. Wang DB, Kinoshita C, Kinoshita Y, Morrison RS. p53 and mitochondrial function in neurons. Biochim Biophys Acta 2014;1842:1186–1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Storz P. Forkhead homeobox type O transcription factors in the responses to oxidative stress. Antioxid Redox Signal 2011;14:593–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Vogel C, Edward M. Insights into the regulation of protein abundance from proteomic and transcriptomic analyses. Nat Rev Genet 2012;13:227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Yeh CC, Wan XS, St CD. Transcriptional regulation of the 5′ proximal promoter of the human manganese superoxide dismutase gene. DNA Cell Biol 1998;17:921–930. [DOI] [PubMed] [Google Scholar]
- 38. Xu Y, Porntadavity S, St CD. Transcriptional regulation of the human manganese superoxide dismutase gene: the role of specificity protein 1 (Sp1) and activating protein‐2 (AP‐2). Biochem J 2002;362:401–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Zhu Y, Park SH, Ozden O, et al. Exploring the electrostatic repulsion model in the role of Sirt3 in directing MnSOD acetylation status and enzymatic activity. Free Radic Biol Med 2012;53:828–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Chen Y, Zhang J, Lin Y, et al. Tumour suppressor SIRT3 deacetylates and activates manganese superoxide dismutase to scavenge ROS. EMBO Rep 2011;12:534–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Tao R, Coleman MC, Pennington JD, et al. Sirt3‐mediated deacetylation of evolutionarily conserved lysine 122 regulates MnSOD activity in response to stress. Mol Cell 2010;40:893–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Paschen W, Mengesdorf T, Althausen S, Hotop S. Peroxidative stress selectively down‐regulates the neuronal stress response activated under conditions of endoplasmic reticulum dysfunction. J Neurochem 2001;76:1916–1924. [DOI] [PubMed] [Google Scholar]
- 43. Kalyanaraman B, Darley‐Usmar V, Davies KJ, et al. Measuring reactive oxygen and nitrogen species with fluorescent probes: challenges and limitations. Free Radic Biol Med 2012;52:1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Greenbaum D, Colangelo C, Williams K, Gerstein M. Comparing protein abundance and mRNA expression levels on a genomic scale. Genome Biol 2003;4:117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kops GJ, Dansen TB, Polderman PE, et al. Forkhead transcription factor FOXO3a protects quiescent cells from oxidative stress. Nature 2002;419:316–321. [DOI] [PubMed] [Google Scholar]
- 46. Furuyama T, Nakazawa T, Nakano I, Mori N. Identification of the differential distribution patterns of mRNAs and consensus binding sequences for mouse DAF‐16 homologues. Biochem J 2000;349:629–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Nemoto S, Finkel T. Redox regulation of forkhead proteins through a p66shc‐dependent signaling pathway. Science 2002;295:2450–2452. [DOI] [PubMed] [Google Scholar]
- 48. Bause AS, Haigis MC. SIRT3 regulation of mitochondrial oxidative stress. Exp Gerontol 2013;48:634–639. [DOI] [PubMed] [Google Scholar]
- 49. Qiu X, Brown K, Hirschey MD, Verdin E, Chen D. Calorie restriction reduces oxidative stress by SIRT3‐mediated SOD2 activation. Cell Metab 2010;12:662–667. [DOI] [PubMed] [Google Scholar]
- 50. Lardinois OM, Mestdagh MM, Rouxhet PG. Reversible inhibition and irreversible inactivation of catalase in presence of hydrogen peroxide. Biochim Biophys Acta 1996;1295:222–238. [DOI] [PubMed] [Google Scholar]
- 51. Tse HM, Milton MJ, Piganelli JD. Mechanistic analysis of the immunomodulatory effects of a catalytic antioxidant on antigen‐presenting cells: implication for their use in targeting oxidation‐reduction reactions in innate immunity. Free Radic Biol Med 2004;36:233–247. [DOI] [PubMed] [Google Scholar]
- 52. Jaramillo MC, Briehl MM, Crapo JD, Batinic‐Haberle I, Tome ME. Manganese porphyrin, MnTE‐2‐PyP5 + , Acts as a pro‐oxidant to potentiate glucocorticoid‐induced apoptosis in lymphoma cells. Free Radic Biol Med 2012;52:1272–1284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Zhao Y, Chaiswing L, Oberley TD, et al. A mechanism‐based antioxidant approach for the reduction of skin carcinogenesis. Cancer Res 2005;65:1401–1405. [DOI] [PubMed] [Google Scholar]