

Summary
Aims
Stroke is a leading cause of disability. However, there is no pharmacological therapy available for promoting recovery. Although treatment of stroke with cystamine has gained increasing interest, the detailed mechanisms underlying this process remain elusive. Thus, our aim is to examine the effect of cystamine on the function recovery after stroke and investigate further cystamine mechanisms.
Methods
Adult male C57BL/6J mice were subjected to photothrombotic model of focal stroke or sham operation. Cystamine or saline was administered intraperitoneally at 24 h after stroke. Functional recovery was analyzed using behavioral tests; axon remodeling was analyzed using magnetic resonance diffusion tensor imaging (DTI) and histological assessment. ANA‐12, an antagonist of tropomyosin‐related kinase B (TrkB), was administrated to examine the mechanisms underlying the neuroprotection mediated by cystamine.
Results
Treatment with cystamine resulted in amelioration of impaired function with concomitant enhancement of axonal remodeling. Cystamine treatment significantly increased brain‐derived neurotrophic factor (BDNF) levels and phosphorylation of TrkB in brain after stroke. Cystamine significantly enhanced neuronal progenitor cell proliferation, neuronal survival, and plasticity through BDNF/TrkB pathway.
Conclusions
These data provide evidence to investigate the promising utility of cystamine for therapy of stroke in a variety of ways, acting principally through BDNF/TrkB pathway.
Keywords: Axonal remodeling, Cystamine, Neuroprotection, Rehabilitation, Stroke
Introduction
Stroke is known to be one of the most devastating neurological disorder leading to high rates of death, physical impairment, and disability in adults 1. Although several clinical trials were undertaken to find effective therapies 2, there is no pharmacological therapy available for promoting brain recovery. Thrombolytic therapy of ischemic stroke is limited by a narrow time window and by side effects of the thrombolytic recombinant tissue plasminogen activator (rt‐PA) 3, 4. Therefore, novel approaches that might overcome these side effects and extend the therapeutic time window of the latter are urgently needed.
Cystamine, as an inhibitor of transglutaminases (TGs) through formation of mixed disulfide 5, has been demonstrated to exert neuroprotection in neurodegenerative diseases, such as Parkinson's disease and Huntington's disease 6, 7. Based on the role of cystamine in the neurodegenerative diseases and the increased expression of tissue‐type transglutaminase (tTG) in the hippocampus after stroke 8, cystamine can be envisioned as a promising candidate for neuroprotection against stroke. While inhibition of tTG for the treatment of stroke has gained increasing interest 9, 10, 11, whether cystamine can improve functional recovery and enhance axon remodeling in animal photothrombotic models of focal stroke remains to be elucidated.
In addition to the role of as an inhibitor of TGs, cystamine also functions though other alternative mechanisms. A previous study indicated that cystamine exerted neuroprotection through upregulation of brain‐derived neurotrophic factor (BDNF) expression 12. BDNF acts via two receptor tropomyosin‐related kinase B (TrkB) and the p75 receptor 13. BDNF has been implicated as a crucial factor in the developing postnatal rat brains and in reversing neuronal toxicity 14. However, whether BDNF and downstream TrkB activation is involved in cystamine‐mediated neuroprotection in stroke remains an enigma.
In this study, we show direct evidence that treatment with cystamine starting at 24 h after stroke resulted in amelioration of impaired behavior with concomitant enhancement of axon remodeling and neuroprotection via the BDNF/TrkB pathway.
Materials and Methods
Animals and Photothrombotic Model of Focal Stroke
This study was approved by Institutional Animal Care and Use Committee (IACUC) of Southeast University (approval ID: SYXK‐2010.4987). Adult male C57BL/6J mice (20.0–25.5 g weight, 8–10 weeks age) were used in this study. Focal stroke was induced by photothrombosis as described previously 15. Briefly, mice were anesthetized by intraperitoneal administration with pentobarbital (50 mg/kg, 1% in sterile saline) and placed in a stereotactic apparatus. The skull was exposed through a midline incision of the skin. Rose Bengal solution (100 mg/kg, 10 mg/mL in normal saline, Sigma‐Aldrich, St. Louis, MO, USA) was administered intraperitoneally 5 min before illumination. For illumination, a cold light source (KL1500 LCD, Zeiss) giving a 4 mm diameter illumination was positioned 2 mm lateral from Bregma. The brain was illuminated through the intact skull for 15 min. Rose Bengal produced singlet oxygen under light excitation resulting in activation of platelets with consequent damages and occludes vascular endothelium, which mimicked the traditional ischemic stroke model. During the experiment, rectal temperature was monitored and maintained between 36.5°C and 37.5°C with a self‐regulating heating pad. The mice were to awaken and returned to their home cage. Sham animals were subjected to the same surgery as stroke animals without administration of Rose Bengal.
In vivo Drug Administration and Experimental Groups
Cystamine (cystamine dihydrochloride, Sigma, St. Louis, MO, USA) was dissolved in 0.9% saline and administered intraperitoneally 100 mg/kg daily 24 h after stroke. The experiment consists of two sets:
For short‐term (7 days) treatment, mice subjected to stroke were randomly separated into three groups (n = 8 per group): vehicle, cystamine, ANA‐12+ cystamine groups. For vehicle group, equal volume of saline was administered intraperitoneally. ANA‐12 (specific antagonist of TrkB, Sigma) was dissolved in dimethylsulphoxide (DMSO) and then diluted 1:10 with 0.9% saline. For blocking TrkB, ANA‐12 (0.5 mg/kg) was injected intraperitoneally 1 h before cystamine administration. For neurogenesis analysis, all mice received daily injection of 5‐Bromo‐2′‐deoxyuridine (BrdU, 50 mg/kg, Sigma) starting at 24 h after stroke and subsequently for seven consecutive days. Mice were killed 30 min after the last cystamine administration at 7 days after stroke and analyzed for BDNF/TrkB levels, cell proliferation, and neuronal apoptosis.
For long‐term (42 days) treatment, mice were divided randomly into the following groups (n = 8 per group): sham + vehicle, stroke + vehicle, stroke + cystamine, and stroke + ANA‐12 + cystamine groups. Mice were administered cystamine or vehicle (equal volume of saline) intraperitoneally starting at 24 h after stroke. For blocking TrkB, ANA‐12 (0.5 mg/kg) was injected intraperitoneally 1 h before cystamine administration. Mice underwent long‐term treatment were analyzed for functional recovery, axon remodeling, BDNF level and neuroplasticity.
Behavioral Testing
Grid‐walking and cylinder/rearing tasks were used to assess functional recovery after stroke by measures of forelimb and hindlimb motor control as previously described 15, 16. Behavioral testing were performed at approximately the same time during the nocturnal period 1 day before stroke, 7, 14, 28 and 42 days after stroke. Behavior videotape was analyzed by investigators blinded to the experimental groups.
In grid‐walking task, each mouse was placed individually on top of the elevated wire grid which was 32 cm × 20 cm × 50 cm (length × width × height) with 12 × 12 mm diameter openings and allowed to freely walk for 5 min. Behavior was recorded using a camera that was placed underneath the grid to assess the animals' stepping errors. A step was considered a foot fault if it was not providing support and the foot went through the grid hole. Furthermore, if an animal was resting with the grid at the level of the wrist, this was also considered a fault. The total number of foot faults for each limb, along with the total number of non‐foot‐fault steps, was counted. Percent foot faults in the grid‐walking were calculated by: number of foot faults/(foot faults + number of non‐foot fault steps) ×100.
Cylinder/rearing task encourages the use of forelimbs for vertical wall exploration in a cylinder (a Plexiglas transparent cylinder; 15 cm in height with a diameter of 10 cm). When placed in the cylinder, the animal rears to a standing position on the cylinder wall, supporting its weight with either one or both of its forelimbs. Animals were videotaped for 5 min. While the videotape was played in slow motion (1/5th real time speed), the total time (seconds) each animal spent on the right forelimb, the left forelimb, and simultaneous use of both forelimbs were calculated. Only rears in which both forelimbs could be clearly seen were timed. Asymmetry index in cylinder/rearing tasks was derived as ([nonimpaired − impaired]/[nonimpaired + impaired + both forepaws]) ×100.
In vivo Magnetic Resonance Imaging (MRI)
MRI was carried out on a 7.0 Tesla small animal magnetic resonance system (Bruker PharmaScan, Ettlingen, Germany) as described previously 17.
In vivo T2‐weighted images were performed at 24 h and 7 days poststroke using a two‐dimensional turbo spin‐echo sequence (repetition time/echo time = 2000/50 ms). Twelve axial slices with a slice thickness of 1 mm, a field of view of 20 × 20 mm, and a matrix of 256 × 256 were positioned over the brain excluding the olfactory bulb.
In vivo diffusion tensor imaging (DTI) was measured at 42 days after stroke using the echo‐planar imaging (EPI) sequence with parameters of 30 different diffusion directions, five reference image, 5000/32.2 ms of repetition time/echo time, 1000 s/mm2 of b‐value, 20 slices, 0.6 mm slice thickness, 20 × 20 mm field of view, 128 × 128 matrix.
A computer‐based analysis of infarct volumes was performed using the software Image J (National Institutes of Health, Bethesda, MD, USA) by subtracting the area of the nonlesioned ipsilateral hemisphere from that of the contralateral side on T2‐weighted image. Infarct sizes were calculated by integration of the lesion areas. Infarct sizes were presented as a volume percentage of the lesion compared with the contralateral hemisphere.
Using Paravision 5.0 software (Bruker Biospin, Ettlingen, Germany), fractional anisotropy (FA) and diffusivity values (mean diffusivity [MD], radial diffusivity [λ⊥], axial diffusivity [λ//]) were derived from the tensor map. These diffusion parameters were used to quantify microstructural axon abnormalities. Regions of interest (ROI) were delimited on FA maps in the cortex and corpus callosum at five image planes of the brain named genu, body 1, body 2, body 3, or splenium by the anatomical location of the corpus callosum, respectively. Fiber tracking was performed using TrackVis (version 0.5.2.1) and Diffusion Toolkit (version 0.6.2.1) software. Diffusion Toolkit was used to generate FA color map. TrackVis, an interactive environment for fiber tracking reconstruction, display, and analysis developed at the Harvard Medical School Martinos Center for Biomedical Imaging at Massachusetts General Hospital was used to generate the tractography and draw ROI (www.trackvis.org). Tractography of the motor cortex and internal capsule was performed by manually drawing regions of interest on each individuals FA color map by a single expert who was blinded to the study. The density of fiber tracks was evaluated by the ratio of the number of fiber tracks to the volume of ROI.
Western Blot
Mice (n = 4/group) were killed 30 min after the last injection with cystamine or vehicle and bilateral cerebral hemisphere was rapidly dissected and homogenized respectively in ice‐cold Nonidet P‐40 (Sigma) containing 1% Protease Inhibitor Cocktail (Sigma) and centrifuged 16000 g for 15 min at 4°C. Supernatant was collected as protein extract and stored at −80°C until further use. For clarification, the ischemic hemisphere homogenate was used for Western blot and ELISA. The expression of TrkB (full‐length) and the ratios of phospho‐TrkB‐Y817/TrkB (full‐length) were measured by Western blot as described previously 18. The following primary antibodies were used: rabbit monoclonal anti‐TrkB (phospho Y817) (1:1000, Abcam, San Francisco, CA, USA) and rabbit polyclonal anti‐TrkB (full‐length) (1:800, Abcam).
Enzyme‐Linked Immunosorbant Assay (ELISA)
The BDNF protein in ischemic brain homogenates was quantified with a conventional sandwich ELISA using the BDNF Emax immunoassay system (Promega) according to the protocol of the manufacturer.
Histological and Immunohistochemical Assessment
At 7 and 42 days after stroke, mice (n = 4 per group) brains were fixed by transcardial perfusion with PBS followed by perfusion and immersion in 4% paraformaldehyde. A standard paraffin block was obtained from the center of the lesion (Bregma 2.5 to +2.5 mm). Every intervals of 1 mm, ten slices of 4 μm thick sections were cut from the block.
BrdU immunostaining was performed as previously described 19. Briefly, the sections were deparaffinized and incubated with 2N HCl at 37°C, washed and treated with 0.1M boric acid (pH 8.5) followed by incubation with H2O2 for 10 min. After blocking sections were incubated with goat polyclonal anti‐BrdU (1:200, Abcam) for 2 h at 37°C, the sections were washed followed by incubation with biotinylated rabbit anti‐goat immunoglobulin G (1:200, Abcam) in immunoblocking buffer at room temperature for 1 h and incubated with an avidin‐biotin‐peroxidase kit for 10 min. Horseradish peroxidase reaction product was visualized with nickel‐enhanced DAB peroxidase substrate kit.
Myelin basic protein (MBP), BDNF, and NeuN immunostaining were performed as previously described 20. Briefly, deparaffinized sections were blocked followed by incubation with mouse monoclonal anti‐MBP (1:200; Abcam), rabbit polyclonal anti‐BDNF (1:100, Abcam), or rabbit polyclonal anti‐NeuN (1:700; Abcam) for 2 h at 37°C. Then, sections were washed and followed by incubation with biotinylated goat anti‐rabbit or rabbit anti‐mouse immunoglobulin G (1:200, Abcam) in immunoblocking buffer at room temperature for 1 h and incubated with an avidin‐biotin‐peroxidase kit for 10 min. Horseradish peroxidase reaction product was visualized with nickel‐enhanced DAB peroxidase substrate kit.
BrdU‐positive cell numbers were counted in subventricular zone (SVZ) and dentate gyrus (DG) area of the ischemic hemisphere. Five sections per brain and eight views per section were digitized using a × 40 objective (Olympus BX 53, Tokyo, Japan) and counted per animal with the number of BrdU‐positive cells averaged. Data were analyzed in a blinded manner and presented as the number of the BrdU‐positive cells per section. The NeuN positive cells were counted five sections per brain, and eight views per section at ×40 objective measured in the peri‐infarct zone and expressed as the mean number of NeuN positive cells/mm2.
For semi‐quantification of MBP and BDNF immunoreactivity, five immunostained coronal sections per brain and eight fields of view in the peri‐infarct of cortex per section were digitized under a × 40 objective. The optical density was measured using Image‐Pro Plus software (vision 6.0, Media Cybernetics, Inc., Bethesda, MD, USA).
Double Immunofluorescent Staining
To specifically identify whether BrdU‐positive cells were colocalized with the neuronal progenitor cell marker, double immunofluorescence staining was performed to detect the expression of BrdU/doublecortin (Dcx, neuronal progenitor marker). As described in the above, sections were incubated with goat polyclonal anti‐BrdU (1:200; Abcam), a polyclonal rabbit antibody against Dcx (1:1000; Abcam) for 2 h at 37°C. Then, the sections were incubated with the secondary antibodies donkey anti‐goat Alexa Fluor‐488 (1:500; Invitrogen, Carlsbad, CA, USA) for BrdU staining, mouse anti‐rat Alexa Fluor‐594 (1:500; Invitrogen) for Dcx. Quantitative analysis was performed in the regions of SVZ and DG. Using double immunohistochemical staining, the number of BrdU‐positive cells in the SVZ per animal was also counted to obtain the percentage of BrdU‐reactive cells colocalized with Dcx.
Terminal Deoxynucleotidyl Transferase–Mediated Fluorescein‐dUTP Nick End Labeling (TUNEL) Staining
Brain sections were stained with an in situ cell death detection kit, fluorescein (Roche Inc, Mannheim, Germany) to assess apoptotic cells in the peri‐infarct zone of brain according to the procedures provided by the manufacturer. The TUNEL‐positive cells were counted eight views in each section at ×40 objective measured in the peri‐infarct zone of brain and expressed as the mean number of TUNEL‐positive cells/mm2.
Statistical Analysis
All values were expressed as mean ± standard error of the mean (SEM). For comparison between two groups, the Student's t‐test was used. Behavioral testing was analyzed using two‐way ANOVA with repeated measures and Newman–Keuls' multiple pair‐wise comparisons. Other multiple comparisons were made by one‐way ANOVA followed with Bonferroni post‐tests. P < 0.05 was considered significant.
Results
Effect of Cystamine on Functional Recovery and Infarct Size after Stroke
To examine whether cystamine promotes functional recovery after stroke, the grid‐walking task and cylinder/rearing task were performed. There was an increase in the number of foot faults in the grid‐walking task and a decrease in forelimb symmetry in the cylinder task from 7 days after stroke; these deficits were ameliorated beginning from at 7 days after stroke by chronic treatment with cystamine starting 24 h after stroke (P < 0.001; Figure 1A–C). To corroborate the role of cystamine in enhancing stroke recovery via the BDNF/TrkB pathway, we tested mice with combined ANA‐12 + cystamine treatment. Pretreatment with TrkB antagonist ANA‐12 abolished functional recovery induced by cystamine and showed similar motor function compared to stroke + vehicle (P > 0.05; Figure 1A–C). MRI was used to examine the effect of cystamine on the infarct size of ipsilesional hemisphere. Animals displayed similarly sized infarcts in similar locations on T2‐weighted images 24 h after photothrombotic stroke (Figure 1D). However, T2‐weighted images indicated no significant differences of infarction volumes between vehicle and cystamine‐treated groups 7 days after stroke (t[15] = 1.25, P = 0.229; Figure 1E,F).
Figure 1.
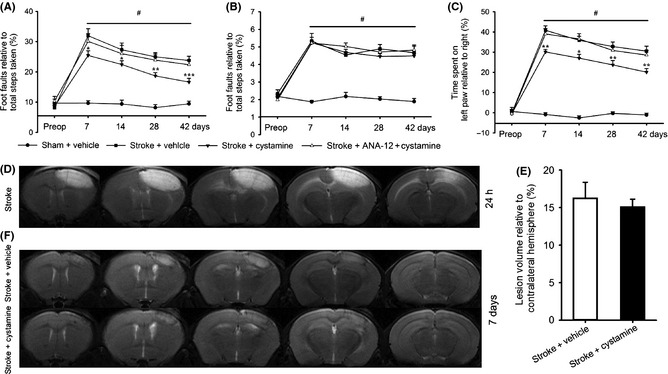
Effect of cystamine on the functional recovery and infarct size after stroke. Cystamine treatment starting from 24 h after stroke resulted in a time‐dependent improvement in functional recovery after stroke. Functional recovery was assessed with the grid‐walking task for forelimb foot faults (A) and hindlimb foot faults (B), and the cylinder task for forelimb asymmetry (C). Data are shown as mean ± SEM for n = 8 per group. #P < 0.001 for stroke + vehicle versus sham, *P < 0.05; **P < 0.01; ***P < 0.001 versus stroke + vehicle using two‐way ANOVA with repeated measures and Newman–Keuls' multiple pair‐wise comparisons. Representative focal infarcts are shown on T2‐weighted images at 24 h after photothrombotic focal stroke (D). Quantification of the lesion volume (E) and representative focal infarcts on T2‐weighted images (F) from the mice treated with or without cystamine groups at 7 days after stroke. Data are shown as mean ± SEM for n = 8 per group. t[15] = 1.25, P = 0.229 versus stroke + vehicle group using Student's t‐test. Preop = preoperation.
Effect of Cystamine on the Axon Remodeling in the Cortex
As cystamine was able to contribute to functional recovery, we hypothesize that cystamine would exert an effect on axon remodeling. Therefore, in vivo DTI was used to further examine effects of chronic cystamine treatment on axon remodeling 42 days after stroke. In the ipsilesional motor cortex, cystamine treatment normalized FA values to the levels seen in vehicle treated sham group, whereas following stroke FA values increased at all levels (P < 0.05; Figure 2A,B). Furthermore, cystamine treatment also normalized λ⊥ in genu, body 1, and body 3 of the motor cortex, which were decreased after stroke, to the levels seen in vehicle treated sham group (P < 0.05; Figure 2C). In addition to the ipsilesional motor cortex, we also examined the effect of cystamine on the contralesional cortex. Cystamine treatment restored the cortex FA values at level genu and body 1 to the level in the vehicle treated sham group (P < 0.05; Figure 2D). These finding encouraged us to further examine the myelination in the motor cortex; therefore, the same set of mice were processed for the expression of MBP in the peri‐infarct zone of the motor cortex. There is decreased MBP at all levels in the stroke group, which was significantly ameliorated by cystamine treatment (P < 0.05; Figure 2E,F).
Figure 2.
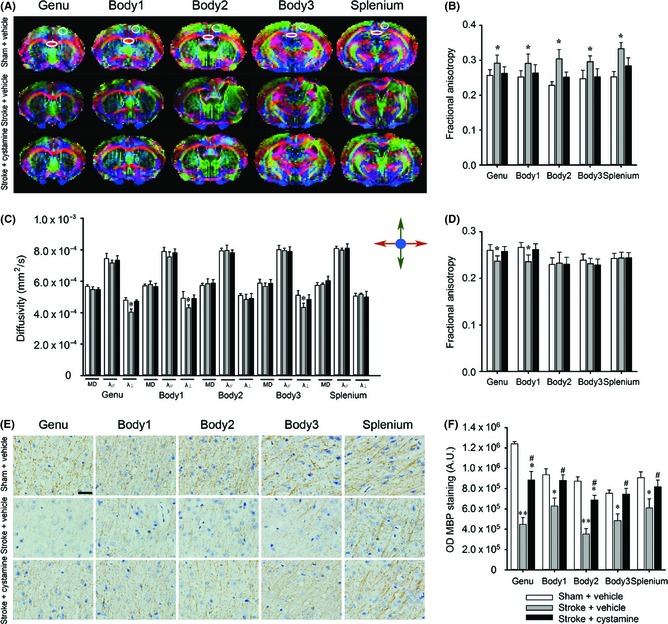
Effect of cystamine on the axon remodeling in the cortex. (A) Representative direction‐encoded FA color maps of a typical brain from the sham or stroke groups with or without cystamine treatment at five different image planes from the genu to the splenium of the corpus callosum. The directions of fiber tracks were color‐coded with red for left–right, blue for superior–inferior, and green for anterior–posterior. (B) FA values were measured in the ipsilesional cortex delimited on photographs (A). (C) Diffusivity values were measured in the ipsilesional cortex delimited on photographs (A). (D) FA values were measured in the contralesional cortex. (E) Representative images of MBP staining at five different image planes in the peri‐infarct zone of the motor cortex. (F) MBP intensity was measured in the peri‐infarct zone of the motor cortex. Data are shown as mean ± SEM of n = 8 mice per group for DTI data, n = 4 mice per group for histology data. Using one‐way ANOVA followed with Bonferroni post‐tests, *P < 0.05, **P < 0.01 versus sham + vehicle; #P < 0.05 versus stroke + vehicle. A.U = absorbance units; λ⊥ = radial diffusivity; λ// = axial diffusivity. Scale bars in (E) = 25 μm.
Next, fiber tracking was used to examine the effect of cystamine on the bilateral motor axons projection (Figure 3A). Compared with the sham control, there was decreased density of fiber tracks emanating from motor cortex at levels genu and body 1 and crossing internal capsule at level body 3, which was recovered by treatment with cystamine (P < 0.05; Figure 3B).
Figure 3.
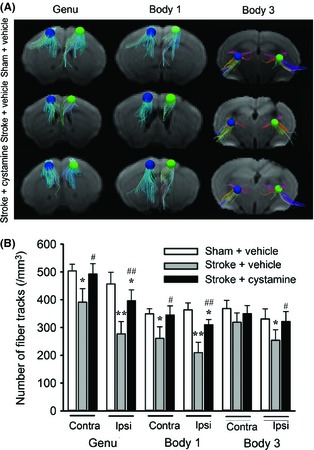
Effect of cystamine on the fiber tracking in motor cortex and internal capsule. (A) Representative photographs of three‐dimensional fiber tracking in the ROI of motor cortex at levels genu and body 1 and internal capsule at level body 3 from the mice treated with or without cystamine 42 days after stroke compared with sham group. The radius of ROI is 0.468 mm. The directions of fiber tracks were color‐coded with red for left–right, blue for superior–inferior, and green for anterior–posterior. (B) Quantification of fiber tracking intensity as measured in ROI in panel (A). Data are shown as mean ± SEM for n = 8 per group. Using one‐way ANOVA followed with Bonferroni post‐tests, *P < 0.05, **P < 0.01 versus sham + vehicle; #P < 0.05, ##P < 0.01 versus stroke + vehicle.
Effect of Cystamine on the BDNF Expression and Activation of TrkB in the Ischemic Brain
We next wanted to dissect the detailed mechanisms in this process. For short‐term study, administration of cystamine increased the BDNF expression in the ischemic brain compared with the vehicle group. Pretreatment with TrkB antagonist ANA‐12 showed no effect on the increase of BDNF expression induced by cystamine (F2,9 = 9.12, P = 0.0068; Figure 4A). This finding was further confirmed by BDNF immunostaining (F2,9 = 8.76, P = 0.0077; Figure 4B–D). Furthermore, cystamine treatment increased the phosphorylation of TrkB at Y817 compared with the vehicle group. ANA‐12 pretreatment significantly decreased cystamine‐induced phosphorylation of TrkB in ischemic brain (P < 0.05; Figure 4E–G). To test the effect of chronic administration of cystamine on BDNF expression, administration of cystamine for 42 days still significantly increased the expression of BDNF compared with vehicle group (F2,9 = 7.32, P = 0.0129; Figure 4H).
Figure 4.
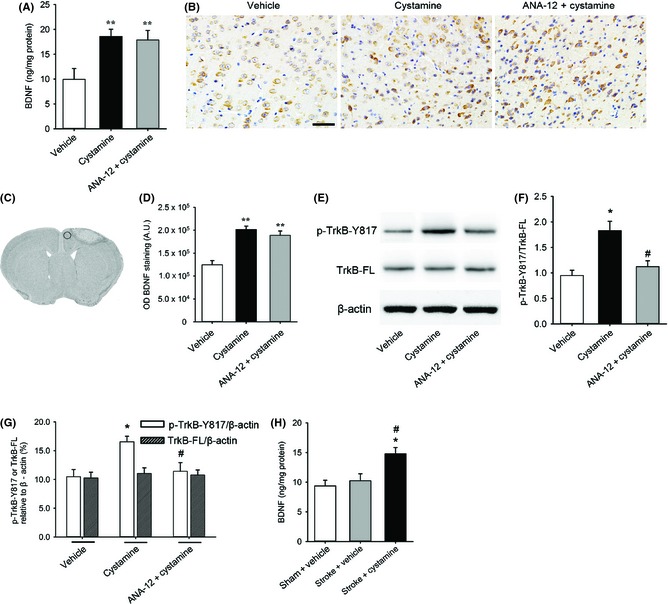
Effect of cystamine on the BDNF expression and activation of TrkB in the ischemic brain. (A) Quantification of the expression of BDNF in ischemic brain homogenates 7 days after stroke from different groups of mice administrated with or without cystamine in the presence or absence of ANA‐12 pretreatment by ELISA. (B) Representative images of BDNF immunostaining in peri‐infarct zone of brain 7 days after stroke from groups administrated with or without cystamine in the presence or absence of ANA‐12. (C) The delimited region indicates the peri‐infarct zone of brain selected for immunohistochemistry. (D) Quantification of the BDNF expression in the peri‐infarct zone of brain. (E) Representative images of Western blot for phosphorylated (pTrkB‐Y817), full‐length TrkB (TrkB‐FL), and β‐actin in ischemic brain homogenates 7 days after stroke from groups treated with or without cystamine in the presence or absence of ANA‐12. (F) Quantification of TrkB phosphorylation (pTrkB‐Y817 relative to TrkB‐FL). (G) Quantification of p‐TrkB‐Y817/β‐actin as well as TrkB‐FL/β‐actin. (H) Quantification of the expression of BDNF in ischemic brain homogenates 42 days after stroke from different groups of mice treatment with or without cystamine by ELISA. Data are shown as mean ± SEM for n = 4 per group; In panels (A), (D), (F) and (G), *P < 0.05, **P < 0.01 versus vehicle, #P < 0.05 versus cystamine using one‐way ANOVA followed with Bonferroni post‐tests. In panel (H), *P < 0.05 versus sham + vehicle; #P < 0.05 versus stroke + vehicle using one‐way ANOVA followed with Bonferroni post‐tests. Scale bars in (B) = 50 μm.
Effect of Cystamine on the Neurogenesis
To investigate whether cystamine affect neurogenesis, we first detected the effect of cystamine on cell proliferation by assessment of BrdU‐positive cells in the SVZ and DG 7 days after stroke. Cystamine treatment resulted in an increase of BrdU‐positive cells in the ipsilateral brain both SVZ (F2,9 = 10.52, P = 0.0044; Figure 5A,C) and DG (F2,9 = 5.75; P = 0.0246; Figure 5B,D) compared with the vehicle group. Intriguingly, pretreatment of ANA‐12 significantly inhibited the increased cell proliferation induced by cystamine. Next, we verified that cystamine affects the proliferation of neuronal progenitor cells by double immunostaining with BrdU and Dcx (Figure 5E). Cystamine treatment increased the percentage of Dcx‐positive cell in BrdU‐reactive cells in the ipsilateral SVZ compared with vehicle group (F2,9 = 6.34, P = 0.0191; Figure 5F); this effect was significantly inhibited by ANA‐12 (Figure 5F).
Figure 5.
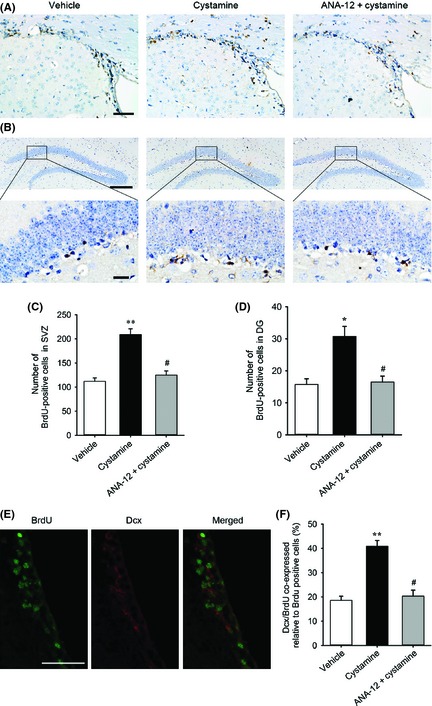
Effect of cystamine on the proliferation of neuronal progenitor cells. Representative brain sections from different groups of mice administrated with or without cystamine in the presence or absence of ANA‐12 pretreatment in SVZ (A) and DG (B) of the ischemic hemisphere by BrdU immunostaining at 7 days after stroke. Quantification of BrdU‐positive cells from groups treated with or without cystamine in the presence or absence of ANA‐12 in the SVZ (C) and DG (D) of the ischemic hemisphere. (E) Representative double immunofluorescent staining images of BrdU (green), Dcx (red) and merged image in the ipsilateral subventricular zone. (F) Quantification of the BrdU‐positive cells coexpressed Dcx in the ipsilateral SVZ. Data are mean ± SEM for n = 4 per group; *P < 0.05, **P < 0.05 versus vehicle, #P < 0.05 versus cystamine using one‐way ANOVA followed with Bonferroni post‐tests. Scale bars in (A) and (E) = 50 μm. Scale bars in (B) = 200 μm (upper) and 20 μm (lower).
Effect of Cystamine on Neuroprotection and Neuroplasticity
Next, we examined apoptotic cells at 7 days after stroke and neuronal density at 42 days after stroke in the peri‐infarct zone. Cystamine treatment decreased TUNEL‐positive cells in the peri‐infarct zone at 7 day after stroke, which was significantly inhibited by ANA‐12 pretreatment (F2,9 = 11.27, P = 0.0034; Figure 6A,B). To examine the effect of cystamine on the neuronal survival, neuronal density was examined by NeuN immunohistochemistry on days 42 poststroke (Figure 6C). Chronic cystamine treatment significantly reduced stroke‐induced loss of neuronal density in the peri‐infarct zone (F2,9 = 15.38, P = 0.0012; Figure 6D).
Figure 6.
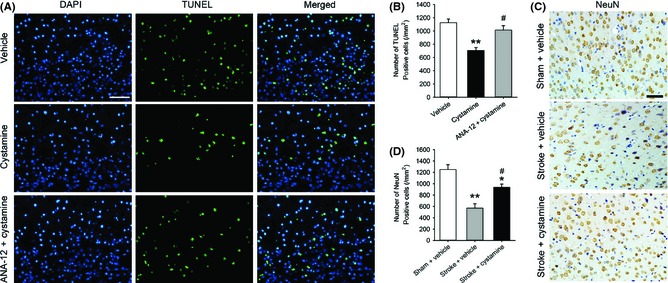
Effect of cystamine on neuroprotection and neuroplasticity. (A) Representative TUNEL staining images 7 days after stroke from different groups of mice administrated with or without cystamine in the presence or absence of ANA‐12 treatment in the peri‐infarct zone of brain. TUNEL‐positive cell (green), DAPI (blue) and merged image. (B) Quantification of TUNEL‐positive cells was shown. (C) Representative NeuN staining images 42 days after stroke from mice in sham or stroke treated with or without cystamine groups in the peri‐infarct zone of brain. (D) Quantification of neuronal density in the peri‐infarct zone of brain was shown. Data are mean ± SEM for n = 4 per group; In panel (B), **P < 0.01 versus vehicle, #P < 0.05 versus cystamine using one‐way ANOVA followed with Bonferroni post‐tests. In panel (D), *P < 0.05, **P < 0.01 versus sham + vehicle, #P < 0.05 versus stroke + vehicle using one‐way ANOVA followed with Bonferroni post‐tests. Scale bars in (A) and (C) = 50 μm.
Discussion
To the best of our knowledge, this is the first study exploring the effect of cystamine on the functional recovery after stroke through an in‐depth analysis of functional recovery‐related axon remodeling. We demonstrate that cystamine treatment improves functional recovery with concomitant enhancement of axon remodeling and neuroprotection. The detailed mechanisms in this process involve the increases of BDNF expression in the brain contributing to enhancement of neuronal progenitor cell proliferation, neuronal survival, and plasticity via the BDNF/TrkB pathway.
Cystamine treatment starting 24 h after stroke promoted functional recovery. As the important element in stroke treatment is the timing of drug delivery, cystamine administrated starting 24 h after stroke was able to exert the functional recovery after stroke. With this study, cystamine may join a short list of small molecules that can promote recovery after stroke when given 24 h after stroke, suggesting it is a promising medicine for stroke therapy. In our study, treatment with cystamine for 7 days has a significant effect on stroke recovery (Figure 1A–C). To assess the necessary of long‐term administration, cystamine treatment was discontinued after 14 days; there is a decrease in functional gains (Figure S1).
Axonal remodeling is a critical aspect of brain repair and contributes to spontaneous improvements in neurological deficits after stroke 21. Current histology of MBP staining was unable to provide quantitative data to delineate the directionality of axonal projection. Thus, using in vivo DTI, we performed an in‐depth analysis of remodeling of axonal in the brain after cystamine treatment following stroke. In the present study, DTI analysis showed increased FA values and decreased λ⊥ after stroke in motor cortex restored by cystamine treatment, suggesting that the increased cortical FA after stroke is due to altered white matter organization rather than increased myelination. In the MBP staining, MBP intensity at the same levels was decreased in the same set of mice after stroke, which was significantly ameliorated by cystamine treatment. The corticospinal tract is the primary transmission route for voluntary forelimb movement from the motor cortex via the internal capsule toward the spinal cord 22. The fiber tracking using DTI was applied to analyze the change of motor axons projection to internal capsule after stroke. DTI analysis showed that fibers in both the ipsi‐ and contralesional internal capsule were disorganized and decreased after stroke. Cystamine treatment increased fiber tracks emitting from motor cortex and crossing internal capsule and restored stroke‐induced subtle white matter abnormalities.
It is well known that BDNF and its receptor TrkB are widely distributed throughout the adult brain in almost all cortical areas, as well as several subcortical and spinal cord regions 13, 23. Exogenous administration of BDNF improves functional recovery and induces widespread neuronal remodeling in the photothrombotic stroke model 24. Thus, agents that promote endogenous BDNF secretion may promote neuroplasticity and functional recovery after stroke. Our study demonstrated that cystamine treatment resulted in an increase of BDNF expression. To examine the mechanisms for neuroprotection by cystamine, we injected ANA‐12, an antagonist of TrkB.ANA‐12 failed to affect the increased expression of BDNF, but was able to decrease cystamine‐mediated TrkB phosphorylation, suggesting the specific effect of cystamine on the TrkB phosphorylation. The effect of cystamine on treatment of stroke was significantly inhibited by pretreatment with ANA‐12, suggesting BDNF/TrkB pathway was involved in cystamine‐mediated therapy of stroke. Our findings are consistent with the previous studies showing that cystamine exerted its neuroprotection in schizophrenia and Huntington's disease by increasing BDNF expression crucial for the neuronal survival 12, 25.
Neural precursors in the SVZ and DG of hippocampal are a potential replacement source for dead neurons after brain injury 26, 27. Administered of BDNF intravenously significantly promotes neurological long‐term recovery in a photothrombotic stroke model in the rat 24, 28. It was reported that cystamine enhanced the BDNF release and TrkB activation, which results in Akt activation and neuronal survival 29. Consistent with our findings, cystamine significantly increased the number of BrdU‐immunoreactive cells in the SVZ and DG with colocalization with Dcx. Interestingly, this effect was significantly inhibited by pretreatment with ANA‐12, suggesting that the BDNF/TrkB pathway was involved in cystamine‐mediated neuronal progenitor cell proliferation. However, we have not demonstrated that the newly produced cells differentiate into neurons or glia; thus, the data associating neurogenesis with functional recovery should be interpreted with caution.
The expression of BDNF by surviving neurons in the ischemic brain has been associated with an early autocrine neuroprotective role that counteracts cell death 30, 31. Consistent with these findings, our study demonstrated that cystamine treatment significantly decreased TUNEL‐positive cells in the peri‐infarct zone of the brain, which was significantly abolished by pretreatment with ANA‐12, suggesting that cystamine‐mediated decrease of neuronal apoptosis contributes to the neuroprotection via the BDNF/TrkB pathway. Furthermore, our study also indicated that cystamine increased neuronal density and plasticity contributing to neuroprotection (Figure 6). This finding was consistent with previous reports showing that synaptic plasticity in the ischemic penumbra region contributes to functional recovery after stroke 32, 33.
Conclusion
We demonstrate that treatment of experimental stroke with cystamine significantly improves functional recovery. This functional benefit may be due to enhancement of axon remodeling, neuronal progenitor cell proliferation, neuronal survival, and plasticity via the BDNF/TrkB pathway.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
Figure S1. Assessment of the necessity of long‐term cystamine treatment after stroke.
Acknowledgments
This work was supported by grants from the Major State Basic Research Development Program of China (Nos. 2013CB733800), National Nature Science Foundation of China (Nos. 81230034, 81101089) and Funded by Jiangsu Provincial Special Program of Medical Science (Nos. BL2013029).
References
- 1. Tu JV. Reducing the global burden of stroke: Interstroke. Lancet 2010;376:74–75. [DOI] [PubMed] [Google Scholar]
- 2. Blanco M, Castillo J. Stroke in 2012: Major advances in the treatment of stroke. Nat Rev Neurol 2013;9:68–70. [DOI] [PubMed] [Google Scholar]
- 3. Orbe J, Barrenetxe J, Rodriguez JA, et al. Matrix metalloproteinase‐10 effectively reduces infarct size in experimental stroke by enhancing fibrinolysis via a thrombin‐activatable fibrinolysis inhibitor‐mediated mechanism. Circulation 2011;124:2909–2919. [DOI] [PubMed] [Google Scholar]
- 4. Lees KR, Bluhmki E, von Kummer R, et al. Time to treatment with intravenous alteplase and outcome in stroke: An updated pooled analysis of ecass, atlantis, ninds, and epithet trials. Lancet 2010;375:1695–1703. [DOI] [PubMed] [Google Scholar]
- 5. Jeitner TM, Delikatny EJ, Ahlqvist J, Capper H, Cooper AJ. Mechanism for the inhibition of transglutaminase 2 by cystamine. Biochem Pharmacol 2005;69:961–970. [DOI] [PubMed] [Google Scholar]
- 6. Gibrat C, Bousquet M, Saint‐Pierre M, et al. Cystamine prevents mptp‐induced toxicity in young adult mice via the up‐regulation of the brain‐derived neurotrophic factor. Prog Neuropsychopharmacolo Biol Psychiatry 2010;34:193–203. [DOI] [PubMed] [Google Scholar]
- 7. Gibrat C, Cicchetti F. Potential of cystamine and cysteamine in the treatment of neurodegenerative diseases. Prog Neuropsychopharmacolo Biol Psychiatry 2011;35:380–389. [DOI] [PubMed] [Google Scholar]
- 8. Hwang IK, Yoo KY, Yi SS, et al. Expression of tissue‐type transglutaminase (ttg) and the effect of ttg inhibitor on the hippocampal ca1 region after transient ischemia in gerbils. Brain Res 2009;1263:134–142. [DOI] [PubMed] [Google Scholar]
- 9. Okauchi M, Xi G, Keep RF, Hua Y. Tissue‐type transglutaminase and the effects of cystamine on intracerebral hemorrhage‐induced brain edema and neurological deficits. Brain Res 2009;1249:229–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Basso M, Berlin J, Xia L, et al. Transglutaminase inhibition protects against oxidative stress‐induced neuronal death downstream of pathological erk activation. J Neurosci 2012;32:6561–6569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Colak G, Johnson GV. Complete transglutaminase 2 ablation results in reduced stroke volumes and astrocytes that exhibit increased survival in response to ischemia. Neurobiol Dis 2012;45:1042–1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Borrell‐Pagès M, Canals JM, Cordelières FP, et al. Cystamine and cysteamine increase brain levels of BDNF in huntington disease via HSJ1b and transglutaminase. J Clin Invest 2006;116:1410–1424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Nagahara AH, Tuszynski MH. Potential therapeutic uses of bdnf in neurological and psychiatric disorders. Nat Rev Drug Discov 2011;10:209–219. [DOI] [PubMed] [Google Scholar]
- 14. Cheng Y, Gidday JM, Yan Q, Shah AR, Holtzman DM. Marked age‐dependent neuroprotection by brain‐derived neurotrophic factor against neonatal hypoxic‐ischemic brain injury. Ann Neurol 1997;41:521–529. [DOI] [PubMed] [Google Scholar]
- 15. Clarkson AN, Huang BS, MacIsaac SE, Mody I, Carmichael ST. Reducing excessive gaba‐mediated tonic inhibition promotes functional recovery after stroke. Nature 2010;468:305–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Baskin YK, Dietrich WD, Green EJ. Two effective behavioral tasks for evaluating sensorimotor dysfunction following traumatic brain injury in mice. J Neurosci Methods 2003;129:87–93. [DOI] [PubMed] [Google Scholar]
- 17. Zhang Y, Fan S, Yao Y, et al. In vivo near‐infrared imaging of fibrin deposition in thromboembolic stroke in mice. PLoS ONE 2012;7:e30262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Schmid DA, Yang T, Ogier M, et al. A trkb small molecule partial agonist rescues trkb phosphorylation deficits and improves respiratory function in a mouse model of rett syndrome. J Neurosci 2012;32:1803–1810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Yao H, Duan M, Yang L, Buch S. Platelet‐derived growth factor‐bb restores human immunodeficiency virus tat‐cocaine‐mediated impairment of neurogenesis: Role of trpc1 channels. J Neurosci 2012;32:9835–9847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Doeppner TR, Mlynarczuk‐Bialy I, Kuckelkorn U, et al. The novel proteasome inhibitor BSc2118 protects against cerebral ischaemia through HIF1A accumulation and enhanced angioneurogenesis. Brain 2012;135:3282–3297. [DOI] [PubMed] [Google Scholar]
- 21. Ueno Y, Chopp M, Zhang L, et al. Axonal outgrowth and dendritic plasticity in the cortical peri‐infarct area after experimental stroke. Stroke 2012;43:2221–2228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. van Velthoven CT, van de Looij Y, Kavelaars A, et al. Mesenchymal stem cells restore cortical rewiring after neonatal ischemia in mice. Ann Neurol 2012;71:785–796. [DOI] [PubMed] [Google Scholar]
- 23. Ploughman M, Windle V, MacLellan CL, White N, Doré JJ, Corbett D. Brain‐derived neurotrophic factor contributes to recovery of skilled reaching after focal ischemia in rats. Stroke 2009;40:1490–1495. [DOI] [PubMed] [Google Scholar]
- 24. Schäbitz W‐R, Berger C, Kollmar R, et al. Effect of brain‐derived neurotrophic factor treatment and forced arm use on functional motor recovery after small cortical ischemia. Stroke 2004;35:992–997. [DOI] [PubMed] [Google Scholar]
- 25. Pillai A, Veeranan‐Karmegam R, Dhandapani KM, Mahadik SP. Cystamine prevents haloperidol‐induced decrease of BDNF/TrkB signaling in mouse frontal cortex. J Neurochem 2008;107:941–951. [DOI] [PubMed] [Google Scholar]
- 26. Thored P, Arvidsson A, Cacci E, et al. Persistent production of neurons from adult brain stem cells during recovery after stroke. Stem Cells 2006;24:739–747. [DOI] [PubMed] [Google Scholar]
- 27. Parent JM, Vexler ZS, Gong C, Derugin N, Ferriero DM. Rat forebrain neurogenesis and striatal neuron replacement after focal stroke. Ann Neurol 2002;52:802–813. [DOI] [PubMed] [Google Scholar]
- 28. Schäbitz W‐R, Steigleder T, Cooper‐Kuhn CM, et al. Intravenous brain‐derived neurotrophic factor enhances poststroke sensorimotor recovery and stimulates neurogenesis. Stroke 2007;38:2165–2172. [DOI] [PubMed] [Google Scholar]
- 29. Kaplan DR, Miller FD. Neurotrophin signal transduction in the nervous system. Curr Opin Neurobiol 2000;10:381–391. [DOI] [PubMed] [Google Scholar]
- 30. Larsson E, Nanobashvili A, Kokaia Z, Lindvall O. Evidence for neuroprotective effects of endogenous brain‐derived neurotrophic factor after global forebrain ischemia in rats. J Cereb Blood Flow Metab 1999;19:1220–1228. [DOI] [PubMed] [Google Scholar]
- 31. Hasegawa Y, Suzuki H, Altay O, Zhang JH. Preservation of tropomyosin‐related kinase B (TrkB) signaling by sodium orthovanadate attenuates early brain injury after subarachnoid hemorrhage in rats. Stroke 2011;42:477–483. [DOI] [PubMed] [Google Scholar]
- 32. Cramer SC. Repairing the human brain after stroke: I. Mechanisms of spontaneous recovery. Ann Neurol 2008;63:272–287. [DOI] [PubMed] [Google Scholar]
- 33. Carmichael ST. Cellular and molecular mechanisms of neural repair after stroke: Making waves. Ann Neurol 2006;59:735–742. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Assessment of the necessity of long‐term cystamine treatment after stroke.