

Summary
Aims
Recently, mutations in COQ2 encoding para‐hydroxybenzoate‐polyprenyl transferase have been identified to increase the risk of multiple system atrophy (MSA) in multiplex families and sporadic cases. The prevalence of COQ2 mutations was showed to be higher in cerebellar subtype (MSA‐C) than parkinsonism subtype (MSA‐P). The aim of this study was to investigate the association between COQ2 mutations and MSA‐C in Chinese patients.
Methods
A Chinese cohort of 116 patients with MSA‐C and 192 healthy control individuals were recruited. Sanger sequencing of COQ2 was performed in all these subjects.
Results
Two missense mutations (p.L402F and p.R173H) and one synonymous mutation (p.A32A) were detected in 3 patients, respectively. They were not found in the 192 controls as well as the 1000 Genomes Database. The p.L402F and p.A32A were novel.
Conclusion
Our results indicated that COQ2 tended to play a population‐specific and subtype‐depended role in conferring susceptibility to MSA.
Keywords: Chinese, COQ2 variants, Multiple system atrophy, Mutation analysis
Introduction
Multiple system atrophy (MSA) is a progressive neurodegenerative disorder characterized by various combinations of autonomic insufficiency, parkinsonism, cerebellar ataxia, and pyramidal dysfunction. The prevalence of MSA is estimated at 3.4 to 4.9 cases per 100,000 individuals, similar to that of motor neuron disease (MND) and Huntington disease (HD) 1. Affecting both genders, the disease usually begins in middle age and progresses slowly, with a median survival of 9.3 years from the onset 2. Clinically, MSA is classified into two subtypes: parkinsonism type (MSA‐P), predominantly featured by parkinsonism, and cerebellar type (MSA‐C), predominantly featured by cerebellar ataxia 2. The MSA‐P is more prevalent among the Caucasian patients 3, 4, while MSA‐C is more prevalent in Asian patients 5. The treatment of MSA is intractable, and there is no adequate approach to halt the progression of the disease so far.
The neuropathologic change of MSA is characterized by cell loss, gliosis, and glial cytoplasmic inclusions (GCI) in many regions of central nervous system (CNS) 6. However, the potential mechanism underlying MSA remains to be elucidated. During the past decades, MSA was widely conceived as a nongenetic disorder, because the vast majority of MSA cases reported were sporadic 7. Nevertheless, several multiplex families with MSA were described over the last few years 8, 9. A breakthrough in our understanding of MSA was the findings that variants within SNCA conferred susceptibility to MSA 10, 11. Recently, a multiple‐center study identified that COQ2 variants were associated with the increased risk of MSA in patients of Japan, Europe, and North America 12. The COQ2 gene was subsequently screened in MSA patients from South Korea, UK, Germany, and southwest China 13, 14, 15, 16, 17. However, these studies presented conflicting results about the association of COQ2 variants with MSA in different populations and different MSA subtypes. In this study, we recruited 116 MSA‐C cases and investigated the association between COQ2 variants and these cases.
Materials and Methods
Subjects
A cohort of 116 unrelated sporadic MSA‐C patients was recruited between June 2008 and December 2013. The cohort consisted of 62 males and 54 females. No consanguineous marriage was found in any family. The mean age of onset was 51.27 ± 7.72 years, and the average age at examination was 53.17 ± 7.81 years. All the patients were neurologically evaluated by at least two senior neurologists. Brain magnetic resonance imaging (MRI) was performed in all the cases. The diagnosis of MSA was made according to the current consensus criteria by Gilman et al. 2. The predominant symptom of all cases in our cohort was cerebellar ataxia including gait ataxia and cerebellar dysarthria. Among them, 79 patients (79/116, 68.1%) were diagnosed as probable MSA‐C, and 37 patients (37/116, 31.9%) were diagnosed as possible MSA‐C. Brain MRI showed severe cerebellar atrophy in all cases and pontine atrophy in 57 cases (57/116, 49.1%). The details of the onset symptoms and follow‐up symptoms were listed in Table S1. All cases were of Han Chinese descent and the majority of them come from southeast of China (Zhejiang, Jiangsu, Shanghai, Jiangxi, and Anhui Province). In addition, 192 healthy individuals matched by gender, age, and ethnicity were enlisted as normal controls. This study was approved by the local ethics committees. Written informed consent was obtained from each participant.
Screening of COQ2 Mutations
Genomic DNA was extracted from peripheral blood using a QIAamp DNA blood minikit (QIAGEN, Hilden, Germany). For mutation analysis of COQ2, fragments encompassing the coding regions and flanking introns were amplified by polymerase chain reaction (PCR). The primers used in this study and the annealing temperatures were listed in the Table S2. The amplified products were purified, followed by Sanger sequencing with ABI 3730 automated DNA sequencer (Applied Biosystems, Foster City, CA, USA). The resulting sequences were compared against the NCBI human reference DNA sequence of COQ2 (NM_015697).
Statistical Analysis
The genotypes in MSA patients and controls were compared using chi‐square test or Fisher's exact test. All statistical analyses were carried out by SPSS version 13.0 (SPSS Inc., Chicago, IL, USA). Difference was considered significant when the P value was less than 0.05 (P < 0.05).
Results
Genetic Investigations
After the Sanger sequencing of COQ2 in 116 unrelated MSA‐C patients, totally 8 variants were identified. Among these variants, c. 196G>T (p.L66V), c. 894T>C (p.D298D), and c. 990C>T (p.S330S) were common and present in 1000 Genomes Database. Furthermore, we found 2 missense mutations including c. 1206G>C (p.L402F) (Figure 1A) and c. 518G>A (p.R173H) (Figure 1B), and one synonymous mutation c. 96G>A (p.A32A) (Figure 1C) in one single patient each. The p.R173H was recently reported in a southwest Chinese study, while the p.L402F and p.A32A were novel and not found in the 192 controls and the 1000 Genomes Database (http://browser.1000genomes.org). The p.L402F mutation was predicted to be deleterious by SIFT software. The frequency of COQ2 mutation in our cohort of patients was consistent with the previous studies (Table 1).
Figure 1.
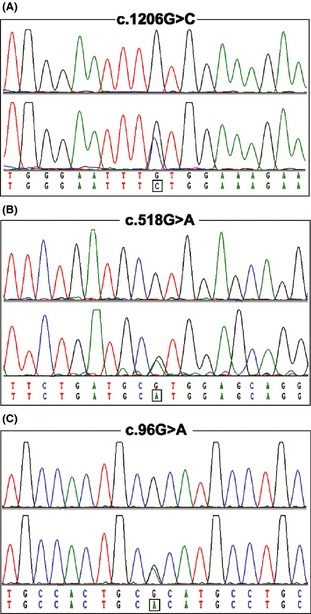
Mutation analysis of 3 COQ2 mutations. Three mutations including c. 1206G>C (p.L402F), c. 518G>A (p.R173H), and c. 96G>A (p.A32A) were confirmed by direct sequencing. The upper panel for each chromatogram depicts the wild‐type sequence, whereas the lower panel represents heterozygous mutated sequence.
Table 1.
Frequencies of COQ2 variants in different cohorts
| Population | Multiple system atrophy | Control | ||||
|---|---|---|---|---|---|---|
| Carrier | Total | Frequency, % | Carrier | Total | Frequency, % | |
| Japanese | 4 | 363 | 1.10 | 2 | 520 | 0.38 |
| European | 4 | 223 | 1.79 | 0 | 315 | 0.00 |
| North American | 1 | 172 | 0.58 | 1 | 294 | 0.34 |
| Korean | 2 | 299 | 0.67 | 0 | 365 | 0.00 |
| European‐GER | 0 | 788 | 0.00 | 1 | 600 | 0.17 |
| European‐UK | 3 | 299 | 1.00 | 1 | 261 | 0.38 |
| American | 2 | 155 | 1.29 | 0 | 500 | 0.00 |
| Chinese‐southwest | 4 | 312 | 1.28 | 0 | 598 | 0.00 |
| Chinese‐southeast | 2 | 116 | 1.72 | 0 | 192 | 0.00 |
The variant c. 1178T>C (p.V393A) (referred to by Mitsui et al. 12 as V343A) was found in our cohort (Figure S1A). The frequency of C allele was 2.2% (5/232) in MSA cases and 0.5% (2/384) in controls, respectively (P = 0.108). We also found another variant c. 73T>G (p.L25V, rs150145464) (Figure 1B). The minor allele frequency (MAF) of G allele was reported to be 0.0046/10 by 1000 Genomes Project (National Center for Biotechnology Information). The frequency of G allele was 3.0% (7/232) in MSA cases and 2.9% (11/384) in controls, respectively (P = 1.000). Both of the results suggested that these two variants were polymorphisms.
Clinical Presentations of the Patients Carrying Missense Mutations
The patient harboring p.R173H mutation was a 59‐year‐old man, who had gait ataxia and dysarthria at age 55 years. In the following one year, he developed a difficulty in handwriting. Three years after the onset, he experienced orthostatic hypotension and urinary frequency. He had no family history of neurological diseases or movement disorders. On physical examination at 58 years old, he showed severe gait disturbance, dysmetria, dysarthria, bradykinesia, and hyperreflexia in all his extremities. There were no muscular atrophy, sensory disturbance, and limitation of ocular movement. Brain MRI showed severe cerebellar and pontine atrophy (Figure 2). The polysomnography revealed an increase in arousal index.
Figure 2.
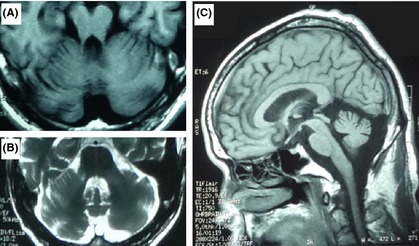
Representative brain MRI of the patient carrying c.518G>A (p.R173H). (A) T1‐weighted image showing atrophy of the cerebellum and pons. (B) T2‐weighted image showing atrophy of the cerebellum and pons with cross sign. (C). T1‐weighted image showing atrophy of the cerebellum and pons.
The case carrying the p.L402F mutation had 4 years history of gait ataxia and dysarthria. When he was 54 years old, he began to exhibit gait unsteadiness, which made him fall to the ground sometimes. In addition, he complained of speech difficulty at the onset. One and a half years later, he suffered from urinary incontinence, constipation, and orthostatic lightheadedness. Neurological examinations at the age 55 revealed a broad‐based ataxic gait, dysarthria, and mild bradykinesia. His muscle strength and tension was normal, but he was diffusely hyperreflexic with extensor plantar responses. Cranial nerve examination was unremarkable. His cognition was normal, based on a MMSE score of 28/30. Brain MRI demonstrated brain atrophy, predominantly cerebellar atrophy.
Discussion
Multiple system atrophy is generally regarded as a nongenetic disorder, because familial aggregation of MSA cases is rarely documented 8, 9. However, emerging evidence suggests that genetic variants increase the susceptibility of MSA. In 2009, Scholz and colleagues demonstrated that variants within SNCA were associated with an increased risk for MSA in Caucasian populations 11, which was subsequently replicated by other studies 10, 18. In addition to SNCA gene, variants within COQ2 were recently identified to increase the risk of familial and sporadic MSA 12. The COQ2 gene was subsequently screened in MSA patients from South Korea, UK, Germany, and southwest China 13, 14, 15, 16, 17. COQ2 encoding Coq2 is essential in the biosynthetic pathway of coenzyme Q10 19. Coenzyme Q10 physiologically functions as electron carrier and participates in the mitochondrial respiratory chain. The deficiency of coenzyme Q10 results in the impairment of mitochondrial respiratory chain and oxidative injury, which was involved in the pathogenesis of MSA.
In the current study, we screened COQ2 variants in 116 sporadic MSA‐C cases, identifying 2 missense mutations (p.R173H and p.L402F) and a synonymous mutation (p.A32A). The p.R173H was recently documented in two cases (one MSA‐P and one MSA‐C) in a cohort of southwest Chinese patients with MSA 17, suggesting that it is common in the Chinese population and may play a distinct role in Chinese MSA patients. The p.L402F and p.A32A variants were novel and not reported previously. All of these three variants were not present in 192 healthy control individuals. Although we did not perform functional experiment to confirm the pathogenicity of p.R173H and p.L402F, both of them were predicted to be deleterious by the prediction software.
In addition, we identified 5 genetic variants in our MSA cases and control individuals. The p.V393A variant associated with MSA in the Japanese population was identified in 5/116 MSA cases and 2/192 controls in our study. The frequency of p.V393A allele in our MSA cases (2.2%) is quite similar to that of a previous Chinese study (2.1% of MSA cases) 17. This implies that the MAF of p.V393A may be approximately 2% in Chinese MSA cases. As shown in Table S3, the association of p.V393A variant with MSA was not significant in our cohort and the study from southwest China 17. In the Korean study, the p.V393A was not associated with MSA either (MAF, 2.7% of cases, 2.6% of controls) 13. Moreover, the p.V393A was not detected in Caucasian MSA cases 12, 14, 15, 16. Thus, p.V393A should be considered as a population‐specific risk factor of Japanese patients with MSA.
Taking all the recent studies together, 24 of 2843 patients with MSA and 5 of 3645 health controls carried COQ2 variants (Table S4). To date, 23 COQ2 variants (Figure 3) have been identified with no hotspot in any population. Overall, the frequency of COQ2 variants in MSA cases varied from 0% to 1.79% in different cohorts (Table 1), and the COQ2 variants are more common in Asian cases than in Caucasian patients (Table S4), indicating a potential population‐specific role of COQ2 in MSA cases. This could be due to the different subtypes of MSA in Asia and Europe, as more COQ2 variants found in MSA‐C than MSA‐P 12, 13, 14, 15, 16, 17.
Figure 3.
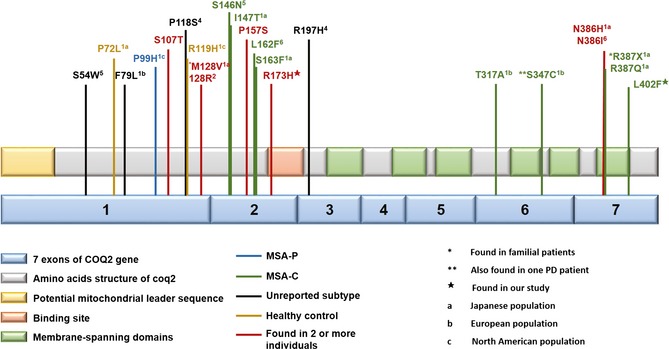
Schematic structure of Coq2 and distribution of identified COQ2 variants. The COQ2 gene consists of 7 coding exons. The coq2 protein is predicted to consist of a mitochondrial leader sequence, an allylic polyprenyl diphosphate substrate‐binding site and 6 membrane‐spanning domains. The p.R173 was located in the binding site and p.L402 in the transmembrane region. All the COQ2 variants reported recently are shown.
In summary, we identified 2 missense COQ2 mutations (p.L402F and p.R173H) and one synonymous mutation (p.A32A) in a cohort of 116 Chinese MSA‐C cases. The frequency of COQ2 mutations (1.72%) was consistent with that of recent studies and demonstrated that COQ2 tended to play a population‐specific and subtype‐depended role in conferring susceptibility to MSA. Owing to the low frequency of COQ2 variants identified in all the MSA cases reported, additional studies will be required to elucidate other genetic factors playing in the pathogenesis of MSA.
Conflict of Interest
The authors declare no conflict of interest.
Supporting information
Figure S1. Chromatograms of two variants including c.1178T>C (p.V393A) and c.73T>G (p.L25V) within COQ2. The upper panel for each chromatogram depicts the wild‐type sequence, whereas the lower panel represents the heterozygous variant.
Table S1. Details of clinical manifestations of 116 MSA‐C patients.
Table S2. PCR forward (F) and reverse (R) primers and annealing temperature designed for COQ2.
Table S3. Genotype and allele frequency of p.V393A allele in different populations.
Table S4. COQ2 variants in MSA cases and controls.
Acknowledgments
The authors thank the participants of this study for their cooperation. This work was supported by a grant from the National Natural Science Foundation of China to Zhi‐Ying Wu (81125009, Beijing).
The first two authors contributed equally to this work.
References
- 1. Alessandra F, Gregor K, Wenning MD. Multiple‐system atrophy. N Engl J Med 2015;372:249–263. [DOI] [PubMed] [Google Scholar]
- 2. Gilman S, Wenning GK, Low PA, et al. Second consensus statement on the diagnosis of multiple system atrophy. Neurology 2008;71:670–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. May S, Gilman S, Sowell BB, et al. Potential outcome measures and trial design issues for multiple system atrophy. Mov Disord 2007;22:2371–2377. [DOI] [PubMed] [Google Scholar]
- 4. Geser F, Seppi K, Stampfer‐Kountchev M, et al. The European multiple system atrophy‐study group (EMSA‐SG). J Neural Transm 2005;112:1677–1686. [DOI] [PubMed] [Google Scholar]
- 5. Tsuji S, Onodera O, Goto J, Nishizawa M. Sporadic ataxias in Japan–a population‐based epidemiological study. Cerebellum 2008;7:189–197. [DOI] [PubMed] [Google Scholar]
- 6. Papp MI, Kahn JE, Lantos PL. Glial cytoplasmic inclusions in the CNS of patients with multiple system atrophy (striatonigral degeneration, olivopontocerebellar atrophy and Shy‐Drager syndrome). J Neurol Sci 1989;94:79–100. [DOI] [PubMed] [Google Scholar]
- 7. Wenning GK, Krismer F. Multiple system atrophy. Handb Clin Neurol 2013;117:229–241. [DOI] [PubMed] [Google Scholar]
- 8. Soma H, Yabe I, Takei A, Fujiki N, Yanagihara T, Sasaki H. Heredity in multiple system atrophy. J Neurol Sci 2006;240:107–110. [DOI] [PubMed] [Google Scholar]
- 9. Hara K, Momose Y, Tokiguchi S, et al. Multiplex families with multiple system atrophy. Arch Neurol 2007;64:545–651. [DOI] [PubMed] [Google Scholar]
- 10. Al‐Chalabi A, Durr A, Wood NW, et al. Genetic variants of the alpha‐synuclein gene SNCA are associated with multiple system atrophy. PLoS ONE 2009;4:e7114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Scholz SW, Houlden H, Schulte C, et al. SNCA variants are associated with increased risk for multiple system atrophy. Ann Neurol 2009;65:610–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Mitsui J, Matsukawa T, Ishiura H, et al. Mutations in COQ2 in familial and sporadic multiple‐system atrophy. N Engl J Med 2013;369:233–244. [DOI] [PubMed] [Google Scholar]
- 13. Jeon BS, Farrer MJ, Bortnick SF. Mutant COQ2 in multiple‐system atrophy. N Engl J Med 2014;371:80. [DOI] [PubMed] [Google Scholar]
- 14. Sharma M, Wenning G, Kruger R. Mutant COQ2 in multiple‐system atrophy. N Engl J Med 2014;371:80–81. [DOI] [PubMed] [Google Scholar]
- 15. Schottlaender LV, Houlden H. Mutant COQ2 in multiple‐system atrophy. N Engl J Med 2014;371:81. [DOI] [PubMed] [Google Scholar]
- 16. Ogaki K, Fujioka S, Heckman MG, et al. Analysis of COQ2 gene in multiple system atrophy. Mol Neurodegener 2014;9:44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Chen YP, Zhao B, Cao B, et al. Mutation scanning of the COQ2 gene in ethnic Chinese patients with multiple‐system atrophy. Neurobiol Aging 2014;36:1222.e7–1222.e11. [DOI] [PubMed] [Google Scholar]
- 18. Ross OA, Vilariño‐Güell C, Wszolek ZK, Farrer MJ, Dickson DW. Reply to: SNCA variants are associated with increased risk of multiple system atrophy. Ann Neurol 2010;67:414–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Forsgren M, Attersand A, Lake S, et al. Isolation and functional expression of human COQ2, a gene encoding a polyprenyl transferase involved in the synthesis of CoQ. Biochem J 2004;382(Pt 2):519–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Chromatograms of two variants including c.1178T>C (p.V393A) and c.73T>G (p.L25V) within COQ2. The upper panel for each chromatogram depicts the wild‐type sequence, whereas the lower panel represents the heterozygous variant.
Table S1. Details of clinical manifestations of 116 MSA‐C patients.
Table S2. PCR forward (F) and reverse (R) primers and annealing temperature designed for COQ2.
Table S3. Genotype and allele frequency of p.V393A allele in different populations.
Table S4. COQ2 variants in MSA cases and controls.