

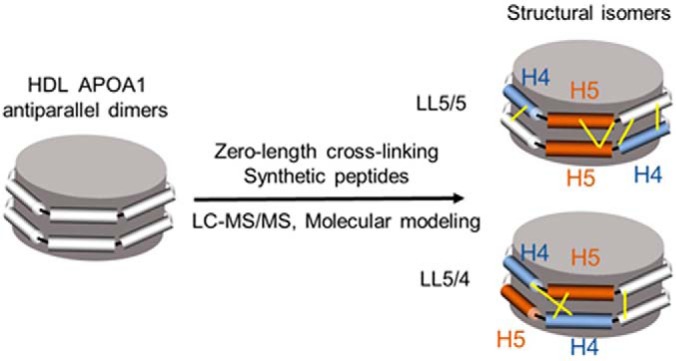
The structural orientation of apolipoprotein A1 (APOA1) dimers in reconstituted high-density lipoprotein (HDL) and human HDL was determined by zero-length chemical cross-linking and MS/MS analysis. The peptide cross-links provide strong biochemical evidence of the LL5/5 and LL5/4 antiparallel APOA1 dimers in human HDL and are in excellent agreement with molecular modeling. We validated our findings using synthetic cross-linked peptides. Our observations indicate that phospholipidated APOA1 forms two distinct antiparallel dimers that likely bind different proteins with important functional consequences.
Keywords: Protein Cross-linking*, Tandem Mass Spectrometry, Protein Conformation*, Computer Modeling, Structural Biology*, APOA1, high density lipoprotein, molecular modeling
Graphical Abstract
Highlights
Zero-length chemical cross-linking of APOA1 peptides in HDL.
Cross-links match antiparallel isomers of APOA dimers in molecular modeling.
Identical MS/MS spectra of native and synthetic cross-linked peptides.
First biochemical evidence of LL5/5 and LL5/4 isomers in human HDL.
Abstract
Apolipoprotein A1 (APOA1), the major protein of high-density lipoprotein (HDL), contains 10 helical repeats that play key roles in protein-protein and protein-lipid interactions. The current structural model for HDL proposes that APOA1 forms an antiparallel dimer in which helix 5 in monomer 1 associates with helix 5 in monomer 2 along a left-left (LL5/5) interface, forming a protein complex with a 2-fold axis of symmetry centered on helix 5. However, computational studies suggest that other orientations are possible. To test this idea, we used a zero-length chemical cross-linking reagent that forms covalent bonds between closely apposed basic and acidic residues. Using proteolytic digestion and tandem mass spectrometry, we identified amino acids in the central region of the antiparallel APOA1 dimer of HDL that were in close contact. As predicted by the current model, we found six intermolecular cross-links that were consistent with the antiparallel LL5/5 registry. However, we also identified three intermolecular cross-links that were consistent with the antiparallel LL5/4 registry. The LL5/5 is the major structural conformation of the two complexes in both reconstituted discoidal HDL particles and in spherical HDL from human plasma. Molecular dynamic simulations suggest that that LL5/5 and LL5/4 APOA1 dimers possess similar free energies of dimerization, with LL5/5 having the lowest free energy. Our observations indicate that phospholipidated APOA1 in HDL forms different antiparallel dimers that could play distinct roles in enzyme regulation, assembly of specific protein complexes, and the functional properties of HDL in humans.
One important risk factor for atherosclerosis is a low level of HDL cholesterol (HDL-C)1, which has been proposed to protect against cardiovascular disease by removing excess cholesterol from artery wall macrophages (1, 2). Thus, the risk of cardiovascular disease is inversely proportional to plasma levels of HDL-C and its major apolipoprotein, APOA1 (3–5). However, alterations in HDL's protein cargo change the lipoprotein's antioxidant and anti-inflammatory properties (6, 7), creating particles that might be less able to protect against cardiovascular disease. Moreover, HDL carries protein families implicated in complement activation and the regulation of proteolysis, raising the possibility that they modulate the innate immune system and alter the cardioprotective properties of HDL (8).
The biogenesis of HDL begins with the secretion and phospholipidation of lipid-free APOA1 by the liver and intestine to form circulating discoidal particles containing two molecules of APOA1 along with free cholesterol and amphipathic phospholipids (9). These immature HDL particles then interact with lecithin: cholesterol acyltransferase (LCAT), which catalyzes the transfer of fatty acyl components from the sn-2 position of phosphatidylcholine to the hydroxyl moiety of free cholesterol (1, 10). The resulting hydrophobic cholesteryl ester migrate to the core, forming mature spherical HDL particles (1, 11).
HDL's biogenesis, metabolism, and function has been extensively investigated (3–5, 12–17). However, surprisingly little is known about the structures of those particles and the relationship between structure and HDL's functions (18–26).
Human APOA1 contains a globular N-terminal domain (residues 1–43) and a lipid-binding C-terminal domain (residues 44–243), which exhibits 10 repeating helical domains (27). Pioneering molecular modeling studies suggested that two molecules of APOA1 in discoidal HDL form an antiparallel dimer that wraps belt-wise in a ring-like amphipathic helix that maximizes intermolecular salt bridges between the proteins (19). Importantly, the proposed docking faces of the two proteins have a left-left orientation (19), in which helix 5 in monomer 1 associates with helix 5 in monomer 2 along an antiparallel interface (LL5/5). The crystal structures of the lipid-free N-terminal and C-terminal truncated forms of APOA1 strongly support the proposed configuration (18, 28), with both structures demonstrating extensive salt bridging between the APOA1 belts and an axis of symmetry in helix 5 in the central region of the dimer centered on the fifth repeated helical domain of APOA1 (18, 28).
In the original description of APOA1 in discoidal HDL, it was noted that two other orientations of the LL ring pair, 5/4 and 5/6, also exhibited high calculated scores for potential intermolecular salt bridging (19). Subsequent work with cysteine mutants of APOA1 (29) supported the possibility that the LL5/4 orientation also exists in reconstituted HDL particles (rHDL). However, no direct biochemical evidence supports the existence of the LL5/4 orientation or other dimers in phospholipidated APOA1 of reconstituted discoidal HDL (rHDL) or human HDL.
To determine whether multiple structurally distinct dimers might form under physiological conditions, we used chemical cross-linking in concert with tandem mass spectrometry (MS/MS) (30–32) to search for intermolecular crosslinks in the central region (helices 4–6) of APOA1 in HDL. Our results strongly support the existence of both LL5/5 and LL5/4 APOA1 dimers in human HDL.
EXPERIMENTAL PROCEDURES
Materials
Recombinant APOA1 (14N- or 15N-labeled) was produced using a bacterial expression system and isolated to greater than 99% purity (33). The recombinant APOA1 is 99.6% identical to human APOA1, with a signal peptide containing a His tag (MHHHHHHGLVPRGSI). The synthetic peptide 1 (AKVQPYLDDFQ[13C]K-amide -THLAPYSDELR) was synthesized by New England Peptide, Inc. (Gardner, MA). The synthetic peptide 2 (AKVQPYLDDFQK-amide-LSPLGEEM[13C15N]R-amide) was synthesized by the Peptide Synthesis Core (University of Alabama at Birmingham). [13C6]K and [13C615N4]R increased the peptide mass by 6 Da and 10 Da, respectively. The change of C-terminal carboxylic acid group to an amide reduced the mass of the peptides by 1 Da. Taken together, the masses of the synthetic peptide 1 and 2 were increased by 5 Da and 8 Da, respectively.
Reconstituted HDL (rHDL)
Discoidal rHDL particles containing two APOA1 molecules were prepared from recombinant human APOA1, palmitoyl-oleoyl-phosphatidylcholine, and free cholesterol by cholate dialysis (34). Light (14N) and heavy (15N) APOA1s were used in a 1:1 ratio. Final molar ratios of POPC:FC:apoA-I (mol/mol/mol) equal to 30:2:1 for 80 Å rHDL (rHDL-2–80), 80:4:1 for 100 Å rHDL (rHDL-2–100), and 160:8:1 for 120 Å rHDL (rHDL-2–120). Particles were further isolated by high-resolution size exclusion chromatography (Superdex 200, 350 μl flow/min).
Human HDL
Plasma was prepared from EDTA-anticoagulated blood from four healthy adults who had fasted overnight. HDL (density 1.125–1.210 g/ml) was isolated from plasma by sequential ultracentrifugation (35) and stored at −80 °C. HDL was further fractionated by high-resolution size exclusion chromatography (Superdex 200, 350 μl flow/min) at 4 °C using PBS. Nine fractions (0.5 ml) with the apparent size of human HDL (7–12 nm diameter) were collected, pooled, and used for analysis.
HDL Particle Concentration and Size (HDL-PIMA)
Analyses were performed on a scanning mobility particle sizer spectrometer (TSI Inc., Shoreview, MN, model 3080N) fitted with a nano-differential mobility analyzer (TSI Inc., model 3085) and a charge-reducing electrospray ionization source (CR-ESI; TSI Inc., model 3480). Monodisperse particles exiting the differential mobility analyzer were detected by a condensation particle counter (TSI Inc., model 3788). Samples were introduced into the electrospray chamber every 15 min by automated loop injections. Particle concentration was determined by external calibration (36).
Chemical Cross-linking of APOA1
HDL was extensively dialyzed against phosphate-buffered saline (PBS, pH 6.5). Cross-linking reactions were carried out in PBS at 4 °C with final concentrations of 20 mm 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (EDC) and 0.56 mg/ml HDL protein. After a 12 h incubation, the reactions were quenched with 1 m ammonium acetate (final concentration 50 mm). Because two HDL particles can be linked together during the cross-linking reaction (which does not provide useful structural information about the structure of APOA1 on individual HDL particles), reaction mixtures were further fractionated by high-resolution size exclusion chromatography (Superdex 200, 350 μl flow/min) to isolate single HDL particles. Purified cross-linked HDL were exchanged into 50 mm ammonium bicarbonate with Amicon Ultra 10K centrifugal filter devices (Millipore, Eschborn, Germany), and stored at 4 °C for MS/MS analysis.
Proteolytic Digestion and Mass Spectrometry Analysis
After cross-linking, HDL was incubated overnight at 37 °C with sequencing grade modified trypsin (Promega, Madison, WI) at a ratio of 20:1 (w/w) protein/trypsin in 100 mm NH4HCO3, pH 8. Digestion was halted by acidification (pH 2–3) with trifluoroacetic acid. Capillary LC-ESI-MS/MS was performed using an IntegraFrit capillary C18 trapping column (Waters XBridge BEH C18, 5 μm, 0.1 × 20 mm), a capillary analytical C18 column packed in-house (Waters XBridge BEH C18, 5 μm, 0.075 × 150 mm) and an LTQ Orbitrap XL mass spectrometer (Thermo Electron, Bremen, Germany). The nanoACQUITY UPLC (Waters, Milford, MA) was used for the separation, with a linear gradient of 0.1% formic acid in water (solvent A) and 0.1% formic acid in acetonitrile (solvent B). In each experiment, 1 μg of tryptic digest was injected onto a trapping column to remove salts and contaminants. Effluent from the trapping column was directed to the capillary analytical column with an 80-min gradient between 2 and 40% mobile phase B at a flow rate of 0.3 μl/min. Eluting peptides were electrosprayed into an LTQ-Orbitrap mass spectrometer operating in the data-dependent mode to acquire a full MS scan (400–2000 m/z) and then subsequent MS/MS scans of the five most intense precursor ions. Isolation width was 2 m/z. MS/MS scans were acquired in the ion trap. Collision-induced dissociation (CID) was performed at 35% normalized collision energy. Charge state rejection was enabled for 1+ and 2+ charge states. As cross-linked peptides formed in these experiments tend to have higher charge states upon ESI than noncross-linked (linear peptide) species, rejection of low-charge states from MS/MS acquisition enhances detection of cross-links. For database searching, MS/MS spectrum lists were extracted from the raw files and converted to mzXML files, using ReAdW (version 4.6.0).
Experimental Design and Statistical Rationale
We performed two replicates in generating rHDL and two technical replicates for the cross-linking MS experiments of both rHDL and human HDL. MS/MS data contained in the mzXML files were subsequently searched against the database for light labeled cross-links, using xQuest (version 2.1.1) (37, 38). For rHDL, the database was built from the sequence of recombinant APOA1. For human HDL, the database contained the sequences of thirty-nine proteins (from Uniprot database) consistently detected in HDL in previous studies (6, 8, 39, 40). xQuest searches for theoretical cross-links whose masses match measured precursor masses, and it subsequently assigns fragment masses from MS/MS spectra if a cross-linked precursor will fragment at only one peptide bond. Spectra were searched using the default settings of xQuest (37) except for the cross-linked residues and the mass shift. The detailed parameters are the following: cross-linked residues: K, D, E; mass shift for cross-linked peptides, −18.010564686; MS ion mass tolerance, 10 ppm; MS2 ion mass tolerance for common-ions: 0.2 Da; MS2 ion mass tolerance for cross-linked ions: 0.3 Da; allow matching second isotopic peak; fixed modification, Cys carbamidomethylation; enzymatic digestion, trypsin; allowed number of missed cleavages, 2 (37). Matches from all searches were required to have less than 5% false-discovery rate (FDR) based on a target-decoy calculation strategy (a reverse decoy database) and more than 15% of the ion current in a given MS/MS spectrum assigned as b- and y-type ions. All matches were manually inspected to ensure that both peptides in the cross-linked product had been sequenced and that most of the abundant fragment ions could be assigned in the MS/MS spectra. The matched cross-links were also identified using two other search engines, Kojak (41) and SIM-XL (42).
Molecular Dynamic Simulations
Initial configuration of the planar double belt structures of LL5/5 (19) and LL5/4 were minimized for 10,000 steps using a conjugate gradient algorithm followed by 1 ns simulation in vacuo. All simulations were performed with using NAMD 2.12 (43) and the CHARMM 36 force field (44). Constant temperature and pressure were maintained at 310 K and 1 atm, respectively, using Langevin thermostat and the Nose-Hoover Langevin piston method (45). A time step of 2 fs was used, and all bond lengths involving H-atoms were subject to constraint using the SHAKE (46) and SETTLE (47) algorithms. Nonbonded interactions were cut off at 12 Å. The particle-mesh Ewald (48) was used to compute full electrostatic interaction.
RESULTS
We used EDC (1-ethyl-3-[3-dimethylaminopropyl]carbodiimide hydrochloride) (49) to generate intramolecular and intermolecular cross-links in APOA1 of rHDL and native HDL. Importantly, all cross-linking reactions were carried out at low concentrations of EDC in phosphate buffered normal saline at pH 6.5, which much more closely mimics physiological conditions than those used for crystallization of proteins. EDC reacts with the amino group of lysine resides near the carboxylic acid group of aspartate and glutamate to form an amide bond. Cross-links formed by EDC represent solvent-accessible salt bridges.
The maximum distance between the α-carbons in the amino acid residues of the cross-linked amino acids is the sum of the length of two side chains plus the length of the amide bond (10.5 Å for the K-D linkage and 12.1 Å for K-E). Considering a 3 Å motion-averaging factor (50, 51), we considered distances ≤ 13.5 Å for K-D linkages and ≤ 15.1 Å for K-E linkages as consistent with a proposed structure of APOA1.
APOA1 Prepared Using Heavy and Light Isotopes Distinguishes Between Intermolecular and Intramolecular Cross-links
To distinguish between inter- and intramolecular cross-links of APOA1 in rHDL, we used a mixture of human [14N]APOA1 (light, L) and [15N]APOA1 (heavy, H) (isotopic purity >99%) to generate rHDL particles (52). When rHDL containing two molecules of APOA1 is generated with a 1:1 molar ratio of L and H APOA1, three combinations theoretically should form: LL, LH, and HH (relative abundance 1:2:1) (Scheme 1). The L and H forms of APOA1 are chemically identical but differ in molecular mass, making intramolecular and intermolecular cross-links readily distinguishable in MS1 scans. For intraprotein cross-links, where the protein is linked to itself, only l-L and H-H forms are detected (Scheme 1A), as documented by two peaks with identical retention times but different masses. In contrast, interprotein cross-links between dimers of APOA1 (termed α and β) generate four species: Lα-Lβ, Lα-Hβ, Hα-Lβ, and Hα-Hβ (Scheme 1B). When the α and β APOA1 peptides contain similar numbers of N atoms, cross-links of Lα-Hβ and Hα-Lβ can have similar m/z values and the two peaks in the center of the mass spectrum for MS1 will merge together.
Scheme 1.
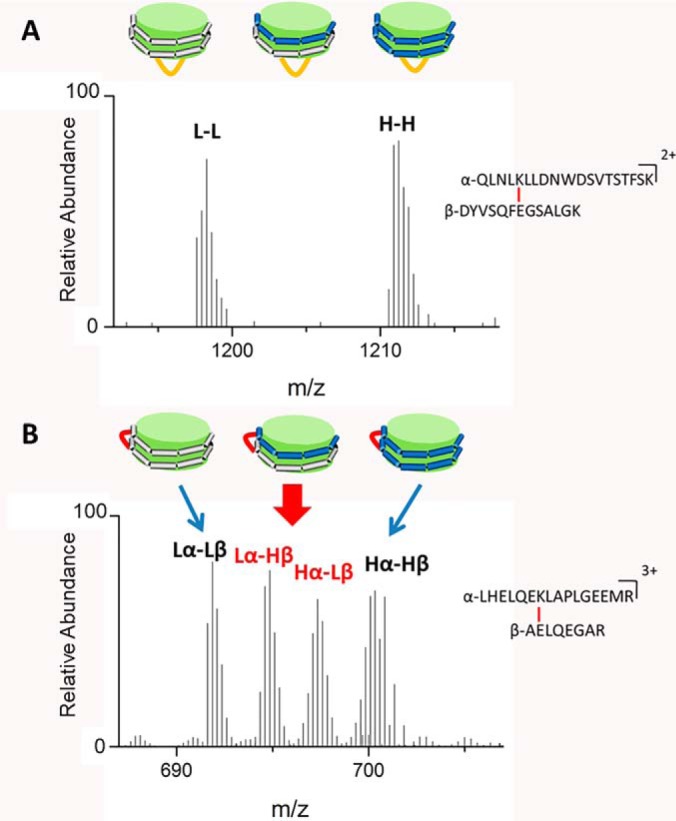
MS1 spectra of (A) intraprotein cross-link GSIDDPPQSPWDR-VKDLATVYVDVLK and (B) interprotein cross-link LHELQEKLAPLGEEMR-AELQEGAR from discoidal rHDL-2–100 using 14N (L) and 15N (H) APOA1.
Chemical Cross-linking in rHDL and MS/MS Analysis Reveal 5/5 and 5/4 APOA1 Dimers
Discoidal rHDL containing 2 molecules of APOA1 were prepared from phospholipid, cholesterol, and APOA1. Following cross-linking with EDC, the reaction mixture was fractionated by high-resolution size exclusion chromatography to eliminate cross-linked HDL particles. The size of the discoidal rHDLs isolated by this procedure was 96 Å, as measured by calibrated ion mobility analysis (Fig. 1B) (36). We term these particles rHDL-2–100, indicating that they contain 2 moles of APOA1 and have a diameter of ∼100 Å. These particles have been extensively characterized (34).
Fig. 1.
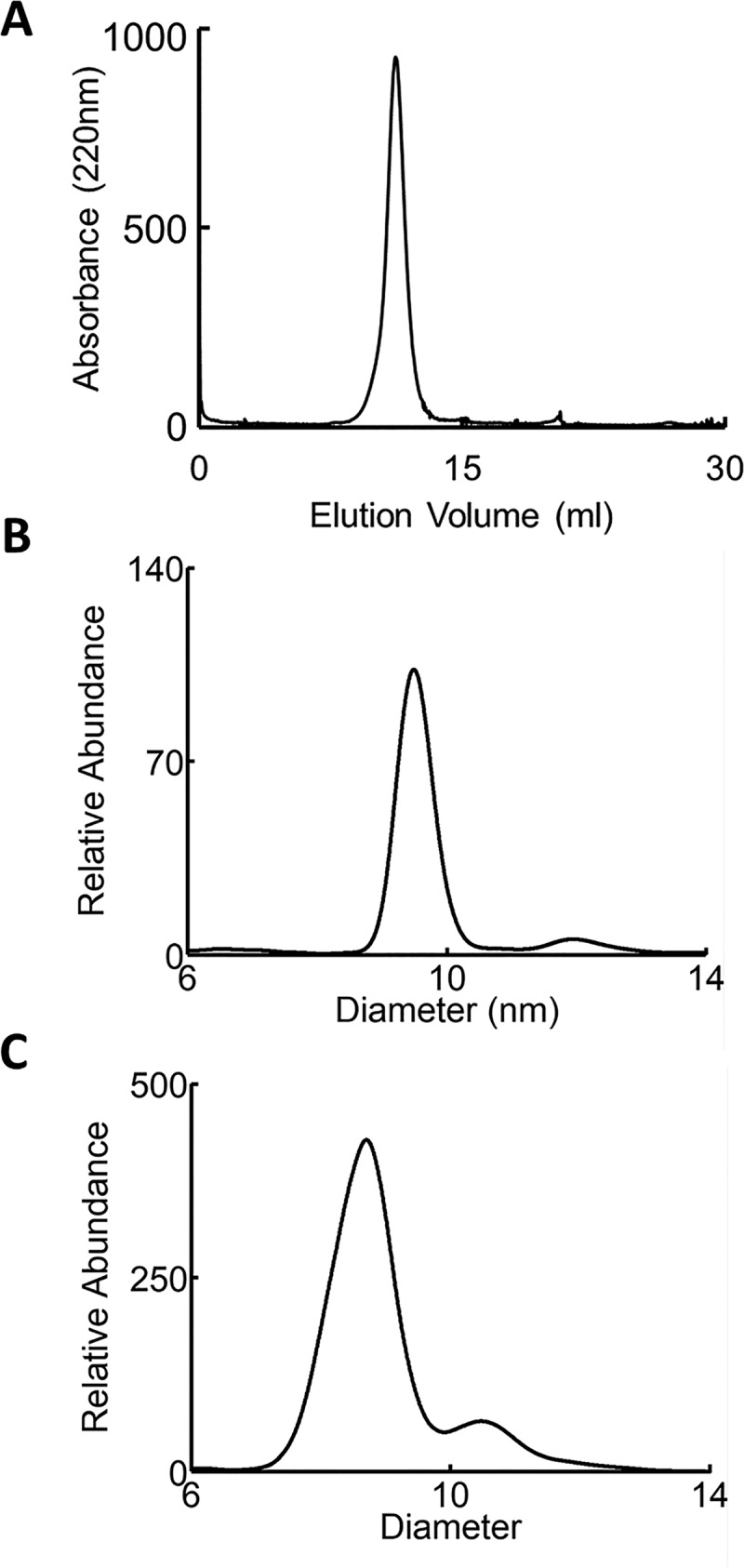
Characterization of rHDL and human HDL particles. A, FPLC size-exclusion chromatogram of rHDL-2–100. B, IMA spectra of rHDL-2–100 and C, isolated human HDL.
Chemical cross-linking and MS/MS analysis of rHDL-2–100 prepared with a 1:1 mol ratio of L and H APOA1 identified 11 interprotein cross-links (Table I). Nine were between amino acids in the central region (helices 3–7) (Scheme 2) of the LL5/5 antiparallel APOA1 dimer in discoidal HDL proposed by Segrest and colleagues (19). The LL5/5 orientation is also present in two different crystal structures of nonlipidated N- and C-terminal truncated forms of APOA1 (18, 28). Distances between the two α carbons in the linked residues (Table I) were determined by mapping the cross-linkers onto the crystal structure of the C-terminal truncated APOA1 dimer and simulated structure of lipid-bound APOA1 dimers (LL5/5 and 5/4) (28). The distances from the nonlipidated crystal structure are larger than the salt bridge distances in the lipid-bound forms of LL5/5 because of a small rotation in contacts between the antiparallel helical domains required for lipid-association (51). Two cross-linking distances were not available because they are involved in the C-terminal end of APOA1, which the crystal structure lacks. Six cross-links were between residues that were 5–15 Å apart in the crystal structure of APOA1, which is consistent with the theoretical maximum distance (15.1 Å) between EDC cross-linked amino acids. The positions of these cross-links are in excellent agreement with those predicted by the crystal structure of the truncated lipid-free APOA1 dimer (28).
Table I. Inter-molecular APOA1 cross-links detected in rHDL-2-100 particles.
| Cross-link | m/z | Charge state | Mass error (ppm)a | Distance in crystal structure (Cα-Cα, Å)b | Distance in LL5/5 (Cα-Cα, Å)c | Distance in LL5/4 (Cα-Cα, Å)3 | Ratio of LL:LH:HH |
|---|---|---|---|---|---|---|---|
| K96-D168 | 684.351 | 4 | −1 | 13.5 | 6.5 | 37.0* | 1:2:1 |
| K96-E169 | 954.831 | 3 | −0.2 | 11.58 | 9.8 | 37.5* | 1:2:1 |
| K96-E147 | 822.092 | 3 | 2.7 | 30.69* | 29.2* | 8.7 | 1:1:1:1 |
| K107-D157 | 725.367 | 3 | 1.8 | 13.74 | 12.1 | 37.1* | 1:1:1:1 |
| K107-E125 | 1133.552 | 2 | 2 | 42.69* | 41.5* | 12.9 | 1:1:1:1 |
| K118-E147 | 471.262 | 4 | 0.7 | 13.02 | 10.2 | 37.7* | 1:2:1 |
| K133-E111 | 637.078 | 4 | 0.5 | 25.75* | 24.3* | 9.8 | 1:2:1 |
| K133-E125 | 669.692 | 3 | −1.4 | 10.31 | 8.0 | 27.7* | 1:1:1:1 |
| K140-E125 | 691.609 | 4 | 1.6 | 14.62 | 10.5 | 37.7* | 1:1:1:1 |
| K195-E70 | 1053.543 | 4 | 2 | N/A | 9.8 | 37.1* | 1:2:1 |
| K195-E78 | 679.101 | 4 | 0.1 | N/A | 19.3* | 47.2* | 1:1:1:1 |
a Relative to theoretical masses of cross-linked peptides.
b Distance between α-carbons of cross-linked amino acids in the crystal structure of lipid-free C-terminal truncated APOA1 dimer (Δ185–243, PDB ID 3R2P) (28).
c Distance between α-carbons of cross-linked amino acids in the simulated structure of the LL5/5 and LL5/4 antiparallel dimers.
* Distance between α-carbons in crystal structure > 15.1 Å.
Scheme 2.

Helical domains in human APOA1 sequence.
We also identified three intermolecular cross-links that exceed the maximum plausible cross-linking distance in the crystal structures of truncated APOA1: K96-E147, K107-E125, and K133-E111. Molecular simulation of APOA1 dimers indicated that all three cross-links are consistent with the LL5/4 antiparallel dimer of APOA1 originally proposed by Segrest and colleagues (29). The distances of these three cross-links in LL5/4 were 8.7 Å, 12.9 Å and 9.8 Å, respectively. Based on the relative intensities of the ion current chromatograms for the intermolecular cross-links, it is clear that the LL5/5 is the major antiparallel conformation of the two dimers in rHDL-2–100 (Fig. 4A).
Fig. 4.

MS and MS/MS spectra of native cross-links from human HDL and synthetic peptides. (A) AKVQPYLDDFQKγÇôTHLAPYSDELR from LL5/5 registry and (B) its synthetic cross-link peptide 1 from LL5/5 registry. (C) AKVQPYLDDFQKγÇôLSPLGEEMR from LL5/4 registry and (D) its synthetic cross-link peptide 2. Inset of B and D show the retention time of native and synthetic cross-links in LL5/5 and LL5/4 registries.
The MS and MS/MS spectra of representative cross-links, K118-E147 from the 5/5 and K133-E111 from the 5/4 registry, are shown in Fig. 2A–2B. In both MS1 spectra, there are three groups of cross-link peaks corresponding to the linkages of l-L, l-H and H-H, indicating that both are intermolecular crosslinks. In the MS/MS spectra of cross-links from the l-L form of APOA1, fragments from both the α and β chains were observed, and the majority of the abundant peaks can be assigned to predicted fragment ions. We also identified three cross-links (K12-E76, K23-D48 and K45-E34) in the N terminus (supplemental Table S1), that are consistent with the configuration in the crystal structure of a C-terminal truncated form of APOA1 (Δ185–243, PDB ID 3R2P, supplemental Fig. S1). For the C terminus, we identified two cross-links: E198-K208 and K208-E223 (Δ1–43, PDB ID 1AV1, supplemental Fig. S2).
Fig. 2.

MS and MS/MS spectra of intermolecular APOA1 cross-link in rHDL-2–100. A, QKVEPLR-LSPLGEEMR consistent with the LL5/5 registry, B, QKLHELQEK-KWQEEMELYR consistent with the LL5/4 registry.
We also observed one intraprotein cross-link, K238-E34, which is strong evidence that the N terminus of APOA1 is in contact with its own C terminus in rHDL-2–100.
To determine if the size of HDL particles influenced the formation of 5/5 and 5/4 antiparallel dimers, we subjected rHDLs of 80 Å diameter (rHDL-2–80) and 120 Å diameter (rHDL-2–120) to cross-linking with EDC, proteolytic digestion, and MS/MS analysis. All of the intermolecular cross-links in the central region of APOA1 detected in rHDL-2–100 were also detected in the smaller and larger rHDL particles (supplemental Table S1). The apparent abundance of the cross-links was like that observed in rHDL-2–100 as monitored by their total ion chromatograms, strongly suggesting that the size of the particles was not a major determinant of the formation of the 5/5 and 5/4 antiparallel APOA1 dimers.
APOA1 in Human HDL Contains Both 5/5 and 5/4 Homodimers
To determine whether APOA1 in human HDL might also form both LL5/5 and LL5/4 antiparallel dimers, we isolated spherical HDL from plasma by ultracentrifugation (d = 1.019 – 1.063 g/ml), cross-linked the protein, digested it proteolytically, and analyzed it with MS/MS. The apparent size of the HDL used in these analyses was 89 Å as determined by calibrated ion mobility analysis (Fig. 1C) (36). This approach identified 10 intermolecular cross-links in the central region of the APOA1 dimer (Table II). Four of the crosslinks were consistent with the LL5/5 antiparallel dimer and two were consistent with the LL5/4 antiparallel orientation. However, we did not detect two cross-linked peptides identified in rHDL-2–100, K107-D157 or K133-E125, in human HDL. We also observed one cross-link, K94-E125, that is consistent with 5/2 registry (19, 53). Based on the relative intensities of the ion current chromatograms of the cross-links, we demonstrate that the LL5/5 is the major antiparallel conformation of the two dimers in native human HDL (Fig. 4B).
Table II. Cross-links detected in helices 3–7 of APOA1 in human HDL.
| Cross-link | m/z | Charge state | Mass Error (ppm)a | Distance (Cα-Cα, Å)b | Orientation |
|---|---|---|---|---|---|
| K94-E125 | 595.984 | 3 | 1.7 | 60.08* | LL5/2 |
| K96-E91 | 459.447 | 5 | −1 | 9.90 | N/A |
| K96-E147 | 616.82 | 4 | 0 | 30.69* | LL5/4 |
| K96-D168 | 477.92 | 6 | 1.5 | 13.5 | LL5/5 |
| K96-E169 | 684.351 | 4 | −0.6 | 11.58 | LL5/5 |
| K106-E92 | 524.276 | 4 | 3.1 | 23.22* | N/A |
| K118-D102 | 526.538 | 4 | 2.1 | 25.46* | N/A |
| K118-E147 | 628.013 | 3 | 1.5 | 13.02 | LL5/5 |
| K133-E110 | 637.071 | 4 | −9.8 | 28.03* | LL5/4 |
| K140-E125 | 604.721 | 5 | 2.6 | 14.62 | LL5/5 |
a Relative to theoretical masses of cross-linked peptides.
b Distance between α-carbons of cross-linked amino acids in the crystal structure of lipid-free C-terminal truncated APOA1 dimer (Δ185–243, PDB ID 3R2P) (28).
* Distance between α-carbons in crystal structure > 15.1 Å.
To further confirm the identity of cross-links, we compared the MS and MS/MS analyses of the cross-links from human HDL with those of two synthetic peptides, K96-E169 (synthetic peptide 1) and K96-E147 (synthetic peptide 2), that mimicked the proposed cross-link between the APOA1 dimers. Except for the anticipated mass shift because of the incorporation of [13C6]Lys-NH2 into the synthetic peptides, the MS and MS/MS spectra of the cross-links from human HDL and the synthetic peptides were indistinguishable (Fig. 3). For K96-E169 from APOA1 with the LL5/5 registry, the m/z of synthetic peptide 1 is shifted from 684.35 ([M+4H]+4, Fig. 3A) to 685.61 ([M+5 + 4H]+4, Fig. 3B). MS/MS analysis of synthetic peptide1 (Fig. 3B) also demonstrated the same fragmentation pattern as that of the native cross-link found in EDC-treated human HDL. Furthermore, the retention time of the synthetic peptide was virtually identical to that of native cross-link (Fig. 3B, Inset). For K96-E147 from APOA1 in the LL5/4 registry, the C-terminal residues Lys and Arg in the synthetic peptide 2 were replace by Lys-NH2 and [13C615N4]Arg-NH2 respectively, which would increase the mass of the peptide by 8 Da. As demonstrated in Fig. 3C and 3D, the expected mass shift was observed from 616.82 [M2 + 4H]4+ to 618.83 [M2 + 8+4H]4+. Importantly, the fragmentation pattern and retention times of the native and synthetic cross-links were indistinguishable. Taken together, these observations strongly support the identification of the cross-link K96-E169 from the LL5/5 registry and K96-E147 from the LL5/4 registry in human HDL.
Fig. 3.

Average ion currents of inter-molecular cross-links identified in APOA1 dimers of (A) rHDL and (B) human HDL.
Molecular Modeling of the 5/5 and 5/4 Antiparallel Dimers in rHDL
We created a closed planar circular model of APOA1 as described previously (54). We manually created the desired dimers and then, to maximize the formation of potential interchain salt bridges and other types of bonds, utilizing the low dielectric constant of vacuum. The idealized circular LL5/5 and LL5/4 structures were minimized for 10,000 steps using the conjugate gradient algorithm. The structures then were subjected to molecular dynamic simulations for 1 ns after fixing all backbones and sidechains, excepting the sidechains located in the wp (wheel position) 2, wp5 and wp9. We previously demonstrated that these can form interchain salt bridges (55).
Fig. 5 shows the structural models of each dimer after 1 ns in vacuo molecular dynamic simulation. The LL5/5 and LL5/4 models each formed 12 solvent-accessible salt bridges, although between different residue pairs. For example, K96-E169, K140-E125 and K118-E147 formed salt bridge pairs in LL5/5, whereas K96-E147 and K133-E111 formed salt bridge pairs in LL5/4.
Fig. 5.

Schematic representation of residues proposed to form salt bridges that stabilize dimerization of LL5/5 and LL5/4 antiparallel double belt structures. This figure is a molecular representation of LL5/5 and LL5/4 highlighting residues positioned to form interchain salt bridges and other types of bonds (such as π-π interactions). To maximize the formation of potential interchain salt bridges and other types of bonds, the idealized circular LL5/5 and LL5/4 structures were subjected to molecular dynamic simulation in vacuo for 1 ns after fixing all backbones and sidechains except sidechains in the wp2, wp5 and wp9 positions (55). A, Schematic molecular representation of LL5/5 centered on the disc edge and pairwise antiparallel H5s to create a bilaterally symmetric view. B, Same view as A rotated 90° in left hand (LH) direction. C, Schematic molecular representation of LL5/4 centered on the disc edge and pairwise antiparallel H5/4s to create a bilaterally symmetric view. D, Same view as C rotated 90° in left hand (LH) direction. E and F, represent an enlargement of the cartoon molecular representation of C focused on the left hand half to illustrate Cα-Cα distances of the observed K96-E147, K107-E125 and K133-E111 cross-links. E, Four residue pairs forming salt bridges. F, Measurement of distances between Cα-Cα atoms of the K96-E147, K107-E125 and K133-E111 cross-links. Solvent-accessible salt bridge residues, stick representation indicated with narrow yellow arrows and smaller labels. Solvent-inaccessible salt bridge residues, space filling representation indicated with wide yellow arrows and larger labels. Tandem helical repeats: D1-N43, blue; H2-H3, gray; H4, light blue; H5, green; H6, light pink H7-H9, gray; H10, red. Basic residues, blue; acidic residues, red; prolines, space filling yellow; aromatic residues, space filling magenta.
Both models also formed solvent-inaccessible salt bridges but between different residue pairs, e.g. H155-E111 in LL5/5 and H155-D89 in LL5/4. However, as noted previously (19). LL5/5 formed 6 pairs of solvent-inaccessible salt bridges whereas LL5/4 only formed 4. There are other potential differences in intermolecular bonds between LL5/5 and LL5/4. For example, aromatic residues appear positioned to form π-π bonds in LL5/5 (Y192-F171 and perhaps Y166-Y100) whereas none can be identified in LL5/4.
The results of the 1 ns in vacuo molecular dynamic simulations suggest that that LL5/5 and LL5/4 APOA1 antiparallel dimers should possess similar—but not identical—free energies of dimerization, with LL5/5 being favored. The relative abundance of the 5/5 and 5/4 dimers (∼90 and ∼10%) as estimated from the relative ions currents of the cross-linked peptides are equivalent to one or two salt bridges more in LL5/5 than in LL5/4.
DISCUSSION
Using zero-length chemical cross-linking, proteolysis, and MS/MS, we identified nine peptide dimers that support the idea of intermolecular cross-linking in the central region (helices 4–6) of rHDL-2–100. Peptides derived from both heavy and light APOA1 provided unequivocal evidence that these cross-links were intermolecular. Six of the cross-links were consistent with the antiparallel LL5/5 APOA1 dimer observed in the crystal structures of the lipid-free C-terminal and N-terminal truncated forms of APOA1 (18, 28). However, the distances between the remaining three intermolecular cross-linked peptides were not consistent with this orientation. Instead, all three cross-links were entirely consistent with the distance constraints of the LL5/4 antiparallel registry first proposed by Segrest et al. (19, 29).
We also identified ten cross-linked peptides derived from helices 3–7 in spherical HDL isolated from human plasma by ultracentrifugation. Four of the cross-linked peptides were consistent with the 5/5 registry of HDL and two were consistent with the LL5/4 registry. However, there were some differences in the cross-linked peptides we detected in rHDL-2–100 and human HDL, likely reflecting differences in structure related to differences in the size and/or shape of discoidal and spherical HDL. Collectively, these observations strongly support the presence of two distinct antiparallel dimers in HDL—structural isomers that appear to be uncommon in proteins based on our search of the literature.
It is important to note that the crystallographic studies of APOA1 used nonlipidated and truncated forms of the protein and that the proteins were crystalized in high concentrations of salt, which can alter proteins' structures. We performed all our cross-linking reactions in physiological buffer at pH 6.5, and the human APOA1 in the reconstituted discoidal and native spherical HDL particles was fully phospholipidated. These observations strongly suggest that the structures we observed are physiologically relevant and likely mimic those of HDL particles in vivo in humans.
Symmetry between subunits is the most common configuration in homodimeric proteins because it maximizes stability while avoiding protein aggregation (56, 57). The basis for the existence of two symmetrical homodimers in APOA1 is likely because of the similar energetics of salt bridging in the LL5/5 and LL5/4 orientations, suggesting that it is an intrinsic property of the protein. Moreover, we observed both orientations in rHDLs of different sizes, where APOA1 is the only protein, and in human HDL, which associates with more than 70 different proteins (8). Thus, neither the size of rHDL, nor the interaction of APOA1 with other proteins is likely to be the driving force for the formation of different dimers.
Molecular dynamic simulations suggested that the LL5/5 and LL5/4 APOA1 antiparallel dimers should possess similar—but not identical—free energies of dimerization, with LL5/5 being favored with one-two additional salt bridges between the antiparallel chains. Consistent with this observation, the cross-linking experiment demonstrates that LL5/5 is the major conformation of the two APOA1 homodimers present in both rHDL-2–100 particles and human HDL. The similarity of the summed ion currents of the cross-linked peptides detected for rHDL and human HDL suggests that the relative abundance of the isomers is similar in the two types of particles. A key question is whether the formation of both LL5/5 and LL5/4 dimers in APOA1 associates with functional advantages as well as with energetic expediency. For example, this behavior might permit the protein to attain multiple conformations with different functional and metabolic properties. It is noteworthy that our molecular modeling demonstrated marked differences in the surface plots of highly charged and aromatic amino acids on the solvent-accessible faces of the LL5/5 and LL5/4 homodimers. For example, aromatic residues appear positioned to form π-π bonds in LL5/5 (Y192-F171 and perhaps Y166-Y100), whereas no π-π bonds can be identified in LL5/4.
These differences might affect APOA1's interactions with other proteins and lipids in ways that modulate HDL's cardioprotective, metabolic, and immune properties. Future studies need to investigate whether differences in APOA1's dimer structure lead to functional differences among HDLs in vivo and if disease states in humans associate with alterations in dimer structure.
In summary, our results strongly support the presence of two antiparallel dimers of APOA1 in HDL, with important implications for the lipoprotein's structure and functional properties.
DATA AVAILABILITY
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium (http://proteomecentral.proteomexchange.org) via the PRIDE partner repository with the dataset identifier PXD009927.
Supplementary Material
Acknowledgments
We thank by Dr. Michael Oda (Children's Hospital Oakland Research Institute) for providing recombinant APOA1 (14N- or 15N-labeled) and Dr. Priska D. von Haller (University of Washington) for technical assistance and helpful discussions. Mass spectrometry experiments were performed in the University of Washington's Proteomics Resource (UWPR95794) and the Quantitative and the Functional Proteomics Core of the Diabetes Research Center, University of Washington (P30 DK017047). Molecular dynamic simulations were performed at the Advanced Computing Center for Research and Education at Vanderbilt University, Nashville, TN.
Footnotes
* This work was supported by grants from the National Institutes of Health (P01HL092969, DP3DK108209 and P01HL128203) and the American Heart Association (15POST22700033).
 This article contains supplemental material.
This article contains supplemental material.
1 The abbreviations used are:
- HDL
- high-density lipoprotein
- APOA1
- apolipoprotein A1
- rHDL
- reconstituted HDL
- EDC
- 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride
- MS
- mass spectrometry.
REFERENCES
- 1. Glomset J. A. (1968) The plasma lecithins:cholesterol acyltransferase reaction. J. Lipid Res. 9, 155–167 [PubMed] [Google Scholar]
- 2. Glomset J. A., and Wright J. L. (1964) Some Properties of a Cholesterol Esterifying Enzyme in Human Plasma. Biochim. Biophys. Acta 89, 266–276 [DOI] [PubMed] [Google Scholar]
- 3. Wilson P. W., Abbott R. D., and Castelli W. P. (1988) High density lipoprotein cholesterol and mortality. The Framingham Heart Study. Arteriosclerosis 8, 737–741 [DOI] [PubMed] [Google Scholar]
- 4. Gordon D. J., and Rifkind B. M. (1989) High-density lipoprotein–the clinical implications of recent studies. N. Engl. J. Med. 321, 1311–1316 [DOI] [PubMed] [Google Scholar]
- 5. Mahdy Ali K., Wonnerth A., Huber K., and Wojta J. (2012) Cardiovascular disease risk reduction by raising HDL cholesterol–current therapies and future opportunities. Br. J. Pharmacol. 167, 1177–1194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Davidson W. S., Silva R. A., Chantepie S., Lagor W. R., Chapman M. J., and Kontush A. (2009) Proteomic analysis of defined HDL subpopulations reveals particle-specific protein clusters: relevance to antioxidative function. Arterioscler. Thromb. Vasc. Biol. 29, 870–876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Gordon S. M., McKenzie B., Kemeh G., Sampson M., Perl S., Young N. S., Fessler M. B., and Remaley A. T. (2015) Rosuvastatin alters the proteome of high density lipoproteins: generation of alpha-1-antitrypsin enriched particles with anti-inflammatory properties. Mol. Cell. Proteomics 14, 3247–3257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Vaisar T., Pennathur S., Green P. S., Gharib S. A., Hoofnagle A. N., Cheung M. C., Byun J., Vuletic S., Kassim S., Singh P., Chea H., Knopp R. H., Brunzell J., Geary R., Chait A., Zhao X. Q., Elkon K., Marcovina S., Ridker P., Oram J. F., and Heinecke J. W. (2007) Shotgun proteomics implicates protease inhibition and complement activation in the antiinflammatory properties of HDL. J. Clin. Inv. 117, 746–756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Oram J. F., and Heinecke J. W. (2005) ATP-binding cassette transporter A1: a cell cholesterol exporter that protects against cardiovascular disease. Physiolog. Rev. 85, 1343–1372 [DOI] [PubMed] [Google Scholar]
- 10. Shih A. Y., Sligar S. G., and Schulten K. (2009) Maturation of high-density lipoproteins. J. R. Soc. Interface 6, 863–871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Soutar A. K., Garner C. W., Baker H. N., Sparrow J. T., Jackson R. L., Gotto A. M., and Smith L. C. (1975) Effect of the human plasma apolipoproteins and phosphatidylcholine acyl donor on the activity of lecithin: cholesterol acyltransferase. Biochemistry 14, 3057–3064 [DOI] [PubMed] [Google Scholar]
- 12. Xiao C., Dash S., Morgantini C., Hegele R. A., and Lewis G. F. (2016) Pharmacological targeting of the atherogenic dyslipidemia complex: the next frontier in CVD prevention beyond lowering LDL cholesterol. Diabetes 65, 1767–1778 [DOI] [PubMed] [Google Scholar]
- 13. Hoekstra M., and Van Berkel T. J. (2016) Functionality of high-density lipoprotein as antiatherosclerotic therapeutic target. Arterioscler. Thromb. Vasc. Biol. 36, e87–e94 [DOI] [PubMed] [Google Scholar]
- 14. Karathanasis S. K., Freeman L. A., Gordon S. M., and Remaley A. T. (2017) The changing face of HDL and the best way to measure it. Clin. Chem. 63, 196–210 [DOI] [PubMed] [Google Scholar]
- 15. Gao X., Yuan S., Jayaraman S., and Gursky O. (2009) Differential stability of high-density lipoprotein subclasses: effects of particle size and protein composition. J. Mol. Biol. 387, 628–638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Movva R., and Rader D. J. (2008) Laboratory assessment of HDL heterogeneity and function. Clin. Chem. 54, 788–800 [DOI] [PubMed] [Google Scholar]
- 17. Rosenson R. S., Brewer H. B. Jr, Chapman M. J., Fazio S., Hussain M. M., Kontush A., Krauss R. M., Otvos J. D., Remaley A. T., and Schaefer E. J. (2011) HDL measures, particle heterogeneity, proposed nomenclature, and relation to atherosclerotic cardiovascular events. Clin. Chem. 57, 392–410 [DOI] [PubMed] [Google Scholar]
- 18. Borhani D. W., Rogers D. P., Engler J. A., and Brouillette C. G. (1997) Crystal structure of truncated human apolipoprotein A-I suggests a lipid-bound conformation. Proc. Natl. Acad. Sci. U.S.A. 94, 12291–12296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Segrest J. P., Jones M. K., Klon A. E., Sheldahl C. J., Hellinger M., De Loof H., and Harvey S. C. (1999) A detailed molecular belt model for apolipoprotein A-I in discoidal high density lipoprotein. J. Biol. Chem. 274, 31755–31758 [DOI] [PubMed] [Google Scholar]
- 20. Brouillette C. G., Anantharamaiah G. M., Engler J. A., and Borhani D. W. (2001) Structural models of human apolipoprotein A-I: a critical analysis and review. Biochim. Biophys. Acta 1531, 4–46 [DOI] [PubMed] [Google Scholar]
- 21. Li H. H., Lyles D. S., Pan W., Alexander E., Thomas M. J., and Sorci-Thomas M. G. (2002) ApoA-I structure on discs and spheres. Variable helix registry and conformational states. J. Biol. Chem. 277, 39093–39101 [DOI] [PubMed] [Google Scholar]
- 22. Carlson J. W., Jonas A., and Sligar S. G. (1997) Imaging and manipulation of high-density lipoproteins. Biophys. J. 73, 1184–1189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Nath A., Atkins W. M., and Sligar S. G. (2007) Applications of phospholipid bilayer nanodiscs in the study of membranes and membrane proteins. Biochemistry 46, 2059–2069 [DOI] [PubMed] [Google Scholar]
- 24. Bhat S., Sorci-Thomas M. G., Tuladhar R., Samuel M. P., and Thomas M. J. (2007) Conformational adaptation of apolipoprotein A-I to discretely sized phospholipid complexes. Biochemistry 46, 7811–7821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Shih A. Y., Sligar S. G., and Schulten K. (2008) Molecular models need to be tested: the case of a solar flares discoidal HDL model. Biophys. J. 94, L87–L89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Marty M. T., Zhang H., Cui W., Blankenship R. E., Gross M. L., and Sligar S. G. (2012) Native mass spectrometry characterization of intact nanodisc lipoprotein complexes. Anal. Chem. 84, 8957–8960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Phillips M. C. (2014) Molecular mechanisms of cellular cholesterol efflux. J. Biol. Chem. 289, 24020–24029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Mei X., and Atkinson D. (2011) Crystal structure of C-terminal truncated apolipoprotein A-I reveals the assembly of high density lipoprotein (HDL) by dimerization. J. Biol. Chem. 286, 38570–38582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Li L., Li S., Jones M. K., and Segrest J. P. (2012) Rotational and hinge dynamics of discoidal high density lipoproteins probed by interchain disulfide bond formation. Biochim. Biophys. Acta 1821, 481–489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Liu F., and Heck A. J. (2015) Interrogating the architecture of protein assemblies and protein interaction networks by cross-linking mass spectrometry. Curr. Opin. Struct. Biol. 35, 100–108 [DOI] [PubMed] [Google Scholar]
- 31. Leitner A., Faini M., Stengel F., and Aebersold R. (2016) Crosslinking and mass spectrometry: an integrated technology to understand the structure and function of molecular machines. Trends Biochem. Sci. 41, 20–32 [DOI] [PubMed] [Google Scholar]
- 32. Hage C., Iacobucci C., Rehkamp A., Arlt C., and Sinz A. (2017) The first zero-length mass spectrometry-cleavable cross-linker for protein structure analysis. Angew Chem. Int. Ed. Engl. 56, 14551–14555 [DOI] [PubMed] [Google Scholar]
- 33. Ryan R. O., Forte T. M., and Oda M. N. (2003) Optimized bacterial expression of human apolipoprotein A-I. Protein Expr. Purif. 27, 98–103 [DOI] [PubMed] [Google Scholar]
- 34. Cavigiolio G., Shao B., Geier E. G., Ren G., Heinecke J. W., and Oda M. N. (2008) The interplay between size, morphology, stability, and functionality of high-density lipoprotein subclasses†. Biochemistry 47, 4770–4779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ronsein G. E., Reyes-Soffer G., He Y., Oda M., Ginsberg H., and Heinecke J. W. (2016) Targeted proteomics identifies paraoxonase/arylesterase 1 (PON1) and apolipoprotein Cs as potential risk factors for hypoalphalipoproteinemia in diabetic subjects treated with fenofibrate and rosiglitazone. Mol. Cell. Proteomics 15, 1083–1093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hutchins P. M., Ronsein G. E., Monette J. S., Pamir N., Wimberger J., He Y., Anantharamaiah G. M., Kim D. S., Ranchalis J. E., Jarvik G. P., Vaisar T., and Heinecke J. W. (2014) Quantification of HDL particle concentration by calibrated ion mobility analysis. Clin. Chem. 60, 1393–1401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Rinner O., Seebacher J., Walzthoeni T., Mueller L. N., Beck M., Schmidt A., Mueller M., and Aebersold R. (2008) Identification of cross-linked peptides from large sequence databases. Nat. Methods 5, 315–318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Walzthoeni T., Claassen M., Leitner A., Herzog F., Bohn S., Forster F., Beck M., and Aebersold R. (2012) False discovery rate estimation for cross-linked peptides identified by mass spectrometry. Nat. Methods 9, 901–903 [DOI] [PubMed] [Google Scholar]
- 39. Gordon S. M., Deng J., Tomann A. B., Shah A. S., Lu L. J., and Davidson W. S. (2013) Multi-dimensional co-separation analysis reveals protein-protein interactions defining plasma lipoprotein subspecies. Mol. Cell. Proteomics 12, 3123–3134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Shah A. S., Tan L., Long J. L., and Davidson W. S. (2013) Proteomic diversity of high density lipoproteins: our emerging understanding of its importance in lipid transport and beyond. J. Lipid Res. 54, 2575–2585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hoopmann M. R., Zelter A., Johnson R. S., Riffle M., MacCoss M. J., Davis T. N., and Moritz R. L. (2015) Kojak: efficient analysis of chemically cross-linked protein complexes. J. Proteome Res. 14, 2190–2198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lima D. B., de Lima T. B., Balbuena T. S., Neves-Ferreira A. G. C., Barbosa V. C., Gozzo F. C., and Carvalho P. C. (2015) SIM-XL: A powerful and user-friendly tool for peptide cross-linking analysis. J. Proteomics 129, 51–55 [DOI] [PubMed] [Google Scholar]
- 43. Phillips J. C., Braun R., Wang W., Gumbart J., Tajkhorshid E., Villa E., Chipot C., Skeel R. D., Kale L., and Schulten K. (2005) Scalable molecular dynamics with NAMD. J. Comput. Chem. 26, 1781–1802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Best R. B., Zhu X., Shim J., Lopes P. E., Mittal J., Feig M., and Mackerell A. D. Jr. (2012) Optimization of the additive CHARMM all-atom protein force field targeting improved sampling of the backbone phi, psi and side-chain chi(1) and chi(2) dihedral angles. J. Chem. Theory Comput. 8, 3257–3273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Feller S. E., Zhang Y., Pastor R. W., and Brooks B. R. (1995) Constant pressure molecular dynamics simulation: The Langevin piston method. J. Chem. Phys. 103, 4613–4621 [Google Scholar]
- 46. Ryckaert J.-P., Ciccotti G., and Berendsen H. J. C. (1977) Numerical integration of the cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J. Computational Phys. 23, 327–341 [Google Scholar]
- 47. Miyamoto S., and Kollman P. A. (1992) Settle: An analytical version of the SHAKE and RATTLE algorithm for rigid water models. J. Computational Chem. 13, 952–962 [Google Scholar]
- 48. Darden T., York D., and Pedersen L. (1993) Particle mesh Ewald: An N·log(N) method for Ewald sums in large systems. J. Chem. Phys. 98, 10089–10092 [Google Scholar]
- 49. Hermanson G. T. (1996) Bioconjugate techniques. San Diego, Academic Press. [Google Scholar]
- 50. Pourmousa M., Song H. D., He Y., Heinecke J. W., Segrest J. P., and Pastor R. W. (2018) Tertiary structure of apolipoprotein A-I in nascent high-density lipoproteins. Proc. Natl. Acad. Sci. USA [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Segrest J. P., Jones M. K., Shao B., and Heinecke J. W. (2014) An experimentally robust model of monomeric apolipoprotein A-I created from a chimera of two X-ray structures and molecular dynamics simulations. Biochemistry 53, 7625–7640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Lima D. B., Melchior J. T., Morris J., Barbosa V. C., Chamot-Rooke J., Fioramonte M., Souza T., Fischer J. S. G., Gozzo F. C., Carvalho P. C., and Davidson W. S. (2018) Characterization of homodimer interfaces with cross-linking mass spectrometry and isotopically labeled proteins. Nat. Protoc. 13, 431–458 [DOI] [PubMed] [Google Scholar]
- 53. Silva R. A. G. D., Hilliard G. M., Li L., Segrest J. P., and Davidson W. S. (2005) A mass spectrometric determination of the conformation of dimeric apolipoprotein A-I in discoidal high density lipoproteins. Biochemistry 44, 8600–8607 [DOI] [PubMed] [Google Scholar]
- 54. Gu F., Jones M. K., Chen J., Patterson J. C., Catte A., Jerome W. G., Li L., and Segrest J. P. (2010) Structures of discoidal high density lipoproteins: a combined computational-experimental approach. J. Biol. Chem. 285, 4652–4665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Bashtovyy D., Jones M. K., Anantharamaiah G. M., and Segrest J. P. (2011) Sequence conservation of apolipoprotein A-I affords novel insights into HDL structure-function. J. Lipid Res. 52, 435–450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Brown J. H. (2006) Breaking symmetry in protein dimers: designs and functions. Protein Sci. 15, 1–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Goodsell D. S., and Olson A. J. (2000) Structural symmetry and protein function. Annu. Rev. Biophys. Biomol. Struct. 29, 105–153 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium (http://proteomecentral.proteomexchange.org) via the PRIDE partner repository with the dataset identifier PXD009927.