

Staphylococcus aureus invades bronchial epithelial cells to reach underlying lung tissue and to escape from the human immune defenses or antibiotic therapy. The internalized pathogen achieves these objectives by differentiation into growing and dormant subpopulations. Here we tracked the dynamic interactions between internalized bacteria and their host over four days by quantitative proteomics. The results highlight metabolic cross-talk between host and pathogen as a key driver for mutual adaptation and the outcome of infection.
Keywords: Energy metabolism, Infectious disease, Apoptosis*, Autophagy, Bacteria, Host-Pathogen Interaction, bronchial epithelial cells, in vivo proteomics, persister, population heterogeneity, Staphylococcus aureus
Graphical Abstract

Highlights
Interplay of epithelial cells and internalized S. aureus was dissected over 96 h.
Surviving host cells contain nonreplicating bacteria that persists in the cytoplasm.
Competition over resources triggers temporal metabolic changes.
Metabolic adaptation of host and bacteria determines the outcome of infection.
Abstract
Staphylococcus aureus is infamous for causing recurrent infections of the human respiratory tract. This is a consequence of its ability to adapt to different niches, including the intracellular milieu of lung epithelial cells. To understand the dynamic interplay between epithelial cells and the intracellular pathogen, we dissected their interactions over 4 days by mass spectrometry. Additionally, we investigated the dynamics of infection through live cell imaging, immunofluorescence and electron microscopy. The results highlight a major role of often overlooked temporal changes in the bacterial and host metabolism, triggered by fierce competition over limited resources. Remarkably, replicating bacteria reside predominantly within membrane-enclosed compartments and induce apoptosis of the host within ∼24 h post infection. Surviving infected host cells carry a subpopulation of non-replicating bacteria in the cytoplasm that persists. Altogether, we conclude that, besides the production of virulence factors by bacteria, it is the way in which intracellular resources are used, and how host and intracellular bacteria subsequently adapt to each other that determines the ultimate outcome of the infectious process.
Staphylococcus aureus is a Gram-positive opportunistic pathogen of humans, but also a commensal of the human body. Specifically, S. aureus is commonly found in the anterior nares of around 30% of the human population (1). Although most S. aureus carriers do not present any clinical symptoms, S. aureus can cause a wide range of diseases such as skin and soft tissue infections, osteomyelitis, septic arthritis and pneumonia (2, 3). This pathogen has gained notoriety in recent years because of its prevalence in nosocomial infections and the rise of methicillin-resistant S. aureus (MRSA) (3–5).
Although S. aureus often acts as an extracellular pathogen, it can evade immune responses and antibiotic therapy by entering human cells. The latter strategy is also used by the bacteria as a mechanism to spread to other tissues and both professional as well as non-professional phagocytic cells are used for internalization (6–8). After the bacteria have been taken up by the host cells, they will initially be localized in vesicles, which subsequently might fuse with lysosomes or be engulfed by an isolation membrane because of autophagy, and the bacteria inside them may prevail or escape into the cytosol. Although the internalization by host cells is potentially lethal for the bacteria, the survivors will have two options: proliferation or persistence. In the first case, the bacteria replicate intracellularly and subsequently induce lysis of the host cells. The released bacteria search for new host cells to be infected and spread into new tissues (6–8). In the second case, the persistent bacteria do not multiply, but adapt to the intracellular environment and may survive intracellularly without causing clinical symptoms for extended time periods. This pattern has been linked to relapse of infections or emergence of small colony variants (SCVs)1 of S. aureus which display reduced metabolic activity (9–11).
Despite strain-specific differences in overall virulence, all S. aureus strains, including laboratory strains, can display proliferative and persistent phenotypes. Although this phenomenon has been known (6), the actual adaptations either enabling active intracellular proliferation or reduced metabolic activity and persistence are still poorly understood. The precise outcome of the interplay between the bacterium and the host depends on the type of host cell involved and, perhaps most importantly, the physiological states of both parties (12, 13). The main challenge in obtaining a detailed understanding of the adaptive behavior of internalized S. aureus lies in the fact that it is essential to study quantitative changes over an extended period, not only in one of the two interacting parties but simultaneously in both. Previous studies have addressed these aspects only partially either by focusing on the internalized bacteria only, or over only short periods of time post infection (p.i.) (11, 12, 14–17). Yet, it is important to get the “complete picture” of such an infection scenario, because the invasion and destruction of lung epithelial cells is representative for some of the most serious staphylococcal diseases possible, especially necrotizing pneumonia.
The present study was designed to close the current knowledge gap on the interplay between S. aureus and lung epithelial cells by a time-resolved analysis of both parties over the longest possible period. The limits for such an analysis are set by the amount of material that can be extracted for bacteria- and host cell-specific analyses, and the parameters to be measured. This led us to a proteomics approach, where adaptations of the bronchial epithelial cell line 16HBE14o- and S. aureus were followed up to 4 days p.i. using a data independent acquisition (DIA) method. Importantly, our findings highlight dynamic adaptive changes, in both the host and the internalized pathogen, and describe the active cross-talk between them at different stages of infection. Additionally, we correlate these adaptations with the intracellular localization of the bacteria p.i. and the epithelial cells' response. The observations suggest that, after a period of violent conflict, both parties reach an equilibrium phase where they are apparently at peace and the bacteria have reached a persister status.
MATERIALS AND METHODS
Bacterial Strains
S. aureus strain HG001 (18) was used to perform all experiments. The bacteria carried plasmid pJL-sar-GFP to constitutively express the green fluorescent protein (GFP; Liese et al., 2013). For the immunostaining protocols, a spa mutant was used to prevent unspecific binding of marker antibodies to protein A. The HG001 Δspa strain was kindly provided by Dr. Jan Pané-Farré, University of Greifswald. Cultivation of bacteria was performed in prokaryotic minimal essential medium (pMEM): 1x MEM without sodium bicarbonate (Invitrogen, Karlsruhe, Germany) supplemented with 1x non-essential amino acids (PAN-Biotech GmbH, Aidenbach, Germany), 4 mm l-glutamine (PAN-Biotech GmbH), 10 mm HEPES (PAN-Biotech GmbH), 2 mm l-alanine, 2 mm l-leucine, 2 mm l-isoleucine, 2 mm l-valine, 2 mm l-aspartate, 2 mm l-glutamate, 2 mm l-serine, 2 mm l-threonine, 2 mm l-cysteine, 2 mm l-proline, 2 mm l-histidine, 2 mm l-phenylalanine and 2 mm l-tryptophan (All from Sigma-Aldrich, Schnelldorf, Germany), adjusted to pH 7.4 and sterilized by filtration. One day before the infection of epithelial cells, bacterial overnight cultures in pMEM supplemented with 0.01% yeast extract (Sigma-Aldrich) and 10 μg/ml erythromycin (Sigma-Aldrich) were prepared by serial dilutions (1×10−6 up to 1×10−10) of a 100 μl glycerol stock of a bacterial culture with an OD600 of 1.2. Incubation was performed at 37 °C and 220 rpm. The following day, the main culture was inoculated from an overnight culture with an OD600 between 0.3 to 0.8. The starting OD600 of the main culture was set to 0.05 and it was incubated for ∼2 h in a shaking water bath at 150 rpm and 37 °C until it reached the mid-exponential phase at an OD600 of ∼0.4 (supplemental Fig. S1). The bacteria were then harvested and used for preparation of the master mix for infection as explained below in the Internalization Experiments” paragraph.
Cell Line
The human epithelial cell line 16HBE14o- is a transformed bronchial epithelial cell line originally derived from a 1-year-old heart-lung transplant patient (20). This cell line is known for its ability to form tight junctions and to differentiate. The cells were cultured at 37 °C in 5% CO2 in eukaryotic minimal essential medium (eMEM): 1× MEM (Biochrom AG, Berlin, Germany) supplemented with 10% (v/v) fetal calf serum (FCS; Biochrom AG), 2% (v/v) l-glutamine 200 mm (PAN-Biotech GmbH) and 1% (v/v) nonessential amino acids 100x (PAN-Biotech GmbH). The splitting of cells was carried out every 3 days with 0.25% trypsin-EDTA (Gibco®, Grand Island, NY). After thawing of frozen stocks (in liquid N2) the cells were maintained for 20 additional passages. The cell lines stocks used are not authenticated.
Experimental Design and Statistical Rationale
Four independent biological replicates of the infection set-up were used for quantification of bacterial and host cell populations, and for mass spectrometry measurements. The number of replicates was selected to ensure that at every time point there were at least three consistent measurements for every protein. The sampling of each independent infection consisted of 8 samples taken over the course of 4 days, including a 0 h sample which is the control condition. In total, 32 samples of the cytosolic proteome of bacteria and 32 samples of the human bronchial epithelial cell proteome were measured. To avoid measuring replicates of the same condition sequentially, the measuring order of each set of samples was determined by assigning a random number between 1 and 32 to each sample (function sample in R version 3.4.4 (21)). Additionally, samples for imaging were collected from three independent infection experiments.
To determine changes over time in protein abundance, an empirical Bayes moderated F-test was conducted for each protein profile. This test also evaluates the similarity of the replicates. The moderated p values were corrected for multiple testing using Benjamini and Hochberg's multiple testing correction.
Internalization Experiments
Internalization experiments were performed essentially as described by Pförtner et al. (22). Briefly, internalization was performed using a confluent 16HBE14o- cell layer seeded at a density of 1×105 cells/cm2 in 12-well plates, 3 days before infection. The infection was carried out at a multiplicity of infection (MOI) of 25 bacteria per host cell. The master mix for infection was prepared from a mid-exponential (OD600 of 0.4) culture of S. aureus HG001 diluted in eMEM, buffered with 2.9 μl sodium hydrogen carbonate (7.5%, PAN-Biotech GmbH) per ml bacterial culture added. The growth medium over the confluent epithelial layer was replaced with the master mix, and the coculture was incubated for 1 h at 37 °C in 5% CO2. Afterward, the medium was collected (non-adherent sample) and replaced with eMEM medium containing 10 μg 47 ml of lysostaphin (AMBI Products LLC, Lawrence, NY). The medium was replaced every 2 days.
For collection of the proteome samples, the culturing medium was aspirated, and the epithelial cell layers were treated for 5 min at 37 °C with UT buffer (8 m urea, 2 m thiourea in MS-grade water; Sigma-Aldrich) to generate samples for analysis by mass spectrometry (MS). If samples were intended for the collection of bacteria, the disruption of epithelial cells was performed for 5 min at 37 °C in 0.05% sodium dodecyl sulfate (SDS; Carl Roth, Karlsruhe, Germany). Samples were collected at 2.5 h, 6.5 h, 24 h, 48 h, 72 h, and 96 h p.i.
To monitor changes in the abundance of human and bacterial cells, counting was performed at the times of sample collection. Epithelial cells were counted after staining with trypan blue dye using a Countess® system (Invitrogen). Quantification of intracellular bacteria and infected epithelial cells was performed with a Guava® easyCyte flow cytometer (Merck Millipore, Darmstadt, Germany) by excitation of the GFP with a 488 nm laser and detection at 510–540 nm.
Preparation of Proteome Samples
After disruption of epithelial cells with 0.05% SDS, two million liberated bacteria were sorted by flow cytometry using a FACSAria IIIu cell sorter (Becton Dickinson Biosciences, Franklin Lakes, NJ) per time point. The recognition of bacteria was carried out by excitation with a 488 nm laser and the emission was detected in the range of 515–545 nm. The bacterial cells were collected on low protein binding filter membranes with a pore size of 0.22 μm (Merck Millipore). These bacteria-containing filters were immediately placed in Eppendorf tubes that were then frozen by transferring them to a −20 °C freezer for the course of the experiment and then kept at −80 °C until use. The bacteria on the filter were lysed by incubation for 30 min at 37 °C with 7.4 μg/ml lysostaphin in 50 mm ammonium bicarbonate (Sigma-Aldrich) (23). Digestion of bacterial proteins on the filter was performed overnight at 37 °C with 0.1% Rapigest SF surfactant (Waters, Eschborn, Germany) and 0.3 μg of trypsin (Promega, Madison, WI).
For human proteome analyses, the protein content of samples was quantified using a Bradford assay (Bio-Rad, Hercules, CA). Four μg of protein per sample were prepared for MS measurements by reduction with 2.5 mmol/L dithiothreitol (Thermo Fisher Scientific, Idstein, Germany) for 1 h at 60 °C and alkylation with 10 mmol/L iodoacetamide (Sigma-Aldrich) for 30 min at 37 °C. Then, the samples were digested overnight with trypsin (protein/trypsin 25:1) at 37 °C.
The following 16HBE14o- samples were used for the construction of the spectral library of the host: a confluent cell layer cultured in a 10 cm dish for 3 days, an apoptotic cell layer in a 10 cm dish cultured for a week, non-polarized cells cultured for 3 days over Transwells®, and lastly polarized cells cultured for 11 days over Transwells® (Corning, Schnelldorf, Germany). The last two conditions were grown over 12 mm inserts with 0.4 μm pores, and with media volumes of 400 μl on the apical side and 1300 μl on the basal side of the cultures. Furthermore, to expand the host proteome library, published reads (24) of the bronchial epithelium cell line S9 were also used. These cells are immortalized cells isolated from a patient with cystic fibrosis that were transformed with a hybrid virus adeno-12-SV40 (ATCC® number CRL-2778) (25). For the construction of the host proteome spectral library, aliquots of the different samples of whole cell lysates of 16HBE14o- in UT buffer were mixed, and then 25 μg of the extract mixture was fractionated by SDS-PAGE. The gel was partitioned into ten protein-containing pieces that were destained by 15 min washes with ammonium bicarbonate solution (200 mm) in 50% acetonitrile (Mallinckrodt Baker, Inc., Deventer, Netherlands) at 37 °C and 500 rpm. Then, the gel pieces were dehydrated by incubation with acetonitrile at 37 °C and 500 rpm. The supernatant was discarded afterward. Proteins in each gel piece were in-gel digested overnight at 37 °C with 20 μl of trypsin (10 ng/μl) and 30 μl ammonium bicarbonate solution (20 mm). Lastly, the peptides were extracted by addition of 0.1% acetic acid (Carl Roth) and incubation in an ultrasound bath for 30 min. Afterward, the supernatant was collected, 50% acetonitrile with 0.05% acetic acid were added to the gel pieces for another 30 min incubation, and both supernatant fractions were united. Two of the supernatants of the ten SDS-PAGE fractions were mixed to generate five final samples with essentially the same protein quantity, which were then used for further processing and DDA-measurements.
The tryptic peptides derived from bacterial or human proteins were concentrated and purified using C18 ZipTip columns (Merck Millipore). All samples were resuspended in a buffer consisting of 2% acetonitrile and 0.1% acetic acid in MS-grade water. Indexed Retention Time (iRT) peptides (Biognosys AG, Schlieren, Switzerland) were added to the samples for feature alignment, peak calibration and signal quantification. The spike in of the samples was carried out according to the manufacturer′s instructions assuring that the injected volumes have one IE (injection equivalent) of iRT peptide mix. The final volume of the samples and the injection volumes were set to 12 μl and 10 μl for S. aureus samples, 20 μl and 5 μl for 16HBE14o- samples, and 20 μl and 4 μl for the spectral library samples, respectively.
Mass Spectrometry Measurements
Tryptic peptides were separated on an Accucore 150-C18 analytical column of 250 mm (25 cm × 75 μm, 2.6 μm C18 particles, 150 Å pore size, Thermo Fischer Scientific, Waltham, MA) using a Dionex Ultimate 3000 nano-LC system (Thermo Fischer Scientific). Peptides were eluted at a constant temperature of 40 °C and a flow rate of 300 nL/min with a 120 min linear gradient (2% to 25%) of buffer (acetonitrile in 0.1% acetic acid).
To design a spectral library MS/MS data were recorded on a Q Exactive mass spectrometer (Thermo Fischer Scientific) in data dependent mode (DDA). The MS scans were carried out in a m/z range of 300 to 1650 m/z. Data was acquired with a resolution of 70,000 and an AGC target of 3×106. The top 10 most abundant isotope patterns with charge ≥2 from the survey scan were selected for fragmentation by high energy collisional dissociation (HCD) with a maximal injection time of 120 ms, an isolation window of 3 m/z, and a normalized collision energy of 27.5 eV. Dynamic exclusion was set to 30 s. The MS/MS scans had a resolution of 17,500 and an AGC target of 2×105.
MS/MS analyses of samples were performed in data independent mode (DIA) on a Q Exactive Plus mass spectrometer (Thermo Fischer Scientific) following the method described by Bruderer et al. (26). Briefly, the data was acquired in the m/z range from 400 to 1220 m/z, the resolution for MS and MS/MS was 35,000, and the AGC target was 5×106 for MS, and 3×106 for MS/MS. The number of DIA isolation windows was 19 with 2 m/z overlap. For further details to the instrumental setup and the parameters for LC-MS/MS analysis in DDA and DIA mode see supplemental Tables S1 and S2.
Immunofluorescent Confocal Microscopy
Time-lapse imaging was carried out with a DeltaVisionRT deconvolution microscope (GE Healthcare Europe GmbH, Freiburg im Breisgau, Germany). To perform the imaging, the actual infection experiment was carried out on a glass bottom 35-mm plate (MatTek, Ashland, MA). After a change of media with lysostaphin, the plate was transferred to the microscope base under incubation conditions. Imaging of the epithelial layer was performed by light microscopy, whereas GFP fluorescent bacteria were observed by excitation with a 490/20 nm mercury vapor lamp and detection of fluorescence at 528/38 nm. Image acquisition was performed every 5 min for the first 48 h, then from 48 h to 72 h and finally from 92 h to 96 h. Picture processing was performed with Fiji (http://fiji.sc/Fiji).
Subcellular localization of microtubule-associated protein 1A/1B-light chain (LC3) and lysosomal-associated membrane protein 1 (LAMP-1) by immunofluorescence microscopy was performed using a Leica TCS SP8 Confocal laser scanning microscope (Leica Microsystems B.V., Amsterdam, Netherlands). The cells were seeded over coverslips of 18 mm diameter 3 days before infection as described above. However, in this case a HG001 Δspa mutant was used to avoid aspecific IgG binding. The samples were collected at 0 h, 1 h, 2.5 h, 6.5 h, 24 h, 48 h, 72 h, and 96 h by fixation with 2% para-formaldehyde (PFA, Merck Millipore) for 20 min at room temperature. Preparation of the samples for the actual microscopy was performed simultaneously after conclusion of the experiment. The samples were permeabilized with 0.5% Tween 20 (Sigma-Aldrich) for 30 min at room temperature and then nonspecific binging sites were blocked with 1% bovine serum albumin, 10% FCS in 0.07% Tween 20 for 120 min at room temperature. All antibodies were diluted in blocking solution. Primary rabbit anti-LC3B (Cat. No. 1384; Novus Biologicals, Oxon, England) and mouse CD107a (LAMP-1; Cat. No. 555798; BD, Drachten, Netherlands) antibodies were used at 1:500 and 1:100 dilutions, respectively. The incubation was carried out simultaneously for 1 h at room temperature in a humidified chamber. The secondary Goat anti-rabbit antibody conjugated with Alexa Fluor 594 (A11012; Invitrogen) and goat anti-mouse antibody conjugated with Alexa Fluor 647 (A-21236; Invitrogen) were used, both, at a 1:500 dilution with incubation for 1 h at room temperature. Lastly, the DNA was stained with 4′,6-diamidino-2-phenylindole (DAPI), the slides were mounted with Mowiol® 4–88 mounting medium (EMD Chemical, Inc., Temecula, CA) and stored at −20 °C until microscopic visualization.
Transmission Electron Microscopy (TEM)
After the invasion assay, 16HBE14o- cells were fixed with 0.2% glutaraldehyde Polyscience, Inc.,Warrington, PA and 2% PFA in 0.1 m sodium cacodylate buffer (pH 7.4; Sigma-Aldrich) for 10 min. Subsequently, the fixative solution was replaced with new fixative solution and incubation was continued for 30 min at room temperature. The cells were rinsed twice for 5 min each in 0.1 m cacodylate buffer at room temperature followed by post-fixation in 1% Osmium tetroxide (Electron Microscopy Sciences, Hatfield, PA)/1.5% potassium ferrocyanide (Merck Millipore) in 0.1 m sodium cacodylate at 4 °C for 30 min. The 16HBE14o- cells were then incubated with 1% tannic acid in 0.05 m of sodium cacodylate buffer for 5 min to enhance the color of the 16HBE14o- cell membranes and demonstrate the internalization of S. aureus. The cells were washed with Milli-Q water, dehydrated through serial incubation in graded ethanol (30%, 50%, 70%, and 100%) and lastly embedded in EPON resin (Hexion, Columbus, OH). Ultrathin sections (80 nm) were cut with an UC7 ultramicrotome (Leica, Vienna, Austria) and contrasted using 5% uranyl acetate for 20 min, followed by Reynolds lead citrate for 2 min. Images were recorded with a FEI CM100 transmission electron microscope operated at 80 KV using a Morada digital camera.
Identification and Quantification of Proteins
Human proteins were identified using SpectronautTM Pulsar 11 (v11.0.18108.11.30271) software (Biognosys AG) against a human bronchial epithelial cell line generated from data-dependent acquisition measurements of 16HBE14o- samples and 12 additional data sets of the cell line S9 that were previously published (24). The spectral library construction was based on a Comet database search using a human protein database in a target-decoy approach using the trans-proteomic-pipeline (TPP) version 4.8.0 PHILAE (27, 28). The raw files were converted to mzML files with msconvert (Proteowizard version 3.0.11537; November 31, 2017) using a vendor peak picker on spectra with MS level 1–2. Then, the mzML files were searched with the Comet search engine (2014.02 rev. 0) against a human data base that comprised 20.217 Uniprot-reviewed entries (February 2018), 102 cRAP common contaminants entries (https://www.thegpm.org/crap/) and 1 entry for the concadenated iRT peptides. The target-decoy version of this database was generated by adding all reverse entries resulting in 40.640 entries in total. The target-decoy search was performed with a parent mass error of ±20 ppm, fragment mass error of 0.01 Da, and allowing full-tryptic peptides (trypsin/P cleavage rule) with up to two internal cleavage sites. The search included fixed modification of +57.021464 for carbamidomethylated cysteine and variable modification of +15.9949 for oxidized methionine. The search results were scored using PeptideProphet (29) and iProphet (30) with a minimal peptide length of 7 amino acids. The global protein FDR calculation of 0.01 was assessed with MAYU (31). The filtered peptide spectrum matches were used to generate the ion library in SpectronautTM v11.0.15038.14.27660 (Asimov; Biognosys AG, Switzerland) setting the m/z mass range from 300 to 1.800, 6 to 10 fragments per peptide, removing fragments smaller than 3 amino acids, no segmented regression, and a minimum root mean square error of 0.5.
The S. aureus ion library used in this study was generated previously (32). Briefly, the data sets used for the construction of this library comprise samples of the cytosolic and exoproteomes of S. aureus HG001 and the isogenic Δrho mutant ST1258. This ion library was constructed with a similar protocol as the one applied for the host library. The Comet database search was based on the fasta file from AureoWiki (33). This database comprises 2852 S. aureus protein entries. The final database used for the target-decoy approach contains 5944 entries in total (including cRAP contaminats). The peptide spectral matching was performed using the trypsin/P digest rule with a number of tolerable termini (NTT) of 2, a parent mass error of ±30 ppm, fragment mass tolerance of 0.01 Da, variable modification of +15.9949 for methionine oxidation and fixed modification of +57.021464 for carbamidomethylation (sample preparation-dependent). The generation of the ion library in SpectronautTM v11.0.15038.14.27660 resulted in a constructed library consisting of 2154 proteins with 38,570 tryptic peptides.
The Spectronaut DIA-MS analysis was carried out using dynamic MS1 and MS2 mass tolerance, dynamic XIC RT extraction window, automatic calibration, dynamic decoy strategy (library size factor = 0.1, minimum limit = 5000), protein Q-value cutoff of 0.01, precursor Q-value cutoff of 0.001. The search included variable modifications of +15.9949 for oxidized methionine, and if reduction and alkylation was used, fixed modifications of +57.021464 for carbamidomethylated cysteine. A local cross run normalization was performed using complete profiles with a Q-value < 0.001. The MS2 peak area was quantified and reported. Missing values were parsed using an iRT profiling strategy with carry-over of exact peak boundaries (minimum Q-value row selection = 0.001). Only nonidentified precursors were parsed with a Q-value > 0.0001. Ion values were parsed when at least 25% of the samples contained high quality measured values. The settings for the SpectronautTM analyses are available in supplemental Table S3. Afterward, parsed values were filtered out again, if they were more than 2-fold higher than the measured values preventing false positives because of parsing. For further analyses proteins with at least two identified peptides were considered. The identified proteins were annotated based on the Uniprot database and the gene names for S. aureus were extracted from AureoWiki (33).
Statistical Testing of Changes in Protein Abundances Over Time
Peptide intensities were normalized by the mean of all time points. The time-course data of each protein were then calculated as the median of all normalized peptide data belonging to that protein. Subsequently, the protein data were analyzed using the LIMMA package version 3.34.9 (34) in R version 3.4.4 (21). First, a natural regression spline with four degrees of freedom was fitted for each protein, considering that expression changes smoothly over time. Then, a linear model (35) with eight parameters was fitted to the data, including an intercept, four spline parameters and three parameters corresponding to comparability of the four replicates. An empirical Bayes moderated F-test was conducted for each protein, which can detect very general changes over time while simultaneously testing for similarity of the replicates. Because of general differences in replicate 1 data at 6.5 h and 24 h, these 16HBE14o- cell samples were excluded for the testing only, but not for calculating the median values. The moderated p values were corrected for multiple testing using Benjamini and Hochberg's multiple testing correction. Finally, proteins with an adjusted p value below α = 0.01 were assumed to be significantly changed during the time course of infection. For further analyses, the data were averaged over the replicates using the median for each protein and shifted to start in zero at time point 0 h.
Afterward the time course data of all significantly changed proteins were clustered using a noise-robust soft-clustering (fuzzy c-means) implemented in the Mfuzz package version 2.38.0 (36, 37) in R version 3.4.4 (21). Because the clustering is performed in Euclidian space, the expression values of proteins were standardized for clustering only to have a standard deviation of 1, so that only the dynamics were considered. The optimal number of clusters was chosen using both a cluster validity index (minimum centroid distance) and a manual validation of biologically meaningful groupings.
RESULTS
S. aureus Population Displays a Dormant State After 48 h p.i. in Human Epithelial Cells
To analyze physiological changes in S. aureus internalized by 16HBE14o- lung epithelial cells over an extended period of time, we applied a previously established infection assay (22) and assessed the longest time span over which bacteria could be reproducibly isolated from the infected cells in sufficient amounts to enable proteome profiling. Pilot experiments showed that this is possible for at least 96 h, which was therefore, selected as the end point for all further analyses. During these 96 h, the fate of the infected cells and the internalized bacteria was recorded through time-lapse microscopy as shown in the three Supplemental movies and illustrated through snapshots in Fig. 1A. Additionally, the abundance of the bacterial population during infection was measured by quantification of the mean fluorescence intensity (Fig. 1B). These data show that the internalized bacteria do not multiply significantly during the first 8 h p.i. Subsequently, most bacteria start to proliferate leading to lysis of their respective host cells. Of note, in the applied experimental setup, host cell lysis is suicidal for the bacteria because of the presence of lysostaphin in the medium. Accordingly, the liberated bacteria disappear and cannot reinfect other cells. After ∼30 h p.i., nonreplicating bacteria remain detectable within the surviving host cells until the end of the experiment at 96 h p.i. These findings were corroborated by flow cytometric counting of the bacterial numbers (Fig. 1C). Notably, two distinctive bacterial phenotypes were observed over time, namely one involving high rates of intracellular bacterial replication and another one involving internalized bacteria in a dormant state. Further, although the number of host cells remains essentially constant during the first 24 h p.i., this number increases during later stages (Fig. 1C). The percentage of host cells that contain bacteria gradually decreases over time, starting with a maximum of ∼27% at 2.5 h p.i. and ending with ∼13% at 96 h p.i.
Fig. 1.

After internalization two subpopulations of S. aureus can be distinguished by differences in replication rate. The progression of the infection was followed by time-lapse microscopy (A; Supplemental videos; scale bar = 20 μm) and the GFP fluorescence intensity was quantified to elucidate the dynamics of the bacterial population (B). Most of the bacteria replicate intracellularly during the first day of infection, but a secondary subpopulation remains in a dormant state during the whole time of observation. The red dots indicate the time points of sample collection for further experiments. Counting of bacterial cells, and infected and uninfected host cells, as jointly presented in (C) confirmed the changes in both populations. Average values of four replicates are presented (B-C). Mass spectrometry quantification of ribosomal proteins of the host (D) or S. aureus (E). The list of proteins presented in the line plots is available in supplemental Table S8. The levels of the proteins were calculated based on the mid-exponential phase or the 0 h time point as reference, for the bacteria and the host, respectively. The SaHPF protein detected in higher amounts is colored in red.
As microscopy and flow cytometry cannot reveal the intricacies of the processes occurring in host cells and internalized bacteria, we employed quantitative proteomics profiling. This approach has the advantage of quantifying many different proteins belonging to the adaptive processes in bacteria and the human host cells with good reproducibility. Thus, changes in each protein's abundance can be followed over time and compared. Through data-independent acquisition, application of an in-house built S. aureus HG001 spectral library (24, 32) and human cell line spectral libraries, we achieved a complete data collection for 3644 human and 930 staphylococcal proteins at all time points during 96 h p.i. (supplemental Tables S6 and S7). Because the bacterial population displays two different phenotypes with different prominence at the different stages of infection, the observed proteome dynamics describe the adaptive changes in the bacteria at each stage and the final “shape” of the persistent bacteria. Likewise, these measurements record the influence of the intracellular bacteria on the host's physiological state over time. To determine the weight of the observed changes, the generated bacterial and host proteome data were fitted to spline lines and tested against constant linearity. This linearity test considers the replicates' variability, rendering it an indicator of the reliability of the observed changes. Furthermore, all proteins that show significant changes were clustered depending on their behavior over time, thereby marking the stages p.i. at which important changes take place (supplemental Fig. S2).
The differences in bacterial and host replication rates as observed by microscopy are mirrored in the abundance of the respective ribosomal proteins (Fig. 1D–1E). Consistent with their unimpaired growth, the host cells do not show major regulatory changes in ribosomal protein abundance over the time course of infection. In contrast, the amounts of bacterial proteins that make up the small and large ribosomal subunits start to decrease after internalization, being detectable at substantially lowered amounts at 48 h p.i. This time point correlates with the absence of bacterial growth observed in the live cell imaging. An exception to this general trend is the “Staphylococcus aureus hibernation promotion factor” (SaHPF), which shows significantly increased levels right from the start of infection (Fig. 1E). This protein represses translation, and therefore, it is important for survival in the stationary phase and during conditions of nutrient deprivation (38, 39).
Nutrient Competition Affects Primarily S. aureus, but Staphylococcal Persistence Triggers Changes in Host Metabolism at 48 h p.i.
The acquisition of metabolites and their subsequent conversion to cellular constituents is a fundamental feature of all living organisms. This implies that different organisms residing in the same system are likely to compete for nutrients. Accordingly, internalized S. aureus will compete with the host for carbon sources. This is clearly reflected in the proteins required for central carbon metabolism, both in the bacteria and the host. At first instance, the proteins related to the bacterial glycolytic pathway remain fairly stable after internalization displaying similar levels as encountered in exponentially growing bacteria (Fig. 2). Yet, at 6.5 h p.i., the pathogen displays changes in the levels of most of the proteins related to the glucose activation phase. Correspondingly, the host presents similar changes, but only from 48 h p.i. onwards, which coincides with transition of the S. aureus population into a dormant state. Remarkably, during the entire course of infection, neither the pathogen nor its host regulates proteins related to the production of acetyl-CoA from pyruvate. During nutrient-rich conditions, S. aureus will produce acetate from acetyl-coA, thereby providing the NAD+ necessary for the glycolysis process (40). Our analysis shows that, after internalization, the bacteria repress some of the respective enzymes, but this regulation is reversed 48 h p.i., suggesting that the dormant bacteria restart the production of acetate. Proteins involved in the TCA cycle do not mirror this behavior, displaying increased levels after internalization and remaining at a constant increased level over the whole duration of the experiment. This strategy would supply the bacteria with enough metabolic energy under conditions of low glucose availability. The strongest level increase takes place around 24 h p.i., which corresponds to the transition in subpopulation predominance from the replicating to the dormant phenotype of the bacteria. The two proteins of this pathway that do not display increased levels (SAOUHSC_01266, and SAOUHSC_01267) are 2-oxoglutarate oxidoreductases, which participate in the reverse TCA cycle and synthetize 2-ketoglutarate. In contrast to the bacteria, most TCA cycle proteins from the human host cells show no changes in abundance, except for those involved in the metabolism of fumarate and α-ketoglutarate. Of note, these compounds are key intermediates in amino acid metabolism.
Fig. 2.

Quantification of proteins related to the central carbon metabolism. Proteins were grouped depending on their main function in the different pathways, and boxes mark selected central metabolites. The proteome data from S. aureus are depicted on the left side of the diagram and the proteins without assigned gene symbol are labeled according to their locus tag without the “SAOUHSC_” identifier. The host data are shown on the right side of the diagram. Protein trends that deviate (p value <0.01) from constant linearity over time were fitted to clusters according to their behavior (supplemental Fig. S2). Clustering assignment is shown in the last column “C.” The color coding is based on the standard deviation of each set of data. σS. aureus = 2.34 and σHBE = 0.33. The ratios were obtained from the measurements of four biological replicates.
After the first 2.5 h p.i., the bacteria activate pathways related to energy acquisition from alternative sources, like fatty acids, as can be inferred from the elevated levels of S. aureus proteins involved in the catabolism of glycerol and fatty acids (Fig. 3). This increased level of fatty acid degrading enzymes becomes even stronger after the bacteria are predominantly in a dormant state, suggesting that the persistent bacteria prefer fatty acids over glycerol. This effect is not observed in the host proteome, where the levels of most proteins related to fatty acid degradation do not show major modifications.
Fig. 3.

Proteins related to alternative carbon sources and respiration. Assignment of proteins to pathogen (S. aureus) and host (HBE) is indicated on top of the respective protein groups. Because of limited carbon sources post infection, alternative pathways for energy consumption are upregulated. Substrates like glycerol and fatty acids are being consumed during intracellular conditions. Another major adjustment is related to oxygen availability which affects the pathways related to fermentation and oxidative phosphorylation. The protein quantities were derived from the mass spectrometry measurements of four biological replicates. Time trends with significant changes (p value < 0.01) were fitted into different clusters depending on their behavior (supplemental Fig. S2). The assigned cluster is presented in the column “C.” The color coding is based on the standard deviation of each set of data. σS. aureus = 2.34 and σHBE = 0.33. HIF: Hypoxia-Inducible Factor.
Oxygen is a key factor limiting bacterial metabolism during infection because of the microaerobic environment they are exposed to after internalization (13, 17). This is clearly reflected by the mild increase in S. aureus proteins related to fermentation after internalization (Fig. 3), but the major rise takes place after 24 h p.i. On the other hand, most proteins related to the electron transport chain present a peak of upregulation at 24 h p.i., after which their abundance decreases. Conversely, the host proteome does not show major changes regarding the possible competition for oxygen. Only four proteins related to fermentation and oxidative phosphorylation present some regulation (Fig. 3). In addition, the proteins Elongin-B (ELOB), Elongin-C (ELOC), and Cullin-5 (CUL2), which are involved in the degradation of the Hypoxia-Inducible Factor (HIFα) remain constant over time, indicating that there is no increase in the abundance of this factor and suggesting that the host cells do not perceive a reduction of the available oxygen.
Amino acids are major alternative sources for carbon and nitrogen, which may play a role in adaptations of a pathogen to the intracellular milieu. Arginine, asparagine, and tryptophan have gained interest because of their relevance in the host's and bacterial defense mechanisms (41, 42). The pathways for biosynthesis and degradation of amino acids are complex because they are interconnected. Nevertheless, the proteomics data purport that S. aureus employs most pathways needed for amino acid metabolism during the entire course of infection. Conversely, the host proteome is geared toward utilization of amino acids that are primarily linked to the TCA cycle (Fig. 4).
Fig. 4.

Progression of the pathways related to amino acid metabolism. Assignment of proteins to pathogen (S. aureus) and host (HBE) is indicated on top of the respective protein groups. The catabolism of amino acids after infection would provide resources to compensate for the lack of carbon and nitrogen sources. Moreover, the anabolism of these molecules is likewise required for some defensive functions of either the bacteria or the host. The represented ratios are the average of four biological replicates. Proteins with significant changes (p value <0.01) were clustered in groups depending on their general behavior and their assigned cluster is showed in column “C.” The color coding is based on the standard deviation of each set of data. σS. aureus = 2.34 and σHBE = 0.33.
Bacterial enzymes linked to the degradation of histidine, serine, cysteine and threonine show increased levels right from the moment of internalization, even preceding the start of intracellular replication. These proteins are related directly with the generation of precursors of pyruvate and the one-carbon metabolism. Nevertheless, because there is no replication of bacteria after 48 h p.i., the degradation of histidine, serine, cysteine, and threonine is most likely used to access an alternative carbon source. On the contrary, the infected host cells induce the GLYM protein at later stages of infection, which is directly related to the conversion of serine into glycine and 5,10-methylenetetrahydrofolate. The latter molecule is an indispensable building block for purine biosynthesis (43, 44). This finding is fully consistent with the continuing growth of the human cell layer once the internalized pathogen predominantly presents a state of dormancy after 48 h p.i.
Branched-chain amino acids (BCAAs; Valine, Leucine, Isoleucine) are essential for human cells and, thus, need to be ingested from the environment. Consequently, the human proteins handling these amino acids are all involved in their degradation. Only at the last time point p.i. the human proteins addressing BCAAs have slightly increased levels (Fig. 4). On the other hand, the BCAAs play a crucial role in the regulation of S. aureus metabolism, because they serve as cofactors for CodY. This regulator also represses the production of the IlvA1, IlvE, and IlvC proteins, which have all increased abundance to participate in the biosynthesis of BCAAs (45). In parallel, the observed upregulation of the Ald1 and Ald2 proteins involved in the degradation of alanine implies that also this amino acid is used as an alternative carbon source during infection.
The amino acid catabolic pathways related to the TCA cycle are mostly needed to generate the intermediate molecules α-ketoglutarate, oxaloacetate and fumarate. S. aureus increases levels of proteins from the urea cycle (RocADF; ArcAC) to degrade arginine and to produce α-ketoglutarate from glutamate (GudB). Therefore, these substrates are used to feed the TCA cycle. Of note, the degradation of arginine is carried out by two pathways. First, the arginase pathway will produce glutamate (46) and presents upregulation right from the beginning of the infection (RocADF). Similarly, the arginine deaminase pathway (ArcAC), converting arginine to ornithine (47), expresses higher levels from the beginning of the infection but its highest quantities are found in the dormant bacterial population. Further, upregulated proteins associated with aspartate metabolism are dedicated to the production of lysine, as was also observed by Michalik et al. and Surmann et al. (13, 24). In contrast to the internalized bacteria, the amino acid metabolism in the infected host cells is geared toward the biosynthesis of amino acids for protein production. Specifically, the glutamate and aspartate metabolic pathways are upregulated in relation to the formation of collagen (PLOD2, PLOD3, P4HA1, P4HA2), and the synthesis of asparagine (ASNS), l-proline (OAT, P5CR1) and spermidine (SPEE). With exception of ASNS and SPEE, all these regulatory events start to take place after 48 h p.i. with a constant increment.
Staphylococcus aureus Escapes Degradative Compartments, Leading to a Persistent Population Residing in the Cytosol
Because S. aureus can reside in different intracellular niches, we determined the subcellular location of the bacteria over time through confocal immunofluorescence microscopy (Fig. 5A; supplemental Fig. S3). Inspection of the colocalization of the GFP-positive bacteria with LAMP-1 and LC3 revealed that, during the first 24 h p.i., most of the bacteria are located inside membranes. Specifically, the replicating population resides inside LAMP-1- or LC3-associated compartments, whereas individual bacteria or small bacterial clusters are detectable in the cytosol from 2.5 h p.i. with increasing frequency over time. Ultimately, at the last time points, most observed bacteria do not colocalize with the membrane markers, suggesting a cytosolic localization of the persistent subpopulation. To substantiate these findings, we performed transmission electron microscopy (Fig. 5B; supplemental Fig. S4), highlighting that the replicating bacteria localize inside so-called “light phagosomal compartments” or “dark degradative compartments,” which are possibly lysosomes, during the first 48 h p.i. Further, at 72 h and 96 h p.i. all bacteria are no longer enclosed by a membrane, ultimately showing that the persister population resides in the cytosol. Of note, all bacterial clusters found inside phagocytic membranes are in dying host cells (Fig. 5B, supplemental Fig. S4).
Fig. 5.

The persistent subpopulation of S. aureus is mostly found in the cytosolic environment of the host. Intracellular localization of S. aureus was examined by colocalization with the protein markers LC3 for phagosomes, and LAMP-1 for lysosomes (A; scale bar: 20 μm). After internalization of S. aureus by the HBE cells, most of the bacterial population is located inside closed compartments. Still, the percentage of bacteria that escapes the vesicles (white arrow heads) increases over time and most of the bacteria are found in a cytosolic environment by the end of the infection. These results were corroborated by electron microscopy (B; scale bar = 2 μm). During the first hours of infection most bacteria are located inside degradative vesicles (dark compartments) or phagosomes (light compartments), but single bacteria escaped the closed compartments as early as 2.5 h p.i. (supplemental Fig. S4). By the end of the time of observation, at 72 h and 96 h p.i., all observed bacterial clusters are cytosolic. The displayed images are representative of different time points, additional images are provided in supplemental Figs. S3 and S4. The escape from the compartments could be induced by proteins related to pathogenesis, including toxins regulated by Agr and SaeRS (C). Moreover, this escape might have an impact on the production of stress proteins related to oxidative stress (D) and DNA repair (E). The list of proteins included in the line plots is available in the supplemental Table S8. A selection of significantly (p value<0.01) regulated proteins is displayed in orange and blue. The mass spectrometry data represents the average of four biological replicates.
Considering that the escape of S. aureus from the vesicles relies on toxins (48), we inspected the possible presence of staphylococcal virulence factors. Consistent with the observed changes in the subcellular localization of bacteria, the toxins HlgB, HlgC, Hla, LukH, and LukG are upregulated after 24 h p.i. (Fig. 5C; supplemental Table S8). These pore-forming proteins have been correlated with escape and subsequent host cell lysis (48). Of note, all these toxins are regulated by the Agr and/or SaeSR systems that positively or negatively regulate many more proteins involved in pathogenesis (49). Accordingly, the surface-bound protein Spa, which is negatively regulated by Agr, is present in decreased amounts after internalization with a minimum at 6.5 h p.i. A similar but less pronounced trend is observed for the surface-bound proteins ClfA and ClfB, the expression of which relies also on other regulators, such as the sigma factor B (SigB) in case of ClfA.
As the physicochemical conditions within different subcellular compartments differ substantially, bacteria within these compartments need to respond appropriately to different stresses. This is particularly true for lysosomes and phagosomes, which represent acidic compartments. In addition, the phagosomes are known to produce massive amounts of reactive oxygen species (ROS). Such conditions have a strong propensity to damage bacterial macromolecules, such as DNA and proteins. Indeed, the levels of many bacterial proteins involved in the prevention of oxidative damage are altered over time. These include KatA, SodA, SodM, AhpC, GapB, Hmp, and Dps (Fig. 5D; supplemental Table S8). In particular, the latter three proteins present a strong upregulation. GapB increases the production of NADPH, which is needed to keep antioxidant proteins in a reduced state to allow them to reduce ROS and keep cytoplasmic proteins in a reduced state. The nitric oxide dioxygenase Hmp is involved in NO detoxification (50), whereas the ferroxidase Dps prevents DNA damage by binding iron and thus inhibiting hydroxyl radical formation (51). Of note, the upregulation of these proteins is triggered at 24 h p.i., a time point at which the growing bacteria are escaping from the cells (Fig. 1A, 1B) and the shift toward dormant/latent cells is taking place. Interestingly, the levels of these proteins remain high at later time points p.i. Similarly, proteins involved in the response to DNA damage, XseA and AddA, are upregulated after internalization (Fig. 5E), whereas other DNA damage-inducible proteins such as UvrB and UvrB2 are upregulated after 24 h. These observations imply that the bacteria are probably exposed to ROS and reactive nitrogen species (RNS) until 96 h p.i. and, remarkably, that this upregulation is not specifically related to the compartment in which they reside.
Intracellular S. aureus Persisters Induce a Nonapoptotic Reaction of the Host
During the live cell imaging, it was observed that host cells lyse as a consequence of the replication of intracellular bacteria. This raised the question whether any indicators related to the mode of cell death could be identified. Indeed, the host cell proteome showed strong regulation of particular proteins implicated in cell death (Fig. 6). Of note, most regulators involved in the so-called apoptotic and pyroptotic pathways are kinases, which require activation to perform their functions, therefore their abundance does not correlate with host responses. Consequently, we investigated the cell death mechanisms by looking at other modulators of these pathways not requiring activation. These include the BAX, BAD, DBLOH, UACA, APAF, and ACS proteins that promote the initiation of apoptosis by stimulating caspase production. However, most of these proteins present no regulation, likely leading to the observed mild abundance changes of pro-apoptotic caspases CASP3, CASP7, and CASP8. However, CASP3 and CASP8 still display moderately increased abundance at early time points until 24 h p.i., hinting at potential apoptotic events occurring during bacterial replication. CASP6, on the other hand, is upregulated during the entire course of infection. Another pathway that can trigger apoptosis of the host cells depends on activation of P53. Its respective activators (BAG6, DDX5) are upregulated at early time points p.i., but their abundance decreases at 48 h p.i. On the other hand, anti-apoptotic proteins, such as TFIP8, 2CL1, NOC2L and BNIP2, display upregulation after infection (Fig. 6). Consistent with the latter observation, the Calpain 1 and 2 proteins, which are regulated in response to apoptosis, show no altered levels over the entire time course of infection. Lastly, it is noteworthy that CASP4 is the only caspase that shows consistent upregulation post internalization. This protein is related with the activation of the inflammasome or pyroptosis. Another protein involved in this pathway is NF-kB, whose upregulation appears synchronized with CASP4. Nonetheless, so-called pro-NF-kB proteins like TF65, ARHG2, TRADD and TRAF2 show no significant regulation. Lastly, proteins related to the activation of the inflammasome on the canonical or non-canonical pathways (52) were present at lower levels at the end of the infection, starting at 48 h p.i., indicating that these pathways are not activated. Altogether, these observations are consistent with apoptotic events that occur exclusively during the early stages of infection, when the bacteria still replicate.
Fig. 6.
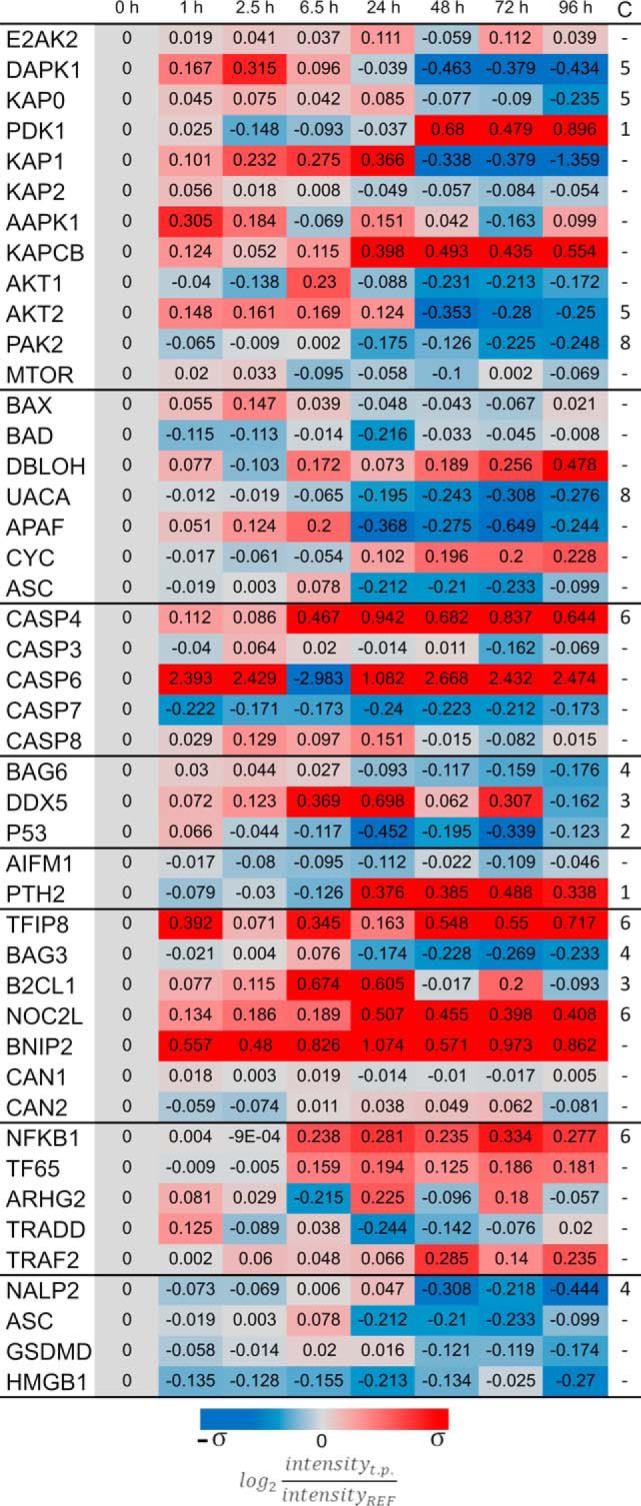
Death of the host cells is not caused by induction of the classical apoptotic pathway. Mass spectrometry data of human proteins associated with different apoptosis-related pathways are presented in the figure. From top to bottom the presented groups of proteins include: kinases, regulators of caspases, the caspases, activators of P53, proteins related to caspase-independent activation of apoptosis, anti-apoptotic proteins, proinflammatory and pro-NF-kB, and lastly pyroptosis. The color coding is based on the standard deviation σHBE = 0.3.
DISCUSSION
During the infectious process, S. aureus has the option to evade the human immune defenses, or to invade nonprofessional phagocytic cells to hide from the host immune system and to evade antibiotic therapy. After internalization, the pathogen needs to adapt to the intracellular conditions so that it can survive, replicate, eventually leave the host cell and spread to other tissues. To optimize its fitness, the internalized pathogen displays population heterogeneity, where a fraction of the internalized bacterial population starts to replicate while another fraction displays low growth rates and reaches a state of dormancy (53). These two populations reflect the two main objectives of the internalized bacteria, where the replicating bacteria will ultimately disrupt epithelial cells to invade and infect the underlying tissue, whereas the dormant bacteria will persist intracellularly for an extended period.
Right from the moment the bacteria are internalized by a host cell, both host and pathogen need to adapt to the new situation, and then they will start to compete for resources. Importantly, such adaptations at the bacterial end are not limited to the production of virulence factors or mounting of defense mechanisms, but they also involve an optimized management of resources and the sensing of changes in the intracellular environment because of host cell adaptations (41). A clear example of this is the observed downregulation of bacterial ribosomal proteins, which relates to the formation of the alarmone ppGpp because of the induction of the stringent response after nutrient starvation and exposure to various stresses. The ppGpp molecule is synthesized from GTP, and the resulting decrease in GTP is sensed by the CodY regulatory system of S. aureus (50). Of note, CodY is a major modulator of S. aureus central carbon and amino acid metabolism and it also influences staphylococcal virulence (54). Consistent with the idea that the observed downregulation of ribosomal proteins is a consequence of ppGpp production, we observed an upregulation of the CodY-regulated proteins PycA, AcsA, ButA, Hom, metE, SerA, ThrC, Asd, DapABD, and ArgG. In this context, the activation of the ilv-leu operon is particularly noteworthy as this will result in the synthesis of BCAAs, which are cofactors of CodY (50). Of note, the inactivation of CodY may also result from reduced amino acid supply, and induction of the biosynthetic operons will counteract this shortage. However, high levels of BCAAs would then lead to repression again, but it remains unclear whether intracellularly such high BCAA levels are reached.
Importantly, the biological pathways in the internalized bacteria and their host cells are not isolated from each other despite their physical separation by the bacterial cell envelope. Accordingly, changes in pathway regulation at either end will impact the whole biological system. Thus, host cell homeostasis will change in response to the presence of internalized bacteria. This is exemplified by the observed optimization of glucose uptake and catabolism in the host cells from 48 h p.i. onwards, once they predominantly carry dormant bacteria. Interestingly, this correlates with the observed shift of the bacterial intracellular localization from an encapsulated state in vesicles to a free state in the cytosol. Therefore, this relocalization event could very well be a trigger for direct competition for nutrients that has a significant impact on the host cells' energy metabolism. Similarly, under nutrient-deprived conditions, the regulation of metabolic processes plays a crucial role in the adaptation of S. aureus to the altered conditions intracellularly. This is underpinned by the observed upregulation of proteins that are crucial for survival of the bacteria, such as PckA, AckA, FumC, SdhA, SucC, SucA and GudB, or proteins that promote their proliferation, such as Ald1, Ald2, Pyc, AspA, Mqo1, GltA, AcnA, Icd, RocA, RocD, and PutA. In fact, this observation is reminiscent of an in vitro study by Halsey et al. (55), who reported that under glucose restriction several of the afore-mentioned proteins have an impact on the fitness of the pathogen.
A fermentative phenotype combined with inactivation of the electron transport chain has been associated with the development of SCVs that are often observed on intracellular persistence of S. aureus (40). Although we did not observe the emergence of such SCVs by plating intracellular bacteria during our experimental setup, the observed changes in the bacterial proteome from 24 h p.i. onwards are indicative of a metabolic shift toward fermentation. Some of the proteins involved in bacterial electron transport become less abundant at late stages p.i compared with their levels during replication, whereas major proteins involved in the fermentation of pyruvate and acetate catabolism are constitutively upregulated throughout till the end of the infection. We observe a shift from terminal oxidases that require high oxygen concentrations to those that need lower concentrations, but do not pump H+ anymore, which is consistent with microaerobic intracellular conditions. Further, it is known that the formation of SCVs is linked to SigB, which is an important factor for intracellular persistence (11). Indeed, a clear role for SigB in reaching the dormancy state is indicated by changes in the abundance of 58 predominantly SigB-dependent proteins during the late stages of infection, among them Asp23, CidC, ClpL, FdhA, GapA, OpuBA, and SpoVG (supplemental Table S6). In this respect, it is noteworthy that S. aureus strains defective in SigB are unable to catabolize acetate (56).
Besides the changes in the central carbon metabolism of the host, the host's amino acid metabolism is also altered, altering the outcome of infection. Contrary to bacteria, which degrade amino acids mostly for the acquisition of carbon and nitrogen, the TCA-related amino acid production by the host cells is not related to an optimization of energy production but rather to the production of other molecules like purines, proline and collagen. However, some of the regulated proteins may also affect other pathways. For example, the observed asparagine synthase (ASNS) upregulation could be related to “infection stress.” This idea would be consistent with a previous study on group A streptococcal (GAS) infection, where the pathogen was shown to induce pores in the host membranes, leading to Ca+2 influx into the cytoplasm. In turn, this led to an upregulated synthesis of asparagine, which was then utilized by the pathogen (57). In our experimental setup, the ASNS abundance peaks at 24 h p.i., which correlates with the highest production of pore-forming toxins and escape of growing bacteria from the host cells, suggesting a similar regulatory mechanism as proposed for GAS.
Importantly, we observed that S. aureus induces its two pathways for arginine degradation right after internalization. It has been reported that arginine depletion by the pathogen induces death of the host cell (42). Moreover, competition for arginine with the host protein iNOS reduces the production of NO, which serves in the host's antibacterial defense. Lastly, the deaminase pathway produces NH3 and ATP, which supports the intracellular survival of the bacteria. In particular, the ammonia produced increases the pH of the intracellular environment thereby preventing fusion of the endosome with the lysosome (41, 42), whereas the production of ATP generates a source of energy during hypoxic conditions or when the electron transport system is deficient (58). The degradation of arginine usually occurs in environments with a high proline concentration (46). Interestingly, proteins related to production of proline are upregulated in the host proteome after 48 h p.i., when the bacterial machinery for arginine catabolism becomes massively upregulated. The host cell death induced by arginine starvation is regulated by activation of AMPK, which suppresses the master regulator mTOR and could lead to autophagy (42). In this context, it is noteworthy that spermidine is implicated in autophagy as well (59), and the SPEE protein, responsible for spermidine synthesis, is part of the same host pathway that also leads to the synthesis of glutamate, glutamine, proline, and arginine. As for ASNS, the abundance of SPEE peaks at 24 h p.i., which correlates well with the localization of the bacteria in phagocytic membranes as observed by fluorescence microscopy and TEM. These results underpin a connection between host amino acid starvation and autophagy, as suggested by a recent study (60). Although the roles of arginine and asparagine metabolism in infectious processes have been studied previously, the presently observed regulation of amino acid metabolism focuses additional attention on proline, glutamate, and alanine as potential modulators of infection.
Membrane-enclosed bacteria can be observed within the host cells up until 48 h p.i. as evidenced by TEM. This time-span covers the bacterial replication phase in the infectious process (i.e. until 24 h p.i.), but also nonreplicating bacteria can escape from lysosomes and phagosomes which is most clearly evident from 48 h p.i. onwards. Consequently, once the bacteria have entered the dormant state, they will persist intracellularly in the cytosol. These findings contrast with what has been found in other models that investigated S. aureus infection of other human cell types, where the cytosolic bacteria proliferated after phagosomal escape (61).
The bacterial escape from vesicles is most likely because of the production of pore-forming toxins that are regulated by SaeRS and Agr (50). The latter regulator is itself regulated by CodY, which also regulates most of the carbon resource management as described above (62). Nevertheless, some bacteria remain in vesicles until the very end of the present experiment. Together, these observations suggest that the bacteria-containing vesicles are possibly not fully functional. This intriguing idea is supported by the observed bacterial behavior, where oxidative stress and DNA-damage-induced responses are observed right from the beginning till the end of the experiment without a detectable peak. In fact, the abundance of some proteins related to these stress responses increased more strongly from 24 h p.i. onwards, when most bacteria have already been liberated from vesicular captivity. A possible vesicle malfunction could be because of the aforementioned production of pore-forming toxins. The latter idea is supported by the fact that the production of these toxins seems to be reduced at the last time points of infection where most intracellular bacteria are no longer enclosed in a membrane.
Lastly, our present proteome analyses reveal that apoptosis of the host cell could occur from 6.5 h until 24 h p.i., suggesting that the replicating bacteria employ this mechanism to escape from their host cells as captured by live cell imaging. In addition, we observed upregulation of Caspase 4 and NF-kB indicating activation of the inflammasome from 6.5 h p.i. Nevertheless, the abundance profile of other proteins implicated in the so-called pyroptosis does not support the activation of this self-destructive pathway. This suggests that the bacterial presence, even in dormancy, somehow leads to suppression of pyroptosis, which would lead to host cell lysis. Whether this is directly related to the bacterial actions, or indirectly to the response of the host to the bacterial presence is unclear.
Taken altogether, the present proteomics dissection of interactions between human bronchial epithelial cells and internalized S. aureus highlights the dynamic adaptive changes in the two interacting systems over an unprecedented period. This was necessary to obtain a proper understanding of the levels at which the two systems collide and eventually reach an equilibrium. Importantly, although in the past years host-pathogen interaction studies have focused attention on the roles of bacterial virulence factors and immune evasion, our present observations place dynamic host-pathogen interactions at the metabolic level in the limelight. Clearly, pore-forming toxins have a crucial role in giving the invading bacteria access to the resources that are hidden within the host cells, but it is the way in which these resources are used by the bacteria and how the host and the bacteria subsequently adapt to each other that determines the ultimate outcome of the infectious process.
DATA AVAILABILITY
The identified peptide ions and their Q-values are included in supplemental Tables S4 and S5. All protein annotations and median abundances are included in supplemental Tables S6 and S7. The raw files from mass spectrometry have been deposited in MassIVE (https://massive.ucsd.edu) with the access codes MSV000082920 (S. aureus) and MSV000082923 (16HBE14o-).
Supplementary Material
Acknowledgments
We thank Jan Pané-Farré for providing the HG001 Δspa, Jeroen Kuipers for his assistance during the electron microscopy imaging, Dr. Muriel Mari for the support in identifying the vesicles in the microscopy images, and Giorgio Gabarrini for his comments that greatly improved the manuscript. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Footnotes
* Funding for this project was received from the Graduate School of Medical Sciences of the University of Groningen [to L.M.P.M., S.A.M., and J.M.v.D.], the Deutsche Forschungsgemeinschaft Grants GRK1870 [to L.M.P.M., S.A.M. and U.V.] and SFBTRR34 [to U.V.], and CEC MSCI-ITN grant 713482 [ALERT, to H.Y., E.J.M.R., A.M.S., and J.M.v.D.]. Part of this work has been performed at the UMCG Imaging and Microscopy Center (UMIC), which is sponsored by NWO-grants 40-00506-98-9021 (TissueFaxs) and 175-010-2009-023 (Zeiss 2p).
 This article contains supplemental Figures and Tables. The authors declare that they have no financial and non-financial competing interests in relation to the documented research.
This article contains supplemental Figures and Tables. The authors declare that they have no financial and non-financial competing interests in relation to the documented research.
1 The abbreviations used are:
- SCVs
- small colony variants
- p.i.
- post infection
- DIA
- data independent acquisition
- GFP
- green fluorescent protein
- pMEM
- protein minimal essential medium
- eMEM
- eukaryotic minimal essential medium
- FCS
- fetal calf serum
- MOI
- multiplicity of infection
- LC3
- microtubule-associated protein 1A/1B-light chain
- LAMP-1
- lysosomal-associated membrane protein 1
- PFA
- para-formaldehyde
- TEM
- transmission electron microscopy
- ROS
- reactive oxygen species
- RNS
- reactive nitrogen species
- BCAAs
- branched-chain amino acids.
REFERENCES
- 1. Wertheim H. F., Melles D. C., Vos M. C., van Leeuwen W., van Belkum A., Verbrugh H. A., and Nouwen J. L. (2005) The role of nasal carriage in Staphylococcus aureus infections. Lancet Infect. Dis. 5, 751–762 [DOI] [PubMed] [Google Scholar]
- 2. Lowy F. D. (1998) Staphylococcus aureus Infections. N. Engl. J. Med. 339, 520–532 [DOI] [PubMed] [Google Scholar]
- 3. Tong S. Y. C., Davis J. S., Eichenberger E., Holland T. L., and Fowler V. G. (2015) Staphylococcus aureus Infections: Epidemiology, Pathophysiology, Clinical Manifestations, and Management. Clin. Microbiol. Rev. 28, 603–661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Breathnach A. S. (2013) Nosocomial infections and infection control. Medicine 41, 649–653 [Google Scholar]
- 5. Appelbaum P. C. (2006) MRSA—the tip of the iceberg. Clin. Microbiol. Infect. 12, 3–10 [DOI] [PubMed] [Google Scholar]
- 6. Fraunholz M., and Sinha B. (2012) Intracellular Staphylococcus aureus: Live-in and let die. Front. Cell. Infect. Microbiol. 2, 43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Garzoni C., and Kelley W. L. (2009) Staphylococcus aureus: new evidence for intracellular persistence. Trends Microbiol. 17, 59–65 [DOI] [PubMed] [Google Scholar]
- 8. Lehar S. M., Pillow T., Xu M., Staben L., Kajihara K. K., Vandlen R., DePalatis L., Raab H., Hazenbos W. L., Hiroshi Morisaki J., Kim J., Park S., Darwish M., Lee B.-C., Hernandez H., Loyet K. M., Lupardus P., Fong R., Yan D., Chalouni C., Luis E., Khalfin Y., Plise E., Cheong J., Lyssikatos J. P., Strandh M., Koefoed K., Andersen P. S., Flygare J. A., Wah Tan M., Brown E. J., and Mariathasan S. (2015) Novel antibody–antibiotic conjugate eliminates intracellular S. aureus. Nature 527, 323–328 [DOI] [PubMed] [Google Scholar]
- 9. Proctor R. A., van Langevelde P., Kristjansson M., Maslow J. N., and Arbeit R. D. (1995) Persistent and relapsing infections associated with small-colony variants of Staphylococcus aureus. Clin. Infect. Dis. Off. Publ. Infect. Dis. Soc. Am. 20, 95–102 [DOI] [PubMed] [Google Scholar]
- 10. Seifert H., Wisplinghoff H., Schnabel P., and von Eiff C. (2003) Small colony variants of Staphylococcus aureus and pacemaker-related infection. Emerg. Infect. Dis. 9, 1316–1318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Tuchscherr L., Bischoff M., Lattar S. M., Noto Llana M., Pförtner H., Niemann S., Geraci J., Van de Vyver H., Fraunholz M. J., Cheung A. L., Herrmann M., Völker U., Sordelli D. O., Peters G., and Löffler B. (2015) Sigma factor SigB is crucial to mediate Staphylococcus aureus adaptation during chronic infections. PLoS Pathog. 11, e1004870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Strobel M., Pförtner H., Tuchscherr L., Völker U., Schmidt F., Kramko N., Schnittler H.-J., Fraunholz M. J., Löffler B., Peters G., and Niemann S. (2016) Post-invasion events after infection with Staphylococcus aureus are strongly dependent on both the host cell type and the infecting S. aureus strain. Clin. Microbiol. Infect. 22, 799–809 [DOI] [PubMed] [Google Scholar]
- 13. Surmann K., Michalik S., Hildebrandt P., Gierok P., Depke M., Brinkmann L., Bernhardt J., Gesell Salazar M., Sun Z., Shteynberg D., Kusebauch U., Moritz R. L., Wollscheid B., Lalk M., Völker U., and Schmidt F. (2014) Comparative proteome analysis reveals conserved and specific adaptation patterns of Staphylococcus aureus after internalization by different types of human non-professional phagocytic host cells. Front. Microbiol. 5, 392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hecker M., Mäder U., and Völker U. (2018) From the genome sequence via the proteome to cell physiology – Pathoproteomics and pathophysiology of Staphylococcus aureus. Int. J. Med. Microbiol. 308, 545–557 [DOI] [PubMed] [Google Scholar]
- 15. Kiedrowski M. R., Paharik A. E., Ackermann L. W., Shelton A. U., Singh S. B., Starner T. D., and Horswill A. R. (2016) Development of an in vitro colonization model to investigate Staphylococcus aureus interactions with airway epithelia. Cell. Microbiol. 18, 720–732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Richter E., Harms M., Ventz K., Nölker R., Fraunholz M. J., Mostertz J., and Hochgräfe F. (2016) Quantitative proteomics reveals the dynamics of protein phosphorylation in human bronchial epithelial cells during internalization, phagosomal escape, and intracellular replication of Staphylococcus aureus. J. Proteome Res. 15, 4369–4386 [DOI] [PubMed] [Google Scholar]
- 17. Surmann K., Simon M., Hildebrandt P., Pförtner H., Michalik S., Stentzel S., Steil L., Dhople V. M., Bernhardt J., Schlüter R., Depke M., Gierok P., Lalk M., Bröker B. M., Schmidt F., and Völker U. (2015) A proteomic perspective of the interplay of Staphylococcus aureus and human alveolar epithelial cells during infection. J. Proteomics 128, 203–217 [DOI] [PubMed] [Google Scholar]
- 18. Herbert S., Ziebandt A.-K., Ohlsen K., Schäfer T., Hecker M., Albrecht D., Novick R., and Götz F. (2010) Repair of global regulators in Staphylococcus aureus 8325 and comparative analysis with other clinical isolates. Infect. Immun. 78, 2877–2889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Liese J., Rooijakkers S. H. M., van Strijp J. A. G., Novick R. P., and Dustin M. L. (2013) Intravital two-photon microscopy of host–pathogen interactions in a mouse model of Staphylococcus aureus skin abscess formation. Cell. Microbiol. 15, 891–909 [DOI] [PubMed] [Google Scholar]
- 20. Cozens A. L., Yezzi M. J., Kunzelmann K., Ohrui T., Chin L., Eng K., Finkbeiner W. E., Widdicombe J. H., and Gruenert D. C. (1994) CFTR expression and chloride secretion in polarized immortal human bronchial epithelial cells. Am. J. Respir. Cell Mol. Biol. 10, 38–47 [DOI] [PubMed] [Google Scholar]
- 21. R Core Team. (2018) R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria [Google Scholar]
- 22. Pförtner H., Wagner J., Surmann K., Hildebrandt P., Ernst S., Bernhardt J., Schurmann C., Gutjahr M., Depke M., Jehmlich U., Dhople V., Hammer E., Steil L., Völker U., and Schmidt F. (2013) A proteomics workflow for quantitative and time-resolved analysis of adaptation reactions of internalized bacteria. Methods 61, 244–250 [DOI] [PubMed] [Google Scholar]
- 23. Depke M., Michalik S., Rabe A., Surmann K., Brinkmann L., Jehmlich N., Bernhardt J., Hecker M., Wollscheid B., Sun Z., Moritz R. L., Völker U., and Schmidt F. (2015) A peptide resource for the analysis of Staphylococcus aureus in host pathogen interaction studies. Proteomics 15, 3648–3661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Michalik S., Depke M., Murr A., Gesell Salazar M., Kusebauch U., Sun Z., Meyer T. C., Surmann K., Pförtner H., Hildebrandt P., Weiss S., Palma Medina L. M., Gutjahr M., Hammer E., Becher D., Pribyl T., Hammerschmidt S., Deutsch E. W., Bader S. L., Hecker M., Moritz R. L., Mäder U., Völker U., and Schmidt F. (2017) A global Staphylococcus aureus proteome resource applied to the in vivo characterization of host-pathogen interactions. Sci. Rep. 7, 9718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zeitlin P. L., Lu L., Rhim J., Cutting G., Stetten G., Kieffer K. A., Craig R., and Guggino W. B. (1991) A cystic fibrosis bronchial epithelial cell line: immortalization by adeno-12-SV40 infection. Am. J. Respir. Cell Mol. Biol. 4, 313–319 [DOI] [PubMed] [Google Scholar]
- 26. Bruderer R., Bernhardt O. M., Gandhi T., Miladinović S. M., Cheng L.-Y., Messner S., Ehrenberger T., Zanotelli V., Butscheid Y., Escher C., Vitek O., Rinner O., and Reiter L. (2015) Extending the limits of quantitative proteome profiling with data-independent acquisition and application to acetaminophen-treated three-dimensional liver microtissues. Mol. Cell. Proteomics 14, 1400–1410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Deutsch E. W., Mendoza L., Shteynberg D., Slagel J., Sun Z., and Moritz R. L. (2015) Trans-proteomic pipeline, a standardized data processing pipeline for large-scale reproducible proteomics informatics. Proteomics Clin. Appl. 9, 745–754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Deutsch E. W., Mendoza L., Shteynberg D., Farrah T., Lam H., Tasman N., Sun Z., Nilsson E., Pratt B., Prazen B., Eng J. K., Martin D. B., Nesvizhskii A., and Aebersold R. (2010) A guided tour of the trans-proteomic pipeline. Proteomics 10, 1150–1159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Keller A., Nesvizhskii A. I., Kolker E., and Aebersold R. (2002) Empirical statistical model to estimate the accuracy of peptide identifications made by MS/MS and database search. Anal. Chem. 74, 5383–5392 [DOI] [PubMed] [Google Scholar]
- 30. Shteynberg D., Deutsch E. W., Lam H., Eng J. K., Sun Z., Tasman N., Mendoza L., Moritz R. L., Aebersold R., and Nesvizhskii A. I. (2011) iProphet: multi-level integrative analysis of shotgun proteomic data improves peptide and protein identification rates and error estimates. Mol. Cell. Proteomics MCP 10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Reiter L., Claassen M., Schrimpf S. P., Jovanovic M., Schmidt A., Buhmann J. M., Hengartner M. O., and Aebersold R. (2009) Protein identification false discovery rates for very large proteomics data sets generated by tandem mass spectrometry. Mol. Cell. Proteomics 8, 2405–2417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Nagel A., Michalik S., Debarbouille M., Hertlein T., Gesell Salazar M., Rath H., Msadek T., Ohlsen K., Dijl J. M. van, Völker U., and Mäder U. (2018) Inhibition of Rho activity increases expression of SaeRS-dependent virulence factor genes in Staphylococcus aureus, showing a link between transcription termination, antibiotic action, and virulence. mBio 9, e01332–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Fuchs S., Mehlan H., Bernhardt J., Hennig A., Michalik S., Surmann K., Pané-Farré J., Giese A., Weiss S., Backert L., Herbig A., Nieselt K., Hecker M., Völker U., and Mäder U. (2018) AureoWiki - The repository of the Staphylococcus aureus research and annotation community. Int. J. Med. Microbiol. 308, 558–568 [DOI] [PubMed] [Google Scholar]
- 34. Ritchie M. E., Phipson B., Wu D., Hu Y., Law C. W., Shi W., and Smyth G. K. (2015) limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Phipson B., Lee S., Majewski I. J., Alexander W. S., and Smyth G. K. (2016) Robust hyperparameter estimation protects against hypervariable genes and improves power to detect differential expression. Ann. Appl. Stat. 10, 946–963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Futschik M. E., and Carlisle B. (2005) Noise-robust soft clustering of gene expression time-course data. J. Bioinform. Comput. Biol. 3, 965–988 [DOI] [PubMed] [Google Scholar]
- 37. Kumar L., and Futschik E. M. (2007) Mfuzz: A software package for soft clustering of microarray data. Bioinformation 2, 5–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Basu A., and Yap M.-N. F. (2016) Ribosome hibernation factor promotes Staphylococcal survival and differentially represses translation. Nucleic Acids Res. 44, 4881–4893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ueta M., Wada C., and Wada A. (2010) Formation of 100S ribosomes in Staphylococcus aureus by the hibernation promoting factor homolog SaHPF. Genes Cells 15, 43–58 [DOI] [PubMed] [Google Scholar]
- 40. Somerville G. A., and Proctor R. A. (2009) At the crossroads of bacterial metabolism and virulence factor synthesis in Staphylococci. Microbiol. Mol. Biol. Rev. 73, 233–248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ren W., Rajendran R., Zhao Y., Tan B., Wu G., Bazer F. W., Zhu G., Peng Y., Huang X., Deng J., and Yin Y. (2018) Amino acids as mediators of metabolic cross talk between host and pathogen. Front. Immunol. 9, 319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Xiong L., Teng J. L. L., Botelho M. G., Lo R. C., Lau S. K. P., and Woo P. C. Y. (2016) Arginine metabolism in bacterial pathogenesis and cancer therapy. Int. J. Mol. Sci. 17, 363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Giardina G., Brunotti P., Fiascarelli A., Cicalini A., Costa M. G. S., Buckle A. M., di Salvo M. L., Giorgi A., Marani M., Paone A., Rinaldo S., Paiardini A., Contestabile R., and Cutruzzolà F. (2015) How pyridoxal 5′-phosphate differentially regulates human cytosolic and mitochondrial serine hydroxymethyltransferase oligomeric state. FEBS J. 282, 1225–1241 [DOI] [PubMed] [Google Scholar]
- 44. Morscher R. J., Ducker G. S., Li S. H.-J., Mayer J. A., Gitai Z., Sperl W., and Rabinowitz J. D. (2018) Mitochondrial translation requires folate-dependent tRNA methylation. Nature 554, 128–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kaiser J. C., King A. N., Grigg J. C., Sheldon J. R., Edgell D. R., Murphy M. E. P., Brinsmade S. R., and Heinrichs D. E. (2018) Repression of branched-chain amino acid synthesis in Staphylococcus aureus is mediated by isoleucine via CodY, and by a leucine-rich attenuator peptide. PLOS Genet. 14, e1007159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Gardan R., Rapoport G., and Débarbouillé M. (1997) Role of the transcriptional activator RocR in the arginine-degradation pathway of Bacillus subtilis. Mol. Microbiol. 24, 825–837 [DOI] [PubMed] [Google Scholar]
- 47. Ryan S., Begley M., Gahan C. G. M., and Hill C. (2009) Molecular characterization of the arginine deiminase system in Listeria monocytogenes: regulation and role in acid tolerance. Environ. Microbiol. 11, 432–445 [DOI] [PubMed] [Google Scholar]
- 48. Jarry T. M., Memmi G., and Cheung A. L. (2008) The expression of alpha-haemolysin is required for Staphylococcus aureus phagosomal escape after internalization in CFT-1 cells. Cell. Microbiol. 10, 1801–1814 [DOI] [PubMed] [Google Scholar]
- 49. Geiger T., Goerke C., Mainiero M., Kraus D., and Wolz C. (2008) The virulence regulator Sae of Staphylococcus aureus: promoter activities and response to phagocytosis-related signals. J. Bacteriol. 190, 3419–3428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Horn J., Stelzner K., Rudel T., and Fraunholz M. (2018) Inside job: Staphylococcus aureus host-pathogen interactions. Int. J. Med. Microbiol. 308, 607–624 [DOI] [PubMed] [Google Scholar]
- 51. Oogai Y., Kawada-Matsuo M., and Komatsuzawa H. (2016) Staphylococcus aureus SrrAB affects susceptibility to hydrogen peroxide and co-existence with Streptococcus sanguinis. PLOS ONE 11, e0159768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Liu X., and Lieberman J. (2017) in Advances in Immunology, ed Alt FW. (Academic Press; ), pp 81–117 [Google Scholar]
- 53. Fisher R. A., Gollan B., and Helaine S. (2017) Persistent bacterial infections and persister cells. Nat. Rev. Microbiol. 15, 453–464 [DOI] [PubMed] [Google Scholar]
- 54. Pohl K., Francois P., Stenz L., Schlink F., Geiger T., Herbert S., Goerke C., Schrenzel J., and Wolz C. (2009) CodY in Staphylococcus aureus: a regulatory link between metabolism and virulence gene expression. J. Bacteriol. 191, 2953–2963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Halsey C. R., Lei S., Wax J. K., Lehman M. K., Nuxoll A. S., Steinke L., Sadykov M., Powers R., and Fey P. D. (2017) Amino acid catabolism in Staphylococcus aureus and the function of carbon catabolite repression. mBio 8, e01434–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Somerville G. A., Saïd-Salim B., Wickman J. M., Raffel S. J., Kreiswirth B. N., and Musser J. M. (2003) Correlation of acetate catabolism and growth yield in Staphylococcus aureus: implications for host-pathogen interactions. Infect. Immun. 71, 4724–4732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Baruch M., Belotserkovsky I., Hertzog B. B., Ravins M., Dov E., McIver K. S., Le Breton Y. S., Zhou Y., Youting C. C., and Hanski E. (2014) An extracellular bacterial pathogen modulates host metabolism to regulate its own sensing and proliferation. Cell 156, 97–108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Makhlin J., Kofman T., Borovok I., Kohler C., Engelmann S., Cohen G., and Aharonowitz Y. (2007) Staphylococcus aureus ArcR controls expression of the arginine deiminase operon. J. Bacteriol. 189, 5976–5986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Minois N. (2014) Molecular basis of the “anti-aging” effect of spermidine and other natural polyamines - a mini-review. Gerontology 60, 319–326 [DOI] [PubMed] [Google Scholar]
- 60. Bravo-Santano N., Ellis J. K., Mateos L. M., Calle Y., Keun H. C., Behrends V., and Letek M. (2018) Intracellular Staphylococcus aureus modulates host central carbon metabolism to activate autophagy. mSphere 3, e00374–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Grosz M., Kolter J., Paprotka K., Winkler A.-C., Schäfer D., Chatterjee S. S., Geiger T., Wolz C., Ohlsen K., Otto M., Rudel T., Sinha B., and Fraunholz M. (2014) Cytoplasmic replication of Staphylococcus aureus on phagosomal escape triggered by phenol-soluble modulin α. Cell. Microbiol. 16, 451–465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Roux A., Todd D. A., Velázquez J. V., Cech N. B., and Sonenshein A. L. (2014) CodY-Mediated Regulation of the Staphylococcus aureus Agr System Integrates Nutritional and Population Density Signals. J. Bacteriol. 196, 1184–1196 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The identified peptide ions and their Q-values are included in supplemental Tables S4 and S5. All protein annotations and median abundances are included in supplemental Tables S6 and S7. The raw files from mass spectrometry have been deposited in MassIVE (https://massive.ucsd.edu) with the access codes MSV000082920 (S. aureus) and MSV000082923 (16HBE14o-).