

An integrated QXL-MS strategy was developed by coupling two-step cross-linking, affinity purification, MSn and quantitative MS to dissect H2O2-induced dynamics of proteasome complexes. In addition, a multi-bait strategy was presented for generating reliable quantitative structural information to infer conformational changes within the complex. These results allowed us to propose an intermediate state of the 26S proteasome that carries weakened interactions between the 19S and 20S. The developed QXL-MS strategy is applicable for studying structural dynamics of other protein complexes.
Keywords: Protein Cross-linking*, Protein-Protein Interactions*, Protein Conformation*, Protein complex analysis, Quantification, Oxidative stress, 26S proteasome, quantitative cross-linking mass spectrometry, structural dynamics
Graphical Abstract

Highlights
Two-step cross-linking coupled with affinity purification to facilitate structural analysis of protein complexes.
Integrated QXL-MS workflow for studying condition-dependent structural changes of protein complexes.
Mechanistic insights on in vivo H2O2-induced conformational dynamics of proteasome complexes.
Abstract
Cytotoxic protein aggregation-induced impairment of cell function and homeostasis are hallmarks of age-related neurodegenerative pathologies. As proteasomal degradation represents the major clearance pathway for oxidatively damaged proteins, a detailed understanding of the molecular events underlying its stress response is critical for developing strategies to maintain cell viability and function. Although the 26S proteasome has been shown to disassemble during oxidative stress, its conformational dynamics remains unclear. To this end, we have developed a new quantitative cross-linking mass spectrometry (QXL-MS) workflow to explore the structural dynamics of proteasome complexes in response to oxidative stress. This strategy comprises SILAC-based metabolic labeling, HB tag-based affinity purification, a 2-step cross-linking reaction consisting of mild in vivo formaldehyde and on-bead DSSO cross-linking, and multi-stage tandem mass spectrometry (MSn) to identify and quantify cross-links. This integrated workflow has been successfully applied to explore the molecular events underlying oxidative stress-dependent proteasomal regulation by comparative analyses of proteasome complex topologies from treated and untreated cells. Our results show that H2O2 treatment weakens the 19S-20S interaction within the 26S proteasome, along with reorganizations within the 19S and 20S subcomplexes. Altogether, this work sheds light on the mechanistic response of the 26S to acute oxidative stress, suggesting an intermediate proteasomal state(s) before H2O2-mediated dissociation of the 26S. The QXL-MS strategy presented here can be applied to study conformational changes of other protein complexes under different physiological conditions.
Oxidative stress has been implicated in a multitude of human pathologies, ranging from neurodegenerative disorders and cancers to the general aging process (1, 2). Reactive oxygen species (ROS)1 can induce oxidative damage of proteins, resulting in aberrant conformational changes in structure that render them functionally impaired or inactive. To avoid cytotoxic aggregation of damaged proteins and jeopardizing cellular functionality, the selective and timely removal of oxidatively damaged proteins is critical for cell survival. Proteasome-mediated protein degradation is the major pathway for the clearance of oxidized proteins that cannot be rescued by redox pathways (3, 4). However, the molecular details describing the effects of oxidative stress on proteasome machinery remains elusive. Further understanding of stress-induced impairment of proteasome structure and function may prove useful in developing new strategies for ameliorating proteasomal down-regulation and preventing cytotoxic aggregation of oxidized proteins.
The 26S proteasome is a 2.5 MDa macromolecular protease responsible for the selective turnover of eukaryotic proteins in the ubiquitin/ATP-dependent protein degradation pathway. The 26S holocomplex consists of a barrel-shaped 20S core particle (CP) that can be flanked on either or both ends by a 19S regulatory particle (RP) (5, 6). In eukaryotes, the 20S CP is comprised of seven α and seven β subunits, arranged in an evolutionarily conserved cylindrical stack of four heptameric rings in the order αββα. The outer α-rings form pores that gate the entrance of substrates to the inner multicatalytic β-rings, which harbor chymotrypsin-, trypsin-, and caspase-like enzymatic activities. In comparison, the 19S RP houses multiple additional functions to facilitate selective ubiquitin/ATP-dependent degradation, including substrate recognition, deubiquitination, unfolding, and assisting gate opening of the 20S for substrate translocation and degradation. Structurally, the regulatory particle can be further divided into the base and lid subcomplexes; the 19S base directly interfaces with the 20S CP and consists of a hexamer ATPase ring (Rpt1–6) and four non-ATPase subunits (Rpn1, 2, 10, and 13), whereas the remaining nine non-ATPase subunits (Rpn3, 5–9, 11, 12, and 15/Sem1) constitute the 19S lid. Compared with the highly ordered and compact structure of the 20S CP, the 19S RP is significantly more flexible and dynamic. Nevertheless, the overall architectures of the 19S, 20S, and 26S are highly conserved from yeast to human (7–10).
During oxidative stress, ubiquitin/ATP-independent degradation by the 20S CP is significantly enhanced to facilitate clearing of oxidized proteins (4, 11), because of increased level of free 20S. The change in relative abundances of 20S CP is not a result of transcriptional control, but instead attributed to oxidative stress-triggered disassembly of the 26S proteasome (12–15). 26S proteasome disassembly was shown to be dependent on the proteasome-interacting protein (PIP) Ecm29 in both yeast (16) and human cells (15). Through affinity purification mass spectrometry (AP-MS) strategies in both studies, Ecm29 was found to be significantly enriched at the 19S in response to H2O2-induced stress. Importantly, Ecm29 deletion in yeast and knockdown in human cells prevented oxidative stress-mediated disassembly of the 26S, suggesting the critical role of Ecm29 in proteasome regulation (15, 16). It seems that the dissociation of the 19S from the 20S is important as the blockage of this event makes cells much more susceptible to acute oxidative stress. To understand how Ecm29 regulates the 26S proteasome disassembly, we have recently employed cross-linking mass spectrometry (XL-MS) technology to identify interactions between Ecm29 and the 26S proteasome upon oxidative stress. In combination with integrative structural modeling, we have proposed a working model describing Ecm29 docking-induced dissociation of the 26S proteasome in response to H2O2-induced oxidative stress (15). In addition to the separation of its two subcomplexes, we suspect that the 26S proteasome undergoes a series of intermediate conformational states during the disassembly process. However, the molecular details underlying stress-mediated structural changes in the 26S remain elusive. Therefore, further studies are needed to define stress-triggered conformational changes of the 26S to fully dissect mechanisms underlying its regulation.
XL-MS studies have been instrumental in the structural determination of large, multi-protein assemblies such as the proteasome (7, 17–20). In recent years, quantitative cross-linking mass spectrometry (QXL-MS) strategies using isotope-labeled cross-linkers have emerged, permitting comparative analyses between multiple conformational states of proteins and protein complexes (20–23). These conformational differences are reflected in changing abundances of cross-links, which are typically quantified based on the relative spectral intensities of isotope-labeled versus non-labeled cross-linked peptides (20). The resulting structural information can then be correlated to various aspects of protein biology, ranging from characterizing perturbations of protein-protein interactions at the systems level (24–26) to identifying structural changes of macromolecular complexes and/or individual proteins for more focused, structural studies (22, 23, 27, 28). As an alternative to isotope-coded cross-linkers, metabolic labeling of cross-linkable residues using SILAC has been employed for QXL-MS analysis to probe in vivo protein interaction dynamics (24). Such SILAC-based methods are advantageous as they bypass the use of isotope-coded cross-linkers, which can be challenging to synthesize. In addition, deuterium is the most commonly incorporated isotope label for cross-linking reagents, which often alters the chromatographic elution of isotope-labeled cross-linked peptides compared with their non-labeled counterparts, leading to complications in automated quantitation.
Previously, we have developed QTAX (quantitative analysis of tandem affinity-purified in vivo cross(x)-linked protein complexes), a method to enable the capture and quantitative identification of stable and transient as well as weak PPIs of protein complexes in a single experiment (29). The QTAX strategy incorporates SILAC-based quantitation, in vivo formaldehyde cross-linking, and HB-tag based tandem affinity purification under fully denaturing conditions to differentiate specific interacting partners from background proteins. To identify interaction contacts of PPIs beyond their identities, we have developed a robust MS-cleavable homobifunctional lysine reactive cross-linker, i.e. DSSO, to enable fast and accurate identification of cross-linked peptides using multistage tandem mass spectrometry (MSn) (30). The DSSO-based XL-MS platform has been successfully applied to characterize the topologies of protein complexes in vivo (19) and in vitro (15, 19, 20, 31). Given the effectiveness of XL-MS methods in their unique ability to provide structural insight on conformational ensembles of protein complexes and quantitatively determine their changes under different physiological conditions, we have developed and employed a QXL-MS strategy by integrating QTAX with DSSO-based XL-MS strategy to delineate in vivo interaction and structural dynamics of the human 26S proteasome in response to H2O2-induced oxidative stress.
EXPERIMENTAL PROCEDURES
Chemicals and Reagents
Regular Dulbecco's modified Eagle's medium (DMEM) and SILAC Dulbecco's modified Eagle's medium (DMEM) (deficient in lysine) were obtained from Thermo Fisher Scientific (Waltham, MA). 13C615N2-Lysine were purchased from Cambridge Isotope Laboratories (Tewksbury, MA). 12C614N2-Lysine was obtained from Sigma (St. Louis, MO). Sequencing grade trypsin was purchased from Promega Corp (Madison, WI), Endoproteinase Lys-C from WAKO chemicals (Irvine, CA). All other general chemicals for buffers and cell culture media were purchased from Fisher Scientific or VWR (Radnor, PA).
Experimental Design and Statistical Rationale
Three types of proteasome expressing cells, i.e. 293Rpn11-HTBH, 293HBTH-Rpt6, and 293α7-HTBH, were selected for proteasome purification (19). At least two biological replicates were performed for each preparation (i.e. Rpn11, 3 biological replicates; Rpt6 and α7, 2 biological replicates each), and each of them was analyzed with at least two technical analyses (i.e. Rpn11, 11 total technical replicates; Rpt6, 10; α7, 9) to maximize identification and quantitation accuracy. Each biological replicate contained a heavy (untreated) control.
Proteasome Purification and DSSO Cross-linking
Briefly, cells were grown to (90%) confluence in light medium supplemented with 73 μg/ml 12C614N2-lysine or heavy medium supplemented with 73 μg/ml 13C615N2-lysine. Cells grown in light medium were treated with 2 mm H2O2 for 30 min, whereas cells grown in heavy medium were untreated as control. Before harvesting, cells were incubated with 0.025% formaldehyde for 10 min at 37 °C (15). Human 26S proteasome was purified from equal amounts (five 15 cm plates, each) of light- and heavy-labeled cell lysates separately by binding to streptavidin-Sepharose resin (19, 32). Bound proteasomes were then mixed and cross-linked on-bead in PBS buffer (pH 7.5) with 0.5 mm DSSO for 1 h at 37 °C. After quenching the cross-linking reaction, the proteins were reduced/alkylated and digested as described (33). Briefly, proteins were digested in 8 m urea buffer using LysC for 4 h at 37 °C, followed by trypsin digestion at 37 °C overnight after diluting urea concentration to <1.5 M. The resulting peptide mixtures were extracted and desalted before MS analyses.
LC MS/MS and Database Searching for Protein Quantification
Cross-linked peptide mixtures were first analyzed to determine the relative abundances of treated and control proteasomes. Samples were subjected to LC MS/MS analysis using an EASY-nLC™ 1000 system (Thermo Fisher Scientific) coupled on-line to an LTQ-Orbitrap XL mass spectrometer (Thermo Fisher Scientific). Reverse-phase separation was performed on a 15 cm x 75 μm I.D. Acclaim® PepMap RSLC column. Peptides were eluted using a gradient of 5% to 30% B over 100 min at a flow rate of 300 nl/min (solvent A: 100% H2O, 0.1% formic acid; solvent B: 100% acetonitrile, 0.1% formic acid). Each cycle consisted of one full Fourier transform scan mass spectrum (350–1800 m/z, resolution of 60,000 at m/z 400) followed by 10 data-dependent MS/MS acquired in the linear ion trap with 29% normalized collision energy. Target ions already selected for MS/MS were dynamically excluded for 30 s.
Protein quantitation of LC MS/MS data was carried out using MaxQuant as described (34). Briefly, raw spectrometric files were searched using MaxQuant (v. 1.5.0.0) against human complete proteome sequences obtained from UniProt (10,100 entries; version from May, 2016). MS/MS spectra were filtered to contain at most eight peaks per 100 mass unit intervals. The first search peptide tolerance was set to 20 ppm, with main search peptide tolerance set to 4.5 ppm. Both peptide spectrum match and protein FDRs were set at 1%, in razor peptide fashion. Trypsin was selected for the protease with up to 2 missed cleavages; no nonspecific cleavage was allowed. For protein quantitation, cysteine carbamidomethylation was set as a fixed modification, whereas methionine oxidation and N-terminal acetylation were selected for variable modifications, maximum of 2 per peptide. Intensities were determined as the full peak volume over the retention time profile. Intensities of different isotopic peaks in an isotope pattern were always summed up for further analysis. “Unique plus razor peptides” was selected as the degree of uniqueness required for peptides to be included in quantification.
LC MSn Analysis, Cross-link Identification and Quantification
LC MSn data for DSSO cross-linked peptides was obtained using an EASY-nLC™ 1200 (Thermo Fisher Scientific) coupled with an Orbitrap Fusion Lumos™ mass spectrometer (Thermo Fisher Scientific) similarly as previously described (35). Briefly, a 25 cm × 75 μm PepMap EASY-Spray Column was used to separate peptides over 210 min acetonitrile gradients of 6% to 30% at a flow rate of 300 nL/min. Two different types of acquisition methods were used to maximize the identification of DSSO cross-linked peptides: (1) top 4 data-dependent MS3; and (2) targeted MS3 acquisition (35). For both methods, only ions with charge of 4+ to 8+ in the MS1 scan were selected for MS2 analysis. MS1 and MS2 scans were acquired in the Orbitrap whereas MS3 scans were detected in the ion trap. For MS1 scans, the scan range was set from 350 to 1800 m/z, and the resolution set to 120,000. For MS2 scans, the resolution was set to 30,000 with precursor isolation width of 2.0 m/z. The CID-MS2 normalized collision energy was 20%. For method 1, HCD-MS3 and/or CID-MS3 acquisitions were triggered for the top 4 most abundant fragment ions in each MS2 scan with a collision energy of 30%. For method 2, mass difference-dependent HCD-MS3 and/or CID-MS3 acquisitions were triggered with a collision energy of 30% if a unique mass difference (Δ = 31.9721) was observed between the two fragment ions in the CID-MS2 spectrum (35). This feature is unique to sulfoxide-containing MS-cleavable cross-linked peptides (e.g. DSSO) during MS2 analysis (30, 36–38).
Raw spectrometric data were converted to MGF files using ProteoWizard MSConvert (v. 3.0.10738). Extracted MS3 spectra were subjected to protein database searching via Batch-Tag within a developmental version of Protein Prospector (v. 5.19.1, University of California, San Francisco) against a decoy database consisting of a normal SwissProt database concatenated with its randomized version (SwissProt.2016.05.09.random.concat with total of 20,200 protein entries). Mass tolerances for parent ions and fragment ions were set as ± 20 ppm and 0.6 Da respectively. Trypsin was set as the enzyme with three maximum missed cleavages allowed. Cysteine carbamidomethylation was selected as a constant modification, whereas protein N-terminal acetylation, methionine oxidation, and N-terminal conversion of glutamine to pyroglutamic acid were selected as variable modifications. For treated (light) peptide searches, three additional defined variable modifications on uncleaved lysines and free protein N termini were selected: alkene (A: C3H2O, +54 Da), sulfenic acid (S: C3H4O2S, +104 Da), and unsaturated thiol (T: C3H2OS, +86 Da) modifications corresponding to remnant moieties for DSSO. For control (heavy) peptide searches, variable modifications corresponding to alkene, sulfenic acid, and unsaturated thiol modifications on heavy lysines were selected. Initial acceptance criteria for all MS3 peptide identifications required an expectation value below 0.05. The FDR of MS3 peptide identification was ≤ 0.65%. MSn data (monoisotopic masses and charges of parent ions and corresponding fragment ions, and ion intensities from cross-linker and peptide fragmentation) and MS3 database search results were integrated via in-house software xl-Tools (35) to automatically generate, summarize and validate identified cross-linked peptide pairs. Experimental cross-link identification FDR was determined to be ≤ 0.33%.
Quantitation and Evaluation of Cross-linked Peptides
Automatic quantitation of cross-linked peptides was performed using xl-Tools and manually verified. Ion chromatograms for cross-linked peptides were extracted from the MS1 full scan using the 4 most abundant isotopes from each cross-linked peptide ion at a mass tolerance of 20 ppm. Spectral noise was estimated by the mean of the spectral intensity over the entire ion chromatogram. In order to most accurately determine peak area for lowly abundant cross-linked peptides, we coupled a simple peak detection algorithm (39) with a model-based strategy to quantify cross-linked peptide abundance. The peak selection model was designed by using a principal component analysis to identify core features later used to classify data via a regression tree. Two components, the dot-product distance between isotopes within a given envelope, and the Pearson's R correlation between the experimental isotope abundance and theoretical envelope of an average peptide, enabled the differentiation of analyte from matrix with high accuracy, enabling fully automated XIC peak selection. Selected peaks were quantified by summing the area of all isotopes using trapezoidal integration. Outliers were detected by the difference in the ratio between technical and biological replicates. The ratio between any two replicates was fit to an elliptical envelope encapsulating 90% of the data, and values outside the envelope were discarded as outliers. The reproducibility of the L/H ratio between technical and biological replicates was calculated using a linear regression.
Normalization of Cross-link Abundance Ratios to Protein Abundance and Data Filtering
To estimate changes in local protein conformations, cross-linked peptide abundances were normalized by the abundances of their comprising proteins. The abundance of each protein was estimated via the mean intensity of MS/MS-quantified peptides calculated by MaxQuant. Three normalization schemes with different subunit groupings were tested; one “coarse” using single normalization values for 19S and 20S subcomplexes, a “medium” categorization based on subcomplex (i.e. 19S base, 19S lid, 20S alpha, 20S beta), and a “fine” scheme using a separate normalization value for every proteasomal subunit. Cross-linked peptides were then normalized to the “limiting” L/H substructure abundance. Bait protein L/Hs were not considered in determining the average L/H ratio for their respective substructures. The p value of each linkage was calculated using a 1-sample, two-sided t test. Linkages with a mean log2 magnitude > 1.00 and a p value < 0.05 were classified as statistically changed, whereas those with a p value < 0.05 but with a mean log2 ≤ 1.00 were classified as unchanged. The threshold for change was justified by calculated σ values for the individual data sets from Rpn11, Rpt6, and α7: 2.04, 2.13, and 1.84, respectively. σ for the entire set of quantitative cross-link values was calculated to be log21.06, or ∼2.08-fold change. Cross-links that did not meet the p value requirement were manually curated and considered if their respective data points could be used qualitatively to describe significant change.
RESULTS
Developing a QXL-MS Strategy for Characterizing In vivo Proteasome Complexes in Response to Oxidative Stress
To explore structural details underlying 26S disassembly during oxidative stress, we have developed a new QXL-MS strategy by integrating QTAX (29, 40) with DSSO cross-linking (30) to enable comparative in vivo structural analyses of the 26S proteasome complex (Fig. 1). Although SILAC-based isotope incorporation is used to distinguish true interactors from nonspecific binding proteins in original QTAX experiments, it is used to label cross-linkable lysines and thus quantify cross-linked peptides for inferring conformational changes within proteasome complexes in this study. To enable pairwise comparisons, one population of cells was grown in light medium (12C14N-Lys) and treated with H2O2, whereas another population of cells was grown in heavy medium (13C15N-Lys) and used as an untreated control. Then, treated and untreated cells were independently subjected to cross-linking before cell lysis. To better preserve the intactness of the 26S proteasome for comparative XL-MS analysis, we have adopted a newly developed 2-step cross-linking strategy that entails mild in vivo formaldehyde cross-linking of intact cells followed by in vitro DSSO cross-linking (15, 20). Mild in vivo formaldehyde cross-linking has proven to be beneficial for stabilizing the wholeness of the proteasome during cell lysis without disturbing its function (41). In addition, this step does not interfere with subsequent DSSO cross-linking for structural characterization of proteasome complexes (15). Therefore, after in vivo formaldehyde cross-linking, proteasome complexes were purified respectively from equal amounts of treated (light-labeled) and untreated (heavy-labeled) cell lysates using 1-step HB-tag based purification by binding to streptavidin beads (19, 32). The bound proteasomes from both types of cells were then mixed and cross-linked on-bead using DSSO. After quenching, the cross-linked proteasomes were enzymatically digested, and analyzed by LC MSn. This QXL-MS platform represents a new strategy to delineate in vivo protein complex dynamics under different physiological conditions.
Fig. 1.

Identification and quantification of DSSO cross-linked peptides of proteasome complexes in response to H2O2 stress. A, The general QXL-MS workflow to dissect condition-dependent conformational changes of protein complexes. B, Representative MSn analysis for the identification of DSSO cross-linked peptides. MS1 spectrum shows the detection of a SILAC labeled pair of DSSO cross-linked peptides (m/z 869.94074+, 874.20374+), of which low energy CID MS2 of the light labeled DSSO cross-linked peptide (m/z 869.94074+) yielded pairs of fragment ions (αA/βT, αT/βA) characteristic to DSSO cross-linked peptides as expected. Subsequent CID-MS3 analysis of individual fragment ions αA and βT (m/z 757.872+, 973.012+) yielded series of b and y ions that accurately identified them as 279APSIIFIDELDAIGTKTR295 and 351KAIEFPMPNEEAR362 of Rpt5, signifying a cross-link between K294 and K351. C, Representative MS1 quantitation of DSSO cross-linked peptides. Quantitation of the cross-link identified in (B), i.e. Rpt5:K294-Rpt5:K351, calculated using spectral abundances of each monoisotopic ion within a single scan XIC (left), and through automated quantitation by xl-Tools calculating the area under each curve (right).
To obtain a comprehensive PPI map of proteasome complexes, we applied this QXL-MS strategy to three stable cell lines each expressing a single HB-tagged proteasome subunit from each respective 26S subcomplex: α7 (20S), Rpt6 (19S base), and Rpn11 (19S lid). These subunits were selected because of their locations within the 26S proteasome and their critical biological functions. While the 20S subunit α7 contributes to the formation of the substrate entrance gate, the 19S subunit Rpt6 is critical for gate opening, substrate translocation, and proteasome assembly (10). In comparison, Rpn11 is the only known 19S subunit that functions as an intrinsic and essential deubiquitinating enzyme for removal of poly-ubiquitin chains before substrate translocation into the 20S proteasome (42). More importantly, the three selected subunits have been successfully applied for proteasome purification and XL-MS analysis in previous studies (19), thus allowing us to perform comparative XL-MS analyses of proteasomes under different conditions.
Identification of Proteasome Cross-links on Oxidative Stress
To examine the structural details of the 26S in response to acute oxidative stress, we have employed DSSO-based XL-MS analysis to identify cross-linked peptides and thus infer the spatial interactions of proteasome subunits in control and H2O2-treated cells. As previously established, MSn analysis workflow is best suited for the analysis of DSSO cross-linked peptides (30) (Fig. 1B). Briefly, during low energy collisional induced dissociation (CID) in MS2 analysis, cleavage of a sulfoxide-containing MS-cleavable bond separates DSSO interlinked peptide α-β into two single peptide chain fragments modified with complementary DSSO remnants (30). Because of the presence of two symmetric MS-cleavable bonds in DSSO, two pairs of predictable peptide fragment ions are detected (αA/βT, αT/βA), which are selected and subjected to MS3 analysis for peptide sequencing (30). As an example, low energy CID-MS2 analysis of a parent ion isolated from MS1 (m/z 869.94074+) yielded pairs of fragment ions characteristic of a DSSO interlinked peptide α-β (30). Subsequent CID-MS3 analysis of individual fragment ions αA (m/z 757.872+) and βT (m/z 973.012+) yielded series of b and y ions identifying them as 279APSIIFIDELDAIGTKTR295 and 351KAIEFPMPNEEAR362, signifying an intrasubunit cross-link between lysines K294 and K351 of Rpt5. Using this approach, we have identified a total of 9097 light- and heavy-labeled cross-linked peptides from 7 QXL-MS experiments (i.e. Rpn11, 3 biological replicates; Rpt6 and α7, 2 biological replicates each), representing 1213 unique lysine-lysine linkages, with 746 corresponding to cross-links resulted only from 26S proteasome subunits (supplemental Tables S1 and S2). Of these, 392 were intersubunit linkages, detailing 87 unique protein-protein contacts within the 26S holocomplex (15 lid-lid, 21 lid-base, 25 base-base, 1 lid-20S, 10 base-20S, 15 20S-20S) (supplemental Table S2A). The remaining 354 intrasubunit identifications spanned 27 proteasome subunits (10 19S base, 8 19S lid, 9 20S) (supplemental Table S2B). Overall, 52% (46/89) of pairwise interprotein interactions were captured commonly across proteasome purifications from all baits, whereas 24% (21/89) overlapped in proteasomes purified by tagged 19S subunits only. The majority of the remaining identified interactions (20/89) were captured by purifications from a single bait (Fig. 2A).
Fig. 2.
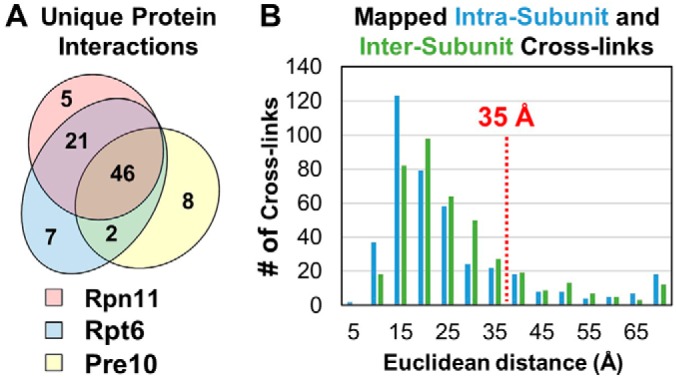
Analyses of the identified DSSO cross-linked peptides of 26S proteasome subunits. A, Summary of identified pair-wise interactions captured by cross-linking from 293HBTH-Rpt6, 293Rpn11-HTBH, and 293α7-HTBH proteasome purifications. B, Distance distribution plots of the identified cross-links to the high-resolution structure of the human 26S proteasome (PDB: 5GJR).
To evaluate the validity of the identified cross-links, we first mapped them to a high-resolution cryo-EM structure (PDB: 5GJR, 3.5 Å) of the human 26S proteasome to assess cross-linked residue proximities (9). Because of missing densities, only 586 out of 746 K-K linkages were mapped, in which 85.2% (499/586) were within the estimated maximum Cα-Cα distance (< 35 Å) spanned by DSSO (Supplemental Table 2) (19). The satisfied interactions comprise 255 out of 302 intersubunit and 244 out of 284 intrasubunit linkages (Fig. 2B, supplemental Table S2A–S2B). The average distances of intrasubunit and intersubunit cross-links were determined to be 22.74 Å and 24.28 Å, respectively. Interestingly, 45 of the 87 violating cross-links (> 35 Å) involved Rpt6, with the majority (31/45) corresponding to intrasubunit Rpt6 cross-links. It is noted that these Rpt6-containing violating cross-links were predominantly identified from Rpt6 but not α7 and Rpn11 purifications, suggesting that they most likely derive from free Rpt6 and/or its subcomplexes. These results are consistent with our previous XL-MS analysis of proteasomes, which also yielded most Rpt6-containing violating cross-links with the majority as intra-Rpt6 linkages (19). Nonetheless, most of the cross-links identified here fit well with the known proteasome structure (PDB: 5GJR) under expected distances.
Quantitation of H2O2-dependent Proteasome Cross-links
To properly assess oxidative stress-dependent changes in cross-link abundance, we have first determined the relative protein abundances of proteasome subunits from control and treated cells based on SILAC ratios (Ltreated/Huntreated) of non-cross-linked peptides using MaxQuant (supplemental Table S3A–S3C and S4). When comparing respective proteasomes purified by Rpn11 and Rpt6 before and after H2O2 treatments, the average relative abundance ratios of 19S subunits were ∼1.0, indicating that their abundances were unaffected by H2O2 stress, as expected (15). In contrast, the relative abundances of all 20S CP subunits decreased substantially following H2O2 treatment, exhibiting SILAC ratios < 0.4 and demonstrating the dissociation of the 20S from the 19S on H2O2 stress as described previously (15). For proteasomes purified by the 20S subunit α7, 20S subunits remained unchanged (with SILAC ratios ∼1.0) and 19S subunits decreased (SILAC ratios < 0.4) on treatment. For 19S and 20S proteasome subunits in Rpn11 and α7 purifications, their respective SILAC ratios before and after in vitro DSSO cross-linking were similar. For Rpt6-purified proteasomes, all subunits except for Rpt6, the bait, preserved their SILAC ratios before and after DSSO cross-linking. In comparison, the L/H ratio for Rpt6 was slightly decreased relative to the rest of the 19S, suggesting that co-purified free Rpt6 and/or Rpt6-containing non-proteasome subcomplexes may be more susceptible to DSSO cross-linking after H2O2 stress. However, this does not interfere with the following quantitative analysis of cross-links within proteasome complexes, as the observed consistency in all other proteasome subunits have indicated that the expected relative abundances of 26S proteasome are maintained during our cross-linking conditions. As such, the SILAC ratios of the bait proteins were not considered when determining the average L/H abundances of their corresponding subcomplexes. Collectively, similar protein abundance ratios of proteasome subunits before and after DSSO cross-linking suggest minimum variance in the cross-linking efficiencies of light- and heavy-labeled proteasomes. Thus, changes in the identified cross-links can be normalized by their protein relative abundances to describe proteasome structural changes.
To determine SILAC ratios of cross-links, we used a peak detection algorithm with a model-based strategy within xl-Tools to measure ion abundances of cross-linked peptides in MS1, like the ratio calculation of non-cross-linked peptides (Fig. 1C). As an example, the L/H ratio of intrasubunit cross-link Rpt5:K294-Rpt5:K351 was determined as 2.78 based on the relative spectral abundances of its light (m/z 869.94074+) and heavy (m/z 874.20374+) labeled cross-linked peptide pair. Therefore, this cross-link was found to be more abundant in proteasomes after H2O2 treatment. Similarly, all cross-links from each QXL-MS experiment were quantified and analyzed accordingly. Of 746 unique proteasome cross-links, 591 were successfully quantified in at least one LC MSn run from any bait purification. To obtain reliable data, only unique cross-links quantified in at least two sample replicates within the same bait were considered for final reporting, yielding 343 quantifiable unique K-K linkages. As discussed above, because of H2O2-induced 26S proteasome disassembly, it is necessary to normalize the quantitative ratios of cross-links to their respective protein abundances. To this end, cross-link ratios were corrected using the abundances of their comprising proteins, as determined by LC MS/MS quantitation of protein abundances through MaxQuant (supplemental Table S4). To avoid overfitting, the relative abundances of each protein was coarsened into 4 substructures: 19S base, 19S lid, 20S alpha, and 20S beta subunits (supplemental Fig. S1). Intersubunit cross-links identified between different substructures were normalized to the “limiting” substructure abundance per cross-link. Lastly, the remaining QXL-MS data were subjected to a filtering step requiring an associated p value below 0.05 within a 1-sample, two-sided t test, further improving the confidence and accuracy of quantified cross-links for subsequent analysis.
To assess the reproducibility of cross-link quantitation determined through XIC-based measurement, we employed regression analyses to compare the normalized SILAC ratios of all cross-links in pairwise fashion between biological replicates. Regression plots for all cross-links quantified from multiple biological replicates were first generated for Rpn11-, Rpt6-, and α7-purified proteasomes, respectively (supplemental Fig. S2A–S2C). In general, all plots displayed a linear relationship between different biological replicates. Regression plot analyses of the same quantitative data following p value filtering resulted in fewer data points, but a noticeable improvement in accuracy of the quantitative values toward a linear equation (Fig. 3A–3C). The narrowing of quantitative data points toward a linear correlation between biological replicates suggests that the used filtering steps were effective in enriching for reliable, reproducible observations of cross-link abundance.
Fig. 3.

Assessment of reproducibility and distribution of quantified 26S cross-links. Following p value filtering, reproducibility of quantitation in separate biological replicates was determined for (A) 293Rpn11-HTBH, (B) 293HBTH-Rpt6, and (C) α7-HTBH. D–F, Volcano plots depicting the distribution of changed (red) and unchanged (blue) cross-links in proteasome purifications from each tagged cell line. Cross-links that did not meet the p value threshold of 0.05 are shown in gray.
In total, 229, 196, and 117 K-K linkages were quantified from Rpn11-, Rpt6-, and α7-purified proteasomes, respectively (supplemental Table S5A–S5C and supplemental Fig. S3). In Rpn11-proteasomes, 47 displayed significant changes (≥ 2-fold) in either direction, whereas 182 were determined as unchanged on oxidative stress. Further filtering with p value ≤ 0.05 reduced the number of significantly changed and unchanged cross-links to 37 and 65, respectively (Fig. 3D). During manual inspection, we have found that several excluded cross-links had p values above 0.05 but consistent significant changes from multiple biological replicates (supplemental Table S5A). For example, the SILAC ratio (log2) for the cross-link Rpt6:K393-α3:K210 was quantified as −1.208 and −1.604 in two biological replicates respectively, resulting in a p value of 0.089. Based on our p value criterion (≤ 0.05), this quantifiable cross-link was omitted. However, these observed changes indeed represent a common directionality and were thus included in the following analysis. Similarly, six additional cross-links with consistent significant changes across biological replicates but with higher p values were manually validated and included for subsequent analyses. Thus, a total of 44 quantifiable K-K linkages were considered with significant changes in Rpn11 purifications; 33 of them were more abundant in untreated proteasome complexes (log2 L/H ≤ −1), whereas 11 were more abundant following oxidative stress (Fig. 3D, supplemental Table 5A). With the same analysis, Rpt6 purifications resulted in a total of 107 quantifiable linkages with 61 unchanged and 46 changed ones. Of those 46, 39 were favored in untreated and 7 were favored in H2O2-treated cells (Fig. 3E, supplemental Table S5B). Finally, α7 purifications yielded 27 unchanged and 43 changed cross-links, all of which were more abundant in untreated proteasomes (Fig. 3F, supplemental Table S5C).
Impaired Connectivity Between the 19S and 20S on Oxidative Stress
To determine the structural regions that reflected H2O2 stress-induced changes within the 26S, we examined the spatial relationships among quantified cross-linked residues on the high-resolution proteasome structure (PDB: 5GJR). We first inspected cross-links bridging residues between the 19S and 20S subunits, which can only derive from intact 26S proteasomes. It is noted that the heterogeneity of purified proteasomes varies with the selected baits. Although the 19S subunits, i.e. Rpn11 and Rpt6, purified proteasomes containing only intact 26S and free 19S RP proteasomes, 20S subunit α7 purifications yielded intact 26S and free 20S proteasomes. Therefore, 19S-20S cross-links from all three purifications can be used to infer conformational changes within the 26S proteasome. In total, nine unique 19S-20S K-K linkages were quantified, representing six proximal subunit interactions between the two subcomplexes. Eight were successfully mapped to the high-resolution proteasome structure, all yielding Cα-Cα distances below 32 Å (Fig. 4). Among them, three K-K linkages describe the interaction of Rpt6 with α3, i.e. Rpt6:K393-α3:K210, Rpt6:K156-α3:K199, and Rpt6:K156-α3:K205, identified from Rpn11, Rpt6 and α7 purifications, respectively. As shown, these interactions significantly decreased in abundance (> 2-fold) on H2O2 treatment—regardless of the bait (Fig. 4A, supplemental Tables S5A–S5C). Although Rpt6:K156 and Rpt6:K393 are distant from each other by 237 residues based on the primary sequence of Rpt6, they are only 28.5Å apart in three-dimensional space and similarly located on the distal surface of the 19S RP abutting the 20S CP. As the maximum distance captured by DSSO cross-linking is estimated at 35 Å, these residues in Rpt6 and α3 are within proximity for cross-linking in normal proteasome structures. Because the reduction of these intersubcomplex cross-links is not attributed to protein abundance changes resulting from 26S disassembly, we suspect that H2O2 induced a conformational change at the interface of 19S and 20S complexes that diminished the cross-linkability between these residues, most likely because of their increased distances and/or lengthened solvent-accessible paths. Similarly, six additional 19S-20S interactions including Rpt2-α4 (Rpt2:K423-α4:K52), Rpt1-α4 (Rpt1:K407-α4:K174), Rpn6-α3 (Rpn6:K159-α3:K54), Rpt5-α7 (Rpt5:K193-α7:K206), and Rpt2-α3 (Rpt2:K387-α3:K176) were determined to decrease significantly following H2O2 treatment—with changes ranging from 2.5∼5-fold. Together, these results strongly suggest that oxidative stress modulates the 26S proteasome structure to form an intermediate state(s) before its full dissociation.
Fig. 4.

Modulation of the 19S-20S interaction by oxidative stress. A, Plot depicting normalized SILAC ratios (i.e. log2 L/H) of eight unique linkages (1–8) bridging the 19S and 20S subcomplexes with significantly decreased abundance in H2O2 treated samples (log2 (L/H) ≤ 1). B, These linkages were mapped to the high-resolution structure of the human 26S proteasome (PDB: 5GJR). Inset shows a cut-away of the rotated 26S complex depicting the distribution of intersubcomplex cross-links along the opposing faces of the ATPase ring and 20S α-ring.
Impact of H2O2 Treatment on 20S Stability
To understand H2O2-induced structural changes within the 20S CP, we examined 20S-20S cross-links, focusing primarily on intersubunit and sequence-distant intrasubunit interactions. In total, 33 unique K-K linkages describing interactions within the 20S CP were identified from all purifications. 16, 8, and 24 were contributed from Rpn11-, Rpt6-, and α7- purifications, respectively. Eight intersubunit cross-links within the 20S CP were cumulatively identified from Rpn11 and Rpt6 purifications and exhibited decreased abundances in treated samples. Among these, seven displayed more than 2-fold changes (i.e. α1:K30-α2:K53, α2:K176-α3:K54, α3:K176-α4:K52, α3:K176-α4:K204, α4:K157-α5:K231, α5:K187-α6:K208, β2:K237-β6:K204) (supplemental Tables S5A–S5B). Like previously discussed 19S-20S cross-links, these 20S-20S cross-linked residues are localized at the surface of the 20S CP, with most comprising the edge interfacing with the 19S RP. In addition to intersubunit cross-links, 10 intrasubunit linkages were identified from Rpn11- and Rpt6- purified proteasomes. These interactions corresponded to six 20S subunits, i.e. α3:K54-α3:K205, α3:K54-α3:K210, α3:K187-α3:K239, α4:K27-α4:K166, α4:K52-α4:K204, α6:K41-α6:K217, α7:K65-α7:K230, α7:K192-α7:K238, β3:K17-β3:K192, and β6:K73-β6:K104 (Supplemental Tables S5A–S5B). These linkages, representing three-dimensional proximity of sequence-distant residues (minimum 30 residues apart), were all found to be decreased in oxidatively stressed cells. In comparison, XL-MS analyses of α7-purified proteasomes yielded 20S-20S cross-links that displayed similar abundances before and after H2O2 stress, indicating that they were unaffected by the treatment. This included four intersubunit (α1:K30-α2:K53, α2:K176-α3:K54 α4:K157-α5:K231, and α5:K187-α6:K208) and five intrasubunit 20S cross-links (α3:K54-α3:K210, α4:K27-α4:K166, α4:K52-α4:K204, α7:K192-α7:K238, and β3:K17-β3:K192). Intriguingly, these unchanged 20S-20S cross-links found in the α7 purifications were also identified but determined to decrease markedly in Rpn11- and Rpt6-purified proteasomes (Fig. 5A–5B, supplemental Tables S5A–S5C). The discrepancies in the SILAC ratios of these 20S-20S cross-links are most likely attributed to the differences in 20S populations purified from the tagged 19S and 20S subunits. Although Rpn11 and Rpt6 purifications can only yield 20S cross-links from intact 26S complexes, α7 purifications can produce two forms of the 20S, one free and the other attached to the 19S RP—both of which can contribute to the identification of 20S-20S cross-links. Thus, quantitative changes in 20S-20S cross-links identified from Rpn11 and Rpt6 purifications reflected H2O2-induced structural changes of the 20S within intact 26S proteasomes, whereas quantification of those identified from the α7 purification represented the average changes from both free and 19S-bound 20S complexes. To better understand the 20S abundance distribution in α7 purifications, we calculated protein iBAQ values and estimated that the relative abundance of the 20S CP and 19S RP is roughly 6 to 1 in untreated samples and 9 to 1 in H2O2 treated samples (data not shown). This indicates that the majority of the 20S-20S cross-links resulting from α7 purifications describe interactions within the free 20S, which appear unchanged following H2O2 stress, suggesting that its structural integrity was undisturbed. This is reasonable as the 20S is the main degradation machine for the removal of oxidized proteins and has been shown to be more resistant to oxidative stress (4).
Fig. 5.

Oxidative stress induces structural changes within the 20S CP. A, Respective distribution of quantified 20S-20S cross-links for proteasomes purified from the three selected baits. The 20S-20S cross-links were decreased significantly in proteasomes from Rpn11 and Rpt6 but remain unchanged in α7 purifications. B, Mapping the 20S-20S cross-links identified from Rpn11 and Rpt6 proteasome purifications to the high-resolution structure of the human 26S proteasome shows distribution of cross-links to the solvent-accessible perimeter of the 20S subcomplex.
H2O2 Stress-induced Changes of the 19S Regulatory Particle
The same analysis was applied to 19S-19S cross-links to infer structural changes within the 19S RP. In this scenario, 19S-19S cross-links obtained from α7 purifications would represent the 19S interactions within the intact 26S complex, whereas those identified from Rpn11 and Rpt6 purifications would describe the interactions of both free and 20S-bound 19S subcomplexes. In total, 263 unique 19S-19S K-K linkages were quantified from all purifications, with 90 from Rpn11, 95 from Rpt6, and 44 from α7. Of the 44 quantified 19S-19S cross-links from α7-purified proteasomes, 41 showed a significant decrease in stress-treated samples (>2.8∼5.7-fold), whereas the remaining three cross-links exhibited log2 L/H values near but below the cut-off for significant changes (1.76∼1.83-fold decrease) (Fig. 6A). The observed decreases in cross-link abundance suggest that H2O2 stress induced conformational changes in the 19S of the 26S holocomplex, resulting in less cross-linkable residues at protein interaction interfaces that would otherwise be captured by DSSO cross-linking under normal conditions (Fig. 6B). This further supports the hypothesis that an intermediate state of the 26S exists before oxidative stress-triggered disassembly.
Fig. 6.

The structure of the 19S RP is impacted by H2O2 stress. A, Respective distribution of quantified 19S-19S cross-links for proteasomes purified from the three selected baits. All cross-links were significantly reduced in α7-purified proteasomes but varied considerably in proteasomes from Rpt6 and Rpn11 purifications. B, 19S-19S cross-links identified from α7-purified proteasomes were mapped to the 26S proteasome high-resolution structure.
Interestingly, quantitation of intra-19S subcomplex cross-links from Rpn11 and Rpt6 purifications revealed a common pattern that was different from the detected changes of the 20S-20S cross-links from α7 purifications (Fig. 6A). XL-MS analyses of Rpn11-purified proteasomes identified 64 unchanged, 15 decreased and 11 increased 19S-19S cross-links, and the same analysis for Rpt6-purified proteasomes yielded 61 unchanged, 27 decreased and 7 increased 19S-19S cross-links (Supplemental Table S5A–S5B). Although 100% of the 20S-20S cross-links from α7 purifications remained unchanged, nearly a third of all 19S-19S cross-links quantified from Rpn11 and Rpt6 purifications displayed oxidative stress-dependent changes in abundances. These results suggest that the 19S RP is more fragile than the 20S and is more prone to change in response to oxidative stress. This observation is certainly plausible as the 19S has been shown to be conformationally heterogeneous, and therefore more dynamic (10, 43, 44). We also evaluated the impact of cross-links derived from free 19S on the quantified L/H ratio of each linkage by estimating the relative amounts of free and 20S-bound 19S RP in proteasomes purified from Rpn11 and Rpt6. As determined through iBAQ calculations, the ratios of 19S to 20S in Rpn11-purified proteasomes was found to be ∼2:1 from control cells. H2O2 treatment increased the relative abundances of 19S and 20S to 4.5:1, corresponding to a 26S dissociation of roughly 55%. In proteasomes purified from Rpt6, the relative abundance ratios of 19S to 20S subunits in control and treated cells were roughly 1:1 and 2.5:1, respectively—representing a dissociation rate of 60% (data not shown). These label-free calculations correlate well with the H2O2-induced dissociation of 26S observed through SILAC quantitation (supplemental Fig. S2 and supplemental Table S4). Altogether, this indicates that the relative abundance of free to 20S-bound 19S RP is significantly lower than that of free to 19S-bound 20S CP. In other words, a higher percentage of cellular 19S RP is complexed to form the 26S proteasome. As a result, quantitative values of cross-links describing oxidative stress-dependent changes within the 19S RP of 26S proteasomes purified by Rpn11 and Rpt6 would be impacted less by the presence of cross-links from co-purified free 19S RP. In total, 25 intra-19S linkages were commonly quantified from all three baits. From α7 purifications, all 25 interactions exhibited decreases greater than 2-fold in favor of untreated proteasomes. In comparison, quantitation in Rpn11- and Rpt6-purified proteasomes exhibited more moderate SILAC ratios, most indicative of no change. Only four K-K linkages were observed to decrease significantly in all three baits; Rpt6:K88-Rpt6:K222, Rpt6:K130-Rpt6:K222, Rpn1:K858-Rpt1:K402, and Rpt2:K178-Rpt6:K287. However, the degree of change was less pronounced in proteasomes from Rpn11 and Rpt6. For the remaining 21 linkages, their quantitative changes observed in Rpn11- and Rpt6-derived proteasomes ranged widely (log2 L/H ≤ 1), implying that the cross-linkability of free 19S played a substantial role in influencing the SILAC ratio describing each linkage. Clearly, the quantitative ratios of intra-19S subcomplex cross-links obtained from 20S subunit-purified proteasomes are best-suited to describe conformational changes in the 19S RP of intact 26S. These results indeed suggest that there are observable conformational changes of the 26S proteasome following H2O2 treatment.
DISCUSSION
In this work, we have developed a new QXL-MS strategy to delineate H2O2-mediated conformational changes of the human 26S proteasome, thus enhancing our understanding of its regulation on oxidative stress. This strategy integrates sequential in vivo formaldehyde and in vitro DSSO cross-linking, HB-tag based affinity purification, multistage tandem mass spectrometry, and SILAC-based quantitation, enabling effective purification of better-preserved proteasome complexes for subsequent quantitative XL-MS analysis to dissect their structural dynamics in response to oxidative stress. DSSO-based XL-MS analyses identified 746 unique K-K linkages (corresponding to 392 intersubunit and 354 intrasubunit interactions), covering ∼54% of previous in vitro and in vivo DSSO XL-MS results. In addition, 55 linkages describe 29 intersubunit interactions that have not been reported before. The differences in cross-link identification are most likely because of the usage of different instruments in these studies, as well as variations in cross-linking strategy. Nonetheless, the substantial overlap suggests the robustness of DSSO cross-linking, and that 2-step cross-linking is as effective as single-step XL-MS analysis. SILAC-based quantitation has quantified 343 unique K-K linkages (corresponding to 180 intersubunit and 163 intrasubunit interactions), permitting determination of interaction regions within the 26S proteasome that are susceptible to change on H2O2 stress. Our results have demonstrated the effectiveness of the integrated method in capturing, identifying and quantifying dynamic interactions of protein complexes, which represents a general QXL-MS strategy for probing interaction and structural dynamics of protein complexes under different physiological conditions.
Our QXL-MS results show that observable conformational changes occur throughout the landscape of the 26S proteasome in the presence of H2O2. The ATPase ring (both the apical surface bordering the 19S lid and the basal surface interacting with the 20S subcomplex) represents one such region that undergoes significant oxidative stress-dependent structural alteration. In particular, the 19S and 20S connectivity is compromised, as evidenced by significant decreases of all quantified 19S-20S cross-links. Cumulatively, these changed cross-links describe at least one intermediary proteasome state in which the 19S and 20S subcomplexes remain tethered as a single complex but display signs of structural remodeling. Interestingly, the complete absence of intersubunit 19S-20S cross-links with increased abundance in H2O2-treated samples implies that the interface between the 19S and 20S is not simply rotated relative to the long axis of the proteasome. One can speculate that such a lateral rotation would result in disfavoring one subset of cross-links while favoring another. Instead, all quantified cross-links between 19S and 20S subunits were observed to decrease following oxidative stress. This indicates that the interaction between the 19S and 20S is most likely weakened, resulting in a uniform reduction of intersubcomplex cross-linking in that region. For instance, divergence of the 19S RP and 20S CP along the proteasomal long axis could increase the distance between normally proximal (and cross-linkable) lysine residues. Mapping of these intersubcomplex interactions shows that cross-linked 20S residues predominantly localized to the edge directly juxtaposed to the 19S. Furthermore, these residues were located near the intersubunit gaps between individual α proteins, which are the reported interaction sites of the α ring with the C-terminal domains of Rpt proteins (45, 46). C-terminal residues of Rpt1, Rpt2, Rpt6 were found participating in these cross-links, suggesting that cross-linking was able to capture the proximal residues near known 19S-20S contact sites. Perturbations of the relative alignment between 19S and 20S subcomplexes because of protein oxidation could play a role in proteasomal down-regulation because of the integral role of 19S in gate opening, unfolding, and transfer of ubiquitinated proteins into the 20S catalytic core. Presumably, dissociation of the 26S would represent a biological response to improve proteasomal activity during oxidative stress conditions, allowing cells to temporarily adapt to oxidative stress and recover when stress conditions are withdrawn, as previously reported (16).
Compared with 19S-20S cross-links, quantitative analysis of cross-links within 19S and 20S subcomplexes is significantly more cumbersome to analyze because of the compositional and conformational heterogeneity of protein species. Nevertheless, we have teased out changed and unchanged cross-links within intact 26S proteasomes by considering bait-dependent proteasome populations. Such analyses have permitted the localization of changed regions based on relative cross-link abundances to areas such as the solvent-accessible surface of the 20S α ring and regions correlating to 19S lid and base connectivity. In α7-purified proteasomes, significant decreases in 19S-19S cross-links were observed in the ATPase ring of the 19S base, suggesting increased conformational heterogeneity within that region. In addition, changes in cross-links containing several non-ATPase subunits of the 19S (i.e. Rpn3, Rpn6, and Rpn7) that are implicated in proteasome assembly were also observed. Rpn3 and Rpn7 comprise different intermediate subcomplexes involved in lid assembly, with Rpn3 playing an integral role in completing the assembly process through its interaction with Rpn5 (47). Other studies have suggested the pivotal role of Rpn6 and Rpn7 in stabilizing the interaction between the 19S RP and 20S CP (48–50). Similarly, decreases in inter- and intrasubunit cross-links within the 20S CP were also identified throughout the α ring in Rpn11- and Rpt6-purified proteasomes. Changes were observed across all α subunits, implying that oxidative stress-dependent conformational changes occurred within and between individual 20S subunits. Although the intersection of these individual topological observations remains to be determined, QXL-MS has nevertheless permitted the detection of oxidative stress-dependent conformational changes of the proteasome from heterogeneous protein complex mixtures. Collectively, our results suggest that H2O2 stress modulates the 19S-20S, 19S-19S and 20S-20S interactions within the 26S proteasome, leading to at least one intermediate state before its full disassembly (15, 16).
Multiple lines of evidence have demonstrated the critical role of Ecm29 in modulating proteasome structure and function (15, 16, 51–55). Specifically, Ecm29 is critical for H2O2-mediated 26S disassembly (15, 16). Our recent XL-MS analyses of Ecm29–26S proteasome complexes have revealed that Ecm29 interacts with five different proteasome subunits (Rpt1, Rpt4, Rpt5, Rpn1 and Rpn10), localizing Ecm29 to the base subcomplex of the 19S regulatory particle (15). It is noted that the cross-links involving Rpn1, Rpt1, Rpt4, and Rpt5 at their Ecm29 binding sites were decreased significantly in H2O2-treated samples, suggesting conformational changes that may attribute to Ecm29 recruitment to the 26S proteasome on oxidative stress. It has been suggested that Ecm29's recruitment to the proteasome relies on its ability to recognize aberrant conformations of the proteasome, specifically those involving misalignment between the 20S core and 19S regulatory particle (45, 53–55). Therefore, our results suggest that H2O2-mediated intermediate state(s) of the 26S proteasome may be the prerequisite for Ecm29 recruitment and subsequent 26S proteasome disassembly.
CONCLUSION
In summary, we have developed and employed a QXL-MS strategy to examine the oxidative stress-triggered structural changes of the human 26S proteasome. Although XL-MS methodologies have been successfully used in conjunction with high-resolution structures in integrative structural biology workflows to delineate protein and protein complex structures, utilization of QXL-MS data to tease apart structural dynamics of complex structures represents a relatively unexplored avenue of research. Here, we have demonstrated the feasibility of the integrated QXL-MS approach, which enabled us to identify various topological events describing the impact of acute oxidative stress on the 26S proteasome structure before its full dissociation. Because of the heterogeneity in protein complexes, quantitation of cross-links must be properly normalized and carefully examined to derive biological relevant information. It is important to note that such structural information cannot be easily assessed by other analytical tools. This QXL-MS strategy presented here can be directly adopted for in vivo studies of other protein complexes, and its throughput can be significantly improved when coupled with isobaric reagent-based multiplexed quantitative strategies (35) in future studies.
DATA AVAILABILITY
Raw data has been deposited at the FTP site: ftp://MSV000083052@massive.ucsd.edu.
Supplementary Material
Acknowledgments
We thank Drs. A.L. Burlingame and Robert Chalkley for the developmental version of Protein Prospector.
Footnotes
* This work was supported by National Institutes of Health grants R01GM074830 to L.H.
 This article contains supplemental Figures and Tables.
This article contains supplemental Figures and Tables.
1 The abbreviations used are:
- ROS
- reactive oxygen species
- 19S RP
- 19S regulatory particle
- 20S CP
- 20S core particle
- PPI
- Protein-protein interaction
- CID
- Collision-induced dissociation
- HCD
- Higher energy collisional dissociation
- DSSO
- Disuccinimidyl sulfoxide
- FDR
- False discovery rate
- HB
- Histidine-biotin
- LC MS
- Liquid chromatography-mass spectrometry
- MS/MS
- Tandem mass spectrometry
- MSn
- Multi-stage tandem mass spectrometry
- QTAX
- Quantitative analysis of tandem affinity-purified in vivo cross(x)-linked protein complexes
- QXL-MS
- Quantitative cross-linking mass spectrometry
- SILAC
- Stable isotope-labeling of amino acids in cell culture
- XL-MS
- Cross-linking mass spectrometry.
REFERENCES
- 1. Uttara B., Singh A. V., Zamboni P., and Mahajan R. T. (2009) Oxidative stress and neurodegenerative diseases: a review of upstream and downstream antioxidant therapeutic options. Curr. Neuropharmacol. 7, 65–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. He J., Cui L, Zeng Y., Wang G., Zhou P., Yang Y., Ji L., Zhao Y., Chen J., Wang Z, Shi T., Zhang P., Chen R., and Li X. (2012) REGgamma is associated with multiple oncogenic pathways in human cancers. BMC Cancer 12, 75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Breusing N., and Grune T. (2008) Regulation of proteasome-mediated protein degradation during oxidative stress and aging. Biol. Chem. 389, 203–209 [DOI] [PubMed] [Google Scholar]
- 4. Aiken C. T., Kaake R. M., Wang X., and Huang L. (2011) Oxidative stress-mediated regulation of proteasome complexes. Mol. Cell Proteomics 10, R110.006924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Voges D., Zwickl P., and Baumeister W. (1999) The 26S proteasome: a molecular machine designed for controlled proteolysis. Annu. Rev. Biochem. 68, 1015–1068 [DOI] [PubMed] [Google Scholar]
- 6. Finley D. (2009) Recognition and processing of ubiquitin-protein conjugates by the proteasome. Annu. Rev. Biochem. 78, 477–513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lasker K., Förster F., Bohn S., Walzthoeni T., Villa E., Unverdorben P., Beck F., Aebersold R., Sali A., and Baumeister W. (2012) Molecular architecture of the 26S proteasome holocomplex determined by an integrative approach. Proc. Natl. Acad. Sci. U.S.A. 109, 1380–1387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Schweitzer A., Aufderheide A., Rudack T., Beck F., Pfeifer G., Plitzko J. M., Sakata E., Schulten K., Förster F., and Baumeister W. (2016) Structure of the human 26S proteasome at a resolution of 3.9 A. Proc. Natl. Acad. Sci. U.S.A. 113, 7816–7821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Huang X., Luan B., Wu J., and Shi Y. (2016) An atomic structure of the human 26S proteasome. Nat. Struct. Mol. Biol. 23, 778–785 [DOI] [PubMed] [Google Scholar]
- 10. Bard J. A. M., Goodall E. A., Greene E. R., Jonsson E., Dong K. C., and Martin A. (2018) Structure and Function of the 26S Proteasome. Annu. Rev. Biochem. 87, 697–724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ben-Nissan G., and Sharon M. (2014) Regulating the 20S proteasome ubiquitin-independent degradation pathway. Biomolecules 4, 862–884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kaake R. M., Wang X., and Huang L. (2010) Profiling of protein interaction networks of protein complexes using affinity purification and quantitative mass spectrometry. Mol. Cell Proteomics 9, 1650–1665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Grune T., Catalgol B., Licht A., Ermak G., Pickering A. M., Ngo J. K., and Davies K. J. (2011) HSP70 mediates dissociation and reassociation of the 26S proteasome during adaptation to oxidative stress. Free Radic. Biol. Med. 51, 1355–1364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Livnat-Levanon N., Kevei É., Kleifeld O., Krutauz D., Segref A., Rinaldi T., Erpapazoglou Z., Cohen M., Reis N., Hoppe T., and Glickman M. H. (2014) Reversible 26S proteasome disassembly upon mitochondrial stress. Cell Rep. 7, 1371–1380 [DOI] [PubMed] [Google Scholar]
- 15. Wang X., Chemmama I. E., Yu C., Huszagh A., Xu Y., Viner R., Block S. A., Cimermancic P., Rychnovsky S. D., Ye Y., Sali A., and Huang L. (2017) The proteasome-interacting Ecm29 protein disassembles the 26S proteasome in response to oxidative stress. J. Biol. Chem. 292, 16310–16320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wang X., Yen J., Kaiser P., and Huang L. (2010) Regulation of the 26S proteasome complex during oxidative stress. Sci. Signal. 3, p. ra88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Beck F., Unverdorben P., Bohn S., Schweitzer A., Pfeifer G., Sakata E., Nickell S., Plitzko J. M., Villa E., Baumeister W., and Förster F. (2012) Near-atomic resolution structural model of the yeast 26S proteasome. Proc. Natl. Acad. Sci. U.S.A. 109, 14870–14875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kao A., Randall A., Yang Y., Patel V.R., Kandur W., Guan S., Rychnovsky S. D., Baldi P., and Huang L. (2012) Mapping the structural topology of the yeast 19S proteasomal regulatory particle using chemical cross-linking and probabilistic modeling. Mol. Cell Proteomics 11, 1566–1577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wang X., Cimermancic P., Yu C., Schweitzer A., Chopra N., Engel J. L., Greenberg C., Huszagh A. S., Beck F., Sakata E., Yang Y., Novitsky E. J., Leitner A., Nanni P., Kahraman A., Guo X., Dixon J. E., Rychnovsky S. D., Aebersold R., Baumeister W., Sali A., and Huang L. (2017) Molecular details underlying dynamic structures and regulation of the human 26S proteasome. Mol. Cell Proteomics 16, 840–854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Yu C., and Huang L. (2018) Cross-linking mass spectrometry: an emerging technology for Interactomics and Structural Biology. Anal. Chem. 90, 144–165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Fischer L., Chen Z. A., and Rappsilber J. (2013) Quantitative cross-linking/mass spectrometry using isotope-labelled cross-linkers. J. Proteomics 88, 120–128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Schmidt M., and Finley D. (2014) Regulation of proteasome activity in health and disease. Biochim. Biophys. Acta 1843, 13–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Yu C., Mao H., Novitsky E. J., Tang X., Rychnovsky S. D., Zheng N., and Huang L. (2015) Gln40 deamidation blocks structural reconfiguration and activation of SCF ubiquitin ligase complex by Nedd8. Nat. Commun. 6, 10053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chavez J. D., Schweppe D. K., Eng J. K., and Bruce J. E. (2016) In vivo conformational dynamics of Hsp90 and its interactors. Cell Chem. Biol. 23, 716–726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Tan D., Li Q., Zhang M. J., Liu C., Ma C., Zhang P., Ding Y. H., Fan S. B., Tao L., Yang B., Li X., Ma S., Liu J., Feng B., Liu X., Wang H. W., He S. M., Gao N., Ye K., Dong M. Q., and Lei X. (2016) Trifunctional cross-linker for mapping protein-protein interaction networks and comparing protein conformational states. Elife 5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zhong X., Navare A. T., Chavez J. D., Eng J. K., Schweppe D. K., and Bruce J. E. (2017) Large-scale and targeted quantitative cross-linking MS using isotope-labeled protein interaction reporter (PIR) cross-linkers. J. Proteome Res. 16, 720–727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Walzthoeni T., Joachimiak L. A., and Rosenberger G. (2015) xTract: software for characterizing conformational changes of protein complexes by quantitative cross-linking mass spectrometry. 12, 1185–1190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Chen Z. A., Pellarin R., and Fischer L. (2016) Structure of complement C3(H2O) revealed by quantitative cross-linking/mass spectrometry and modeling 15, 2730–2743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Guerrero C., Tagwerker C., Kaiser P., and Huang L. (2006) An integrated mass spectrometry-based proteomic approach: quantitative analysis of tandem affinity-purified in vivo cross-linked protein complexes (QTAX) to decipher the 26 S proteasome-interacting network. Mol. Cell Proteomics 5, 366–378 [DOI] [PubMed] [Google Scholar]
- 30. Kao A., Chiu C. L., Vellucci D., Yang Y., Patel V. R., Guan S., Randall A., Baldi P., Rychnovsky S. D., and Huang L. (2011) Development of a novel cross-linking strategy for fast and accurate identification of cross-linked peptides of protein complexes. Mol. Cell Proteomics 10, M110.002212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Liu F., Rijkers D. T., Post H., and Heck A. J. (2015) Proteome-wide profiling of protein assemblies by cross-linking mass spectrometry. Nat. Methods 12, 1179–1184 [DOI] [PubMed] [Google Scholar]
- 32. Wang X., Chen C. F., Baker P. R., Chen P. L., Kaiser P., and Huang L. (2007) Mass spectrometric characterization of the affinity-purified human 26S proteasome complex. Biochemistry 46, 3553–3565 [DOI] [PubMed] [Google Scholar]
- 33. Wang X., and Huang L. (2018) dissecting dynamic and heterogeneous proteasome complexes using in vivo cross-linking-assisted affinity purification and mass spectrometry. Methods Mol. Biol. 1844, 401–410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Cox J., Matic I., Hilger M., Nagaraj N., Selbach M., Olsen J. V., and Mann M. (2009) A practical guide to the MaxQuant computational platform for SILAC-based quantitative proteomics. Nat. Protoc. 4, 698–705 [DOI] [PubMed] [Google Scholar]
- 35. Yu C., Huszagh A., Viner R., Novitsky E. J., Rychnovsky S. D., and Huang L. (2016) Developing a multiplexed quantitative cross-linking mass spectrometry platform for comparative structural analysis of protein complexes. Anal. Chem. 88, 10301–10308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Yu C., Kandur W., Kao A., Rychnovsky S., and Huang L. (2014) Developing new isotope-coded mass spectrometry-cleavable cross-linkers for elucidating protein structures. Anal. Chem. 86, 2099–2106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kaake R. M., Wang X., Burke A., Yu C., Kandur W., Yang Y., Novtisky E. J., Second T., Duan J., Kao A., Guan S., Vellucci D., Rychnovsky S. D., and Huang L. (2014) A new in vivo cross-linking mass spectrometry platform to define protein-protein interactions in living cells. Mol. Cell Proteomics 13, 3533–3543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Gutierrez C. B., Yu C., Novitsky E. J., Huszagh A. S., Rychnovsky S. D., and Huang L. (2016) Developing an acidic residue reactive and sulfoxide-containing MS-cleavable homobifunctional cross-linker for probing protein-protein interactions. Anal. Chem. 88, 8315–8322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Aoshima K., Takahashi K., Ikawa M., Kimura T., Fukuda M., Tanaka S., Parry H. E., Fujita Y., Yoshizawa A. C., Utsunomiya S., Kajihara S., Tanaka K., and Oda Y. (2014) A simple peak detection and label-free quantitation algorithm for chromatography-mass spectrometry. BMC Bioinformatics 15, 376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Guerrero C., Milenkovic T., Przulj N., Kaiser P., and Huang L. (2008) Characterization of the proteasome interaction network using a QTAX-based tag-team strategy and protein interaction network analysis. Proc. Natl. Acad. Sci. U.S.A. 105, 13333–13338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Yu C., Yang Y., Wang X., Guan S., Fang L., Liu F., Walters K.J., Kaiser P., and Huang L. (2016) Characterization of dynamic UbR-proteasome subcomplexes by in vivo cross-linking (X) assisted bimolecular tandem affinity purification (XBAP) and label-free quantitation. Mol. Cell Proteomics 15, 2279–2292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Verma R., Aravind L., Oania R., McDonald W. H., Yates J. R. 3rd, Koonin E. V., and Deshaies R. J. (2002) Role of Rpn11 metalloprotease in deubiquitination and degradation by the 26S proteasome. Science 298, 611–615 [DOI] [PubMed] [Google Scholar]
- 43. Wehmer M., Rudack T., Beck F., Aufderheide A., Pfeifer G., Plitzko J.M., Förster F., Schulten K., Baumeister W., and Sakata E. (2017) Structural insights into the functional cycle of the ATPase module of the 26S proteasome. Proc. Natl. Acad. Sci. U.S.A. 114, 1305–1310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Zhu Y., Wang W. L., Yu D., Ouyang Q., Lu Y., and Mao Y. (2018) Structural mechanism for nucleotide-driven remodeling of the AAA-ATPase unfoldase in the activated human 26S proteasome. Nat. Commun. 9, 1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Gillette T. G., Kumar B., Thompson D., Slaughter C. A., and DeMartino G. N. (2008) Differential roles of the COOH termini of AAA subunits of PA700 (19 S regulator) in asymmetric assembly and activation of the 26 S proteasome. J. Biol. Chem. 283, 31813–31822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Tian G., Park S., Lee M. J., Huck B., McAllister F., Hill C. P., Gygi S. P., and Finley D. (2011) An asymmetric interface between the regulatory and core particles of the proteasome. Nat. Struct. Mol. Biol. 18, 1259–1267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Estrin E., Lopez-Blanco J. R., Chacón P., and Martin A. (2013) Formation of an intricate helical bundle dictates the assembly of the 26S proteasome lid. Structure 21, 1624–1635 [DOI] [PubMed] [Google Scholar]
- 48. Isono E., Saeki Y., Yokosawa H., and Toh-e A. (2004) Rpn7 Is required for the structural integrity of the 26 S proteasome of Saccharomyces cerevisiae. J. Biol. Chem. 279, 27168–27176 [DOI] [PubMed] [Google Scholar]
- 49. Isono E., Saito N., Kamata N., Saeki Y., and Toh-E A. (2005) Functional analysis of Rpn6p, a lid component of the 26 S proteasome, using temperature-sensitive rpn6 mutants of the yeast Saccharomyces cerevisiae. J. Biol. Chem. 280, 6537–6547 [DOI] [PubMed] [Google Scholar]
- 50. Pathare G. R., Nagy I, Bohn S, Unverdorben P, Hubert A, Körner R, Nickell S, Lasker K, Sali A, Tamura T, Nishioka T, Förster F, Baumeister W, and Bracher A. (2012) The proteasomal subunit Rpn6 is a molecular clamp holding the core and regulatory subcomplexes together. Proc. Natl. Acad. Sci. U.S.A. 109, 149–154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Leggett D. S., Hanna J., Borodovsky A., Crosas B., Schmidt M., Baker R.T., Walz T., Ploegh H., and Finley D. (2002) Multiple associated proteins regulate proteasome structure and function. Mol. Cell 10, 495–507 [DOI] [PubMed] [Google Scholar]
- 52. Kleijnen M. F., Roelofs J., Park S., Hathaway N. A., Glickman M., King R. W., and Finley D. (2007) Stability of the proteasome can be regulated allosterically through engagement of its proteolytic active sites. Nat. Struct. Mol. Biol. 14, 1180–1188 [DOI] [PubMed] [Google Scholar]
- 53. Park S., Kim W., Tian G., Gygi S. P., and Finley D. (2011) Structural defects in the regulatory particle-core particle interface of the proteasome induce a novel proteasome stress response. J. Biol. Chem. 286, 36652–36666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Lee S. Y., De la Mota-Peynado A., and Roelofs J. (2011) Loss of Rpt5 protein interactions with the core particle and Nas2 protein causes the formation of faulty proteasomes that are inhibited by Ecm29 protein. J. Biol. Chem. 286, 36641–36651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. De La Mota-Peynado A., Lee S. Y., Pierce B. M., Wani P., Singh C. R., and Roelofs J. (2013) The proteasome-associated protein Ecm29 inhibits proteasomal ATPase activity and in vivo protein degradation by the proteasome. J. Biol. Chem. 288, 29467–29481 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw data has been deposited at the FTP site: ftp://MSV000083052@massive.ucsd.edu.