

Abstract
Abstract. Objectives: To generate non‐haematopoietic tissues from mobilized haematopoietic CD133+ stem cells. Materials and methods: Mobilized peripheral blood CD133+ cells from adult healthy donors were used. In vitro ability of highly enriched CD133+ cells from mobilized peripheral blood to generate multipotent cells, and their potential to give rise to cells with characteristics of neuroectoderm, endoderm and mesoderm layers was investigated. Results: We found that a recently identified population of CD45+ adherent cells generated in vitro after culture of highly purified CD133+ cells for 3–5 weeks with Flt3/Flk2 ligand and interleukin‐6 can, in presence of the appropriate microenvironmental cues, differentiate into neural progenitor‐like cells (NPLCs), hepatocyte‐like cells and skeletal muscle‐like cells. We have termed them to be adult multipotent haematopoietic cells (AMHCs). AMHC‐derived NPLCs expressed morphological, phenotypic and molecular markers associated with primary neural progenitor cells. They can differentiate into astrocyte‐like cells, neuronal‐like cells and oligodendrocyte‐like cells. Moreover, AMHC‐derived NPLCs produced 3,4‐dihydrophenylalanine and dopamine and expressed voltage‐activated ion channels, suggesting their functional maturation. In addition, AMHC‐derived hepatocyte‐like cells and skeletal muscle‐like cells, showed typical morphological features and expressed primary tissue‐associated proteins. Conclusion: Our data demonstrate that AMHCs may therefore serve as a novel source of adult multipotent cells for autologous replacement cell therapies.
INTRODUCTION
Developmental studies over the last few decades have provided a large body of evidence for the existence of stem cells in different tissues of the organism even when still an embryo (Thomson et al. 1998), adult skin (Toma et al. 2001), nervous system (Stemple & Anderson 1992), pancreas (Seaberg et al. 2004), heart (Beltrami et al. 2003), liver (Yang et al. 2002), bone marrow (Jiang et al. 2002) and umbilical cord blood (Kogler et al. 2004). Odorico et al. (2001) showed that embryonic stem cells can differentiate into derivatives of all three embryonic germ layers when transferred to an in vivo environment. In addition, they demonstrated that human embryonic stem cells can initiate lineage‐specific differentiation of many tissue and cell types in vitro. Based on this property, it is likely that human embryonic stem cells will provide a useful differentiation culture system to study the mechanisms underlying many facets of human development. Until recently, it has been thought that segregation of cells into particular germ layers during embryogenesis is an enduring event, and that with few exceptions, all subsequently arising cells, including mature cells, progenitor cells and stem cells of each of the resulting tissue lineages, irreversibly maintained this specification into and throughout adulthood. However, recent reports have challenged this notion and called into question the lineage commitment of various adult stem cell populations by suggesting that under certain circumstances these cells may transdifferentiate to contribute to a much wider spectrum of differentiated progeny than previously anticipated (Wagers & Weissman 2004). Krause et al. (2001) showed multiorgan, multilineage engraftment after transplantation of a single bone marrow‐derived stem cell into primary and secondary recipients. These adult bone marrow cells have tremendous differentiational capacity as they can also differentiate into epithelial cells of the liver, lung, gastrointestinal tract and skin. Verfaillie and colleagues (Jiang et al. 2002) demonstrated that the bone marrow contains a population of cells designated multipotential adult progenitor cells (MAPCs). These adherent CD45− glycophorin A− cells in common with the previously discovered bone marrow‐derived mesenchymal stem cells are capable of differentiating into mesodermal derivatives (bone, cartilage, muscle and fat), neuroectodermal cells (neurons) and endodermal cells (hepatocytes). D’Ippolito et al. (2004) isolated a class of pluripotent mesenchymal CD45− cell (MIAMI cells) that differentiated in vitro into mesodermal derivatives as well as into neural cells, and can be directed to express genes associated with pancreatic islets. MIAMI cells were recovered from human donors of ages ranging from 3 to 72 years. Miller's group (Toma et al. 2001) identified a skin progenitor cell, which is able to differentiate into neuroectodermal and mesodermal derivatives, including smooth muscle, neurons, glia and adipocytes. Unrestricted somatic stem cells have been obtained from placental cord blood at 34–42 weeks gestational age in approximately 40% of pregnancies examined and were found to be pluripotent in vitro. In addition, they were examined in a non‐injury, pre‐immune foetal sheep model; on inoculation into a sheep embryo, these cells contributed to the liver parenchyma and heart muscle (Kogler et al. 2004). These studies have shown that non‐haematopoietic cells can give rise to one or more germ layer tissues. However, there are controversial data on the issue of whether haematopoietic cells are able to cross tissue boundaries and differentiate from a mesodermal lineage into tissues of the other germ layers. It has been shown that purified haematopoietic stem cells are the only bone marrow cells to give rise to hepatocytes in vivo (Lagasse et al. 2000). In a following report, the same group demonstrated that cell fusion was the principal source of bone marrow‐derived hepatocytes and not the differentiation of haematopoietic stem cells into hepatocytes (Wang et al. 2003). In contrast, Jang et al. (2004) showed that mouse haematopoietic stem cells converted in vivo into hepatocytes, within days, without fusion.
We recently identified a novel population of adherent cells generated in vitro after culture of highly purified CD133+ cells, for 3–5 weeks, with Flt3/Flk2 ligand (FL) and interleukin‐6 (IL‐6) (Handgretinger et al. 2003; Kuci et al. 2003). In that study, adherent cells gave rise to a non‐adherent cell fraction, which after transplantation into non‐obese diabetic/severe combined immunodeficiency mice, generated multilineage human haematopoiesis. Having in mind all controversial data on differentiation potential of haematopoietic stem cells, we asked whether these haematopoietic adherent cells would be able to differentiate into cells of other germ layers. We addressed this question by culturing them in appropriate induction media and characterizing their morphology, gene expression profile, phenotype and functional maturation of cells generated. Here, we show that these haematopoietic adherent cells possess the potential to differentiate in vitro into non‐haematopoietic tissues with characteristics of neuroectodermal (neural progenitor‐like cells), mesodermal (skeletal muscle‐like cells) and endodermal (hepatocyte‐like cells) layers. In addition to their typical morphology and activation of tissue‐specific sets of genes, these cells expressed tissue‐specific proteins and in the case of neural progenitor‐like cells, they showed functional maturation by producing significant amounts of neurotransmitters 3,4‐dihydrophenylalanine (DOPA) and dopamine, and expression of voltage‐activated ion channels. Because of their potential to give rise to different cell types in vitro, we termed them ‘adult multipotent haematopoietic cells’, and thus propose that they may represent a novel source of autologous multipotent cells for cell replacement therapy.
MATERIALS AND METHODS
Positive selection of CD133+ cells
Mobilized peripheral blood mononuclear cells were obtained from healthy donors who gave informed consent according to the guidelines of the Ethics Committee of the University of Tübingen, Tübingen, Germany. CD133+ cells were positively selected from the light‐density mononuclear cell population by magnetic‐activated cell sorting with the AC133 cell isolation kit (Miltenyi Biotec, Bergisch Gladbach, Germany). Purity of the CD133+ cell population was evaluated using a FACSCalibur flow cytometer (Becton Dickinson, Heidelberg, Germany). No CD45− cells in the initial CD133+ cell population were observed.
Generation of AMHCs from highly purified CD133+ cells
We generated adult multipotent haematopoietic cells (AMHCs) as previously described (Kuci et al. 2003). Briefly, 2 × 105 highly purified CD133+ cells per cm2 were cultured in expansion medium consisting of RPMI‐1640 supplemented with 10% foetal bovine serum and 100 ng/mL FL and IL‐6 (R&D Systems, Wiesbaden‐Nordenstadt, Germany). On day 7 of culture, half the expanded cell population was transferred to a further flask containing fresh RPMI‐1640 supplemented with 10% foetal bovine serum and cells were cultured at 37 °C, 5% CO2 for 2–4 weeks. AMHCs were generated from 20 enriched CD133+ cell samples (100%) from 20 healthy blood cell‐mobilized donors, suggesting that CD133+ cells represent a very consistent source of multipotent cells. After a monolayer of adherent cells was generated, cells were treated with trypsin, phenotyped by flow cytometry for the expression of CD45 and other antigens and were further used for differentiation studies.
Banding cytogenetics
Metaphase spreads were prepared from AMHCs and standard GTG‐banding was performed. Chromosomes of 17 AMHCs were karyotyped by using the Ikaros system (Metasystems, Altlussheim, Germany).
Generation of neural progenitor‐like cells from AMHCs
After adherent AMHCs were trypsinized and washed twice with phosphate‐buffered saline, they were cultured at a density of 2.5 × 104 cells per cm2 in a Lab‐Tek Slide Flaskette (Nunc, Wiesbaden, Germany) for 7–14 days in serum‐free neural progenitor basal medium (NPBM) (Cambrex, Biozym Scientific GmbH, Hess Oldendorf, Germany) supplemented with recombinant epidermal growth factor (EGF), human recombinant basic fibroblast growth factor and neural survivor factor‐1. Every 3 days, medium was changed and catecholamine production was assayed by high pressure liquid chromatography (HPLC) in a sample of supernatant. At specific time points, cells were trypsinized and were used for reverse transcriptase‐polymerase chain reaction (RT‐PCR) analysis.
For electrophysiological studies, day 7 AMHC‐derived NPLCs were cultured in the presence of brain‐derived neurotrophic factor (50 ng/mL) and glial‐derived neurotrophic factor (10 ng/mL) for another 3–7 days. To confirm the cells’ neural progenitor potential, day 3 AMHC‐derived NPLCs were trypsinized, washed, and were further cultured in growth media for neurons and astrocytes. To generate more neurons, we cultured 5–7 day AMHC‐derived NPLCs in NPBM supplemented with brain‐derived neurotrophic factor (50 ng/mL) and glial‐derived neurotrophic factor (10 ng/mL). To generate astrocytes, we used commercially available astrocyte growth medium (Cambrex; Biozym Scientific GmbH, Hess Oldendorf, Germany).
Generation of hepatocyte‐like cells from AMHCs
Adult multipotent haematopoietic cells (2.5 × 104/cm2) were cultured for 1 week in hepatocyte basal medium (Cambrex, Biozym Scientific GmbH) supplemented with growth factors [ascorbic acid, bovine serum albumin‐fatty acid free (BSA‐FAF), transferrin, insulin, hEGF, GA‐1000]. Cells were then cultured for 2 weeks in hepatocyte maintenance medium (Cambrex; Biozym Scientific GmbH). For immunocytochemical analysis, AMHCs were cultured on Lab‐Tek (Nunc, Wiesbaden, Germany) chamber slides.
Generation of skeletal muscle‐like cells from AMHCs
Adult multipotent haematopoietic cells (2.5 × 104/cm2) were cultured for 7–14 days in human skeletal muscle medium (Cambrex; Biozym Scientific GmbH) supplemented with hEGF, insulin, fetuin, dexamethasone, l‐glutamine, BSA and gentamicin/amphotericin B. Medium was changed every 3 days.
RT‐PCR analysis of catecholamine gene expression in AMHC‐derived NPLCs
Total RNA was extracted from 14 day AMHC‐derived NPLCs using RNeasy spin columns (Qiagen, Hilden, Germany) according to the manufacturer's instructions. cDNAs reverse were transcribed from 2 µg of total RNA using 0.5 µg oligo(dT)12–18 primer and 200 units SuperScript™ II RNase H reverse transcriptase (Invitrogen, Karlsruhe, Germany) according to the supplier's protocol. PCR reactions were performed in 50 µL mixture containing 2 µL cDNA transcribed from 200 ng RNA, 10 mm Tris (pH 8.3), 50 mm KCl, 1.5 mm MgCl2, 200 µm dATP, dGTP, dTTP and dCTP (Invitrogen, Karlsruhe, Germany), 20 pmol of amplimers for glyceraldehyde‐3‐phosphate dehydrogenase (GAPDH), dopamine‐β‐hydroxylase, DOPA‐decarboxylase, tyrosine hydroxylase, noradrenaline transporter (NAT) and dopamine transporter (DAT), and 1.25 U AmpliTaq Gold polymerase (Applied Biosystems, Kassel, Germany). Amplification conditions for the first PCR were: 96 °C for 15 s, 55 °C for 30 s, and 72 °C for 90 s. Number of cycles for the first PCR reaction were: GAPDH, 28; dopamine‐β‐hydroxylase, tyrosine hydroxylase and NAT, 38; DOPA‐decarboxylase, 33; and DAT, 45.
Nested PCR
Two microlitres of product generated by the first PCR were used in nested PCR for all genes except the DAT gene. Nested PCR was performed under the same conditions as the first PCR. Number of cycles was 30 for GAPDH and 38 for all other genes. The following primers were used:
GAPDH PCR1, 5′‐GACAACAGCCTCAAGATCATC‐3′, 5′‐GACGGCAGGTCAGGTCCACCA‐3′, 325 bp; GAPDH nested, 5′‐AATGCCTCCTGCACCACC‐3′, 5′‐ATGCCAGTGAGCTTCCCG‐3′, 247 bp; tyrosine hydroxylase PCR1, 5′‐TGTCAGAGCTGGACAAGT‐3′, 5′‐GATATTGTCTTCCCGGTAGC‐3′, 299 bp; tyrosine hydroxylase nested, 5′‐GTTCGACCCTGACCTGGACT‐3′, 5′‐TGTACTGGAAGGCGATCTCA‐3′, 95 bp. Both internal and external primers for the tyrosine hydroxylase gene were generated from sequences of different exons of the constant region (exons 3–14), in order to avoid interference with possible genomic DNA contamination; dopamine β‐hydroxylase PCR1, 5′‐ATCCACATCTTCGCCTCTCAGC‐3′, 5′‐TCTTCCGTGTTGTACGTGCAGG‐3′, 206 bp; dopamine β‐hydroxylase nested, 5′‐ACACACCTGACTGGGAGAAAGG‐3′, 5′‐CACGACCTTCTTCAACATGCGG‐3′, 127 bp; DOPA‐decarboxylase PCR1, 5′‐ACTGGACCCCACTTACCTGA‐3′, 5′‐GCTGGCTTCACTCCTACTCC‐3′, 476 bp; DOPA‐decarboxylase nested, 5′‐CAGGATTCAGGGCTTATCAC‐3′, 5′‐GCGAGAACAGATGGCAAAG‐3′, 348 bp; dopamine transporter PCR1, 5′‐ATAGACGGCATCAGAGCATACC‐3′, 5′‐ACTTGTTGTAGCTGGAGAAGGC‐3′, 142 bp; noradrenaline transporter PCR1, 5′‐TCTTCTCCTCCTTCACCCTCAACC‐3′, 5′‐GACACCACGCTCATAAAACTCGGC‐3′, 294 bp; noradrenaline transporter nested, 5′‐TCTTCTCCTCCTTCACCCTCAACC‐3′, 5′‐GACACCACGCTCATAAAACTCGGC‐3′, 167 bp; vesicular monoamine transporter 1 PCR1, 5′‐CTGAAGGAGGGGAGAGC‐3′, 5′‐GGGCCACAAAGAGTAGAGT‐3′, 553 bp; vesicular monoamine transporter 1 nested, 5′‐TGTGCCCACCTTCCTATA‐3′, 5′‐TCCAAGAAACCTGTGCCT‐3′, 267 bp; vesicular monoamine transporter 2 PCR1, 5′‐CTTTGGAGTTGGTTTTGC‐3′, 5′‐GCAGTTGTGATCCATGAG‐3′, 301 bp; vesicular monoamine transporter 2 nested, 5′‐TGCAATTGGAATGGTGGAT‐3′, 5′‐GAGAGGGGCAAAAAGAATA‐3′, 220 bp. After the second PCR, 16 µL of amplicons were applied to polyacrylamide gel (8% acrylamide, 0.25% bis acrylamide). Post‐electrophoresis, gels were digitally imaged using a CS‐1 Videoimager (Cybertech, Berlin, Germany).
HPLC determination of DOPA and dopamine production by AMHC‐derived NPLCs
Supernatants of 106 AMHC‐derived NPLCs were collected on days 5, 10 and 14 of culture. HClO4 was added to final concentration of 0.5 M. After centrifugation, supernatants were frozen at –80 °C until HPLC analysis. Two hundred microlitres of each sample were mixed with 10 µL internal standard (3,4‐dihydroxybenzylamine, DHBA; Sigma, Deisenhofen, Germany), loaded on alumina columns, and further treated as instructed by HPLC system manufacturer (Chromsystems, Munich, Germany). Finally, 50 µL of eluted sample were analysed by HPLC, HP 1100 with an HP 1049 electrochemical detection unit (BIORAD, Munich, Germany). Specially prepared mobile phase was created (11.36 g/L trilithium citrate, 1.97 g/L citric acid, 0.3 g/L 1‐octanesulfonic acid, 3% v/v acetonitrile, pH 2.55, adjusted with ortho‐phosphoric acid) that permitted simultaneous determination of DOPA and dopamine.
Electrophysiological measurements
The whole‐cell patch‐clamp method was used to measure electrophysiological events in AMHC‐derived NPLCs. Neural basal medium supplemented with specific neural growth factors (NPBM) was replaced with a HEPES‐buffered artificial cerebrospinal fluid containing 150 mm NaCl, 5 mm KCl, 2 mm CaCl2, 1 mm MgCl2, 10 mm HEPES and 10 mm glucose. Patch electrodes were fashioned from borosilicate capillary tubes (Science Products, Hofheim, Germany) and were filled with solutions containing 130 mm potassium gluconate, 5 mm KCl, 0.6 mm EGTA, 10 mm HEPES, 2 mm MgCl2, pH 7.2 (KOH), 270–290 mOsm. Electrode resistance was 2–5 Mω. Data were not corrected for junction potential of 9 mV. Measurements were made in the voltage clamp configuration at –70 mV by using an Axopatch 200B amplifier, and digitized with a Digidata 1320 (both from Axon Instruments, Union City, CA, USA). Data were filtered with a 10‐kHz low‐pass filter; sampling rate 20 kHz; pClamp 8.0.2 software (Axon Instruments) was used for data acquisition and analysis. Series resistance, membrane capacity and seal quality were thoroughly controlled during recordings.
Immunostaining of AMHC‐derived NPLCs
Seven‐ to ten‐day‐old AMHC‐derived NPLCs were stained with neural‐specific antibodies. Rabbit polyclonal antibody to glial fibrillary acidic protein (GFAP) (1 : 100) was purchased from DAKO (Hamburg, Germany). Monoclonal antineuron‐specific class III β‐tubulin antibody (mouse IgG2a; Clone TuJ‐1, 7.5 µg/mL) was purchased from R&D Systems and polyclonal rabbit antihuman erythropoietin antibody (1 : 500) from DAKO. Pre‐diluted monoclonal mouse antihuman myelin basic protein antibody (Clone B103.1, IgG1) was purchased from Biomeda (Natutec, Frankfurt, Germany), and monoclonal antibody to tyrosine hydroxylase (mouse IgG1, 1 : 100) was purchased from Chemicon (Hofheim, Germany). Secondary antibodies were goat antimouse IgG‐Cy3 (1 : 500), goat antimouse IgG1 FITC (1 : 100), mouse antirabbit IgG FITC (1 : 100), goat antirabbit FITC (1 : 100) and goat antirabbit Cy3 (1 : 500) from Jackson Immuno‐Research Laboratories Inc. (Dianova, Hamburg, Germany). Negative controls were performed by staining cells with secondary antibodies only. Nuclei were stained with 4′,6‐diamidino‐2‐phenylindole (DAPI)‐containing Vectashield mounting medium (Vector Laboratories Inc., Burlingame, CA, USA). Cells were examined by fluorescence microscopy using an Olympus BX60 fluorescence microscope attached to an F‐View digital camera (Olympus Optical Co., Hamburg, Germany). One hundred to three hundred cells per culture were evaluated under low‐power magnification to determine proportions of reactive cells.
Immunostaining of AMHC‐derived hepatocyte‐like cells
Differentiated AMHC‐derived hepatocyte‐like cells were fixed with methanol for 10 min at –20 °C and were permeabilized with 0.1% Triton‐X 100 (Sigma‐Aldrich, Taufkirchen, Germany) for 10 min before assays for nuclear ligands. Slides were incubated for 45 min each with primary antibodies and then with antimouse, antigoat or antirabbit IgG Ab labelled with FITC or Cy3, as appropriate. Between incubations, slides were washed with phosphate‐buffered saline supplemented with 1% BSA. Antibodies to human hepatocyte nuclear factor‐3β (HNF‐3β) and hepatocyte nuclear factor‐1α (HNF‐1α) (Santa Cruz Biotechnology Inc., Heidelberg, Germany) were used at a 1 : 50 dilution. Antibodies to human albumin (mouse IgG2a, 1 : 500), human α‐fetoprotein (αFP, 1 : 500), CK18 (1 : 800), and CK19 (1 : 50) were purchased from Sigma‐Aldrich. Secondary rabbit antigoat IgG FITC (1 : 50) was obtained from DAKO, and goat antimouse IgG Cy3 (1 : 500) from Jackson Immuno‐Research Laboratories. Negative controls were performed by staining cells with secondary antibodies only, at the same dilution as that used for the sample staining. Nuclei were stained with DAPI‐containing Vectashield mounting medium (Vector Laboratories Inc.). Cells were examined by fluorescence microscopy using an Olympus BX60 (Olympus Optical Co.).
RT‐PCR for albumin gene expression in AMHC‐derived hepatocyte‐like cells
Total RNA was extracted from 106 AMHCs cultured for 14 or 23 days in hepatocyte‐specific medium and from HepG2 hepatoma cells (positive control), by using the RNeasy Micro Kit and digestion with DNase I. First‐strand cDNA was synthesized by using SuperScript II reverse transcriptase (Invitrogen) as instructed by the manufacturer. PCR reactions were performed in 50 µL mixture containing 2 µL cDNA transcribed from 200 ng RNA, 10 mm Tris (pH 8.3), 50 mm KCl, 1.5 mm MgCl2, 200 µm dATP, dGTP, dTTP and dCTP (Invitrogen), 20 pmol of amplimers for GAPDH, human albumin and 1.25 U AmpliTaq Gold polymerase (Applied Biosystems). For PCR analysis, GAPDH was used as internal standard. Gene‐specific oligonucleotide primers based on human sequences encoding albumin (162 bp) (Schwartz et al. 2002) and GAPDH (5′‐GACAACAGCCTCAAGATCATC‐3′, 5′‐GACGGCAGGTCAGGTCCACCA‐3′, 325 bp) were used for amplification. A pre‐PCR heat step (95 °C, 5 min) provided a PCR hot start by partially activating AmpliTaq Gold DNA polymerase (Applied Biosystems, Foster City, CA, USA). Each amplification cycle (GAPDH, 27 cycles; albumin, 40 cycles for cultured AMHC‐derived hepatocyte‐like cells and 22 cycles for HepG2) consisted of denaturation for 15 s at 95 °C, annealing for 30 s at 56 °C (GAPDH) or 60 °C (albumin), and elongation for 90 s at 72 °C followed by a final extension step of 7 min at 72 °C. PCR products were separated by polyacrylamide gel electrophoresis and were stained with ethidium bromide.
Immunostaining of AMHC‐derived skeletal muscle‐like cells
Adult multipotent haematopoietic cell‐derived skeletal muscle‐like cells were stained with tissue‐specific antibodies. Primary antibodies to MyoD (rabbit polyclonal, 1 : 200), myogenin (mouse IgG3, 1 : 500) and desmin (goat polyclonal, 1 : 100) were purchased from Santa Cruz Biotechnologies Inc. Antibody to α‐sarcomeric actin (mouse IgM, 1 : 100) was from Sigma‐Aldrich. Secondary antibodies were mouse antirabbit FITC (1 : 100) and goat antimouse Cy3 (1 : 100) (Jackson Immuno‐Research Laboratories) and rabbit antigoat FITC (1 : 100) was purchased from DAKO. Negative controls were performed by staining the cells with secondary antibodies only, at the same dilution as that used for sample staining. Nuclei were stained with DAPI‐containing Vectashield mounting medium (Vector Laboratories Inc.).
RESULTS
The adherent cells used in this study were derived from highly enriched granulocyte colony‐stimulating factor‐mobilized CD133+ cells from healthy donors, which co‐expressed CD34 (Fig. 1a) and CD45 (Fig. 1b). AMHCs derived from CD133+ cells through their culture with FL and IL‐6 for 3–5 weeks, possess distinctive morphology (Fig. 1c), and because of their size (range from 15 to 55 µm) show a typical scatter of large cells (Fig. 1d). AMHCs do not express any of the well‐known haematopoietic stem cell markers, such as CD34, CD133, CD90 and KDR. However, they express considerable levels of vascular endothelial cadherin, CD73, high levels of CD45 and the adhesion molecules CD54 and CD44, as well as high levels of HLA class I and class II molecules plus CD105 (Fig. 1e). Limiting dilution analysis showed that 1 of every 25 CD133+ cells (95% confidence interval, 1 of 20 to 1 of 34 CD133+ cells) under the same culture conditions gave rise to an AMHC (n = 3) (Kuci et al. 2003). Additional analysis of the proportion of CD133+ cells that adhered and gave rise to typical AMHCs through culture with FL and IL‐6 after 3 weeks (AMHC‐progenitor cells) (n = 10) revealed a frequency of 3.6 ± 1.4% (range 2–5%), which very closely correlated with the frequency estimated by limiting dilution analysis. Moreover, no correlation was detected between the age of donors and success of generation of AMHCs. The unique feature of AMHCs in comparison to bone marrow‐derived MAPCs (Reyes et al. 2001) and MIAMI cells (D’Ippolito et al. 2004), embryonic stem cells (Odorico et al. 2001) and unrestricted somatic stem cells (Kogler et al. 2004) that do not express CD45 is that AMHCs are derived from adult mobilized peripheral blood CD133+ cells through their culture for 3–5 weeks; they do not need presence of a feeder cell monolayer and can be generated from each CD133+ sample, and most strikingly expressing high levels of the haematopoietic marker CD45. After in vitro culture with tissue‐specific media, however, these cells down‐regulate CD45 and thus give rise to nonhaematopoietic tissues. For differentiation studies, we used 6–8‐week‐old AMHCs. However, single‐cell experiments have shown that even 6–12‐month‐old AMHCs retain the potential to differentiate into non‐haematopoietic cells (data not shown), and the cells could be cultured and passaged for more than a year without observing chromosomal aberration (Fig. 1f).
Figure 1.

Adult multipotent haematopoietic cells derived from highly enriched CD133+ cells. (a) Flow cytometric analysis of highly enriched CD133+ cells. The purity of the enriched cells was higher than 98%, as evaluated by flow cytometry. (b) All enriched CD133+ cells express CD45. (c) Phase contrast micrograph of 6‐week‐old AMHCs. Because of their size (range from 15 to 55 µm) they show typical scatter of large cells (d). (e) Flow cytometric analysis of the AMHCs. Cells were analysed using a FACSCalibur. Black lines represent isotype controls and red lines indicate specific cell staining. (f) Cytogenetic analysis of 6‐month‐old AMHCs. GTG‐banded chromosome analysis (400 bands per haploid genome) showed normal 46, XY karyotype in all cells analysed.
AMHCs give rise to cells with neuroectodermal characteristics
Because in vitro generation of neural progenitor cells might provide an on‐demand source of cells for grafts in patients with various neurodegenerative diseases, we began by examining the capacity of AMHCs to differentiate into cells with neuroectodermal characteristics. From 10 different donors, we cultured AMHCs under serum‐free conditions in the presence of recombinant human epidermal growth factor (hEGF), basic fibroblast growth factor (bFGF) and neural survivor factor‐1 (NSF‐1) for 7–14 days. Cultured AMHCs from all donors under these conditions gave rise to a neural progenitor‐like cell population with neuroectodermal characteristics (Fig. 2a), suggesting the high potential for AMHCs to adopt neural fate. During the first week of culture, AMHCs extended neurofibrillar processes and expressed markers of neural development. Notably, morphologically distinct neuron‐like cells were readily detected by day 5 in culture, and oligodendrocyte‐like and astrocyte‐like cells were prominent after 7–10 days. By immunocytochemical analysis, most 7–10‐day‐old AMHC‐derived NPLCs (70 ± 15%) expressed β‐tubulin III (found in early neuronal development), and approximately 15 ± 4% of cells expressed GFAP (Fig. 2b). By day 14, 27 ± 8%β‐tubulin III positive cells expressed tyrosine hydroxylase (Fig. 2c). Most GFAP+ cells had the morphology of oligodendrocytes and expressed GFAP and myelin basic protein, which are associated with the astroglial lineage (Fig. 2d).
Figure 2.

AMHC‐derived neural progenitor‐like cells (NPLCs). (a) Phase‐contrast micrograph of 10‐day‐old AMHC‐derived NPLCs (magnification ×100). (b) AMHC‐derived NPLCs immunostained with anti‐β‐tubulin III and anti‐GFAP (magnification ×200). (c) AMHC‐derived dopaminergic‐like neurons stained with anti‐β‐tubulin III and antityrosine hydroxylase antibody (magnification ×100). White arrows denote cells or cell clusters positive for tyrosine hydroxylase. (d) AMHC‐derived oligodendrocyte‐like cells stained with anti‐GFAP and anti‐MBP antibody (×200).
Reverse transcriptase‐polymerase chain reaction analysis revealed that in day 14 AMHC‐derived NPLCs expressed DOPA‐decarboxylase, dopamine β‐hydroxylase and the most important rate‐limiting enzyme in catecholamine synthesis, tyrosine hydroxylase. Remarkably, these cells expressed NAT and DAT (Fig. 3a), which in vertebrates is found exclusively in dopaminergic neurons. This finding suggested that these cells would be capable of both the outwards transport and the re‐uptake of dopamine. Specific transporter proteins (vesicular monoamine transporters) package classical neurotransmitters into vesicles so that their release can be regulated by neural activity. We asked whether AMHC‐derived NPLCs express vesicular monoamine transporter 1 (VMAT1) and VMAT2. RT‐PCR analysis (Fig. 3a) revealed expression of both transporters, suggesting that these cells are able to transport the monoamines from cytoplasm into vesicles, prior to their secretion. These findings demonstrate that neural precursor gene expression can be induced when human haematopoietic stem cells are exposed to a suitable microenvironment.
Figure 3.

Functional maturation of AMHC‐derived NPLCs. (a) RT‐PCR analysis of catecholamine genes in day 14 AMHC‐derived NPLCs. Lane 1, molecular weight marker; lane 2, GAPDH gene expression; lane 3, dopamine transporter; lane 4, noradrenaline transporter; lane 5, vesicular monoamine transporter 1; lane 6, vesicular monoamine transporter 2; lane 7, tyrosine hydroxylase; lane 8, dopamine β‐hydroxylase; lane 9, DOPA‐decarboxylase. (b) A representative chromatogram of DOPA and dopamine production by 10‐day‐old AMHC‐derived NPLCs (IST, internal standard: 3,4‐dihydroxybenzylamine). Day 5 AMHC‐derived NPLCs cultured in neural progenitor basal medium supplemented with BDNF and GDNF differentiated mainly into β‐tubulin III‐positive cells red (c), same cells cultured in astrocyte growth medium differentiated into GFAP+ astrocyte‐like cells (red), that expressed erythropoietin (green) around the nucleus (d) plus GFAP+ (green) and MBP+ oligodendrocyte‐like cells (red) (overlay: yellow) (e). All AMHC‐derived NPLCs down‐regulated CD45 expression almost completely by week 4 in culture (n = 4) (f, i–iii). Black histograms indicate isotype control, white histograms the positivity for CD45.
Functional maturation of AMHC‐derived NPCs
As suggested by the PCR data, AMHC‐derived NPLCs produced DOPA by day 5 in culture [concentration in the supernatant, 23 ± 6.2–174.7 ± 69.2 pM/mL (106 NPLCs); n = 10]. Most importantly, by days 7–10, significant levels of dopamine were detected [8 pM/mL (106 NPLCs)] (Fig. 3b). Concentration of DOPA in supernatants became proportionally diminished, suggesting dynamic conversion of catecholamines. Neither DOPA nor dopamine was detected in the incubated culture medium alone (data not shown). To our knowledge, this is the first time to show that human blood‐derived neural progenitor‐like cells are able to produce these vital neurotransmitters. Remarkably, synthesis and release of DOPA and dopamine occurred without exogenous stimulation of AMHC‐derived NPLCs, although in other studies, bone marrow stromal cells were transduced with the tyrosine hydroxylase gene to allow for production of these neurotransmitters (Schwarz et al. 1999). Low efficiency of generation of dopaminergic neurons from primary cultures of foetal, neonatal and adult stem cells limits their therapeutic potential as donor cells. However, the AMHCs efficiently generated neural precursor‐like cells that contained approximately 30% dopaminergic‐like neurons.
To confirm the potential of these cells as neural progenitors, we cultured day 5 AMHC‐derived NPLCs (n = 5) in appropriate induction media for neuronal and astrocytic lineages. Under these conditions, all samples of NPLCs differentiated into cells with characteristics of adult mammalian central nervous system cells: neuron‐like cells (Fig. 3c), astrocyte‐like cells (Fig. 3d) and oligodendrocyte‐like cells (Fig. 3e). Most strikingly, the initially CD45+ AMHCs down‐regulated this antigen by week 3 (Fig. 3f, ii) and by week 4 it disappeared (Fig. 3f, iii).
To determine whether AMHC‐derived NPLCs acquire functional voltage‐gated channels, we performed whole‐cell patch‐clamp recordings after 10–14 days in culture (n = 6) (Fig. 4a). Nine of 18 cells showed a sustained rectifying inwards current when hyperpolarized for 200 ms to potentials less than –80 mV. The same cells also showed delayed rectifying outwards current when depolarized to more positive potentials than 50 mV (Fig. 4b). Resting membrane potentials of these nine cells ranged from –70 to –22 mV (mean, –42 mV); current amplitudes reached several hundred picoamperes. One of these cells also displayed a small transient inwards current, with maximal amplitude of 100 picoamperes, that resembled a voltage‐activated Na+‐current (Fig. 4c). Four of 18 cells (22%) showed only a small delayed rectifying outwards current. The resting membrane potential of these four cells was –36 mV to 0 mV (mean, –17 mV). Five of 18 cells (28%) showed no voltage‐activated currents, and their resting membrane potential was close to 0 mV. A subpopulation of cells within AMHC‐derived NPLCs appears to establish a negative resting potential and expresses voltage‐activated ion channels. Our findings are consistent with those of Hogg et al. (2004) who reported that undifferentiated neural progenitor cells from the adult rat hippocampus were electrically unexcitable but became excitable after differentiation to neurons in vitro, expressing voltage‐dependent Na+, Ca2+ and delayed rectifier K+ channels.
Figure 4.
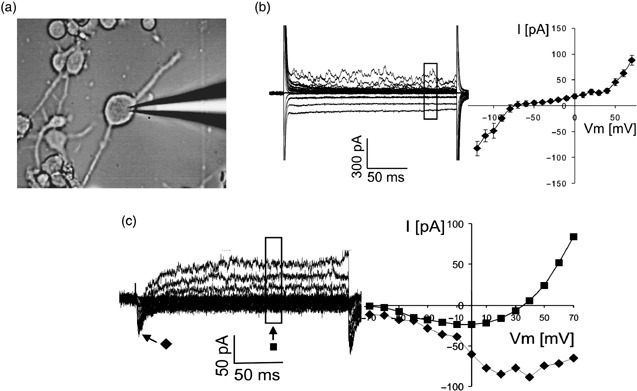
Electrophysiological maturation of AMHC‐derived NPLCs. (a) Electrophysiological properties of 14‐day‐old AMHC‐derived NPLCs. Typical electrophysiological recording in a cell by patch clamp electrode. (b) Cells were voltage‐clamped at –70 mV and hyperpolarized and depolarized for 200 ms in 10 mV steps. Whole cell current elicited by this protocol was measured (left). A plot of the amplitude of the sustained currents depending on membrane potential reveals inwards and outwards rectifying properties of the current (right, n = 6). (c) Voltage‐activated currents of one cell after leak subtraction (left). Amplitudes of transient inwards current ( ) and sustained outwards current (
) and sustained outwards current ( ) of this cell, shown at different membrane potentials.
) of this cell, shown at different membrane potentials.
AMHCs possess the potential to differentiate into cells with endodermal and mesodermal characteristics
Many recent reports have demonstrated that non‐haematopoietic multipotent progenitor cells are able to give rise to tissues of endodermal origin. Schwartz et al. (2002) have shown that multipotent CD45− adult progenitor cells from adult bone marrow differentiate into functional hepatocyte‐like cells. To test whether CD45+ AMHCs have this capacity, we cultured them in hepatocyte basal medium supplemented with growth factors (ascorbic acid, BSA‐FAF, transferrin, insulin, hEGF, GA‐1000) for 2–3 weeks (n = 5). After 2 weeks, 70 ± 18% of the AMHCs from all donors differentiated into epithelial cells showing morphological features similar to those of primary hepatocytes and expressed liver‐associated proteins such as hepatocyte nuclear factor 3β (HNF‐3β) and albumin (Fig. 5a), hepatocyte nuclear factor 1α (HNF‐1α), α‐fetoprotein (AFP) (Fig. 5b) and cytokeratins (CK) 18 (Fig. 5c) and 19 (Fig. 5d). A small percentage of these cells were binucleate by week 3 in culture (Fig. 5d). RT‐PCR analysis showed greater expression of human albumin mRNA on day 23 than on day 14, suggesting functional maturation of these cells (Fig. 5e).
Figure 5.

AMHC‐derived hepatocyte‐like cells and skeletal muscle‐like cells. (a) Immunostaining with anti‐HNF‐3β (green) and anti‐albumin (red) antibodies. (b–d) Immunostaining with (b) anti‐HNF‐α (green) and anti‐AFP (red), (c) anti‐cytokeratin 18 (red) and (d) anti‐cytokeratin 19 (red) antibodies. (e) RT‐PCR analysis of albumin gene expression after 14 and 23 days in culture. Lane 1, molecular weight marker; lane 2, GAPDH gene expression in day 14 AMHC‐derived hepatocyte‐like cells; lane 3, GAPDH gene expression in day 23 AMHC‐derived hepatocyte‐like cells; lane 4, albumin gene expression in day 14 AMHC‐derived hepatocyte‐like cells; lane 5, albumin gene expression in day 23 AMHC‐derived hepatocyte‐like cells; lane 6, molecular weight marker; lane 7, GAPDH gene expression in HepG2 hepatoma cell line; lane 8, albumin gene expression in HepG2 hepatoma cell line (positive control). (f) Immunostaining of day 7 AMHC‐derived skeletal muscle‐like cells with anti‐MyoD (green) and anti‐myogenin (red) (magnification ×100) (overlay: yellow) antibodies. (g) Immunostaining of day 14 AMHC‐derived skeletal muscle‐like cells with anti‐desmin (green) and anti‐actin (red) antibodies (magnification ×200) (overlay: yellow).
When AMHCs (n = 5) were cultured in skeletal muscle medium supplemented with hEGF, dexamethasone, l‐glutamine, foetal bovine serum and gentamicin/amphotericin B, 95 ± 5% of them differentiated into skeletal muscle‐like cells. Their morphology changed during the first week, and after 5–7 days they expressed transcription factors MyoD and myogenin (Fig. 5f), which are characteristic of young muscle cells. After the first week, they expressed desmin and actin (Fig. 5g), usually associated with the phenotype of more mature skeletal muscle cells. AMHCs of all donors showed this kind of differentiation, suggesting a high potential of these cells to adopt skeletal muscle‐like fate in culture.
DISCUSSION
It is generally accepted that embryonic stem cells are able to generate tissues of all three germ layers either in vitro or in vivo (Thomson et al. 1998; Odorico et al. 2001). A great number of recent studies has demonstrated that even tissue‐specific stem cells (which like embryonic stem cells do not express CD45 and therefore are non‐haematopoietic, for example, skin, adipose, cord blood and bone marrow‐derived stem cells) under appropriate conditions could change their fate and give rise to cells of the other embryonic germ layers (Stemple & Anderson 1992; Krause et al. 2001; Toma et al. 2001; Jiang et al. 2002; Yang et al. 2002; Kogler et al. 2004; Seaberg et al. 2004). This ability is known as stem cell plasticity, or according to some authors as ‘non‐orthodox differentiation’ (Bianco & Cossu 1999) or ‘transdifferentiation’ (Lagasse et al. 2000; Anderson et al. 2001; Rutenberg et al. 2004). Further research has suggested that instead of seemingly mature cells reprogramming, cell fusion could be responsible for this phenomenon (Terada et al. 2002; Wang et al. 2003). According to these findings, it may be possible that transdifferentiation can be a partial phenomenon only in certain circumstances. On the other hand, there is a much greater controversy on the issue whether haematopoietic stem cells could give rise to cells of other embryonic germ layers. Some reports have demonstrated that both cell fusion and transdifferentiation can account for transformation of human peripheral blood CD34+ cells into cardiomyocytes in vivo (Zhang et al. 2004). Ishikawa et al. (2006) reported that human cord blood‐derived Lin(–)CD34(+)CD38(–) haematopoietic stem cells, after transplantation into neonatal NOD/SCID/IL2r(null) mice contribute to generation of cardiomyocytes exclusively through cell fusion and not through transdifferentiation. Other studies have shown that haematopoietic cells contribute little to hepatocyte formation either in vitro (Lian et al. 2006) or in vivo (Thorgeirsson & Grisham 2006), while Jang et al. (2004) showed that mouse haematopoietic stem cells in vivo convert into hepatocytes within days, without fusion. Fairly recently, two controversial reports appeared concerning neural potential of haematopoietic cells. Chen et al. (2005) found that within the haematopoietic cell fraction of human umbilical cord blood, a subpopulation of cells exists that seems to have the potential to become neural cells. In contrast, Roybon et al. (2006) reported that bone marrow‐derived mouse adult haematopoietic stem cells do not have the capacity to leave the haematopoietic lineage and differentiate into neurons. Having these very controversial data in mind, here we have asked whether the haematopoietic cells, that we have termed as AMHCs and express CD45, are able to differentiate into non‐haematopoietic tissues. We show that these cells under appropriate microenvironmental cues are able to give rise to cells with characteristics of neuroectoderm, endoderm and mesoderm. AMHC‐derived neural‐like progenitor cells expressed neural tissue specific markers at mRNA and protein levels. The cells expressed all genes for enzymes important for catecholamine synthesis such as DOPA‐decarboxylase and most importantly the rate‐limiting enzyme, tyrosine hydroxylase. Immunocytochemical analysis showed that 27 ± 8% of the β‐tubulin III positive cells expressed tyrosine hydroxylase, suggesting the presence of a subpopulation of dopaminergic‐like neurons within AMHC‐derived neural progenitor‐like cells. In addition, these cells express noradrenaline and dopamine transporter VMAT1 and VMAT2, suggesting that they possess mechanisms for both the outwards transport and the re‐uptake of dopamine. Most strikingly, AMHC‐derived neural progenitor‐like cells produce DOPA and dopamine, without any exogenous stimulation such as with highly concentrated KCl solution (over 50 mm) or without transduction of tyrosine hydroxylase gene, which is a usual procedure in almost all reports published so far. To our knowledge, this is the first time that a haematopoietic cell‐derived neural‐like cell population has been shown that can produce DOPA and dopamine. In addition, by day 14 in culture AMHC‐derived NPLCs established a negative resting potential and expressed functional voltage‐gated channels, suggesting functional maturation of these cells. Therefore, differentiation of AMHCs into neural progenitor‐like cells may constitute a source of autologous cells for treatment of different central nervous system disorders. Interestingly, AMHCs under these specific conditions had the capacity, not only to change their surface phenotype and express a series of neural tissue‐specific genes, but also to lose their initial haematopoietic identity. They down‐regulated expression of CD45 through the culture and at week 4 they lost it completely. If we take into account that this antigen is an exclusive property of haematopoietic cells, then our data provide the first direct evidence for conversion of haematopoietic cells into non‐haematopoietic tissues. Furthermore, culture of AMHCs in a specific medium supplemented with growth factors for primary hepatocytes for 2 weeks, induced conversion of more than 80% of these cells into cells with morphological features similar to those of primary hepatocytes. They expressed liver‐associated proteins such as HNF‐3β, HNF‐1α, α‐fetoprotein and cytokeratins 18 and 19. A small percentage of the cells were binucleate by week 3 in culture, suggesting the presence of hepatocyte‐like oval cells. RT‐PCR analysis of AMHC‐derived hepatocyte‐like cells showed greater expression of human albumin mRNA on day 23 than on day 14, suggesting the capacity of AMHCs to give rise to more mature cells with characteristics of endodermal lineage. A fairly recent study has illustrated the myogenic potential of CD45+ stem cells purified from regenerating skeletal muscle (Polesskaya et al. 2003). These cells were able to express the myogenic determination factor Myf5 in injured muscle. Nevertheless, they continued to express CD45 and thus did not complete the transdifferentiation process. In our study, AMHC‐derived skeletal muscle‐like cells in 5–7 days of culture changed their morphology and expressed transcription factors MyoD and myogenin, which are characteristic of young muscle cells. After the first week, they expressed desmin and actin, which are usually associated with the phenotype of more mature skeletal muscle cells. Moreover, immunocytochemical analysis of CD45 expression, revealed down‐regulation and thereafter the loss of this antigen in both AMHC‐derived hepatocyte‐like cells and AMHC‐derived skeletal muscle‐like cells (data not shown). Ongoing in vitro clonal studies in our laboratory, and transplantation of AMHCs into immunodeficient non‐obese diabetic/severe combined immunodeficiency mice plus their transplantation into mouse blastocysts will provide further insight into the whole potential of these haematopoietic cells, at a single‐cell level, in generation of non‐haematopoietic tissues in vivo. In addition, in order to determine whether these cells possess functional capacity to correct diseases of different tissues, AMHCs or AMHC‐derived neural‐like progenitor cells, hepatocyte‐like cells and skeletal muscle‐like cells would be transplanted into mice with specific defects such as mouse model of Parkinson's disease, mice with induced liver injury and myodystrophic mice, respectively.
In conclusion, our data provide direct evidence that adult multipotent haematopoietic cells under the influence of appropriate microenvironmental cues activate a set of tissue‐specific genes and consequently give rise to non‐haematopoietic cells with characteristics of neuroectodermal, endodermal and mesodermal lineages. These cells may therefore serve as a novel alternative source of multipotent autologous cells for cell replacement therapies in various diseases.
ACKNOWLEDGEMENTS
We gratefully acknowledge Drs Diane Krause and James Morgan for critical reading of the manuscript and Mrs Sharon Naron for professional editing of the manuscript. We thank also Mrs Sieglinde Haug, Johanna Backes, Birgit Kustermann and Silke Singer for excellent technical assistance. This work was supported by a grant from Wilhelm‐Sander Stiftung (Munich, Germany).
Research grants: Wilhelm‐Sander Stiftung grant 2000.063.1 (Munich, Germany).
REFERENCES
- Anderson DJ, Gage FH, Weissman IL (2001) Can stem cells cross lineage boundaries? Nat. Med. 7, 393–395. [DOI] [PubMed] [Google Scholar]
- Beltrami AP, Barlucchi L, Torella D, Baker M, Limana F, Chimenti S, Kasahara H, Rota M, Musso E, Urbanek K, Leri A, Kajstura J, Nadal‐Ginard B, Anversa P (2003) Adult cardiac stem cells are multipotent and support myocardial regeneration. Cell 114, 763–776. [DOI] [PubMed] [Google Scholar]
- Bianco P, Cossu G (1999) Uno, nessuno e centomila: searching for the identity of mesodermal progenitors. Exp. Cell Res. 251, 257–263. [DOI] [PubMed] [Google Scholar]
- Chen N, Hudson JE, Walczak P, Misiuta I, Garbuzova‐Davis S, Jiang L, Sanchez‐Ramos J, Sanberg PR, Zigova T, Willing AE (2005) Human umbilical cord blood progenitors: the potential of these hematopoietic cells to become neural. Stem Cells 23, 1560–1570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Handgretinger R, Gordon PR, Leimig T, Chen X, Buhring HJ, Niethammer D, Kuci S (2003) Biology and plasticity of CD133+ hematopoietic stem cells. Ann. N. Y. Acad. Sci. 996, 141–151. [DOI] [PubMed] [Google Scholar]
- Hogg RC, Chipperfield H, Whyte KA, Stafford MR, Hansen MA, Cool SM, Nurcombe V, Adams DJ (2004) Functional maturation of isolated neural progenitor cells from the adult rat hippocampus. Eur. J. Neurosci. 19, 2410–2420. [DOI] [PubMed] [Google Scholar]
- D’Ippolito G, Diabira S, Howard GA, Menei P, Roos BA, Schiller PC (2004) Marrow‐isolated adult multilineage inducible (MIAMI) cells, a unique population of postnatal young and old human cells with extensive expansion and differentiation potential. J. Cell Sci. 117, 2971–2981. [DOI] [PubMed] [Google Scholar]
- Ishikawa F, Shimazu H, Shultz LD, Fukata M, Nakamura R, Lyons B, Shimoda K, Shimoda S, Kanemaru T, Nakamura KI, Ito H, Kaji Y, Perry AC, Harada M (2006) Purified human hematopoietic stem cells contribute to the generation of cardiomyocytes through cell fusion. FASEB J. 20, 950–952. [DOI] [PubMed] [Google Scholar]
- Jang YY, Collector MI, Baylin SB, Diehl AM, Sharkis SJ (2004) Hematopoietic stem cells convert into liver cells within days without fusion. Nat. Cell Biol. 6, 532–539. [DOI] [PubMed] [Google Scholar]
- Jiang Y, Jahagirdar BN, Reinhardt RL, Schwartz RE, Keene CD, Ortiz‐Gonzalez XR, Reyes M, Lenvik T, Lund T, Du Blackstad MJ, Aldrich S, Lisberg A, Low WC, Largaespada DA, Verfaillie CM (2002) Pluripotency of mesenchymal stem cells derived from adult marrow. Nature 418, 41–49. [DOI] [PubMed] [Google Scholar]
- Kogler G, Sensken S, Airey JA, Trapp T, Muschen M, Feldhahn N, Liedtke S, Sorg RV, Fischer J, Rosenbaum C, Greschat S, Knipper A, Bender J, Degistirici O, Gao JZ, Caplan AI, Colletti E, Almeida‐Porada G, Muller HW, Zanjani E, Wernet P (2004) A new human somatic stem cell from placental cord blood with intrinsic pluripotent differentiation potential. J. Exp. Med. 200, 123–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krause DS, Theise ND, Collector MI, Henegariu O, Hwang S, Gardner R, Neutzel S, Sharkis SJ (2001) Multi‐organ, multi‐lineage engraftment by a single bone marrow‐derived stem cell. Cell 105, 369–377. [DOI] [PubMed] [Google Scholar]
- Kuci S, Wessels JT, Buhring HJ, Schilbach K, Schumm M, Seitz G, Loffler J, Bader P, Schlegel PG, Niethammer D, Handgretinger R (2003) Identification of a novel class of human adherent CD34− stem cells that give rise to SCID‐repopulating cells. Blood 101, 869–876. [DOI] [PubMed] [Google Scholar]
- Lagasse E, Connors H, Al Dhalimy M, Reitsma M, Dohse M, Osborne L, Wang X, Finegold M, Weissman IL, Grompe M (2000) Purified hematopoietic stem cells can differentiate into hepatocytes in vivo . Nat. Med. 6, 1229–1234. [DOI] [PubMed] [Google Scholar]
- Lian G, Wang C, Teng C, Du Zhang CL, Zhong Q, Miao C, Ding M, Deng H (2006) Failure of hepatocyte marker‐expressing hematopoietic progenitor cells to efficiently convert into hepatocytes in vitro . Exp. Hematol. 34, 348–358. [DOI] [PubMed] [Google Scholar]
- Odorico JS, Kaufman DS, Thomson JA (2001) Multilineage differentiation from human embryonic stem cell lines. Stem Cells 19, 193–204. [DOI] [PubMed] [Google Scholar]
- Polesskaya A, Seale P, Rudnicki MA (2003) Wnt signaling induces the myogenic specification of resident CD45+ adult stem cells during muscle regeneration. Cell 113, 841–852. [DOI] [PubMed] [Google Scholar]
- Reyes M, Lund T, Lenvik T, Aguiar D, Koodie L, Verfaillie CM (2001) Purification and ex vivo expansion of postnatal human marrow mesodermal progenitor cells. Blood 98, 2615–2625. [DOI] [PubMed] [Google Scholar]
- Roybon L, Ma Z, Asztely F, Fosum A, Jacobsen SE, Brundin P, Li JY (2006) Failure of transdifferentiation of adult hematopoietic stem cells into neurons. Stem Cells. 24, 1594–1604. [DOI] [PubMed] [Google Scholar]
- Rutenberg MS, Hamazaki T, Singh AM, Terada N (2004) Stem cell plasticity, beyond alchemy. Int. J. Hematol. 79, 15–21. [DOI] [PubMed] [Google Scholar]
- Schwartz RE, Reyes M, Koodie L, Jiang Y, Blackstad M, Lund T, Lenvik T, Johnson S, Hu WS, Verfaillie CM (2002) Multipotent adult progenitor cells from bone marrow differentiate into functional hepatocyte‐like cells. J. Clin. Invest. 109, 1291–1302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz EJ, Alexander GM, Prockop DJ, Azizi SA (1999) Multipotential marrow stromal cells transduced to produce l‐DOPA: engraftment in a rat model of Parkinson disease. Hum. Gene Ther. 10, 2539–2549. [DOI] [PubMed] [Google Scholar]
- Seaberg RM, Smukler SR, Kieffer TJ, Enikolopov G, Asghar Z, Wheeler MB, Korbutt G, Van Der Kooy D (2004) Clonal identification of multipotent precursors from adult mouse pancreas that generate neural and pancreatic lineages. Nat. Biotechnol. 22, 1115–1124. [DOI] [PubMed] [Google Scholar]
- Stemple DL, Anderson DJ (1992) Isolation of a stem‐cell for neurons and glia from the mammalian neural crest. Cell 71, 973–985. [DOI] [PubMed] [Google Scholar]
- Terada N, Hamazaki T, Oka M, Hoki M, Mastalerz DM, Nakano Y, Meyer EM, Morel L, Petersen BE, Scott EW (2002) Bone marrow cells adopt the phenotype of other cells by spontaneous cell fusion. Nature 416, 542–545. [DOI] [PubMed] [Google Scholar]
- Thomson JA, Itskovitz‐Eldor J, Shapiro SS, Waknitz MA, Swiergiel JJ, Marshall VS, Jones JM (1998) Embryonic stem cell lines derived from human blastocysts. Science 282, 1145–1147. [DOI] [PubMed] [Google Scholar]
- Thorgeirsson SS, Grisham JW (2006) Hematopoietic cells as hepatocyte stem cells: a critical review of the evidence. Hepatology 43, 2–8. [DOI] [PubMed] [Google Scholar]
- Toma JG, Akhavan M, Fernandes KJL, Barnabe‐Heider F, Sadikot A, Kaplan DR, Miller FD (2001) Isolation of multipotent adult stem cells from the dermis of mammalian skin. Nat. Cell Biol. 3, 778–784. [DOI] [PubMed] [Google Scholar]
- Wagers AJ, Weissman IL (2004) Plasticity of adult stem cells. Cell 116, 639–648. [DOI] [PubMed] [Google Scholar]
- Wang X, Willenbring H, Akkari Y, Torimaru Y, Foster M, Al Dhalimy M, Lagasse E, Finegold M, Olson S, Grompe M (2003) Cell fusion is the principal source of bone marrow‐derived hepatocytes. Nature 422, 897–901. [DOI] [PubMed] [Google Scholar]
- Yang L, Li S, Hatch H, Ahrens K, Cornelius JG, Petersen BE, Peck AB (2002) In vitro transdifferentiation of adult hepatic stem cells into pancreatic endocrine hormone‐producing cells. Proc. Natl. Acad. Sci. USA 99, 8078–8083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S, Wang D, Estrov Z, Raj S, Willerson JT, Yeh ET (2004) Both cell fusion and transdifferentiation account for the transformation of human peripheral blood CD34‐positive cells into cardiomyocytes in vivo . Circulation 110, 3803–3807. [DOI] [PubMed] [Google Scholar]