

Summary
In the past decade, the significant contribution of the spleen to ischemic brain damage has gained considerable attention in stroke research. As the largest natural reservoir of immune cells, the spleen establishes critical connections with the ischemic brain during the progression of stroke and mobilizes its cells to the site of injury. Multiple “alarm” signals released from the injured brain are essential for the initiation of brain–spleen communication. Spleen‐derived cells, including neutrophils, lymphocytes, and monocytes/macrophages, are known to contribute significantly to ischemic brain damage. Understanding the dynamic splenic responses to stroke will not only provide insights into the evolvement of ischemic brain injury but will also identify potential targets for stroke treatment. Here, we review recent studies on the functions of the spleen in ischemic stroke. We have included a discussion of several therapeutic strategies that target splenic responses and reduce acute ischemic brain damage in preclinical studies. Future investigations on the effects of the spleen on long‐term stroke recovery are highly warranted.
Keywords: Immune response, Spleen, Stroke, Therapeutic strategies
Introduction
In the past few decades, the importance of immune responses in the pathobiology of ischemic stroke has been highlighted in the literature 1. A variety of immune cells, both within the central nervous system (CNS) and in the periphery, are activated soon after stroke onset and contributes significantly to brain damage or repair. The spleen is a major reservoir of blood cells and a key lymph organ that releases immune cells into the circulation for self‐defense against infections and injuries, including damage to the CNS 2. Therefore, splenic responses after stroke and their contribution to ischemic brain damage have gained considerable attention. Early studies in animals revealed that the spleen shrinks dramatically during the acute phase after middle cerebral artery occlusion (MCAO) 3, 4. This morphological change is accompanied by the long‐distance trafficking of splenocytes all the way from the spleen into the ischemic area 5. Remarkably, the MCAO‐induced reduction in spleen size is correlated with the extent of ischemic damage 6. Similarly, recent clinical data suggest that the size of the spleen is quite dynamic in stroke patients, with an early contraction followed by a subsequent re‐expansion. This splenic volume change might be associated with the early release of splenocytes into the circulation, which in turn may contribute to the poststroke inflammatory cascade 7. Despite our awareness of these splenic alterations, the exact functions of the spleen in ischemic brain injury are not completely clear. Although most animal studies have documented that splenectomy in stroke animals is protective to the ischemic brain, a recent study demonstrated that removal of the spleen after MCAO does not reduce the brain infarct. These discrepancies may be attributed to differences in the timing of spleen removal and in the severity of the stroke (Table 1). Furthermore, these discrepancies probably reflect the complexity and dynamism of splenic functions during the course of ischemic injury. Therefore, a through characterization of the evolution of splenic responses to stroke would not only provide insights into the progression of ischemic brain injury but also identify potential targets for stroke treatment.
Table 1.
Effect of spleen removal on brain infarct
| Stroke model | Infarct | Time of spleen removal | Species | Duration of occlusion | Time of observation | Citations | |
|---|---|---|---|---|---|---|---|
| 1 | MCAO | ↓ | 2 weeks before MCAO | Rat | Permanent | 96 h | 13 |
| 2 | MCAO | ↓ | 2 weeks before MCAO | Rat | Permanent | 24 h | 45 |
| 3 | MCAO | ↓ | 2 weeks before MCAO | Rat | Permanent | 96 h | 12 |
| 4 | MCAO | ↓ | 2 weeks before MCAO | Rat | Permanent | 48 h | 4 |
| 5 | MCAO | ↓ | 2 weeks before MCAO | Rat | Permanent | 96 h | 40 |
| 6 | MCAO | ↓ | Splenic irradiation 3, 4, 6, or 8 h after start of cerebral ischemia | Rat | 2 h | 2 and 7 days | 35 |
| 7 | MCAO | ↓ | 2 weeks before MCAO | Mouse | 60 min | 5 days | 66 |
| 8 | MCAO | — | Immediately before MCAO | Mouse | 30 min | 3 and 7 days | 36 |
| 9 | Hypoxia–ischemia (HI) | ↓ | 3 days before HI | Neonatal rat | Permanent followed by 2 h of hypoxia | 72 h | 67 |
MCAO, middle cerebral artery occlusion.
Splenic Changes after Stroke
Changes in the Cellular Components of the Spleen after Stroke
Over the course of ischemic injury, the spleen can establish long‐distance connections with the brain by mobilizing its cellular constituents. As mentioned above, the total number of cells inside the spleen reduces dramatically within several days after stroke. This reduction probably reflects the increased efflux of immune cells from the spleen into the blood as well as increased cell death within the spleen 3, 5. Many of the released immune cells subsequently infiltrate into the ischemic brain and contribute to brain injury. Using carboxyfluorescein diacetate succinimidyl ester (CFSE) to track splenocyte migration following MCAO, it has been shown that splenocytes such as lymphocytes, monocytes, neutrophils, and NK cells can enter the systemic circulation and migrate into the brain 5. The reduction of immune cells in the spleen, especially of T lymphocytes and B lymphocytes, can last for several months, resulting in a persistent immunosuppressed status in stroke victims and increasing their susceptibility to poststroke infections 8.
Interestingly, despite the general reduction of lymphocytes in the spleen, the numbers of several specialized cell populations have been shown to increase after stroke. For example, there is an increase in CD4+FoxP3+ regulatory T cells (Tregs) in the spleen at 96 h after stroke 3. Given the evidence in support of protective effects of Tregs in the ischemic brain 9, 10, the natural upregulation of the Treg population in the spleen after ischemia might represent an endogenous protective response that mitigates stroke‐induced injury.
Cytokine Production in the Spleen after Stroke
Immune cells in the spleen are known to contribute to stroke‐induced elevations in the levels of cytokines in the blood and subsequently in the brain. For example, spleen cells collected from stroke‐afflicted mice display a higher capacity to secrete inflammatory cytokines upon activation, including tumor necrosis factor‐α (TNF‐α), interleukin‐6 (IL‐6), monocyte chemoattractant protein‐1 (MCP‐1), and interferon γ (IFNγ), compared with splenocytes collected under physiological conditions 11. Furthermore, many of these splenocyte‐derived inflammatory cytokines and chemokines, such as IFNγ and interferon‐inducible protein‐10 (IP‐10), have been shown to be critical for stroke‐induced neurodegeneration 12, 13.
How are Splenic Responses Triggered after Stroke?
Although the exact mechanisms underlying the initiation of splenocyte responses to the ischemic brain have yet to be identified, several events, including chemokine/chemokine receptor interactions, autonomic nervous system activation, and the release of CNS antigen have been documented to be essential for efficient brain–spleen communication.
CNS Antigens
In response to cerebral ischemic injury, the damaged brain may secrete a variety of antigens that activate adaptive immune responses and recruit immune cells from the spleen. Neo‐antigens, such as microtubule‐associated protein 2 (MAP2), NMDA receptor subunit NR‐2A, myelin basic protein (MBP), and myelin oligodendrocyte glycoprotein (MOG), can all be released into the periphery in acute stroke patients and captured by antigen‐presenting cells (APCs), especially dendritic cells (DCs) and macrophages. This response is thought to eventually initiate the activation of T‐cell‐dependent adaptive immune responses in the T‐cell zone 14, 15.
Chemokine/Chemokine Receptor System
A variety of chemokines, including the chemokine (C‐C motif) ligand 2 (CCL2), CCL3, CCL5, CX3CL1, CXCL8, CXCL12, etc., are secreted by damaged CNS cells to recruit inflammatory cells into the injured brain (Figure 1). During this process, the corresponding chemokine receptors are also upregulated in splenocytes after stroke 3.
Figure 1.
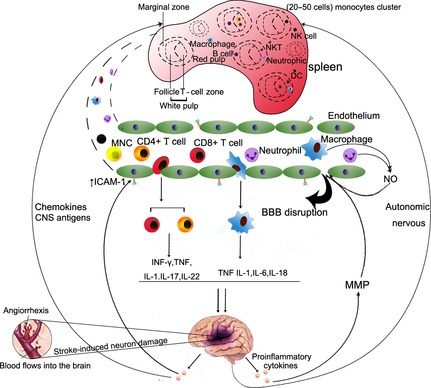
Spleen–brain communication after stroke. The ischemic brain stimulates the autonomic nervous system and releases chemotactic factors or CNS antigens to trigger the efflux of immune cells from the spleen to the site of brain injury. At the site of injury, inflammatory cells migrate through the compromised blood–brain barrier and enter the brain with a large amount of chemokines/cytokines, free radicals, and other inflammatory mediators. These factors are thought to exacerbate brain injury in the acute phases of stroke.
CCL2 is a potent mediator of monocyte/macrophage and neutrophil infiltration in cerebral ischemic injury 16. The deficiency of CCR2 expression resulted in reduced monocyte and neutrophil infiltration into the ischemic brain and subsequent reduction in cerebral inflammation and brain infarct. Furthermore, it is suggested that the CCL2–CCR2 interaction might be important for the deployment of monocyte from the spleen and their movement to the injured brain 17. The inhibition of CCR2 expression on monocytes by moxifloxacin treatment significantly attenuates infarct size after stroke.
The CXCR4–CXCL12 axis is implicated in the pathology of ischemic stroke. Cerebral ischemia results in a prompt and long‐lasting elevation of CXCL12 in the ischemic penumbra. Transplanted GFP+ bone marrow cells are recruited in proximity to these CXCL12+ vessels and display the characteristics of activated microglial cells. These results suggest that CXCL12 is important in the homing of bone marrow‐derived monocytes, which transform into microglia at the site of ischemic injury 18. Interestingly, a recent study demonstrated that the blocking of CXCR4–CXCL12 interaction by CXCR4 antagonist AMD3100 not only reduce cerebral inflammation and brain infarct, but also prevent the poststroke spleen atrophy, suggesting a role of CXCR4‐CXCL12 in regulation splenic responses after stroke 19.
Many other cytokines have been shown to be important for the recruitment of immune cells to the ischemic brain. For example, CCL3 is associated with monocyte and neutrophil accumulation in injured brain tissue 20, 21. CCL5 is involved in leukocyte infiltration after stroke 22. CX3CR1 knockout mice display reduced neuroinflammation after focal cerebral ischemia, suggesting that CX3CL1 promotes inflammation after stroke, an effect that may be related to chemotaxis in monocyte, T cells and NK cells 23, 24, 25. CXCL8 is thought to be an important chemoattractant for neutrophils after ischemic stroke 26. The functions of these cytokines on the mobilization of splenocytes warrant to be further investigated.
Autonomic Nervous System
Recent studies have demonstrated a link between autonomic nervous system activity and the peripheral immune response (Figure 1) 27, 28. The sympathetic nervous system is one of the major pathways whereby the CNS communicates with the immune system 29. Shortly after an ischemic insult, there is an increase in norepinephrine and epinephrine in the systemic circulation. Both norepinephrine and epinephrine have been shown to induce significant splenic atrophy 30. Blockade of α‐ and β‐adrenergic receptors significantly inhibits the reduction in spleen size and reduces infarct volume, while α1‐receptor antagonism blocks the shrinkage of the spleen. These findings indicate that activation of both α‐ and β‐adrenergic receptors mediates the splenic response to stroke and that both receptor subtypes are potential targets for therapeutic treatments of ischemic injury 4.
Other Factors
Lymphocytes are important players in the pathophysiology of acute ischemic stroke. Sphingosine‐1‐phosphate (S1P) and S1P‐receptor‐1 (S1PR1) are vital for lymphocyte efflux from white to red pulp and back into the circulation 31. Furthermore, the S1PR1 modulator FTY720 has been shown to block the egress of lymphocytes from lymphoid organs, resulting in reduced ischemic neurodegeneration 32.
The Spleen Contributes to Neurodegeneration after Stroke
Various immune cells of the innate and adaptive immune systems contribute to the pathogenesis of ischemic injury and profoundly affect stroke outcomes 33. After the initial ischemic injury, alarm signals are released that promote an influx of peripheral leukocytes into the brain 34. The trafficking of peripheral immune cells into the brain is thought to exacerbate the local brain inflammatory response, thereby enhancing neurodegeneration. Dramatic changes in splenic cellular components after stroke have long suggested that the splenic response may play a pathological role in ischemic injury. Splenic leukocytes such as various subsets of T and B cells, DCs, and macrophages may participate in the systemic inflammatory response to stroke and contribute to neurodegeneration. Removal of the largest pool of peripheral immune cells before MCAO or neonatal hypoxia–ischemia by splenectomy has been shown to reduce brain infarct volume in rodents. As expected, the splenectomy‐induced protection is accompanied by a decrease in the number of immune cells in brain tissue (Table 1). Poststroke splenic irradiation can also significantly reduce brain infarct size and inflammation after MCAO 35. Thus far, the majority of studies are in agreement that inhibition of splenic function prior to or soon after MCAO is neuroprotective and significantly reduces neuroinflammation during the acute phases of stroke. However, Kim et al. 36 recently observed that splenectomy does not alter stroke‐induced acute brain injury when performed immediately before 30 min of MCAO. This finding contradicts other studies of the beneficial effects of splenectomy (Table 1). The discrepancy may be due to the differences in the timing of spleen removal and the severity of the stroke insult. Further investigations are needed to explain the outcome differences using different stroke models and systematically changing the timing of spleen removal.
Immune Cells from the Spleen are Involved in the Progression of Ischemic Brain Damage
As summarized above, the spleen exerts a profound influence on the ischemic brain by mobilizing its resident cells, including lymphocytes, monocytes, neutrophils, and natural killer cells into the brain. In this section, we discuss the roles of immune cells from the spleen in the pathogenesis of ischemic brain damage.
Neutrophils
Neutrophils are the first responders to ischemic injury and infiltrate into the injured brain within a few hours. Neutrophil infiltration peaks at 1–3 days after cerebral ischemia in animal models of stroke 37. In patients with stroke, neutrophils are recruited within 24 h of symptom onset 38. A number of studies indicate that neutrophil infiltration may contribute to postischemic brain injury 38, 39, 40. Moreover, neutrophils may cause the breakdown of the BBB by releasing matrix metalloproteinase‐9 (MMP‐9), resulting in the infiltration of even more leukocytes and the exacerbation of neuroinflammation 41, 42. As expected, the protective effects of splenectomy 2 weeks before stroke are associated with a reduction in the number of neutrophils in the injured area 40.
Monocyte/Macrophages
Peripheral monocytes/macrophages (MMs) have been shown to infiltrate the ischemic brain in a CCL‐2 dependent manner and contribute to poststroke cerebral inflammation and brain injury 17. Interestingly, both pro‐inflammatory Ly‐6Chigh and antiinflammatory Ly‐6Clow monocyte subsets are mobilized from the spleen and migrate toward the brain. A recent study showed that complete removal of spleen‐derived MMs by splenectomy does not provide any protection to the ischemic brain 36. Therefore, selective depletion of pro‐inflammatory Ly‐6Chigh and antiinflammatory Ly‐6Clow MM subsets would be useful to determine the contribution of individual subsets to stroke outcomes.
Lymphocytes
Several studies have confirmed that T lymphocytes play a detrimental role in ischemia/reperfusion injury 43, 44. The protective effects of splenectomy on the brain are accompanied by reductions in the number of T cells in the injured brain 45. Interestingly, recent studies suggest the some specific T‐cell subsets, such as Tregs, may exert beneficial roles in stroke 9, 10. The number of Tregs in the spleen is known to increase after stroke 3, which might reflect an endogenous protective mechanism.
B cells are a major type of splenic lymphocyte. However, there are few studies investigating the role of B lymphocytes in cerebral ischemia/reperfusion injury. Mice lacking only B cells fail to show improvements against ischemic injury, suggesting that endogenous B cells do not play a detrimental role following cerebral ischemia. Furthermore, a special population of regulatory B lymphocytes secretes IL10 and is protective in cerebral ischemia/reperfusion injury 46. Further studies are warranted to verify whether these protective B cells are released from the spleen after stroke.
Natural Killer Cells
Natural killer (NK) cells, which also travel from the spleen into the ischemic brain 5, are a type of cytotoxic cell and form part of the innate immune system. Recent studies have shown that ischemic neuron‐derived chemokines recruit NK cells into the brain and that these NK cells subsequently promote brain injuries 25, 47.
Dendritic Cells
Splenic T lymphocytes are known to be activated by antigen‐presenting cells (APCs), especially DCs. An increase in the number of DCs is observed in the brain in animal models of permanent/transient focal cerebral ischemia 48. Immature DCs patrol the blood and invade injured tissues, where they pick up antigens and acquire the ability to stimulate T cells in lymphoid tissues, such as the lymph nodes and spleen 49, 50. Thus, DCs may present antigens to T lymphocytes in the spleen to activate adaptive immunity after stroke. The exact role of splenic DCs on stroke outcome remains to be explored.
Therapeutic Strategies that Impact Spleen Responses to Stroke
Although splenectomy soon after ischemic stroke might weaken the pro‐inflammatory immunologic response and reduce cerebral injury, it is obviously not a clinically practical approach for stroke treatment. Permanent loss of the spleen will significantly impair immune defense against infections and may be catastrophic over the long term. Therefore, alternative approaches have to be developed to target splenic responses after stroke. Recent promising studies have shown that some treatment strategies can indeed modulate splenic responses and ameliorate stroke‐induced brain injury.
Stem Cells
Human Umbilical Cord Blood Cells
Human umbilical cord blood (HUCB) cells have been shown to reduce brain infarcts after stroke and improve functional outcomes 51, 52, 53. In additional to their capacity to differentiate into neuronal cells 51, HUCB cells can protect the ischemic brain through immunomodulation. Transplantation of HUCB cells after stroke can reduce the number of infiltrating immune cells in the ischemic areas and inhibit the increase in inflammatory cytokines 54. Moreover, some studies have shown that HUCB cells move from the site of the injection to the spleen, altering immune cell profiles and cytokine production in the spleen 6. Specifically, HUCB cell transplantation reduces splenic atrophy and increases the number of CD8+ T cells in the spleen after MCAO. By increasing IL‐10 and decreasing IFN‐γ release, HUCB cell treatment can inhibit the proliferation of MCAO‐associated T cells 6. These studies suggest that HUCB cells directly target the spleen and may achieve therapeutic effects in stroke victims at least partially by modulating immune cell activation inside the spleen.
Other Stem Cells
Other stem cells, such as mesenchymal stromal cells (MSCs), embryonic stem cells (ESCs), induced pluripotent stem cells (iPSs), neural stem cells (NSCs), and human amniotic epithelial cells (hAECs), have all been applied toward the treatment of experimental stroke. The mechanism of protection by these cell types may all be related to immune modulation 55. For example, MSCs have been shown to protect the brain against ischemic injury by modulating inflammatory responses, as manifested in reduced cytokine/chemokine levels in the blood and mitigated glial activation in the brain 56. Whether these stem cells can target the spleen is still unknown.
Gamma Ray Radiation
Immune cells derived from spleen are highly sensitive to irradiation. A low dose of Gamma ray (Gy) can induce cell apoptosis in the spleen 57. Ostrowski and colleagues have used gamma irradiation with 8 Gy of Cobalt 60 to irradiate the splenic area at different time points after cerebral ischemia. The cerebral infarct was significantly reduced when the spleen was irradiated at 3 or 4 h after the onset of ischemia. The authors also observed reduction of microglial activation, T‐cell infiltration, and neuronal apoptosis 35. Irradiation at later time points failed to elicit protection of the ischemic brain, suggesting that the therapeutic temporal window for strategies targeting splenic inflammatory responses is fairly narrow.
Catecholamines and Other Products Targeting the Sympathetic Nervous System
As mentioned above, catecholamine levels in the spleen are raised by activation of the sympathetic nervous system after MCAO. Catecholamines can modulate the splenic response to stroke by binding to splenic α‐ and β‐adrenoreceptors. Ajmo and colleagues have observed that carvedilol, an α‐ and β‐adrenergic receptor repressor, significantly reduces spleen atrophy and brain infarct volume. Their findings demonstrate that activation of both α‐ and β‐adrenergic receptors by catecholamines can regulate the splenic response to stroke and represents a promising target for stroke therapies 4.
Agmatine is an α2‐ adrenergic and imidazoline receptor agonist and an N‐methyl‐D‐aspartate receptor antagonist 58. Uranchimeg et al. 59 have shown that agmatine ameliorates immune responses in the spleen after brain ischemia, as shown by reduced contraction of the white pulp and decreases in the number of macrophages inside the spleen. As a consequence, agmatine can reduce the size of the cerebral infarct and lower the risk for poststroke infection 59.
Statins
Preclinical studies have shown that statins are beneficial against ischemic brain injury 60, 61. For example, pretreatment with simvastatin remarkably reduces infarct size and improves neurological recovery after stroke 62. Delayed simvastatin treatment until 24 h after stroke fails to decrease infarct volume, but still improves neurological recovery 63. Clinical studies have also confirmed that statins exert protective effects on patients with ischemic stroke 64. The protective effect of statins is associated with their antiinflammatory properties 65. Simvastatin can reduce spleen atrophy and inhibit splenocyte apoptosis after MCAO through an antiapoptotic mechanism involving increased expression of Bcl‐2 and decreased expression of Bax 66. Simvastatin treatment also inhibits IFN‐γ, which may be critical for Simvastatin‐afforded neuroprotection 66.
Conclusions
The past decade has witnessed significant achievements in our understanding of the splenic response after ischemic stroke. The release and migration of immune cells from the spleen into the circulation and their subsequent infiltration into the ischemic brain is thought to exacerbate brain damage during the acute stages of stroke. However, there are still many unsolved questions in this area. For example, the direct interaction between infiltrated splenocyte and CNS cells and how such interaction impact the pathogenesis of stroke have not been addressed. The advances in cell labeling and two‐photon imaging techniques would help to address this question in stroke animals. In addition, it is still not clear whether and how the spleen contributes to brain repair after stroke. Given the evidence favoring an increase in the release and the numbers of neuroprotective immune cells such as Tregs from the spleen after stroke, we can certainly envision potentially protective roles for the spleen in long‐term recovery from stroke. Although some experimental strategies that target splenic responses have shown therapeutic effects during acute phases of stroke models, translating these studies into the clinic for the long term will be far more challenging and demand a more comprehensive understanding of splenic responses in stroke patients.
Conflict of Interest
The authors declare no conflicts of interest.
Acknowledgments
Liu Z and Chen C contribute equally to this work. Hu X is supported by the American Heart Association (13SDG14570025). Liu Z is supported by the Beijing Postdoctoral Foundation (No. 2014ZZ).
References
- 1. Iadecola C, Anrather J. The immunology of stroke: From mechanisms to translation. Nat Med 2011;17:796–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Mebius RE, Kraal G. Structure and function of the spleen. Nat Rev Immunol 2005;5:606–616. [DOI] [PubMed] [Google Scholar]
- 3. Offner H, Subramanian S, Parker SM, et al. Splenic atrophy in experimental stroke is accompanied by increased regulatory T cells and circulating macrophages. J Immunol 2006;176:6523–6531. [DOI] [PubMed] [Google Scholar]
- 4. Ajmo CT Jr, Collier LA, Leonardo CC, et al. Blockade of adrenoreceptors inhibits the splenic response to stroke. Exp Neurol 2009;218:47–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Seifert HA, Hall AA, Chapman CB, et al. A transient decrease in spleen size following stroke corresponds to splenocyte release into systemic circulation. J Neuroimmune Pharmacol 2012;7:1017–1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Vendrame M, Gemma C, Pennypacker KR, et al. Cord blood rescues stroke‐induced changes in splenocyte phenotype and function. Exp Neurol 2006;199:191–200. [DOI] [PubMed] [Google Scholar]
- 7. Sahota P, Vahidy F, Nguyen C, et al. Changes in spleen size in patients with acute ischemic stroke: A pilot observational study. Int J Stroke 2013;8:60–67. [DOI] [PubMed] [Google Scholar]
- 8. Prass K, Meisel C, Hoflich C, et al. Stroke‐induced immunodeficiency promotes spontaneous bacterial infections and is mediated by sympathetic activation reversal by poststroke T helper cell type 1‐like immunostimulation. J Exp Med 2003;198:725–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Li P, Gan Y, Sun BL, et al. Adoptive regulatory T‐cell therapy protects against cerebral ischemia. Ann Neurol 2013;74:458–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Liesz A, Suri‐Payer E, Veltkamp C, et al. Regulatory T cells are key cerebroprotective immunomodulators in acute experimental stroke. Nat Med 2009;15:192–199. [DOI] [PubMed] [Google Scholar]
- 11. Offner H, Subramanian S, Parker SM, et al. Experimental stroke induces massive, rapid activation of the peripheral immune system. J Cereb Blood Flow Metab 2006;26:654–665. [DOI] [PubMed] [Google Scholar]
- 12. Seifert HA, Leonardo CC, Hall AA, et al. The spleen contributes to stroke induced neurodegeneration through interferon gamma signaling. Metab Brain Dis 2012;27:131–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Seifert HA, Collier LA, Chapman CB, et al. Pro‐Inflammatory Interferon Gamma Signaling is Directly Associated with Stroke Induced Neurodegeneration. J Neuroimmune Pharmacol 2014;9:679–689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Planas AM, Gomez‐Choco M, Urra X, et al. Brain‐derived antigens in lymphoid tissue of patients with acute stroke. J Immunol 2012;188:2156–2163. [DOI] [PubMed] [Google Scholar]
- 15. Tsuchida T, Parker KC, Turner RV, et al. Autoreactive CD8+ T‐cell responses to human myelin protein‐derived peptides. Proc Natl Acad Sci U S A 1994;91:10859–10863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Dimitrijevic OB, Stamatovic SM, Keep RF, Andjelkovic AV. Absence of the chemokine receptor CCR2 protects against cerebral ischemia/reperfusion injury in mice. Stroke 2007;38:1345–1353. [DOI] [PubMed] [Google Scholar]
- 17. Bao Y, Kim E, Bhosle S, Mehta H, Cho S. A role for spleen monocytes in post‐ischemic brain inflammation and injury. J Neuroinflammation 2010;7:92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hill WD, Hess DC, Martin‐Studdard A, et al. SDF‐1 (CXCL12) is upregulated in the ischemic penumbra following stroke: Association with bone marrow cell homing to injury. J Neuropathol Exp Neurol 2004;63:84–96. [DOI] [PubMed] [Google Scholar]
- 19. Ruscher K, Kuric E, Liu Y, et al. Inhibition of CXCL12 signaling attenuates the postischemic immune response and improves functional recovery after stroke. J Cereb Blood Flow Metab 2013;33:1225–1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kim JS, Gautam SC, Chopp M, et al. Expression of monocyte chemoattractant protein‐1 and macrophage inflammatory protein‐1 after focal cerebral ischemia in the rat. J Neuroimmunol 1995;56:127–134. [DOI] [PubMed] [Google Scholar]
- 21. Reichel CA, Rehberg M, Lerchenberger M, et al. Ccl2 and Ccl3 mediate neutrophil recruitment via induction of protein synthesis and generation of lipid mediators. Arterioscler Thromb Vasc Biol 2009;29:1787–1793. [DOI] [PubMed] [Google Scholar]
- 22. Terao S, Yilmaz G, Stokes KY, et al. Blood cell‐derived RANTES mediates cerebral microvascular dysfunction, inflammation, and tissue injury after focal ischemia‐reperfusion. Stroke 2008;39:2560–2570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bazan JF, Bacon KB, Hardiman G, et al. A new class of membrane‐bound chemokine with a CX3C motif. Nature 1997;385:640–644. [DOI] [PubMed] [Google Scholar]
- 24. Denes A, Ferenczi S, Halasz J, Kornyei Z, Kovacs KJ. Role of CX3CR1 (fractalkine receptor) in brain damage and inflammation induced by focal cerebral ischemia in mouse. J Cereb Blood Flow Metab 2008;28:1707–1721. [DOI] [PubMed] [Google Scholar]
- 25. Gan Y, Liu Q, Wu W, et al. Ischemic neurons recruit natural killer cells that accelerate brain infarction. Proc Natl Acad Sci U S A 2014;111:2704–2709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Domac FM, Misirli H. The role of neutrophils and interleukin‐8 in acute ischemic stroke. Neurosciences (Riyadh) 2008;13:136–141. [PubMed] [Google Scholar]
- 27. Ay I, Sorensen AG, Ay H. Vagus nerve stimulation reduces infarct size in rat focal cerebral ischemia: An unlikely role for cerebral blood flow. Brain Res 2011;1392:110–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Rosas‐Ballina M, Olofsson PS, Ochani M, et al. Acetylcholine‐synthesizing T cells relay neural signals in a vagus nerve circuit. Science 2011;334:98–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kohm AP, Sanders VM. Norepinephrine and beta 2‐adrenergic receptor stimulation regulate CD4+ T and B lymphocyte function in vitro and in vivo. Pharmacol Rev 2001;53:487–525. [PubMed] [Google Scholar]
- 30. Mignini F, Streccioni V, Amenta F. Autonomic innervation of immune organs and neuroimmune modulation. Auton Autacoid Pharmacol 2003;23:1–25. [DOI] [PubMed] [Google Scholar]
- 31. Arnon TI, Cyster JG. Blood, sphingosine‐1‐phosphate and lymphocyte migration dynamics in the spleen. Curr Top Microbiol Immunol 2014;378:107–128. [DOI] [PubMed] [Google Scholar]
- 32. Kraft P, Gob E, Schuhmann MK, et al. FTY720 ameliorates acute ischemic stroke in mice by reducing thrombo‐inflammation but not by direct neuroprotection. Stroke 2013;44:3202–3210. [DOI] [PubMed] [Google Scholar]
- 33. An C, Shi Y, Li P, et al. Molecular dialogs between the ischemic brain and the peripheral immune system: Dualistic roles in injury and repair. Prog Neurobiol 2014;115:6–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Emsley HC, Smith CJ, Gavin CM, et al. An early and sustained peripheral inflammatory response in acute ischaemic stroke: Relationships with infection and atherosclerosis. J Neuroimmunol 2003;139:93–101. [DOI] [PubMed] [Google Scholar]
- 35. Ostrowski RP, Schulte RW, Nie Y, et al. Acute splenic irradiation reduces brain injury in the rat focal ischemic stroke model. Transl Stroke Res 2012;3:473–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kim E, Yang J, Beltran CD, Cho S. Role of spleen‐derived monocytes/macrophages in acute ischemic brain injury. J Cereb Blood Flow Metab 2014;34:1411–1419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Jin R, Yang G, Li G. Inflammatory mechanisms in ischemic stroke: Role of inflammatory cells. J Leukoc Biol 2010;87:779–789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Price CJ, Menon DK, Peters AM, et al. Cerebral neutrophil recruitment, histology, and outcome in acute ischemic stroke: An imaging‐based study. Stroke 2004;35:1659–1664. [DOI] [PubMed] [Google Scholar]
- 39. Kim J, Song TJ, Park JH, et al. Different prognostic value of white blood cell subtypes in patients with acute cerebral infarction. Atherosclerosis 2012;222:464–467. [DOI] [PubMed] [Google Scholar]
- 40. Ajmo CT Jr, Vernon DO, Collier L, et al. The spleen contributes to stroke‐induced neurodegeneration. J Neurosci Res 2008;86:2227–2234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Amantea D, Nappi G, Bernardi G, Bagetta G, Corasaniti MT. Post‐ischemic brain damage: Pathophysiology and role of inflammatory mediators. FEBS J 2009;276:13–26. [DOI] [PubMed] [Google Scholar]
- 42. Justicia C, Panes J, Sole S, et al. Neutrophil infiltration increases matrix metalloproteinase‐9 in the ischemic brain after occlusion/reperfusion of the middle cerebral artery in rats. J Cereb Blood Flow Metab 2003;23:1430–1440. [DOI] [PubMed] [Google Scholar]
- 43. Hurn PD, Subramanian S, Parker SM, et al. T‐ and B‐cell‐deficient mice with experimental stroke have reduced lesion size and inflammation. J Cereb Blood Flow Metab 2007;27:1798–1805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Yilmaz G, Arumugam TV, Stokes KY, Granger DN. Role of T lymphocytes and interferon‐gamma in ischemic stroke. Circulation 2006;113:2105–2112. [DOI] [PubMed] [Google Scholar]
- 45. Zhang BJ, Men XJ, Lu ZQ, et al. Splenectomy protects experimental rats from cerebral damage after stroke due to anti‐inflammatory effects. Chin Med J (Engl) 2013;126:2354–2360. [PubMed] [Google Scholar]
- 46. Bodhankar S, Chen Y, Vandenbark AA, Murphy SJ, Offner H. IL‐10‐producing B‐cells limit CNS inflammation and infarct volume in experimental stroke. Metab Brain Dis 2013;28:375–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Zhang Y, Gao Z, Wang D, et al. Accumulation of natural killer cells in ischemic brain tissues and the chemotactic effect of IP‐10. J Neuroinflammation 2014;11:79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Gelderblom M, Leypoldt F, Steinbach K, et al. Temporal and spatial dynamics of cerebral immune cell accumulation in stroke. Stroke 2009;40:1849–1857. [DOI] [PubMed] [Google Scholar]
- 49. Alvarez D, Vollmann EH, von Andrian UH. Mechanisms and consequences of dendritic cell migration. Immunity 2008;29:325–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Banchereau J, Briere F, Caux C, et al. Immunobiology of dendritic cells. Annu Rev Immunol 2000;18:767–811. [DOI] [PubMed] [Google Scholar]
- 51. Chen J, Sanberg PR, Li Y, et al. Intravenous administration of human umbilical cord blood reduces behavioral deficits after stroke in rats. Stroke 2001;32:2682–2688. [DOI] [PubMed] [Google Scholar]
- 52. Vendrame M, Cassady J, Newcomb J, et al. Infusion of human umbilical cord blood cells in a rat model of stroke dose‐dependently rescues behavioral deficits and reduces infarct volume. Stroke 2004;35:2390–2395. [DOI] [PubMed] [Google Scholar]
- 53. Willing AE, Vendrame M, Mallery J, et al. Mobilized peripheral blood cells administered intravenously produce functional recovery in stroke. Cell Transplant 2003;12:449–454. [DOI] [PubMed] [Google Scholar]
- 54. Vendrame M, Gemma C, de Mesquita D, et al. Anti‐inflammatory effects of human cord blood cells in a rat model of stroke. Stem Cells Dev 2005;14:595–604. [DOI] [PubMed] [Google Scholar]
- 55. Hao L, Zou Z, Tian H, et al. Stem cell‐based therapies for ischemic stroke. Biomed Res Int 2014;2014:468748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Yang M, Wei X, Li J, et al. Changes in host blood factors and brain glia accompanying the functional recovery after systemic administration of bone marrow stem cells in ischemic stroke rats. Cell Transplant 2010;19:1073–1084. [DOI] [PubMed] [Google Scholar]
- 57. Takahashi A, Ohnishi K, Asakawa I, et al. Radiation response of apoptosis in C57BL/6N mouse spleen after whole‐body irradiation. Int J Radiat Biol 2001;77:939–945. [DOI] [PubMed] [Google Scholar]
- 58. Li G, Regunathan S, Barrow CJ, et al. Agmatine: An endogenous clonidine‐displacing substance in the brain. Science 1994;263:966–969. [DOI] [PubMed] [Google Scholar]
- 59. Uranchimeg D, Kim JH, Kim JY, et al. Recovered changes in the spleen by agmatine treatment after transient cerebral ischemia. Anat Cell Biol 2010;43:44–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Chen J, Zhang ZG, Li Y, et al. Statins induce angiogenesis, neurogenesis, and synaptogenesis after stroke. Ann Neurol 2003;53:743–751. [DOI] [PubMed] [Google Scholar]
- 61. Zhang L, Zhang ZG, Ding GL, et al. Multitargeted effects of statin‐enhanced thrombolytic therapy for stroke with recombinant human tissue‐type plasminogen activator in the rat. Circulation 2005;112:3486–3494. [DOI] [PubMed] [Google Scholar]
- 62. Cakmak A, Yemisci M, Koksoy C, et al. Statin pre‐treatment protects brain against focal cerebral ischemia in diabetic mice. J Surg Res 2007;138:254–258. [DOI] [PubMed] [Google Scholar]
- 63. Zacharek A, Chen J, Cui X, Yang Y, Chopp M. Simvastatin increases notch signaling activity and promotes arteriogenesis after stroke. Stroke 2009;40:254–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Tziomalos K, Giampatzis V, Bouziana SD, et al. Effect of prior treatment with different statins on stroke severity and functional outcome at discharge in patients with acute ischemic stroke. Int J Stroke 2013;8:E49. [DOI] [PubMed] [Google Scholar]
- 65. Jain MK, Ridker PM. Anti‐inflammatory effects of statins: Clinical evidence and basic mechanisms. Nat Rev Drug Discov 2005;4:977–987. [DOI] [PubMed] [Google Scholar]
- 66. Jin R, Zhu X, Liu L, et al. Simvastatin attenuates stroke‐induced splenic atrophy and lung susceptibility to spontaneous bacterial infection in mice. Stroke 2013;44:1135–1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Fathali N, Ostrowski RP, Hasegawa Y, et al. Splenic immune cells in experimental neonatal hypoxia‐ischemia. Transl Stroke Res 2013;4:208–219. [DOI] [PMC free article] [PubMed] [Google Scholar]