

Abstract
Objectives: 17β‐oestradiol interacts with growth factors to modulate lactotroph cell population. However, contribution of isoforms of the oestrogen receptor in these activities is not fully understood. In the present study, we have established participation of α and β oestrogen receptors in effects of 17β‐oestradiol on lactotroph proliferation induced by insulin and shown involvement of the NO/sGC/cGMP pathway.
Materials and methods: Cell cultures were prepared from anterior pituitaries of female rats to evaluate lactotroph cell proliferation using bromodeoxyuridine (BrdUrd) detection, protein expression by western blotting and cGMP by enzyme immunoassay.
Results: In serum‐free conditions, 17β‐oestradiol and α and β oestrogen receptor agonists (PPT and DPN) failed to increase numbers of lactotroph cells undergoing mitosis. Co‐incubation of 17β‐oestradiol/insulin and PPT/insulin significantly decreased lactotroph mitogenic activity promoted by insulin alone. Both ICI 182780 and NOS inhibitors (L‐NMMA and L‐NAME) induced reversal of the anti‐proliferative effect promoted by 17β‐oestradiol/insulin and PPT/insulin. Moreover, 17β‐oestradiol, PPT and insulin increased sGC α1 protein expression and inhibited β1, whereas co‐incubation of 17β‐oestradiol/insulin or PPT/insulin induced increases of the two isoforms α1 and β1. 17β‐oestradiol and insulin reduced cGMP production, while 17β‐oestradiol/insulin co‐incubation increased this cyclic nucleotide.
Conclusions: Our results suggest that 17β‐oestradiol is capable of arresting lactotroph proliferation induced by insulin through ER α with participation of the signalling NO/sGC/cGMP pathway.
Introduction
17β‐oestradiol is an important regulator of anterior pituitary function, by stimulating prolactin (PRL) secretion and modulation of lactotroph cell population size (1, 2). These effects are mediated by the two isoforms, α and β, of the oestrogen receptor (ER). Both of these have been identified in different pituitary cell types including lactotrophs (3). Furthermore, variants of ER, termed truncated ER products (TERP‐1 and TERP‐2), have been detected in rat pituitary cells (4, 5).
The biological role of these two ER subtypes is poorly understood, with its elucidation being the aim of much present research. Effects of oestrogen are mainly mediated through ER α. However, in different tissues, several studies have shown that ER β has the capacity to repress transcriptional activity of ER α by acting as a dominant negative regulator (6, 7). In the pituitary gland, some evidence suggests that ER α mediates oestradiol action on lactotroph cell population growth (8, 9) and PRL secretion (10, 11). However, although other studies indicate that the biological action of oestradiol in the pituitary may involve both isoforms of ER (12), specific contribution of ER α and ER β to control of lactotroph proliferation has still not been completely resolved.
17β‐oestradiol binds to the two ER subtypes with very similar affinity, thus regulating expression of key proteins at the genomic level. Moreover, 17β‐oestradiol can rapidly modulate cell functions through signalling pathways that begin at cell membrane level and involve phosphatidylinositol‐3 kinase (PI3K), mitogen‐activated protein kinase (MAPK) (13, 14) and nitric oxide (NO) (15). In a previous study, we have reported that 17β‐oestradiol inhibited insulin‐induced lactotroph proliferation via membrane ER, with protein kinase C/MAPK involvement (14). However, mediation of the NO/soluble guanylyl cyclase (sGC)/3′,5′‐cyclic guanosine monophosphate (cGMP) signalling pathway on anti‐mitogenic effects of oestradiol remains unknown.
NO is a versatile molecule with a wide spectrum of effects on numerous tissues (16, 17). As a signalling molecule, synthesis of NO is catalysed by NO synthase (NOS). Three isoforms of this have been well characterized, neuronal NOS, inducible NOS and endothelial NOS (18), which are widely expressed in most cell types including those of the pituitary (19, 20). NO acts mainly through its major intracellular receptor, heterodimeric enzyme sGC (21), which is constituted by α and β subunits, with four types existing (α1, α2, β1, and β2). α1/β1 is the most abundant heterodimer, also showing the greatest activity (22). Binding of NO to sGC leads to formation of cGMP, which binds to target proteins such as cGMP‐dependent protein kinase, cGMP‐regulated ion channels and several families of phosphodiesterases, to subsequently alter cell responses (23, 24).
It has been reported that NO modulates proliferation of different cell types (25, 26). However, its participation in mitogenic activity of lactotrophs has not previously been evaluated. Moreover, NO regulates secretion of various anterior pituitary hormones (27) including that of PRL (28, 29).
17β‐oestradiol influences NO/sGC/cGMP pathway in different tissues (15, 30) including in the pituitary gland, where it has been demonstrated to have an inhibitory role. It has been demonstrated that oestrogen down‐regulates nNOS mRNA and protein expression (31), while castration reverts these effects by inducing increase in NOS enzyme activity (32). In addition, oestrogen decreases sensitivity of anterior pituitary cells to inhibitory effects of NO on PRL release by down‐regulating the NO/cGMP pathway (33). Moreover, acute treatment with 17β‐oestradiol has been shown to have opposite effects on sGC subunits, increasing α1 while decreasing the β1 subunit protein and mRNA expression as well as sGC activity, in immature rats (34). Additionally, these sGC subunits fluctuate through the oestrous cycle, with these changes being directly related to oestradiol level fluctuations rather than to NO level variations (35).
To increase the body of knowledge concerning molecular mechanisms responsible for the modulatory role of 17β‐oestradiol in expansion of lactotroph population, we focused the present work on establishing participation of α and β ER in the effects of 17β‐oestradiol on insulin‐induced lactotroph proliferation and also on determining involvement of the NO/sGC/cGMP pathway.
Materials and methods
Animals
Three‐month‐old female Wistar rats of were used. Large pools of these animals were assigned to provide each culture, taken at random cycle stages. All rats were raised in our laboratory under controlled temperature (21 ± 3 °C) and lighting conditions (14 h light/10 h dark), with free access to commercial laboratory chow and tap water. Animal conditions were in compliance with the Guidelines on the Handling and Training of Laboratory Animals, published by the Universities Federation for Animal Welfare and the local Institutional Animal Care Committee.
Dissociation of anterior pituitary cells
Protocol for dissociation of pituitary cells was as described previously (36). Briefly, anterior pituitaries excised from female rats were placed in minimal essential medium for suspension culture (SMEM) before being minced, digested with 0.4% trypsin, and dispersed using Pasteur pipettes. Cell yield was 1.5–2 × 106 per pituitary and cell viability (tested by trypan blue exclusion) was always better than 90%. Final suspensions were adjusted to 1 × 106 cells/ml of medium. For proliferation assays, cells were seeded on glass coverslips (13 mm) density 2 × 105 cells/well, and placed at the bottom of 24‐well culture plates (Corning, New York, NY, USA). For other techniques, cells were plated in six‐well culture plates (Corning) at density of 5 × 105 cells/well. Then, cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM) without phenol red, supplemented with 4% foetal calf serum and 8% horse serum (Gibco, New York, NY, USA), in an incubator with humidified atmosphere of 5% CO2 and 95% air at 37 °C for 3 days.
All culture media were filtered through 0.2 μm Nalgene membranes (Nalge Company, New York, NY, USA). Cell culture grade reagents were obtained from the Sigma Chemical Co. (St Louis, MO, USA).
Cell treatments
After 3 days culture, medium was discarded and replaced with serum‐free DMEM, supplemented with hydrocortisone (100 μg/l), 3,3′‐triiodothyronine (400 ng/l), transferrin (10 mg/l) and sodium selenite (5 μg/l) for 24 h. Then, cells were stimulated for 60 min with 17β‐oestradiol (1 nm, Sigma) and insulin (1000 ng/ml, from bovine pancreas, Sigma) either alone or in combination.
Selective ER agonists: α agonist, 4,4′,4′′‐(4‐propyl‐[1H]‐pyrazole‐1,3,5‐tryl) trisphenol (PPT; 1, 10 and 100 nm) and β agonist, 2,3‐bis (4‐hydroxyphenyl)‐propionitrile (DPN; 1, 10 and 100 nm; Tocris Cookson Inc, Ellisville, MO, USA) were added separately to culture media for 60 min. These concentrations were chosen based on previous reports (37, 38).
ER inhibitor ICI 182780 (100 nm, Sigma) was added 30 min before other agents to be assayed, and then cultures were incubated for 60 min.
To study possible involvement of NOS/NO via 17β‐oestradiol, and insulin effects on lactotroph proliferation, other batches of cells were treated with NOS inhibitors. As NOS activity is competitively inhibited by l‐arginine analogues, including Nω‐nitro‐l‐arginine methyl ester (L‐NAME) and Ng‐monomethyl‐l‐arginine (L‐NMMA) (Sigma) (16), L‐NMMA (0.1 mm) and L‐NAME (1 mm) were added 30 min prior to addition of further agents, then cell cultures were maintained for 60 min.
At the end of each experimental condition, anterior pituitary cells were processed to evaluate lactotroph proliferation using bromodeoxyuridine (BrdUrd) detection, protein expression by western blotting and cGMP by enzyme immunoassay.
Immunocytochemical detection of lactotroph proliferation
Cells at DNA‐synthesizing stage and lactotrophs were individualized by the dual‐immunocytochemical detection of BrdUrd and PRL, according to Oomizu et al. (39) with modifications (2). After 60‐min stimulation with the different reagents, culture medium was replaced and BrdUrd (100 nm) was added for an additional 24 h. Then cells attached to coverslips were fixed in 4% formaldehyde in phosphate‐buffered saline (PBS) for 30 min at room temperature, washed in PBS and permeabilized with 0.5% Triton X‐100 for 10 min. Non‐specific immunoreactivity was blocked with 1% PBS–BSA for 30 min at room temperature. Cells were then incubated overnight with monoclonal antibody to BrdUrd (Amersham, Buckinghamshire, UK) at 4 °C in a wet chamber. After washing in PBS, cells were incubated in biotinylated anti‐mouse IgG, diluted 1:100, for 30 min at room temperature. Coverslips were washed again in PBS and incubated in avidin–biotin–peroxidase complex (ABC). Immunoreactivity of BrdUrd was visualized using 3,3‐diaminobenzidine tetrahydrochloride (DAB) as chromogen that leaves nuclei of proliferating cells stained brown. PRL immunocytochemistry for lactotroph detection was then performed on the same coverslip, and cells were incubated with rabbit anti‐rat PRL at 1:3000 dilution (NIH Hormone Program) in a wet chamber for 1 h at 37 °C. This was followed by washing in PBS and incubation in biotinylated anti‐rabbit IgG 1:150 for 30 min. After a further wash in PBS, avidin–biotin–peroxidase complex was applied for 30 min at room temperature. Immunoreactivity of PRL was then detected using chloronaphthol, by which immunostained lactotrophs acquired purplish blue colour. Finally, coverslips were mounted on glass slides using glycerol. Controls were also performed, by applying the same protocols, but omitting BrdUrd or PRL antibodies.
Totals of 1000 PRL‐immunoreactive cells were examined by light microscopy in randomly chosen fields, on each glass slide, to establish percentage of immunoreactive pituitary cells for both PRL and BrdUrd. Three slides were analysed for each experimental group.
Preparation of cell homogenates for immunoblot analysis
Once the experimental protocols were completed, cells were rinsed in PBS and lysed on ice by addition of 200 μl of cold PBS containing 1.25% Igepal CA‐630, 1 mm ethylenediaminetetraacetic acid, 2 mm phenylmethylsulphonyl fluoride, 10 μg/ml leupeptin and 10 μg/ml aprotinin. This was followed by scraping off and transfer of lysate to centrifuge tubes placed on ice. After 30 min, lysates were centrifuged at 14 000 g for 10 min at 4 °C to pellet Igepal CA‐630‐insoluble material, and supernatants were stored in aliquots and frozen at −40 °C until required.
Protein measurement
Protein content was measured using a Bio‐Rad kit (Bio‐Rad Protein Assay; Bio‐Rad Laboratories, Hercules, CA, USA).
Western blot analysis
Thirty micrograms of total homogenate was run in 12% acrylamide gel (Sigma Chemical Co.). To estimate corresponding molecular weights, Full Range Rainbow Molecular Weight Marker was run in parallel (Amersham‐Life Science). Proteins were transferred to a nitrocellulose membrane, and non‐specific binding was blocked by PBS containing 5% non‐fat dried milk and 0.1% Tween 20 (blocking buffer), at room temperature. Membranes were rinsed and incubated for 2 h with the following appropriate primary antibodies: guanylyl cyclase α1 rabbit antibody (1:1700) and guanylyl cyclase β1 rabbit antibody (1:700) (Sigma‐Aldrich). These antibodies were raised against amino acid residues 673–690 and 189–207 of rat guanylyl cyclases α1 and β1 respectively. Blots were incubated with peroxidase‐conjugated (HRP) goat anti‐rabbit secondary antibody (Jackson Immunoresearch Labs Inc, West Grove, PA, USA) diluted in blocking buffer (1:5000). These blots were thoroughly rinsed in PBS/0.1% Tween‐20, and HRP‐coupled secondary antibody was revealed with ECL western blot detection reagents (Amersham Biosciences), according to manufacturer’s instructions. Emitted light was captured on Hyperfilm (Amersham‐Pharmacia‐Biotech). Signals were scanned and quantified using Scion Image software (V. beta 4.0.2; Scion Image Corp., Frederick, MD, USA) at three different exposure times. Expression of β‐actin (1:5000, mouse monoclonal antibody; Sigma‐Aldrich) was used as internal control to confirm equivalent total protein loading.
Intracellular cGMP determination
Cyclic GMP was measured in triplicate using cGMP enzyme immunoassay Biotrak System (Amersham), according to manufacturer’s instructions. After the different experimental protocols, culture media were decanted, lysis reagent added and plates were shaken for 10 min to facilitate cell lysis. Then, acetylation reagent was added to all sample wells and mixed on a shaker for 5 min before antiserum was added and incubated at 3–5 °C for exactly 2 h. This was followed by addition of diluted conjugated, and plates were incubated at 3–5 °C for 60 min in the dark. All wells were washed, enzyme substrate was added to the plates and then mixed for 30 min at room temperature. Colour intensity was first read at 630 nm, and then again at 450 nm after addition of sulphuric acid.
Intracellular cGMP concentrations were expressed as fmol/μg protein, with total protein content in pellets being measured as described above.
Statistical analysis
Statistical analysis was carried out on three replicates measured on three independent cell cultures using ANOVA. This was followed up using Tukey test of InfoStat software package. Significance levels were chosen at P < 0.05.
Results
Lactotroph cell proliferation
ER α mediated anti‐proliferative effects of 17β‐oestradiol in interaction with insulin As we previously reported in serum‐free pituitary cell cultures, although 17β‐oestradiol did not induce a proliferative effect on lactotrophs, it was able to reverse mitogenic activity promoted by insulin (14). With the aim of investigating whether this anti‐mitogenic effect was mediated by ER α or β, PPT and DPN (α and β agonists) were used. First, we performed dose–response analysis for both agonists at 1–10 and 100 nm. BrdUrd‐labelling index was determined in these pituitary cells cultured in serum‐free DMEM and subjected to double‐labelling immunocytochemistry. All doses of PPT and DPN failed to increase numbers of lactotrophs undergoing mitosis (Fig. 1).
Figure 1.
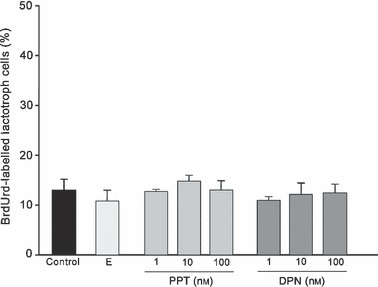
Effects of 17β‐oestradiol (E; 1 nm), PPT and DPN on lactotroph proliferation. None of the concentrations tested increased numbers of BrdUrd‐labelled lactotrophs. The ANOVA‐Tukey test was performed on three independent experiments. Data are expressed as mean ± SEM of triplicate determinations from a representative experiment.
On the other hand, increased numbers of BrdUrd‐labelled lactotrophs induced by insulin (150%) was significantly reversed with all doses of PPT. Moreover, doses of DPN tested were unable to induce anti‐mitogenic effects, resulting in a similar number of BrdUrd‐labelled lactotrophs observed after insulin treatment (Fig. 2). Taking into account these results, and considering that high concentration of DPN (100 nm) provokes loss of ERβ selectivity and activates ERα as well (37, 38), lowest doses of both agonists (1 nm) were selected for the following experiments.
Figure 2.
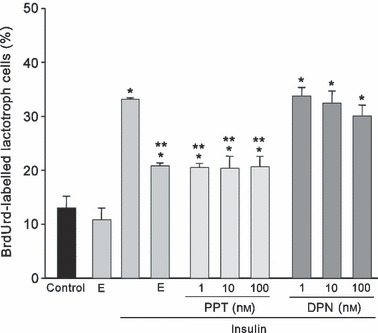
17β‐oestradiol (E; 1 nm) and all doses tested of α agonist, PPT, reversed mitogenic activity induced by insulin (1000 ng/ml) on lactotrophs. DPN failed to induce any anti‐mitogenic effect. The ANOVA‐Tukey test was performed on three independent experiments. *P < 0.001 versus control, **P < 0.001 versus insulin. Data are expressed as the mean ± SEM of triplicate determinations from a representative experiment.
ER inhibitor ICI 182780 reverted effects of 17β‐oestradiol and PPT. To confirm that both 17β‐oestradiol and PPT act through specific ER, experimental protocols with inhibitor ICI 182780 were tested. Addition of 100 nm of ICI 182780 did not modify lactotroph proliferation induced by insulin. Pre‐incubation with ICI blocked anti‐proliferative effects promoted by 17β‐oestradiol and also induced partial reversal of PPT effects on prolactin cell proliferation stimulated by insulin (P < 0.001) (Fig. 3).
Figure 3.
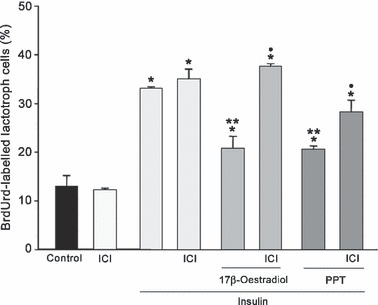
Effects of ICI 182780 (100 nm) on lactotroph cell proliferation. This inhibitor reversed anti‐proliferative effects promoted by 17β‐oestradiol (1 nm) or PPT (1 nm) on lactotroph proliferation induced by insulin (1000 ng/ml). The ANOVA‐Tukey test was performed on three independent experiments. *P < 0.001 versus control, **P < 0.001 versus insulin, •P < 0.01 versus 17β‐oestradiol/insulin or PPT/insulin. Data are expressed as mean ± SEM of triplicate determinations from a representative experiment.
NOS inhibitors attenuated anti‐mitogenic effects induced by 17β‐oestradiol on insulin‐stimulated lactotrophs. To investigate whether the NO/GC signalling pathway was involved in the anti‐mitogenic effect induced by 17β‐oestradiol/insulin co‐incubation, action of NOS inhibitors (L‐NMMA and L‐NAME) was evaluated. These inhibitors were able to revert 17β‐oestradiol‐induced inhibition of insulin‐dependent lactotroph mitogenic activity reaching similar values to those produced by insulin alone (P < 0.001). These results could suggest that co‐incubation of 17β‐oestradiol/insulin increased NOS activity inducing NO‐mediated anti‐mitogenic effects on lactotrophs. Addition of L‐NMMA or L‐NAME alone did not modify BrdUrd labelling index compared to basal values (Fig. 4).
Figure 4.
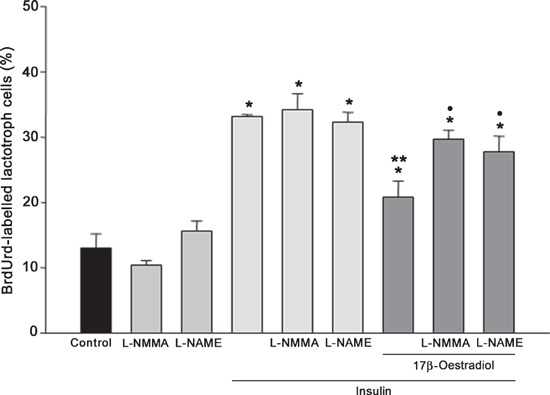
Effects of NOS inhibitors on lactotroph proliferation induced by 17β‐oestradiol (1 nm) and insulin (1000 ng/ml). Inhibition of NOS activity by L‐NMMA (0.1 mm) and L‐NAME (1 mm) reversed anti‐proliferative effects induced by 17β‐oestradiol/insulin. The ANOVA‐Tukey test was performed on three independent experiments. *P < 0.001 versus control, **P < 0.001 versus insulin and •P < 0.01 versus 17β‐oestradiol/insulin. Data are expressed as mean ± SEM of triplicate determinations from a representative experiment.
L‐NMMA and L‐NAME attenuated anti‐proliferative effects induced by PPT/insulin on lactotrophs. NOS inhibitors L‐NMMA and L‐NAME were added for 30 min prior to addition of ER α agonist then cell cultures were maintained for 60 min. These inhibitors partially reverted anti‐mitogenic effects induced by PPT on insulin‐stimulated cells. As shown in Fig. 5, lactotroph proliferation increased by around 33% due to the effect of L‐NMMA and by 25% after addition of L‐NAME in cell cultures treated with PPT/insulin (P < 0.01). These results show that ER α activation by its agonist increased NOS activity, which could be involved in modulating lactotroph proliferation induced by insulin.
Figure 5.
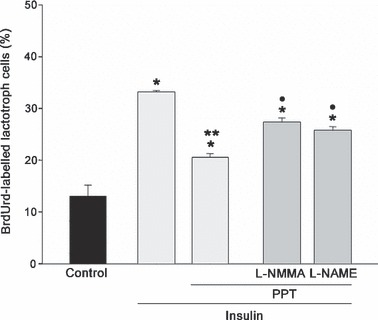
Effects of L‐NMMA (0.1 mm) and L‐NAME (1 mm) on lactotroph proliferation induced by 17β‐oestradiol agonists and insulin. Both inhibitors reversed only anti‐proliferative effects exerted by PPT/insulin. The ANOVA‐Tukey test was performed on three independent experiments. *P < 0.001 versus control, **P < 0.01 versus insulin, •P < 0.01 versus PPT/insulin. Data expressed as mean ± SEM of triplicate determinations from a representative experiment.
Guanylyl cyclase expression by western blotting
17β‐oestradiol and insulin modified sGC α1 and β1 expression. The two sGC isoforms α1 and β1 were detected by immunoblotting using specific antibodies, as bands of approximately 80 and 70 kDa respectively. 17β‐oestradiol applied for 60 min in serum‐free culture medium increased sGC α1 protein expression but inhibited β1, compared to respective control model. Insulin was able to induce similar dual effects on the two sGC isoforms, with significant increase noted in α1 protein level, and reduction occurring in β1 expression. However, co‐incubation of 17β‐oestradiol/insulin induced an increase in the two isoforms α1 and β1, with respect to controls (P < 0.01) (Fig. 6). Expression of β‐actin was used as internal control to confirm equivalent total protein loading.
Figure 6.
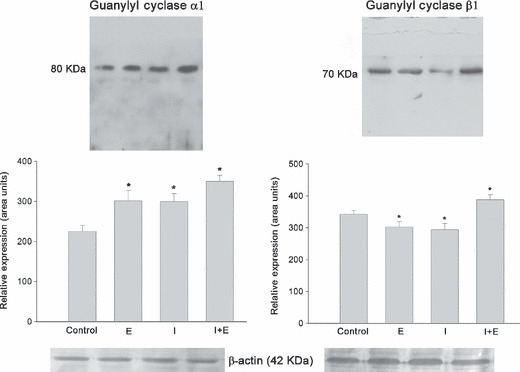
sGC α1 and β1 expression in total pituitary extract from cell cultures stimulated with 17β‐oestradiol (E; 1 nm) or insulin (I; 1000 ng/ml) alone or as co‐incubations. Both E or I increased protein expression of sGC α1 and decreased β1. In contrast, E + I induced increases in both isoforms. Bands correspond to representative experiment from a total of three with similar results. The ANOVA‐Tukey test was performed on three independent cell cultures: *P < 0.01 versus control. Data shown as mean ± SEM of triplicate determinations from a representative experiment.
PPT/insulin increased both sGC isoforms. As shown in Fig. 7, ER α agonist PPT applied for 60 min in pituitary cultures induced increases in sGC α1 and decrease in β1 protein levels in a similar way to that of 17β‐oestradiol (P < 0.001). As expected, co‐incubation of PPT/insulin provoked significant increase in both sGC isoforms α1 and β1. These results suggest that 17β‐oestradiol effects on GC expression are mediated by ER α. Expression of β‐actin was used as internal control to confirm equivalent total protein loading.
Figure 7.
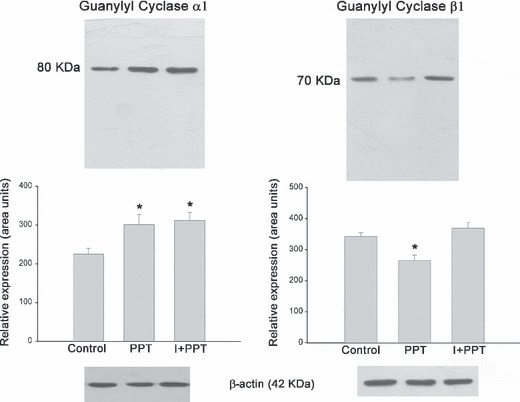
sGC α1 and β1 expression in total pituitary extract from cell cultures stimulated with PPT (1 nm) or insulin (I; 1000 ng/ml) alone or co‐incubations. Bands correspond to a representative experiment from a total of three which had similar results. The ANOVA‐Tukey test was performed on three independent cell cultures: *P < 0.001 versus control. Data are shown as the mean ± SEM of triplicate determinations from a representative experiment.
ICI 182780 reverted 17β‐oestradiol and PPT effects on sGC isoforms. To confirm the role of ER in modulating sGC α1 and β1 expression, other pituitary cell batches were pre‐incubated with ICI 182780 (100 nm) for 30 min and then cultures were stimulated for 60 min with 17β‐oestradiol or PPT. Addition of ICI 182780 blocked effects promoted by 17β‐oestradiol and PPT on sGC α1 and β1 expression (P < 0.001) (Fig. 8). Expression of β‐actin was used as internal control to confirm total equivalent protein loading.
Figure 8.
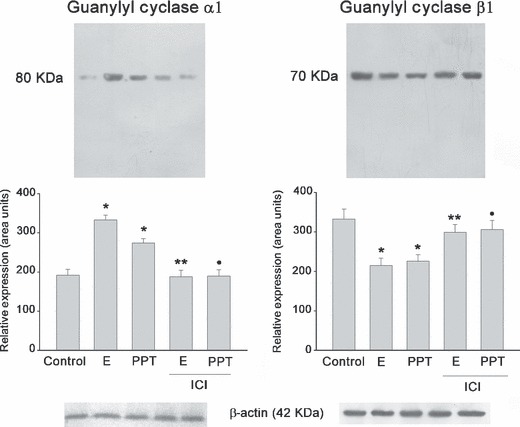
ICI 182780 (100 nm) blocked effects promoted by 17β‐oestradiol (1 nm) or PPT (1 nm) on sGC α1 and β1 expression. Bands correspond to a representative experiment from a total of three which had similar results. The ANOVA‐Tukey test was performed on three independent cell cultures: *P < 0.01 versus control, **P < 0.01 versus 17β‐oestradiol, •P < 0.01 versus PPT. Data are shown as the mean ± SEM of triplicate determinations from a representative experiment.
Intracellular cGMP determination
With the purpose of investigating sGC activity, its intracellular product cGMP was quantified in these pituitary cell cultures after all different treatments. 17β‐oestradiol or insulin stimulus for 60 min induced reduction in cGMP production of around 44% (415 ± 1.64 fmol/μg protein) and 26% (550 ± 37 fmol/μg protein), respectively, when compared to controls (745 ± 15.26 fmol/μg protein). However, 17β‐oestradiol/insulin co‐incubation provoked a significant increase in cGMP with respect to 17β‐oestradiol or insulin alone (670 ± 21.64 fmol/μg protein).
Discussion
Our current data provide evidence that 17β‐oestradiol modulates proliferation of lactotrophs induced by insulin. The isoform α of ER seems to participate in this effect, which in turn, could regulate NO/sGC/cGMP signalling pathway.
In this investigation, we used ER isoform‐selective ligands, which specifically activate each one of ER subtype, in physiological presence of the other. This is important in the study of ER signalling pathways, given that several isoforms may contribute as regulators in target tissues. Thereby, this method has an advantage over knockout models, where either α or β forms of ER are selectively eliminated. Although such knockout models are very instrumental, activation of compensatory mechanisms in the absence of one receptor may partially hamper interpretation of the results.
In the present study, insulin treatment increased numbers of BrdUrd‐labelled PRL cells, thus demonstrating that this hormone is a growth‐promoting factor for this cell type. These results concur with previous reports demonstrating that this hormone stimulates replication of lactotrophs in vivo and in vitro (40, 41, 42). Interactions with oestradiol have also been previously shown, with cooperative crosstalk between oestradiol and insulin signalling pathways playing a critical role in cell proliferation in different tissues (43). In our study, ICI 182780 did not modify percentages of BrdUrd‐labelled PRL cells stimulated with insulin, suggesting that effects of this hormone on lactotroph proliferation are independent of ERs.
It has been described that effects of insulin and IGF‐1 were mediated by either insulin or IGF‐1 receptors (44). Related to this in pituitary glands, insulin at high doses may interact with IGF‐1 receptor (45). However, in previous studies, we demonstrated that the lowest dose of insulin tested (10 ng/ml) was effective in augmenting mitogenic activity of lactotrophs (42) through PKC, ERK 1/2 and Pit‐1 (14) pathways. Therefore, the possibility in our study that insulin at high concentrations might be stimulating lactotroph proliferation via the IGF‐1 receptor is not ruled out.
Our findings have shown that the α agonist, PPT, attenuated mitogenic effects exerted by insulin on lactotrophs. Moreover, the data allowed us to infer that ER α participated in this anti‐mitogenic effect of 17β‐oestradiol on lactotrophs. Previous studies have indicated that most of oestradiol activity on the pituitary is exerted through ER α, with this isoform being involved in stimulatory action and cell proliferation, as well as PRL release (10, 46). However, it remains unknown whether ER α is involved in negative effects in tissues such as the pituitary, in which mitogenic and anti‐mitogenic effects of 17β‐oestradiol have been demonstrated previously. In this way, 17β‐oestradiol inhibited lactotroph proliferation via ER, although the isoform of ER involved in this result was not explored (1). In addition, in a previous report, we have demonstrated that 17β‐oestradiol induced anti‐proliferative effects through membrane‐bound ER (14). It is possible that in our system, 17β‐oestradiol activated ER α, whereas ER β exerted competitive blocking of ER α binding to DNA sites, thus inhibiting ER α transcriptional activity, as has been proposed previously (7).
Results obtained with L‐NMMA and L‐NAME showed that inhibition of NOS reversed anti‐mitogenic effects exerted by 17β‐oestradiol, reaching similar values to those observed with insulin alone. These data suggest that 17β‐oestradiol stimulates NOS activity, which could be responsible for arrest of lactotroph proliferation induced by insulin. It has been demonstrated previously that NO inhibits proliferation of different cell types, such as in neuronal and endothelial cell proliferation (26, 47, 48). Moreover, a similar effect of endogenous NO, acting as a negative signal for proliferation, has been shown to occur in FRTL‐5 thyroid cells (16). NOS inhibition has been reported to reduce mitogenic activity in adult olfactory epithelium (25). However, in tissues such as vascular smooth muscle, oestradiol induces an anti‐proliferative effect that could be mediated by a NO independent mechanism (49). Our findings support that NO in response to 17β‐oestradiol arrests lactotroph proliferation induced by insulin, thus suggesting that this mechanism might regulate expansion of lactotroph population. NOS inhibitors reversed anti‐mitogenic effects induced by PPT in interaction with insulin, indicating that effects of 17β‐oestradiol on NOS were mediated by ER α. In agreement with our results, Scordalakes et al. (50) demonstrated that ER α regulates nNOS in both male and female brains. In contrast, in hypothalamic neurons, oestradiol stimulates the NO system through ER β (51, 52).
In addition, we observed an opposite effect of 17β‐oestradiol on the two sGC subunits, with α1 protein levels being elevated and β1 levels lowered. Furthermore, our results showed decrease in sGC activity, as evidenced by reduction in endogenous cGMP production. However, both results are consistent, because the two sGC isoforms are required in 1:1 stoichiometry to form an active cGMP‐producing enzyme. A previous study has demonstrated that 17β‐oestradiol regulates α1 and β1 subunits of NO receptor sGC, modifying mRNA and protein levels in rat uterus (53). In agreement with our findings, it has previously been observed that 17β‐oestradiol exerts acute inhibitory effects on sGC in the pituitary gland by down‐regulating sGC β1 subunits and sGC activity in a specific ER‐dependent manner (34). The effect of 17β‐oestradiol on GC isoform protein expression shown in this study was mediated by ER α, as indicated by increase in GCα1 and decrease in GCβ1 induced by ER α agonist PPT. In addition, blocking of these effects by ICI 182780 strengthens this conclusion. This result is further evidence that 17β‐oestradiol binds to ER α to affect the NO pathway.
Insulin used alone induced similar antagonistic effects on two sGC subunits and inhibited endogenous cGMP production. Several studies have suggested that NO mediates many central and peripheral effects of insulin (54, 55). In our study, co‐incubation with 17β‐oestradiol/insulin significantly increased both sGC subunits and induced increase in endogenous cGMP, which could be involved in arrest of the lactotroph proliferation. These results suggest that the NO signalling pathway is mediated by 17β‐oestradiol/insulin and could explain anti‐mitogenic effects of 17β‐oestradiol on lactotroph population.
Our results strengthen the inhibitor role of NO/sCG/cGMP pathway, which mediates anti‐mitogenic oestrogen effects on insulin‐induced lactotroph proliferation. 17β‐oestradiol, through ER α, was able to regulate NOS activity, protein expression of sGC subunits and cGMP production.
Activation of the NO/sGC/cGMP pathway as possible mediator of anti‐proliferative effects of 17β‐oestradiol in interaction with insulin on lactotrophs, here provides additional data for our previous study that showed inhibition of PKC/ERK 1/2 signalling pathway under the same experimental conditions (14). Both signalling mechanisms were able to participate in the regulatory effect of 17β‐oestradiol on the lactotroph population in these pituitary glands. From these and previous results, it could be proposed that 17β‐oestradiol is capable of arresting sustained activation of lactotroph proliferation induced by growth factors, thereby counterbalancing this response and modulating pituitary cell population.
Acknowledgements
The authors thank Ms Mercedes Guevara and Ms Elena Pereyra for their excellent technical assistance. We also thank native speaker Dr Paul Hobson for revising the English of the manuscript. This study was supported by grants from Consejo Nacional de Investigaciones Científicas y Técnicas (CONICET), Fondos para la Investigación Científica y Tecnológica (FONCyT) and the Secretaría de Ciencia y Tecnología de la Universidad Nacional de Córdoba (SECyT).
References
- 1. Kawashima K, Yamakawa K, Takahashi W, Takizawa S, Yin P, Sugiyama N et al. (2002) The estrogen‐occupied estrogen receptor functions as a negative regulator to inhibit cell proliferation induced by insulin/IGF‐1: a cell context‐specific antimitogenic action of estradiol on rat lactotrophs in culture. Endocrinology 143, 2750–2758. [DOI] [PubMed] [Google Scholar]
- 2. Gutiérrez S, Petiti JP, De Paul AL, Mukdsi JH, Aoki A, Torres AI et al. (2005) Antagonic effects of estradiol in interaction with IGF‐1 on proliferation of lactotroph cells in vitro. Histochem. Cell Biol. 124, 291–301. [DOI] [PubMed] [Google Scholar]
- 3. Mitchner NA, Garlick C, Ben‐Jonathan N (1998) Cellular distribution and gene regulation of estrogen receptors α and β in the rat pituitary gland. Endocrinology 139, 3976–3983. [DOI] [PubMed] [Google Scholar]
- 4. Friend KE, Ang LW, Shupnik MA (1995) Estrogen regulates the expression of several different estrogen receptor mRNA isoforms in rat pituitary. Proc. Natl. Acad. Sci. USA 92, 4367–4371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Shupnik MA (2002) Oestrogen receptors, receptor variants and oestrogen actions in the hypothalamic‐pituitary axis. J. Neuroendocrinol. 14, 85–94. [DOI] [PubMed] [Google Scholar]
- 6. Pettersson K, Grandien K, Kuiper GG, Gustafsson JA (1997) Mouse estrogen receptor beta forms estrogen response element‐binding heterodimers with estrogen receptor α. Mol. Endocrinol. 11, 1486–1496. [DOI] [PubMed] [Google Scholar]
- 7. Hall JM, McDonnell DP (1999) The estrogen receptor β‐isoform (ERβ) of the human estrogen receptor modulates ERα transcriptional activity and is a key regulator of the cellular response to estrogens and antiestrogens. Endocrinology 140, 5566–5578. [DOI] [PubMed] [Google Scholar]
- 8. Scully KM, Gleiberman AS, Lindzey J, Lubahn DB, Korach KS, Rosenfeld MG (1997) Role of estrogen receptor‐alpha in the anterior pituitary gland. Mol. Endocrinol. 11, 674–681. [DOI] [PubMed] [Google Scholar]
- 9. Tena‐Sempere M, Navarro VM, Mayen A, Bellido C, Sánchez‐Criado JE (2004) Regulation of estrogen receptor (ER) isoform messenger RNA expression by different ER ligands in female rat pituitary. Biol. Reprod. 70, 671–678. [DOI] [PubMed] [Google Scholar]
- 10. Bulayeva NN, Wozniak AL, Lash LL, Watson CS (2005) Mechanisms of membrane ER‐alpha‐mediated rapid stimulation of Ca2+ levels and PRL release in a pituitary cell line. Am. J. Physiol. Endocrinol. Metab. 288, E388–E397. [DOI] [PubMed] [Google Scholar]
- 11. Ben‐Jonathan N, Chen S, Dunckley JA, LaPensee C, Kansra S (2009) Estrogen receptor‐alpha mediates the epidermal growth factor‐stimulated prolactin expression and release in lactotrophs. Endocrinology 150, 795–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sánchez‐Criado JE, Martín De Las Mulas J, Bellido C, Tena‐Sempere M, Aguilar R, Blanco A (2004) Biological role of pituitary estrogen receptors ERalpha and ERbeta on progesterone receptor expression and action and on gonadotropin and prolactin secretion in the rat. Neuroendocrinology 79, 247–258. [DOI] [PubMed] [Google Scholar]
- 13. Dominguez R, Liu R, Baudry M (2007) 17‐beta‐estradiol‐mediated activation of extracellular‐signal regulated kinase, phosphatidylinositol 3‐kinase/protein kinase B‐Akt and N‐methyl‐d‐aspartate receptor phosphorylation in cortical synaptoneurosomes. J. Neurochem. 101, 232–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gutiérrez S, De Paul AL, Petiti JP, Del Valle Sosa L, Palmeri CM, Soaje M et al. (2008) Estradiol interacts with insulin through membrane receptors to induce an antimitogenic effect on lactotroph cells. Steroids 73, 515–527. [DOI] [PubMed] [Google Scholar]
- 15. Jaubert AM, Mehebik‐Mojaat N, Lacasa D, Sabourault D, Giudicelli Y, Ribiere C (2007) Nongenomic estrogen effects on nitric oxide synthase activity in rat adipocytes. Endocrinology 148, 2444–2452. [DOI] [PubMed] [Google Scholar]
- 16. Fozzatti L, Vélez ML, Lucero AM, Nicola JP, Mascanfroni ID, Macció DR et al. (2007) Endogenous thyrocyte‐produced nitric oxide inhibits iodide uptake and thyroid‐specific gene expression in FRTL‐5 thyroid cells. J. Endocrinol. 192, 627–637. [DOI] [PubMed] [Google Scholar]
- 17. Virdis A, Colucci R, Fornai M, Polini A, Daghini E, Duranti E et al. (2009) Inducible nitric oxide synthase is involved in endothelial dysfunction of mesenteric small arteries from hypothyroid rats. Endocrinology 150, 1033–1042. [DOI] [PubMed] [Google Scholar]
- 18. Knowles RG, Moncada S (1994) Nitric oxide synthases in mammals. Biochem. J. 298, 249–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kostic TS, Andric SA, Stojilkovic SS (2001) Spontaneous and receptor‐controlled soluble guanylyl cyclase activity in anterior pituitary cells. Mol. Endocrinol. 15, 1010–1022. [DOI] [PubMed] [Google Scholar]
- 20. Qian X, Jin L, Lloyd RV (1998) Percoll density gradient‐enriched populations of rat pituitary cells: interleukin 6 secretion, proliferative activity, and nitric oxide synthase expression. Endocr. Pathol. 9, 339–346. [DOI] [PubMed] [Google Scholar]
- 21. Cary SP, Winger JA, Derbyshire ER, Marletta MA (2006) Nitric oxide signaling: no longer simply on or off. Trends Biochem. Sci. 31, 231–239. [DOI] [PubMed] [Google Scholar]
- 22. Koglin M, Behrends S (2004) Native human nitric oxide sensitive guanylyl cyclase: purification and characterization. Biochem. Pharmacol. 67, 1579–1585. [DOI] [PubMed] [Google Scholar]
- 23. Hofmann F (2005) The biology of cyclic GMP‐dependent protein kinases. J. Biol. Chem. 280, 1–4. [DOI] [PubMed] [Google Scholar]
- 24. Bredt DS (1999) Endogenous nitric oxide synthesis: biological functions and pathophysiology. Free Radic. Res. 31, 577–596. [DOI] [PubMed] [Google Scholar]
- 25. Sülz L, Astorga G, Bellette B, Iturriaga R, Mackay‐Sim A, Bacigalupo J (2009) Nitric oxide regulates neurogenesis in adult olfactory epithelium in vitro. Nitric Oxide 20, 238–252. [DOI] [PubMed] [Google Scholar]
- 26. Torroglosa A, Murillo‐Carretero M, Romero‐Grimaldi C, Matarredona ER, Campos‐Caro A, Estrada C (2007) Nitric oxide decreases subventricular zone stem cell proliferation by inhibition of epidermal growth factor receptor and phosphoinositide‐3‐kinase/Akt pathway. Stem Cells 25, 88–97. [DOI] [PubMed] [Google Scholar]
- 27. Rubinek T, Rubinfeld H, Hadani M, Barkai G, Shimon I (2005) Nitric oxide stimulates growth hormone secretion from human fetal pituitaries and cultured pituitary adenomas. Endocrine 28, 209–216. [DOI] [PubMed] [Google Scholar]
- 28. Yen SH, Pan JT (1999) Nitric oxide plays an important role in the diurnal change of tuberoinfundibular dopaminergic neuronal activity and prolactin secretion in ovariectomized, estrogen/progesterone‐treated rats. Endocrinology 140, 286–291. [DOI] [PubMed] [Google Scholar]
- 29. Russell JM, Murphree E, Janik J, Callahan P (2005) Effect of steroids and nitric oxide on pituitary hormone release in ovariectomized, peripubertal rats. Reproduction 129, 497–504. [DOI] [PubMed] [Google Scholar]
- 30. D’Anglemont de Tassigny X, Campagne C, Steculorum S, Prevot V (2009) Estradiol induces physical association of neuronal nitric oxide synthase with NMDA receptor and promotes nitric oxide formation via estrogen receptor activation in primary neuronal cultures. J. Neurochem. 109, 214–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Qian X, Jin L, Lloyd RV (1999) Estrogen downregulates neuronal nitric oxide synthase in rat anterior pituitary cells and GH3 tumors. Endocrine 11, 123–130. [DOI] [PubMed] [Google Scholar]
- 32. Shi Q, LaPaglia N, Emanuele NV, Emanuele MA (1998) Castration differentially regulates nitric oxide synthase in the hypothalamus and pituitary. Endocr. Res. 24, 29–54. [DOI] [PubMed] [Google Scholar]
- 33. Velardez MO, Del Carmen Díaz M, Lasaga M, Franchi AM, Duvilanski BH (2003) Estrogen decreases the sensitivity of anterior pituitary to the inhibitory effect of nitric oxide on prolactin release. Horm. Res. 60, 111–115. [DOI] [PubMed] [Google Scholar]
- 34. Cabilla JP, Díaz Mdel C, Machiavelli LI, Poliandri AH, Quinteros FA, Lasaga M et al. (2006) 17 beta‐estradiol modifies nitric oxide‐sensitive guanylyl cyclase expression and down‐regulates its activity in rat anterior pituitary gland. Endocrinology 147, 4311–4318. [DOI] [PubMed] [Google Scholar]
- 35. Cabilla JP, Ronchetti SA, Nudler SI, Miler EA, Quinteros FA, Duvilanski BH (2009) Nitric oxide sensitive‐guanylyl cyclase subunit expression changes during estrous cycle in anterior pituitary glands. Am. J. Physiol. Endocrinol. Metab. 296, E731–E737. [DOI] [PubMed] [Google Scholar]
- 36. De Paul A, Pons P, Aoki A, Torres A (1997) Different behavior of lactotroph cell subpopulations in response to angiotensin II and thyrotrophin‐releasing hormone. Cell. Mol. Neurobiol. 17, 245–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Harrington WR, Sheng S, Barnett DH, Petz LN, Katzenellenbogen JA, Katzenellenbogen BS (2003) Activities of estrogen receptor alpha‐ and beta‐selective ligands at diverse estrogen responsive gene sites mediating transactivation or transrepression. Mol. Cell. Endocrinol. 206, 13–22. [DOI] [PubMed] [Google Scholar]
- 38. Gorosito SV, Lorenzo AG, Cambiasso MJ (2008) Estrogen receptor alpha is expressed on the cell‐surface of embryonic hypothalamic neurons. Neuroscience 154, 1173–1177. [DOI] [PubMed] [Google Scholar]
- 39. Oomizu S, Takeuchi S, Takahashi S (1998) Stimulatory effect of insulin‐like growth factor I on proliferation of mouse pituitary cells in serum‐free culture. J. Endocrinol. 157, 53–62. [DOI] [PubMed] [Google Scholar]
- 40. Takahashi S, Osawa T (1994) Decreased proliferation of pituitary cells of streptozotocin induced diabetic rats in response to estradiol‐ 17‐beta. Acta Anat. 151, 239–244. [DOI] [PubMed] [Google Scholar]
- 41. Oomizu S, Takahashi S (1996) Insulin stimulates the proliferation of mouse anterior pituitary cells in vitro. Biomed. Res. 17, 365–371. [Google Scholar]
- 42. Gutiérrez S, Mukdsi JH, Aoki A, Torres AI, Soler AP, Orgnero EM (2007) Ultrastructural immunolocalization of IGF‐1 and insulin receptors in rat pituitary culture: evidence of a functional interaction between gonadotroph and lactotroph cells. Cell Tissue Res. 327, 121–132. [DOI] [PubMed] [Google Scholar]
- 43. Lanzino M, Morelli C, Garofalo C, Panno ML, Mauro L, Andò S et al. (2008) Interaction between estrogen receptor alpha and insulin/IGF signaling in breast cancer. Curr. Cancer Drug Targets 8, 597–610. [DOI] [PubMed] [Google Scholar]
- 44. LeRoith D, Bondy C, Yakar S, Liu JL, Butler A (2001) The somatomedin hypothesis. Endocr. Rev. 22, 53–74. [DOI] [PubMed] [Google Scholar]
- 45. Houslay MD, Wakelam MJO (1988) Structure and function of the receptor for insulin In: Cooke BA, King RJB, Van Der Molen HJ, eds. Hormones and their Actions, part II, pp. 321–348, Amsterdam: Elsevier. [Google Scholar]
- 46. Kansra S, Yamagata S, Sneade L, Foster L, Ben‐Jonathan N (2005) Differential effects of estrogen receptor antagonists on pituitary lactotroph proliferation and prolactin release. Mol. Cell. Endocrinol. 239, 27–36. [DOI] [PubMed] [Google Scholar]
- 47. Matarredona ER, Murillo‐Carretero M, Moreno‐López B, Estrada C (2004) Nitric oxide synthesis inhibition increases proliferation of neural precursors isolated from the postnatal mouse subventricular zone. Brain Res. 995, 274–284. [DOI] [PubMed] [Google Scholar]
- 48. Cartwright JE, Johnstone AP, Whitley GS (2000) Endogenously produced nitric oxide inhibits endothelial cell growth as demonstrated using novel antisense cell lines. Br. J. Pharmacol. 131, 131–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Odenlund M, Ekblad E, Nilsson BO (2008) Stimulation of oestrogen receptor‐expressing endothelial cells with oestrogen reduces proliferation of cocultured vascular smooth muscle cells. Clin. Exp. Pharmacol. Physiol. 35, 245–248. [DOI] [PubMed] [Google Scholar]
- 50. Scordalakes EM, Shetty SJ, Rissman EF (2002) Roles of estrogen receptor alpha and androgen receptor in the regulation of neuronal nitric oxide synthase. J. Comp. Neurol. 453, 336–344. [DOI] [PubMed] [Google Scholar]
- 51. Gingerich S, Krukoff TL (2005) Estrogen modulates endothelial and neuronal nitric oxide synthase expression via an estrogen receptor beta‐dependent mechanism in hypothalamic slice cultures. Endocrinology 146, 2933–2941. [DOI] [PubMed] [Google Scholar]
- 52. Gingerich S, Krukoff TL (2008) Activation of ERbeta increases levels of phosphorylated nNOS and NO production through a Src/PI3K/Akt‐dependent pathway in hypothalamic neurons. Neuropharmacology 55, 878–885. [DOI] [PubMed] [Google Scholar]
- 53. Krumenacker JS, Hyder SM, Murad F (2001) Estradiol rapidly inhibits soluble guanylyl cyclase expression in rat uterus. Proc. Natl. Acad. Sci. USA 98, 717–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Canabal DD, Song Z, Potian JG, Beuve A, McArdle JJ, Routh VH (2007) Glucose, insulin and leptin signaling pathways modulate nitric oxide synthesis in glucose‐inhibited neurons in the ventromedial hypothalamus. Am. J. Physiol. Regul. Integr. Comp. Physiol. 292, R1418–R1428. [DOI] [PubMed] [Google Scholar]
- 55. Shankar RR, Wu Y, Shen HQ, Zhu JS, Baron AD (2000) Mice with gene disruption of both endothelial and neuronal nitric oxide synthase exhibit insulin resistance. Diabetes 49, 684–687. [DOI] [PubMed] [Google Scholar]