

Summary
There is an increasing evidence to support a role of inflammatory processes in epilepsy. However, most clinical and experimental studies have been conducted in adult patients or using adult rodents. The pediatric epilepsies constitute a varied group of diseases that are most frequently age specific. In this review, we will focus on the possible role of inflammation in pediatric epilepsy syndromes. We will first describe the clinical data available and provide an overview of our current understanding of the role of inflammation in these clinical situations. We will then review experimental data regarding the role of inflammation in epilepsy in the developing brain. To summarize, inflammation contributes to seizure precipitation, and reciprocally, prolonged seizures induce inflammation. There is also a relationship between inflammation and cell injury following status epilepticus, which differs according to the developmental stage. Finally, inflammation seems to contribute to epileptogenesis even in the developing brain. Based on the available data, we highlight the need for further studies dissecting the exact role of inflammation in epilepsy during development.
Keywords: Developing Brain, Epilepsy, Epileptogenesis, IL‐1, Inflammation
Introduction
The immune system represents all the biological processes that protect an organism against external aggression, including infectious disease. The immune response is usually divided into the innate immune response and the adaptive immune response. While the innate immune system is responsible for a nonspecific response with an immediate maximal response resulting from cell‐mediated and humoral components, the adaptive immune system is antigen‐specific and takes time to build up to a maximal response after exposure. Although the adaptive immune response also results from cell‐mediated and humoral components, it leads to an immunological memory. Inflammation is not a static but a dynamic process, involving a cascade of mediators that can be either proinflammatory or antiinflammatory, as well as molecules that resolve inflammation.
Despite the fact that the brain is considered an immune‐privileged organ, both types of immune responses (innate and adaptive) are observed in the central nervous system (CNS) following exposure to pathogens or injury of varying etiologies. In addition, an inflammatory response in the CNS can be induced in the absence of infection 1. This phenomenon is observed after various types of brain injury, including status epilepticus.
There is now much clinical and experimental evidence concerning the multiple links between brain inflammation and epilepsy. Brain inflammation might be a consequence as well as a cause of seizure/epilepsy: 1. seizures and their related cellular injury might result in inflammation; 2. inflammation might precipitate seizures; 3. inflammation might participate in cell injury resulting from prolonged seizures; 4. inflammation might contribute to epileptogenesis. The involvement of inflammation in epilepsy is thus of great interest because pharmacological interventions targeting inflammation might be of use in treating patients with epilepsy 2, 3. However, the relationship between inflammation and epilepsy should not be regarded as a link between two separate pathophysiological entities. When we think about the role of inflammation in epilepsy, it is first important to distinguish the exact type of epilepsy syndrome as well as the subparts of the immune response that are involved. While most of the available data in the field of inflammation and epilepsy come from clinical studies performed in adults or experimental studies conducted with mature rodents, most pediatric epilepsy syndromes differ from the epilepsy syndromes observed in adults. Here, we review clinical and experimental evidence of the links between inflammation and epilepsy focusing on the data from pediatric studies and from experimental studies performed in the developing brain.
Clinical Evidence
The range of epilepsy syndromes, in particular during childhood and adolescence, is very wide 4. Focusing on syndromes involving inflammation in seizure occurrence, we can distinguish many clinical circumstances: febrile seizures, fever as a trigger for seizures in patients with epilepsy, acute encephalopathy with inflammation‐related status epilepticus, infectious encephalitis, autoimmune encephalitis, and Rasmussen syndrome. There are also other epileptic disorders with some clinical data pointing to the role of inflammation: epileptogenesis following febrile status epilepticus and brain inflammation in structural focal epilepsy. Table 1 summarizes our current understanding of the contribution of inflammation in these clinical entities.
Table 1.
Epilepsy syndromes and clinical conditions with possible role of inflammation
| Clinical entities | Age of the clinical entity | Role of inflammation | Part of immune response involved | Established Mechanisms | Hypothesis |
|---|---|---|---|---|---|
| Febrile seizures | Only in infants and children | Probable role on ictogenesis |
Cytokines Mostly IL1β |
No | IL1β from blood decreases seizure threshold |
| Fever triggering of seizures in epilepsy patients | Mostly children | Probable role on ictogenesis | Unknown | No |
Contribution of proinflammatory cytokines Role of hyperthermia cannot be excluded |
| Acute encephalopathy with inflammation‐mediated status epilepticus | Mostly children |
Role on cerebral injury Probable role on epileptogenesis |
Cytokines Mostly IL1β |
No | Inflammation increases cell injury related to initial status epilepticus |
| Infectious encephalitis | Children and Adults |
Role on seizures at acute phase Probable role on epileptogenesis following the acute episode |
Cytokines Microglia/Marcophages |
No |
Cytokines Microglia/Macrophages Virus‐induced brain injury |
| Autoimmune encephalitis |
Mostly Adults Also in children |
Role on ictogenesis |
Adaptative immunity Production of autoantibody Anti‐NMDA Anti‐VGKC |
No |
Auto‐Antibody Maybe proinflammatory cytokines also contribute to seizure occurrence Anti‐NMDA antibodies induce NMDA‐R internalization causing a decrease in inhibitory synapse density onto excitatory hippocampal neurons |
|
Rasmussen encephalitis |
Mostly children | Role on ictogenesis |
Activated microglial cells and proinflammatory mediators, and are infiltrated by lymphocytes presence of autoantibodies |
No |
Microglia Proinflammatory cytokines might contribute to seizure occurrence |
| Epileptogenesis following febrile status epilepticus | Only in infants and children | Possible role on epileptogenesis |
Cytokines Mostly IL1 β |
No |
Precipitating initial injury increased by inflammation lead to epileptogenesis Prolonged brain inflammation following the initial episode contributes to epileptogenesis |
| Focal epilepsy | Children and Aduts | Role on ictogenesis | Cytokines | No |
‐ Inflammation in the structural brain abnormalities facilitate seizure occurrence ‐ Possible role in refractoriness |
Febrile Seizures
Febrile seizures (FSs) are the most common form of childhood seizures, affecting 2–5% of all children. A febrile seizure is a seizure accompanied by fever, without CNS infection, occurring in children between 6 months and 5 years. The peak occurrence of FSs is at 18 months 5. The International League Against Epilepsy defined FS in 1993 as “an epileptic seizure occurring in childhood after the age of 1 month, associated with a febrile illness not caused by an infection of the central nervous system, without previous neonatal seizures or a previous unprovoked seizure, and not meeting criteria for other acute symptomatic seizures.” Despite its predominantly benign nature, a febrile seizure is a terrifying experience for most parents 6, 7.
The mechanisms underlying the occurrence of FSs remain poorly understood 8. It appears that several factors need to be associated, including an immature brain, an increase in body temperature, an inflammatory response, genetics, and a viral infection (Figure 1). There is no evidence to support the common belief that the rate of rise in temperature is more important for the development of FSs than the actual temperature achieved 9, 10.
Figure 1.
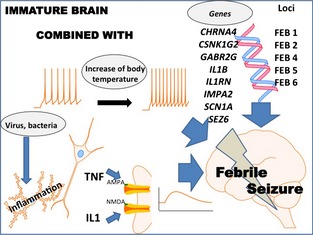
Mechanisms of FSs. The association of several factors is involved in each patient, among which are an immature brain, an increase in body temperature, inflammatory responses, genetics, and viral infection. FSs are genetically complex disorders believed to be influenced by variations in several susceptibility genes. Six FS susceptibility loci have been identified: FEB1 (8q13‐q21), FEB2 (19p13.3), FEB4 (5q14‐q15), FEB5 (6q22‐q24), FEB6 (18q11.2), and FEB7 (21q22). Recently, several association studies on FSs have been reported, but the results vary among groups and no consistent or convincing FS susceptibility gene has emerged. IL‐1β increases NMDA receptor function through the activation of tyrosine kinases and subsequent phosphorylation of the NR2A/B (GluN2A/B) subunit, resulting in an increase in NMDA‐induced calcium influx. TNF α causes an increase in the surface expression of neuronal AMPA receptors, responsible for an increase in synaptic efficacy. Viral infections are the most common causes of fever in cases of FSs. Human herpesvirus‐6 and influenza are frequently discussed as possible inducing factors for FSs because of their neurotropic properties, but the role of the viruses and the related inflammatory response remain poorly understood.
Regarding the cause of infections inducing fever in the case of FSs, it has been established that the fever is generally of viral origin 11, 12. Severe bacterial infection is usually infrequent but remains a concern in developing countries. Several studies have failed to determine whether a particular type of virus is responsible for the occurrence of FSs 13. In recent years, attention has focused on FSs due to infection with human herpesvirus (HHV) 6 or 7, because they are both frequent causes of fever in children in addition to being neurotropic 14.
Numerous clinical studies investigating cytokine levels in the blood or CSF of children with FSs suggest the involvement of inflammatory responses in the occurrence of FSs. However, the heterogeneity of the methods used in these studies (type of cytokines studied or control populations) makes it difficult to draw any clear conclusion. However, despite these limitations, the involvement of IL‐1β has been suggested 8. The experimental studies discussed below contribute to our understanding of the role of inflammation in FSs.
Epileptogenesis Following Febrile Status Epilepticus
There is accumulating evidence to suggest that inflammation plays an important role in both the development and the long‐term consequences of FSs. The long‐term risk of developing epilepsy is greater in cases of prolonged FSs. FEBSTAT, an ongoing prospective multicenter study to follow children who have experienced prolonged febrile status epilepticus, will definitively determine whether there is a relationship between this initial event and the occurrence of temporal lobe epilepsy (TLE) and hippocampal sclerosis. In this study, about a third of the patients have been identified so far as having had an HHV6 infection 14. Of 226 patients displaying prolonged febrile seizures, hippocampal T2 hyperintensity and increased volume have been observed acutely in 22 children. At follow‐up, 14 children of the 22 with acute T2 hyperintensity, hippocampal sclerosis was seen in 10, and a reduction of hippocampal volume in 12 children 15. In the future, the follow‐up of this cohort may reveal for the first time the link between FSs and TLE in humans.
Looking at genetic studies that link FSs and the occurrence of TLE, an association between the interleukin‐1β 511T allele, prolonged FSs, and the emergence of TLE was first reported in the Japanese population 16. These data have been challenged by other studies. However, a recent meta‐analysis tends to confirm the link between IL‐1β gene polymorphism and TLE 17.
Fever as a Trigger for Seizures in Children and Adolescents with Epilepsy
It is well known that some factors precipitate seizures in patients with epilepsy. Fever seems to be one of them, in particular in children. A study conducted in children with intractable epilepsy reports that 62% (of 120 children with intractable epilepsy) cited at least one precipitating factor for seizures. The three most common precipitants were illness or fever (32%), sleep deprivation (13%), and menstruation (10%) 18. The frequency of patients reporting fever as a precipitating factor is lower in adults (14% of 400 patients with epilepsy) 19.
In addition to this general observation, it is now well established that some epilepsy syndromes are more strongly linked with seizure precipitation by fever. Evidence in this regard has been published for patients with Dravet syndrome or Panayiotopoulos syndrome.
Dravet syndrome or severe myoclonic epilepsy is a refractory epilepsy. A mutation in the gene for the alpha‐1 subunit of the voltage‐gated sodium channel (SCN1A) is observed in most of these patients (70–80%). The seizure history usually starts with a febrile partial status epilepticus occurring before the age of 1. This first seizure is often considered a complex febrile seizure, but shortly thereafter, both FSs and seizures without fever occur, leading to diagnosis. It is now well established that several triggers may precipitate seizures in Dravet syndrome. Fever is the most frequent trigger, remaining throughout the course of the disease. Immunizations as well as hot baths may also trigger seizures 20. It is currently difficult to draw any conclusion as to the mechanisms leading to seizure precipitation in Dravet syndrome. Both hyperthermia, as reported by patients taking hot baths, and inflammation per se seem able to trigger seizures. Immunizations are also a frequent trigger although hyperthermia is not always observed 21, 22. Recent studies exclude the contribution of immunization as well as of the number of prolonged seizures to the long‐term outcome of Dravet syndrome 23, 24, 25.
Panayiotopoulos syndrome is an age‐related focal seizure disorder occurring in early and mid‐childhood. It is characterized by seizures, often prolonged, with predominantly autonomic symptoms. EEG recordings usually show shifting and/or multiple foci, often with occipital predominance. Autonomic seizures in Panayiotopoulos syndrome consist of episodes characterized by emetic symptoms, usually accompanied by other autonomic features (color changes, especially pallor, pupillary changes, cardiorespiratory, and thermoregulatory alterations) and more conventional ictal clinical manifestations such as the unilateral deviation of the eyes and convulsions 26. Panayiotopoulos syndrome starts at 3–6 years of age in most cases and affects about 6% of children aged 1–15 years with seizures 27. Both an increased rate of febrile seizures 27 and the occurrence of autonomic seizures during a febrile illness 28 have been reported in patients with Panayiotopoulos syndrome.
These reports point to the role of fever as a precipitating factor. However, there are no data regarding the involvement of inflammation in these observations.
Acute Encephalopathy with Inflammation‐Mediated Status Epilepticus
Acute encephalopathy with inflammation‐mediated status epilepticus has been suggested to include two conditions that occur in specific age groups. These conditions, hemiconvulsion–hemiplegia syndrome (HHS) and fever‐induced refractory epileptic encephalopathy in school‐aged children (FIRES), share common clinical characteristics. They probably also share similar underlying mechanisms 29.
Both HHS and FIRES involve severe seizures evolving into status epilepticus and are triggered by fever but without an identifiable cause 30, 31, 32, 33. Fever might have preceded the onset of neurological symptoms and may no longer be present. Whether fever itself or some other factor is the trigger remains to be determined. The outcome is very severe with strong motor and cognitive involvement as well as refractory epilepsy 33. These conditions were formerly called “encephalitis”. Apart from fever, no humoral or cellular expression of inflammation or microorganism has been found. In these conditions, the epileptic phenomenon itself seems to play a larger part than the infectious trigger (in contrast to encephalitis with virus‐induced brain damage). Acute encephalopathy with inflammation‐mediated status epilepticus seems to result from the synergy of inflammation and status epilepticus, the latter resulting partly from inflammation itself, and contributing to the induction of inflammation, thus generating a vicious cycle that leads to major neurological consequences 29. Brain maturation might be the key factor explaining the age dependence of the clinical presentations. HHS mostly occurs between 1 and 4 years while FIRES is reported in children between 4 years and adolescence 29.
Infectious Encephalitis
Infectious encephalitides are defined by pronounced brain inflammation in response to a CNS infection. They are most often associated with seizures during the infection period 34, 35 and are established risk factors for the development of epilepsy at a later time 34, 36, 37. Although many infectious encephalitides are of unknown origin, the majority of clinical studies point to a viral origin in most of them (Whitley, 1990; Davison, 2003). Common viruses associated with encephalitis‐induced seizures are herpes simplex and cytomegalovirus (CMV). Children are three times more prone to viral encephalitis than adults 38, 39, and in the pediatric population, infants under 1 year are particularly affected 38. Both mechanisms leading to increased seizure occurrence at the time of infection and mechanisms of encephalitis‐induced epileptogenesis are poorly understood. Virus‐induced cell injury, brain inflammation, and initial prolonged seizures seem to be the key factors contributing to epileptogenesis after the acute phase of encephalitis.
Autoimmune Encephalitis
Autoimmune encephalitis is caused by the presence of antibodies binding to the neuronal surface. There are now more than six types of antibodies that have been described. In children, the most frequent types also occur with limbic encephalitis related to antibodies targeting the voltage‐gated potassium channel (VGKC) complex and encephalitis related to antibodies targeting the NMDA receptor (NMDAR) 40.
Limbic encephalitis consists of an alteration of mental status associated with cognitive involvement. There are also seizures that usually originate in the temporal lobe. The antibody most strongly associated with LE is the VGKC antibody, which in fact consists of three different categories targeting three different proteins: contactin‐associated protein‐like 2 (CASPR2), leucine‐rich glioma inactivated 1 (LGI1), and contactin‐2. In recent years, clinically distinct features have been described for the various subtypes of limbic encephalitis. This encephalopathy appears to be reversible when treated with immunotherapy, correlating with a decrease in the elevated serum antibody titer.
In the case of anti‐NMDAR encephalitis, the symptoms are characterized by psychiatric symptoms, dyskinesias, and seizures. This autoimmune encephalitis is among the most common in children and young adults. On MRI, the cortex might be involved and this involvement might be restricted to the temporal lobe. CSF analyses confirm the intrathecal synthesis of anti‐NMDAR antibodies. In adult patients, this disorder is associated with ovarian teratoma in 40% of cases, and 76% develop seizures in the acute stages 41.
The exact mechanisms underlying autoimmune encephalitis are under investigation. It is still unclear whether the autoantibodies are responsible for all the symptoms or whether nonspecific inflammation also contributes to them, for example, through the release of proinflammatory cytokines. The most recent study shows that the antibodies are responsible for receptor internalization. This downregulation of surface NMDARs modifies the homeostasis of synaptic plasticity mechanisms, contributing to disease progression 42.
Rasmussen Syndrome or Rasmussen's Encephalitis
Rasmussen syndrome is a rare progressive disease with drug‐resistant focal epilepsy (epilepsia partialis continua) followed by progressive hemiplegia and cognitive decline. The progression of the disease is associated with unilateral hemispheric brain atrophy.
Neuropathological studies of the involved hemisphere demonstrate the presence of cortical inflammation, neuronal loss, and gliosis. The progression of this immune‐mediated disease is associated with both an adaptive immune reaction characterized by a T‐lymphocyte response, and an innate immune reaction facilitated by both microglia and astroglia 43. It is interesting to know that recently case series of surgical specimens have also revealed a dual pathology with both focal cortical dysplasia (FCD) and Rasmussen syndrome 44, 45. This suggests that Rasmussen's encephalitis might be a form of inflammation superposed on FCD.
Careful analysis of the association between the histopathology and clinical presentation suggests that T cells and microglia mediate the initial damage to the brain. Most of the inflammatory T cells are CD8+, and about 10% of these are granzyme‐B+. Such granzyme‐B+ cells have been found in apposition to neurons and astrocytes, with a polarization of the cytotoxic granules to face the target cell membrane 46. Microglia, by releasing proinflammatory cytokines, might contribute to the occurrence of seizures but this needs to be further investigated 3.
Some rare cases of patients with autoantibodies have been described (against GluR3, the alpha‐7 nicotinic acetylcholine receptor or Munc‐18‐1, a neuronal protein essential for synaptic vesicle release). However, these reports are limited and do not provide any evidence that these autoantibodies are responsible for Rasmussen's encephalitis 47.
Advances in neuroimaging suggest that the progression of the inflammatory process as visualized by MRI might be a good biomarker of disease progression. Immunomodulatory treatments seem to slow rather than halt disease progression, without changing the eventual outcome. Thus the real challenge in clinical management is to determine the right time to shift from medical management to surgery 47.
Brain Inflammation in Structural Focal Epilepsy
Focal epilepsy accounts for at least half the new‐onset epilepsy cases in children. The etiologies are diverse, and range from “benign” epilepsy syndromes with normal neuroimaging to focal malformations of cortical development or hippocampal sclerosis with intractable seizures persisting throughout life. Various types of structural changes may be associated with epilepsy, some of which are more commonly associated with intractability. FCD, an abnormality of cortical development, is one of the most common abnormalities reported in children undergoing surgery. FCDs are classified into three major types, with Types I and II referring to isolated FCD, and Type III referring to dysplasia associated with other epileptogenic lesions, such as hippocampal sclerosis or tumors 48. Hippocampal sclerosis is also a pathological finding that is observed in children with refractory TLE, but mesial TLE with hippocampal sclerosis is mostly observed in adult patients.
Inflammation in focal epilepsy has been investigated in surgical samples from patients with refractory epilepsy. Inflammation in structural focal epilepsy has been documented in FCD and in tubers from tuberous sclerosis patients (Table 2). There are more reports on inflammation in TLE with hippocampal sclerosis, but most of these come from studies in adult patients. Both microglial activation and IL‐1β are commonly observed in TLE (Table 2). The inflammation observed in focal epilepsy can be clearly differentiated from inflammation in Rasmussen's encephalitis, where cells of the adaptive immune system are strongly represented in the lesional tissue.
Table 2.
Studies investigating inflammation in surgical samples from patients with refractory epilepsies
| References | Age | Population | Findings |
|---|---|---|---|
| 83 | Adults | 18 TLE with HS | NFkB |
| 84 | Adults | TLE (8 HS + 6 non‐HS) | Activated microglia and activation of the classical complement pathway in cellular and perivascular elements in HS |
| 85 | Adults | TLE (12 HS + 6 non‐HS) | Neuronal and glial activation of the IL1β system |
| 86 | Children | 5 TLE | IL1b and miR‐146 |
| 87 | Adults | 6 TLE | COX‐2, TGF‐β, NF‐κB |
| 88 | Adults | TLE | TLR‐4 related to seizure frequency |
| 51 | Mostly adults | 9 FCD, 9 DNT, 9 Ganglioglioma | IL‐1β, IL 1‐RA and IL‐1β receptors correlated with clinical course |
| 49 | Adolescents | 20 FCD | Activated microglia within the dysplastic cortex |
| 89 | Children and Adults | 9 TSC | Microglial activation, IL1b, complement cascade activation, focal changes in BBB permeability |
| 53 | Children | 6 FCD, 5 encephalomalacia, 1 TLE |
Astrocyte and Microglial activation IL‐1b IL‐8, IL‐12p70 and MIP‐1β |
| 50 | Adults | 17 FCD (8 IB and 9 IIA) |
Microglia activation mTOR only in FCDIIA IL‐1β and MCP1 mainly in FCD II |
| 52 | Mostly adults | 6 FCD, 6 TSC, 6 Ganglioglioma | Intralesional overexpression and cellular distribution of HMGB1 and its cognate receptors TLR2, TLR4 and RAGE |
BBB, blood–brain barrier; COX‐2, cyclo‐oxygenase 2; DNT, dysembryoplastic neuroepithelial tumor, FCD, focal cortical dysplasia; HMGB1, high‐mobility group box 1; HS, hippocampal sclerosis; IL1β, InterLeukin 1β, IL1‐RA, IL1 receptor antagonist; IL6, Interleukin 6; mi146, microRNA; mTOR, mammalian Target Of Rapamycin; NFkB, nuclear factor‐kappa B; MCP1, Monocyte chemoattractant protein 1; MIP‐1β, Macrophage inflammatory protein‐1β; TGFβ, Transforming growth factor β; TLE, temporal lobe epilepsy; TSC, tuberous sclerosis complex.
Proinflammatory signaling is also activated in developmental lesions associated with epilepsy 49, 50, 51. The activation of astrocytes and microglia‐macrophages has been described in FCD specimens 49, 52, 53. IL‐1β is frequently observed in focal malformations of cortical development (Table 2). IL‐1β has been shown to have proconvulsant activity in different models 3, 51. Activated glia in surgical FCD specimens, tubers from patients with tuberous sclerosis and gangliogliomas also display the expression of Toll‐like receptors (TLR) 2 and 4, receptors for advanced glycation end products (RAGEs) and their endogenous ligand, cytoplasmic high‐mobility group box (HMGB) 1. TLR4 and RAGE have also been described in dysplastic neurons 52. Moreover, the activation of HMGB1‐TLR4 and HMGB1‐RAGE signaling has been described in experimental models of epilepsy in adult rodents, suggesting that this pathway represents a possible mechanism of epileptogenesis 54, 55.
Experimental Models
As for humans, in whom the incidence of epilepsy is highest in infancy, in experimental animal models the immature brain shows increased susceptibility to seizures 56. However, the immature brain seems less vulnerable to status epilepticus‐induced injury and the ensuing epileptogenesis 56. In the lithium‐pilocarpine model of status epilepticus, a significant level of status epilepticus‐induced cell injury is observed at P14. In 2‐ and 3‐week‐old pups, cell injury is mainly observed in the CA1. At P21, the injuries to the hilus and CA3 attain an adult‐like pattern, whereas neuronal injury in the amygdala increases progressively with age 57. Lithium‐pilocarpine‐induced status epilepticus at P14 results in significant epileptogenesis with spontaneous recurrent seizures in approximately 10 to 20% of rat pups 57, 58. When status epilepticus is induced at P21 or P28, the percentage of animals with spontaneous recurrent seizures is close to the rate of epileptic animals generated by the same model in adult rats 57.
Experimental models of febrile seizures or induced inflammation in models of status epilepticus have been instrumental in deciphering the role of inflammatory pathways in seizure occurrence and epileptogenesis in the immature brain.
Inflammation as a Result of Seizure in the Developing Brain
Inflammation Activation After Prolonged Seizures in the Immature Brain
As in adult rodent models, prolonged seizures induce an inflammatory response in the developing brain. This inflammatory reaction is variably associated with neuronal loss, depending on age. In adult animals, studies have demonstrated that the innate inflammatory response following seizure comprises the generation of inflammatory mediators including proinflammatory cytokines, prostaglandins, complements, and free radicals, as well as the activation of microglia, astrocytes, and endothelial cells of the blood–brain barrier. Immune cells from the blood are also recruited. More recently, the interaction of HMGB1 with TLR4 has been described as a crucial event for the initiation of brain inflammation and the lowering of the seizure threshold 55. HMGB1 is considered to be a danger signal released by injured or stressed cells to alert the microenvironment of an immediate or ongoing injury. Once activated, glial cells sustain the inflammatory response in injured regions by releasing more inflammatory mediators and chemokines and modulating adhesion molecule expression 3.
In the developing brain, there is less information available and the investigation of inflammatory pathways has been conducted in a less extensive manner. Using kainate‐induced status epilepticus in the developing brain, it has been shown that 4 h after the start of status epilepticus, glial activation occurs, predominating in the hippocampus. This process affects all parts of the hippocampus indifferently (the CA1, CA3, and dentate gyrus) and increases with age 59. A developmental profile of cellular activation has been described, with limited microglial and astrocytic activation in P9 rats, but strong activation in P15 and P21 rats 59, 60. In P9 rats, no cytokines are detectable. In P15 rats, IL‐1β mRNA is upregulated in the hippocampus. In P21 rats, IL‐1β, IL‐6, TNFα, and IL‐1RA were all upregulated 60.
In P11 rats, prolonged hyperthermic seizures and hyperthermia‐induced status epilepticus (HSE) lead to strong astrocytic activation and mild microglial activation 6 h after seizure onset. At 24 h after HSE onset, glial activation is accompanied by IL‐1β production in the hippocampus. At 48 h, the upregulation of IL‐1β is no longer visible. However, the resolution of glial activation has not been studied 61.
Few studies have examined the inflammatory response induced by status epilepticus in the developing brain, although an astrocytic activation by IL‐1β of microglial origin has been observed after seizures in both the mature and immature brain 61, 62. It would be interesting to know whether the inflammatory response, when present, follows the same pattern in adult and in immature animals. There is currently no study that explores the role of HMGB1 in the developing brain, and it is unknown whether there is any difference in the expression or release of this molecule during development.
Relationship between Status Epilepticus‐Induced Cell Injury and Inflammation
In the developmental study using kainate‐induced status epilepticus, Rizzi et al. showed a correlation between the levels of upregulated cytokines and the occurrence of status epilepticus‐induced neuronal injury 60. In P9 rats, no neuronal injury is observed while scattered injured neurons are detected in the CA3 and subiculum in P15 rats. At P21, the neuronal damage is more pronounced, with FluoroJade‐B‐positive neurons in the CA1 and CA3 and in other regions in 40% of the animals. The age dependence of neuronal injury seems to be correlated with glial activation (microglia and astrocytes) and the level of proinflammatory cytokines. The activation of the inflammatory response is observed before cell injury, suggesting the involvement of cytokines in seizure‐induced injury. Due to the developmental characteristics of the kainate model of status epilepticus, different doses of kainate were used in this study, limiting the interpretation of the results.
In the lithium‐pilocarpine model of status epilepticus, it is well established that rat pups display a lower level of neuronal injury than adult rats. An age‐dependent pattern of neuronal injury has been described, but the inflammatory profile has not been well described. One week after pilocarpine‐induced status epilepticus, no IL‐1β immunoreactivity is detected in P9 rats, while it is clearly observed in P21 rats 63.
When inflammation is induced before the induction of status epilepticus by a systemic injection of lipopolysaccharide (LPS), an increase in the degree of cell injury is observed in models of status epilepticus in the developing brain. Neuronal injury is enhanced with a discernible increase of cell injury in P7 rats and increases of cell injury in P14 rats compared to the lithium‐pilocarpine model 64.
These data strongly converge to support a role of cytokines in enhancing neuronal death. However, the underlying mechanisms need to be studied in the field of epilepsy. Microglial activation might be a key player and contribute to excitotoxicity. Activated microglia and neurons take part in a reciprocal relationship where microglia release neurotoxic factors and damaged neurons further activate microglia through microglial NMDAR. IL‐1 appears to be one of these factors 65.
Inflammation Increases Seizure Susceptibility
To examine the experimental data on inflammation and epilepsy, we will arbitrarily distinguish between the role of inflammation in the occurrence of seizures (ictogenesis) and its role in epileptogenesis. From clinical observations, the effect of inflammation on seizure susceptibility has been suspected for a long time (See Table 1).
Inflammation Triggers Ictogenesis
Animal Models Mimicking Febrile seizures
As mentioned in the section on human data, several factors seem to contribute to the occurrence of febrile seizures. By definition, a child who experiences a febrile seizure presents an increase in body temperature. This increase is the result of a change in the thermoregulatory system induced by an inflammatory response, which is in turn induced by an infection. The causes of fever in cases of febrile seizures are mainly of viral origin 11, 12. Based on clinical observations, two main types of animal models of febrile seizure have been established. The first group is based on the induction of hyperthermia while the second primarily induces peripheral inflammation in a model of seizure or status epilepticus.
Models of Hyperthermic Seizure
The main model of hyperthermic seizure consists of increasing body temperature in P10‐P11 rats by a heated airstream 61, 66, 67, 68. The mean increase of core temperature is around 2.9°C, which has been claimed to be in agreement with the increase in temperature observed in children experiencing FS (Berg 1992; el‐Radhi 1986). In rat pups, seizures are characterized by motor arrest, chewing, forelimb clonus, and in some cases, body flexion with biting. Generalized tonic seizures are rarely observed 66. The seizure threshold temperature and seizure duration are usually the parameters used to study ictogenesis. The role of IL‐1β in ictogenesis has been investigated using this model in mice. An i.c.v. injection of IL‐1β lowered the threshold for hyperthermic seizures in mouse pups. A high‐dose i.c.v. injection of IL‐1β was even capable of inducing seizures without a thermal trigger 69.
Fever Model
To induce fever in animal models, injections of LPS have been used. In adult rats, the injection of LPS results in a biphasic increase in body temperature driven by an inflammatory response. The use of LPS in rat pups does not result in such an increase in body temperature when at room temperature. However, an increase in body temperature is observed when the room temperature is about 30°C, corresponding to thermoneutrality in rat pups 70.
When LPS is injected i.p. 2 h before a subconvulsant dose of kainate (1.75 mg/kg), P14 rat pups experience an increase in body temperature and seizures. They exhibit face scratching, lying on one side, and wet‐dog shakes 70. At the onset of seizures, IL‐1β is significantly increased in the hippocampus but not in the hypothalamus or cortex when compared to rats that do not undergo seizures, despite the fact that they are also treated with LPS and kainate. When IL‐1β is given i.c.v., the number of animals seizing after a subconvulsant dose of kainate is significantly increased and the time to seizure onset is reduced, while the opposite occurs after i.c.v. injection of IL‐1RA. Thus, IL‐1β clearly contributes to seizure occurrence in P14 rats in this model 71, 72.
In contrast, LPS at low doses, which does not increase core temperature, also does not change acute susceptibility to short hyperthermic seizures in either P11 or P16 rats 73. However, these animals develop a lower threshold for pentylenetetrazol (PTZ)‐induced seizures when they are adults. A similar nonfebrile dose of LPS does not exacerbate lithium‐pilocarpine‐induced status epilepticus at P7 or P14, although it does induce a delay in the onset time of status epilepticus; however, these rats display an increase in seizure‐induced hippocampal damage 64. It appears from these findings that relatively low doses of LPS in immature rats, which do not increase core temperature, do not alter acute susceptibility to seizures although they increase seizure‐induced cell loss (described above) and long‐term predisposition to seizures, described later in this article. The effects of LPS in immature and adult animals are clearly different, as nonfebrile doses also exacerbate seizures in adults 74, 75.
The mechanisms underlying these effects, and the role played by fever and brain inflammation in determining the threshold to acute seizures, remain to be elucidated. However, these studies provide evidence that inflammation contributes to the occurrence of seizures in the developing brain.
Role of Inflammation in Epileptogenesis
In addition to its role in seizure precipitation, inflammation has been implicated in epileptogenesis 2. Several studies show that an inflammatory episode during gestation or early life has a long‐term impact on seizure susceptibility or even a pro‐epileptogenic effect.
Double‐hit models with status epilepticus‐kainate show that a first hit due to an early status epilepticus predisposes the subject to increased seizure susceptibility in response to a second hit. This is accompanied by greater glial activation with the two consecutive hits than with a single hit. The administration of minocycline, which effectively reduces glial activation, between the first and second hits protects from the long‐term epileptogenic effect of early‐life seizures 76. Treatment with minozac, an experimental molecule that targets proinflammatory cytokine production, attenuates the enhanced microglial and cytokine responses, resulting in decreased seizure susceptibility in adulthood 77. Both studies suggest that microglial activation and proinflammatory signaling occurring in the developing brain can contribute to the long‐term modification of brain excitability. In the model of prolonged hyperthermic seizures in P11 rat pups, IL‐1β is chronically upregulated in rats developing spontaneous limbic seizures compared to rats exposed to prolonged FS without spontaneous seizure development 61.
Inflammation by itself occurring during a particular window of maturation also has a long‐term effect on brain excitability. When LPS is administered to P7 or P14 rats, but not younger (P1) or older animals (P20), seizure susceptibility is increased when these rats become adults, as assessed using PTZ‐, lithium‐pilocarpine‐ and kainate‐induced seizures 78. Viral‐like inflammation induced in P14 rats by the i.c.v. injection of polyinosinic:polycytidylic acid (Poly I:C), a TLR3 agonist, is also responsible for increased seizure susceptibility in adulthood, as shown using PTZ‐ and lithium‐pilocarpine‐induced seizures 79. Moreover, the early immune challenge also has an impact on cognitive function and anxiety 79, 80. When Poly I:C is given i.c.v., IL‐1β, and TNFα are upregulated at P14 but the expression levels of these cytokines are not different in adulthood. However, an increase in the mRNA levels of NMDAR subunits, GluN2A, GluN2C, and GluA1 has been noted 79. There is currently no evidence that an immune challenge results in spontaneous recurrent seizures even whether the inflammation occurred during a specific period of time during development.
Inflammation in the immature brain also acts as a disease modifier when it is coupled to a second hit. Inflammation aggravates the consequences in all models of seizure, but it does not modify the main characteristics of each model. These multiple‐hit models highlight the importance of inflammation in epileptogenesis (Table 3). Moreover, these models are of great value from a translational point of view. Thus, the systemic injection of LPS in P14 rats before lithium‐pilocarpine‐induced status epilepticus does not change the severity of acute status epilepticus but results in more severe spontaneous seizures in adulthood 81. The number of epileptic animals is also increased. However, no significant changes in cell number in the CA1 sector are observed in LPS‐pretreated rats, although in some animals, FluoroJade‐positive cells are detected, suggesting ongoing neurodegeneration, which is absent in rats not preexposed to LPS. The CA1 of rats pretreated with LPS also displays more intense reactive gliosis 81. Using rapid kindling in P14 rats, it has been found that LPS enhances epileptogenesis and increases hippocampal excitability after the completion of kindling. These effects are prevented by IL‐1RA suggesting the involvement of IL‐1β in the mechanisms underlying hyperexcitability 58. More recently, an increase in epileptogenesis assessed by rapid kindling has been shown in an animal model of autism, consisting of prenatal (E12 to E16 embryonic days) immune activation by Poly I:C. The use of neutralizing antibodies to either IL‐1β or IL‐6 counteracts the pro‐epileptogenic effect of Poly I:C, implicating these two cytokines in the pro‐epileptogenic effect of maternal infection 82.
Table 3.
Summary of the modifying effects of inflammation on seizure/Status epilepticus model in the developing brain
| Consequences of seizure‐inducing agents in naïve animals | Consequences of seizure‐inducing agents after an immune challenge | |||
|---|---|---|---|---|
| Seizure occurrence / cell injury | Epileptogenesis | Type of immune challenge | Inflammation‐induced modification | |
| Short Hyperthermic Seizure |
Hypethermia‐induced seizures No cell injury |
No Only for Prolonged HS or HSE |
LPS |
Absence of cell injury Decrease of Sz threshold in adulthood 73. |
| Kainic acid |
Dose dependence on seizure precipitation No cell injury in immature brain |
No Epileptogenesis | LPS |
Increased cell injury in P17 90
Induced hyperthermic seizure with subconvulsant dose of kainate in P14 70 Decreased seizure susceptibility in adulthood |
| Lithium pilocarpine |
P7: Very little P14: CA1 Yes in 10–20% P21: Hilus‐CA3 Yes 57 |
After P7 SE: No After P14 SE: 10–20% SRS After P21 SE: about 100% SRS 57 |
LPS |
Increased SE‐induced cell injury 64
Increased epileptogenesis after SE at P14 81 |
| IL1β |
Increase SE‐induced cell injury (Auvin, unpublished) |
|||
| Rapid Kindling | No |
Epileptogenesis 91, 92, 93 |
LPS | Increased Epileptogenesis without change in hippocampal excitability 81 |
HS, Hyperthermic Seizures; HSE, Hyperthermic Status Epilepticus; IL1β, InterLeukin 1β; LPS, Lipopolysaccharides; P7, 7th postnatal day.
In summary, studies conducted in immature animals show that inflammation contributes to both ictogenesis and epileptogenesis, as has been established in adult experimental models.
Conclusion
It is clear that inflammation and epilepsy are linked in various manners even in the developing brain. However, there is a need for more experimental studies in the developing brain. First, there are some epilepsy syndromes for which there is no available model (e.g., Rasmussen's encephalitis). Moreover, the available animal models might help to further understand the role of inflammation in some clinical conditions (the contribution of inflammation to seizure precipitation in the Dravet mouse model, the role of inflammation in epileptogenesis in models of focal cortical dysplasia). Finally, it is still unclear why FS occurs only in infants and children. This is also a question that could be addressed by a comparison of the role of inflammation in ictogenesis and epileptogenesis at various ages using the appropriate models of seizure/epileptogenesis.
More importantly, experimental studies are needed to better dissect the mechanisms underlying inflammation in the developing brain. As shown in this review, the mechanisms linking inflammation to ictogenesis/epileptogenesis have been described in detail using experiments performed in adult animals or by analyzing brain samples from adult patients. A particular evaluation of components contributing to inflammatory responses that evolve during development (e.g., the blood–brain barrier, microglia, etc.) might be of particular interest.
Conflict of Interest
The authors declare no conflict of interest.
Acknowledgments
Stéphane Auvin is partially supported by an INSERM Grant (Contrat Interface INSERM 2010). This work was supported by the INSERM, Université Paris‐Diderot and the Association INJENO.
References
- 1. Rivest S. Regulation of innate immune responses in the brain. Nat Rev Immunol 2009;9:429–439. [DOI] [PubMed] [Google Scholar]
- 2. Vezzani A, Friedman A, Dingledine RJ. The role of inflammation in epileptogenesis. Neuropharmacology 2013;69:16–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Vezzani A, Aronica E, Mazarati A, Pittman QJ. Epilepsy and brain inflammation. Exp Neurol 2013;244:11–21. [DOI] [PubMed] [Google Scholar]
- 4. Berg AT, Berkovic SF, Brodie MJ, et al. Revised terminology and concepts for organization of seizures and epilepsies: Report of the ILAE Commission on Classification and Terminology, 2005‐2009. Epilepsia 2010;51:676–685. [DOI] [PubMed] [Google Scholar]
- 5. Shinnar S, Glauser TA. Febrile seizures. J Child Neurol 2002;17:S44–S52. [DOI] [PubMed] [Google Scholar]
- 6. Sakai R, Nujima S, Marui E. Parental knowledge and perceptions of fever in children and fever management practices differences between parents of children with and without a history of febrile seizures. Pediatr Emerg Care 2009;25:231–237. [DOI] [PubMed] [Google Scholar]
- 7. Kolahi AA, Tahmooreszadeh S. First febrile convulsions: Inquiry about the knowledge, attitudes and concerns of the patients’ mothers. Eur J Pediatr 2009;168:167–171. [DOI] [PubMed] [Google Scholar]
- 8. Auvin S, Vallée L. Febrile seizures: Current understanding of pathophysiological mechanisms. Arch Pediatr 2009;16:450–456. [DOI] [PubMed] [Google Scholar]
- 9. Berg AT. Are febrile seizures provoked by a rapid rise in temperature. Am J Dis Child 1993;147:1101–1103. [DOI] [PubMed] [Google Scholar]
- 10. Minchom PE, Wallace SJ. Febrile convulsions ‐ electroencephalographic changes related to rectal temperature. Arch Dis Child 1984;59:371–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Millichap JG, Millichap JJ. Role of viral infections in the etiology of febrile seizures. Pediatr Neurol 2006;35:165–172. [DOI] [PubMed] [Google Scholar]
- 12. Millichap JJ, Millichap JG. Methods of investigation and management of infections causing febrile seizures. Pediatr Neurol 2008;39:381–386. [DOI] [PubMed] [Google Scholar]
- 13. Chung B, Wong V. Relationship between five common viruses and febrile seizure in children. Arch Dis Child 2007;92:589–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Epstein LG, Shinnar S, Hesdorffer DC, et al. Human herpesvirus 6 and 7 in febrile status epilepticus: The FEBSTAT study. Epilepsia 2012;53:1481–1488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lewis DV, Shinnar S, Hesdorffer DC, et al. Hippocampal sclerosis after febrile status epilepticus: The FEBSTAT study. Ann Neurol 2014;75:178–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kanemoto K, Kawasaki J, Yuasa S, et al. Increased frequency of Interleukin‐1 beta‐511T allele in patients with temporal lobe epilepsy, hippocampal sclerosis, and prolonged febrile convulsion. Epilepsia 2003;44:796–799. [DOI] [PubMed] [Google Scholar]
- 17. Kauffman MA, Moron DG, Consalvo D, Bello R, Kochen S. Association study between interleukin 1 beta gene and epileptic disorders: A HuGe review and a meta‐analysis. Genet Med 2008;10:83–88. [DOI] [PubMed] [Google Scholar]
- 18. Fang PC, Chen YJ, Lee IC. Seizure precipitants in children with intractable epilepsy. Brain Dev 2008;30:527–532. [DOI] [PubMed] [Google Scholar]
- 19. Frucht MM, Quigg M, Schwaner C, Fountain NB. Distribution of seizure precipitants among epilepsy syndromes. Epilepsia 2000;41:1534–1539. [DOI] [PubMed] [Google Scholar]
- 20. Desnous B, Goujon E, Bellavoine V, Merdariu D, Auvin S. Perceptions of fever and fever management practices in parents of children with Dravet syndrome. Epilepsy Behav 2011;21:446–448. [DOI] [PubMed] [Google Scholar]
- 21. McIntosh AM, McMahon J, Dibbens LM, et al. Effects of vaccination on onset and outcome of Dravet syndrome: A retrospective study. Lancet Neurol 2010;9:592–598. [DOI] [PubMed] [Google Scholar]
- 22. Verbeek NE, van der Maas NAT, Jansen FE et al. Prevalence of SCN1A‐related dravet syndrome among children reported with seizures following vaccination: A population‐based ten‐year Cohort Study. PLoS ONE, 2013; 8:e65758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Villeneuve N, Laguitton V, Viellard M, et al. Cognitive and adaptive evaluation of 21 consecutive patients with Dravet syndrome. Epilepsy Behav 2014;31:143–148. [DOI] [PubMed] [Google Scholar]
- 24. Nabbout R, Chemaly N, Chipaux M et al. Encephalopathy in children with Dravet syndrome is not a pure consequence of epilepsy. Orphanet J Rare Dis 2013; 8:176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Auvin S. Should we still consider Dravet syndrome an epileptic encephalopathy? Epilepsy Behav 2014;36:80–81. [DOI] [PubMed] [Google Scholar]
- 26. Panayiotopoulos CP, Michael M, Sanders S, Valeta T, Koutroumanidis M. Benign childhood focal epilepsies: Assessment of established and newly recognized syndromes. Brain 2008;131:2264–2286. [DOI] [PubMed] [Google Scholar]
- 27. Covanis A. Panayiotopoulos syndrome: A benign childhood autonomic epilepsy frequently imitating encephalitis, syncope, migraine, sleep disorder, or gastroenteritis. Pediatrics 2006;118:E1237–E1243. [DOI] [PubMed] [Google Scholar]
- 28. Cordelli DM, Aldrovandi A, Gentile V, et al. Fever as a seizure precipitant factor in Panayiotopoulos syndrome: A clinical and genetic study. Seizure 2012;21:141–143. [DOI] [PubMed] [Google Scholar]
- 29. Nabbout R, Vezzani A, Dulac O, Chiron C. Acute encephalopathy with inflammation‐mediated status epilepticus. Lancet Neurol 2011;10:99–108. [DOI] [PubMed] [Google Scholar]
- 30. Auvin S, Bellavoine V, Merdariu D, et al. Hemiconvulsion‐hemiplegia‐epilepsy syndrome: Current understandings. Eur J Paediatr Neurol 2012;16:413–421. [DOI] [PubMed] [Google Scholar]
- 31. Auvin S, Devisme L, Maurage CA, et al. Neuropathological and MRI findings in an acute presentation of hemiconvulsion‐hemiplegia: A report with pathophysiological implications. Seizure 2007;16:371–376. [DOI] [PubMed] [Google Scholar]
- 32. Howell KB, Katanyuwong K, Mackay MT, et al. Long‐term follow‐up of febrile infection‐related epilepsy syndrome. Epilepsia 2012;53:101–110. [DOI] [PubMed] [Google Scholar]
- 33. Kramer U, Chi CS, Lin KL, et al. Febrile infection‐related epilepsy syndrome (FIRES): Pathogenesis, treatment, and outcome A multicenter study on 77 children. Epilepsia 2011;52:1956–1965. [DOI] [PubMed] [Google Scholar]
- 34. Getts DR, Balcar VJ, Matsumoto I, Muller M, King NJ. Viruses and the immune system: Their roles in seizure cascade development. J Neurochem 2008;104:1167–1176. [DOI] [PubMed] [Google Scholar]
- 35. Misra UK, Tan CT, Kalita J. Viral encephalitis and epilepsy. Epilepsia 2008;49(Suppl 6):13–18. [DOI] [PubMed] [Google Scholar]
- 36. Annegers JF, Hauser WA, Beghi E, Nicolosi A, Kurland LT. The risk of unprovoked seizures after encephalitis and meningitis. Neurology 1988;38:1407–1410. [DOI] [PubMed] [Google Scholar]
- 37. Rocca WA, Sharbrough FW, Hauser WA, Annegers JF, Schoenberg BS. Risk factors for complex partial seizures: A population‐based case‐control study. Ann Neurol 1987;21:22–31. [DOI] [PubMed] [Google Scholar]
- 38. Davison KL, Crowcroft NS, Ramsay ME, Brown DW, Andrews NJ. Viral encephalitis in England, 1989‐1998: What did we miss? Emerg Infect Dis 2003;9:234–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Nicolosi A, Hauser WA, Beghi E, Kurland LT. Epidemiology of central nervous system infections in Olmsted County, Minnesota, 1950‐1981. J Infect Dis 1986;154:399–408. [DOI] [PubMed] [Google Scholar]
- 40. Armangue T, Petit‐Pedrol M, Dalmau J. Autoimmune Encephalitis in Children. J Child Neurol 2012;27:1460–1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Mann AP, Grebenciucova E, Lukas RV. Anti‐N‐methyl‐D‐aspartate‐receptor encephalitis: Diagnosis, optimal management, and challenges. Ther Clin Risk Manag 2014;10:517–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Moscato EH, Peng XY, Jain A, et al. Acute mechanisms underlying antibody effects in anti‐N‐methyl‐D‐aspartate receptor encephalitis. Ann Neurol 2014;76:108–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Pardo CA, Vining EPG, Guo L, et al. The pathology of Rasmussen syndrome: Stages of cortical involvement and neuropathological studies in 45 hemispherectomies. Epilepsia 2004;45:516–526. [DOI] [PubMed] [Google Scholar]
- 44. Hart YM, Andermann F, Robitaille Y, et al. Double pathology in Rasmussen's syndrome ‐ A window on the etiology? Neurology 1998;50:731–735. [DOI] [PubMed] [Google Scholar]
- 45. Takei H, Wilfong A, Malphrus A, et al. Dual pathology in Rasmussen's encephalitis: A study of seven cases and review of the literature. Neuropathology 2010;30:381–391. [DOI] [PubMed] [Google Scholar]
- 46. Schwab N, Bien CG, Waschbisch A, et al. CD8 T‐cell clones dominate brain infiltrates in Rasmussen encephalitis and persist in the periphery. Brain 2009;132:1236–1246. [DOI] [PubMed] [Google Scholar]
- 47. Varadkar S, Bien CG, Kruse CA, et al. Rasmussen's encephalitis: Clinical features, pathobiology, and treatment advances. Lancet Neurol 2014;13:195–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Blumcke I, Thom M, Aronica E, et al. The clinicopathologic spectrum of focal cortical dysplasias: A consensus classification proposed by an ad hoc Task Force of the ILAE Diagnostic Methods Commission. Epilepsia 2011;52:158–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Boer K, Spliet WG, van Rijen PC, et al. Evidence of activated microglia in focal cortical dysplasia. J Neuroimmunol 2006;173:188–195. [DOI] [PubMed] [Google Scholar]
- 50. Iyer A, Zurolo E, Spliet WG, et al. Evaluation of the innate and adaptive immunity in type I and type II focal cortical dysplasias. Epilepsia 2010;51:1763–1773. [DOI] [PubMed] [Google Scholar]
- 51. Ravizza T, Boer K, Redeker S, et al. The IL‐1beta system in epilepsy‐associated malformations of cortical development. Neurobiol Dis 2006;24:128–143. [DOI] [PubMed] [Google Scholar]
- 52. Zurolo E, Iyer A, Maroso M, et al. Activation of Toll‐like receptor, RAGE and HMGB1 signalling in malformations of cortical development. Brain 2011;134:1015–1032. [DOI] [PubMed] [Google Scholar]
- 53. Choi J, Nordli DR, Alden TD et al. Cellular injury and neuroinflammation in children with chronic intractable epilepsy. J Neuroinflammation 2009;6:38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Iori V, Maroso M, Rizzi M, et al. Receptor for Advanced Glycation Endproducts is upregulated in temporal lobe epilepsy and contributes to experimental seizures. Neurobiol Dis 2013;58:102–114. [DOI] [PubMed] [Google Scholar]
- 55. Maroso M, Balosso S, Ravizza T, et al. Toll‐like receptor 4 and high‐mobility group box‐1 are involved in ictogenesis and can be targeted to reduce seizures. Nat Med 2010;16:413–419. [DOI] [PubMed] [Google Scholar]
- 56. Auvin S, Pineda E, Shin D, Gressens P, Mazarati A. Novel Animal Models of Pediatric Epilepsy. Neurotherapeutics 2012;9:245–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Sankar R, Shin DH, Liu HT, et al. Patterns of status epilepticus‐induced neuronal injury during development and long‐term consequences. J Neurosci 1998;18:8382–8393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Auvin S, Shin D, Mazarati A, Sankar R. Inflammation induced by LPS enhances epileptogenesis in immature rat and may be partially reversed by IL1RA. Epilepsia 2010;51:34–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Ravizza T, Rizzi M, Perego C, et al. Inflammatory response and glia activation in developing rat hippocampus after status epilepticus. Epilepsia 2005;46(Suppl 5):113–117. [DOI] [PubMed] [Google Scholar]
- 60. Rizzi M, Perego C, Aliprandi M, et al. Glia activation and cytokine increase in rat hippocampus by kainic acid‐induced status epilepticus during postnatal development. Neurobiol Dis 2003;14:494–503. [DOI] [PubMed] [Google Scholar]
- 61. Dube CM, Ravizza T, Hamamura M, et al. Epileptogenesis provoked by prolonged experimental febrile seizures: Mechanisms and biomarkers. J Neurosci 2010;30:7484–7494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Balosso S, Maroso M, Sanchez‐Alavez M, et al. A novel non‐transcriptional pathway mediates the proconvulsive effects of interleukin‐1beta. Brain 2008;131:3256–3265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Marcon J, Gagliardi B, Balosso S, Maroso M, Noé F, Morin M, Lerner‐Natoli M, Vezzani A, Ravizza T. Age‐dependent vascular changes induced by status epilepticus in rat forebrain: implications for epileptogenesis. Neurobiol Dis. 2009;34:121–132. [DOI] [PubMed] [Google Scholar]
- 64. Auvin S, Shin D, Mazarati A, et al. Inflammation exacerbates seizure‐induced injury in the immature brain. Epilepsia 2007;48:27–34. [DOI] [PubMed] [Google Scholar]
- 65. Kaindl AM, Degos V, Peineau S, et al. Activation of microglial N‐methyl‐D‐aspartate receptors triggers inflammation and neuronal cell death in the developing and mature brain. Ann Neurol 2012;72:536–549. [DOI] [PubMed] [Google Scholar]
- 66. Baram TZ, Gerth A, Schultz L. Febrile seizures: An appropriate‐aged model suitable for long‐term studies. Brain Res Dev Brain Res 1997;98:265–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Bender RA, Dube C, Gonzalez‐Vega R, Mina EW, Baram TZ. Mossy fiber plasticity and enhanced hippocampal excitability, without hippocampal cell loss or altered neurogenesis, in an animal model of prolonged febrile seizures. Hippocampus 2003;13:399–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Dube C, Chen K, Eghbal‐Ahmadi M, et al. Prolonged febrile seizures in the immature rat model enhance hippocampal excitability long term. Ann Neurol 2000;47:336–344. [PMC free article] [PubMed] [Google Scholar]
- 69. Dube C, Vezzani A, Behrens M, Bartfai T, Baram TZ. Interleukin‐1 beta contributes to the generation of experimental febrile seizures. Ann Neurol 2005;57:152–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Heida JG, Boisse L, Pittman QJ. Lipopolysaccharide‐induced febrile convulsions in the rat: Short‐term sequelae. Epilepsia 2004;45:1317–1329. [DOI] [PubMed] [Google Scholar]
- 71. Heida JG, Teskey GC, Pittman QJ. Febrile convulsions induced by the combination of lipopolysaccharide and low‐dose kainic acid enhance seizure susceptibility, not epileptogenesis, in rats. Epilepsia 2005;46:1898–1905. [DOI] [PubMed] [Google Scholar]
- 72. Heida JG, Pittman QJ. Causal links between brain cytokines and experimental febrile convulsions in the rat. Epilepsia 2005;46:1906–1913. [DOI] [PubMed] [Google Scholar]
- 73. Auvin S, Porta N, Nehlig A, et al. Inflammation in rat pups subjected to short hyperthermic seizures enhances brain long‐term excitability. Epilepsy Res 2009;86:124–130. [DOI] [PubMed] [Google Scholar]
- 74. Sayyah M, Javad‐Pour M, Ghazi‐Khansari M. The bacterial endotoxin lipopolysaccharide enhances seizure susceptibility in mice: Involvement of proinflammatory factors: Nitric oxide and prostaglandins. Neuroscience 2003;122:1073–1080. [DOI] [PubMed] [Google Scholar]
- 75. Kovacs Z, Dobolyi A, Juhasz G, Kekesi KA. Lipopolysaccharide induced increase in seizure activity in two animal models of absence epilepsy WAG/Rij and GAERS rats and Long Evans rats. Brain Res Bull 2014;104:7–18. [DOI] [PubMed] [Google Scholar]
- 76. Abraham J, Fox PD, Condello C, Bartolini A, Koh S. Minocycline attenuates microglia activation and blocks the long‐term epileptogenic effects of early‐life seizures. Neurobiol Dis 2012;46:425–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Somera‐Molina KC, Robin B, Somera CA, et al. Glial activation links early‐life seizures and long‐term neurologic dysfunction: Evidence using a small molecule inhibitor of proinflammatory cytokine upregulation. Epilepsia 2007;48:1785–1800. [DOI] [PubMed] [Google Scholar]
- 78. Galic MA, Riazi K, Heida JG, et al. Postnatal inflammation increases seizure susceptibility in adult rats. J Neurosci 2008;28:6904–6913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Galic MA, Riazi K, Henderson AK, Tsutsui S, Pittman QJ. Viral‐like brain inflammation during development causes increased seizure susceptibility in adult rats. Neurobiol Dis 2009;36:343–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Harre EM, Galic MA, Mouihate A, Noorbakhsh F, Pittman QJ. Neonatal inflammation produces selective behavioural deficits and alters N‐methyl‐D‐aspartate receptor subunit mRNA in the adult rat brain. Eur J Neurosci 2008;27:644–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Auvin S, Mazarati A, Shin D, Sankar R. Inflammation enhances epileptogenesis in the developing rat brain. Neurobiol Dis 2010;40:303–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Pineda E, Shin D, You SJ, et al. Maternal immune activation promotes hippocampal kindling epileptogenesis in mice. Ann Neurol 2013;74:11–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Crespel A, Coubes P, Rousset MC, et al. Inflammatory reactions in human medial temporal lobe epilepsy with hippocampal sclerosis. Brain Res 2002;952:159–169. [DOI] [PubMed] [Google Scholar]
- 84. Aronica E, Boer K, van Vliet EA, et al. Complement activation in experimental and human temporal lobe epilepsy. Neurobiol Dis 2007;26:497–511. [DOI] [PubMed] [Google Scholar]
- 85. Ravizza T, Gagliardi B, Noe F, et al. Innate and adaptive immunity during epileptogenesis and spontaneous seizures: Evidence from experimental models and human temporal lobe epilepsy. Neurobiol Dis 2008;29:142–160. [DOI] [PubMed] [Google Scholar]
- 86. Omran A, Peng J, Zhang CL, et al. Interleukin‐1 beta and microRNA‐146a in an immature rat model and children with mesial temporal lobe epilepsy. Epilepsia 2012;53:1215–1224. [DOI] [PubMed] [Google Scholar]
- 87. Das A, Wallace GC, Holmes C, et al. Hippocampal tissue of patients with refractory temporal lobe epilepsy is associated with astrocyte activation, inflammation, and altered expression of channels and receptors. Neuroscience 2012;220:237–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Pernhorst K, Herms S, Hoffmann P, et al. TLR4, ATF‐3 and IL8 inflammation mediator expression correlates with seizure frequency in human epileptic brain tissue. Seizure 2013;22:675–678. [DOI] [PubMed] [Google Scholar]
- 89. Boer K, Troost D, Jansen F, et al. Clinicopathological and immunohistochemical findings in an autopsy case of tuberous sclerosis complex. Neuropathology 2008;28:577–590. [DOI] [PubMed] [Google Scholar]
- 90. Lee SH, Han SH, Lee KW. Kainic acid‐induced seizures cause neuronal death in infant rats pretreated with lipopolysaccharide. NeuroReport 2000;11:507–510. [DOI] [PubMed] [Google Scholar]
- 91. Lothman EW, Hatlelid JM, Zorumski CF, et al. Kindling with rapidly recurring hippocampal seizures. Brain Res 1985;360:83–91. [DOI] [PubMed] [Google Scholar]
- 92. Michelson HB, Lothman EW. An ontogenic study of kindling using rapidly recurring hippocampal seizures. Dev Brain Res 1991;61:79–85. [DOI] [PubMed] [Google Scholar]
- 93. Sankar R, Auvin S, Kwon YS, et al. Evaluation of development‐specific targets for antiepileptogenic therapy using rapid kindling. Epilepsia 2010;51:39–42. [DOI] [PMC free article] [PubMed] [Google Scholar]