

Abstract
The caspase family is well characterized as playing a crucial role in modulation of programmed cell death (PCD), which is a genetically regulated, evolutionarily conserved process with numerous links to many human diseases, most notably cancer. In this review, we focus on summarizing the intricate relationships between some members of the caspase family and their key apoptotic mediators, involving tumour necrosis factor receptors, the Bcl‐2 family, cytochrome c, Apaf‐1 and IAPs in cancer initiation and progression. We elucidate new emerging types of cross‐talk between several caspases and autophagy‐related genes (Atgs) in cancer. Moreover, we focus on presenting several PCD‐modulating agents that may target caspases‐3, ‐8 and ‐9, and their substrates PARP‐1 and Beclin‐1, which may help us harness caspase‐modulated PCD pathways for future drug discovery.
The most important hallmarks of cancer cells comprise ten biological characteristics, acquired during the multi‐step development of human tumours. Amongst these ten ‘weapons’, are in humans, tumour mutations that may render themselves resistant to programmed cell death (PCD) assisting defective cell survival 1. The complex processes of programmed cell death, such as apoptosis (type I PCD), are orchestrated by caspases, a family of cysteine proteases with unique substrate specificities. Accumulating evidence has recently revealed that phosphorylation of caspases can finely regulate PCD signalling, and that caspase‐mediated cleavage of kinases can terminate pro‐survival signalling or generate pro‐death peptide fragments, execute the death program, and thus ultimately determine the fate of the cancer cells 2.
Hitherto, the official nomenclature has named fourteen caspases in mammals, of which two‐thirds may function in apoptosis 3. Caspases are involved in two different apoptotic pathways: (i) the death‐receptor pathway (extrinsic), which is triggered by ligation of death receptors and subsequent caspase‐8 activation and (ii) the mitochondrial pathway (intrinsic), which is initiated by intracellular stress, and subsequently activated by caspase‐9. Both extrinsic and intrinsic apoptotic pathways can activate downstream caspase‐3, which is responsible for execution and cancer cell demise 4, 5.
However, caspases are not only executioners of PCD in cancer, but they can also modulate several apoptosis‐related proteins including the TNFRs, Bcl‐2 family, cytochrome c, Apaf‐1 and inhibitors of apoptosis (IAPs) 6, 7. Intriguingly, caspases‐3, ‐7 and ‐8 are involved in autophagy (type II PCD), a multi‐step lysosomal degradation process in which a cell degrades long‐lived proteins and damaged organelles 8, 9. Of note, the process of autophagy is highly regulated by a limited number of autophagy‐related genes (Atgs) that may play a pro‐survival or a pro‐death role for regulation of several cancer‐related signalling pathways 10. To date, some PCD‐modulating agents have been known to target caspase members (for example caspases‐3, ‐8, ‐9) and their substrates (for example PARP‐1 and Beclin‐1) and these could be potential therapeutic purposes 11. In this review, we focus on highlighting recent data concerning the multifaceted roles of caspases in apoptosis and autophagy, which may in turn provide a new perspective for targeting caspase‐mediated PCD pathways, in future cancer therapy.
Caspase‐mediated apoptosis in cancer
Tumour necrosis factor receptors
Tumour necrosis factor receptor (or death receptor), which is a trimeric cytokine receptor that binds tumour necrosis factor (TNF), is important for ‘decisions’ of progress towards apoptosis. Of the death receptors there are TNFR1 (also there are further TNFRs) and Fas that can kill many types of cancer cells, and thus induce apoptosis by binding to their cognate receptors that are capable of transmitting a caspase‐activating signal 12. During this process, Fas ligand (FasL), which triggers trimerization of receptors and aggregation of intracellular death domains (DDs), leads to formation of a death‐inducing signalling complex (DISC) 13. The death adaptor protein FADD and proteolytic enzyme caspase‐8 are recruited to Fas. In a homotypic interaction, the DD of Fas binds to a C‐terminal FADD, and subsequently, the death effector domain (DED) of FADD can interact with the DED of the pro‐domain of caspase‐8; thereby, recruiting this proenzyme to the DISC 14. Moreover, pro‐caspase‐8 is proteolytically cleaved and activated at the DISC. DISC‐activated caspase‐8 then directly cleaves the cascade of downstream effector caspases, thus culminating in cell death.
So far, at least nine members of the TNFR family possess a cytosolic DD, included in development of DR‐targeted pro‐apoptotic therapies 15. Moreover, Fas is frequently present in most tumour tissues; however, the capacity for caspase‐8 to elicit PCD following Fas ligation varies dramatically in cancer cells, suggesting that some additional factors may impact on this pathway 16. In contrast to Fas, TNF‐related apoptosis‐inducing ligand (TRAIL), identified by sequence homology to FasL and TNF, induces apoptosis in around 60% of cancer cells. DR4, DR5 and DR5 are the key regulators in DISC formation for caspase‐8/10‐mediated apoptosis in cancer 17, 18 (Fig. 1a).
Figure 1.
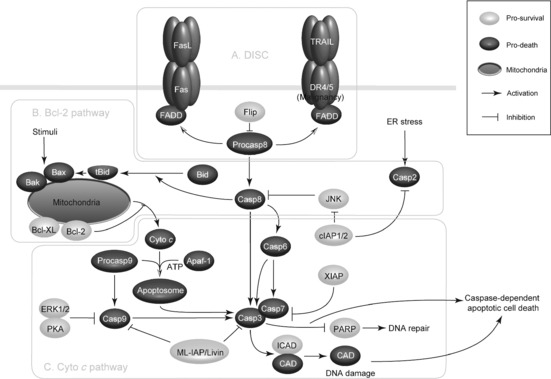
Caspase‐mediated apoptotic pathways in cancer. In the extrinsic apoptotic pathway, Fas and DR4/5 are ligated by FasL and TRAIL respectively, recruiting FADD and binding pro‐caspase‐8 in a homotypic interaction to form a DISC. Once pro‐caspase‐8 is activated, it cleaves the execution caspases (caspases‐3, ‐6, ‐7) to promote DNA damage and apoptosis. Both caspase‐8 and caspase‐2 can cleave and activate Bid, which sequesters anti‐apoptotic Bcl‐2 family members and promotes oligomerization of Bax/Bak, leading to release of cytochrome c from mitochondria to induce intrinsic apoptosis. Cytochrome c binds to Apaf‐1, which recruits pro‐caspase‐9 through CARD–CARD interactions to form the apoptosome. Subsequently, caspase‐9 is activated and Apaf‐1 recruits and cleaves caspase‐3 and caspase‐7 to bring about apoptosis. During this process, ERK1⁄2 and PKA can inactivate caspase‐9, while IAPs (Livin, XIAP, cIAP1/2) can inhibit activity of some caspase members, acting as the key pro‐survival regulators in cancer.
Bcl‐2 family
The Bcl‐2 family is pivotal for regulation of apoptosis, and includes both anti‐ and pro‐apoptotic proteins, and with slight changes in the dynamic balance of these members, there is either inhibition or promotion of cell death 19. Of note, the Bcl‐2 family consists of some pro‐apoptotic members such as Bax, Bak, Bad, Bcl‐Xs, Bid, Bik, Bim and Hrk, as well as anti‐apoptotic members such as Bcl‐2, Bcl‐XL, Bcl‐W, Bfl‐1 and Mcl‐1. Pro‐apoptotic Bcl‐2 members enhance apoptosis by facilitating cytochrome c release, whereas anti‐apoptotic members (Bcl‐2, Bcl‐XL and Mcl‐1) may counteract this effect by sequestering pro‐apoptotic members 20.
In response to apoptotic stimuli, upstream regulators undergo post‐translational modifications such as dephosphorylation and cleavage, resulting in their activation and translocation to mitochondria, thereby triggering apoptosis 15. In this process, pro‐apoptotic BH3‐only proteins (Bim, Bid and Bad) are activated and thus promote oligomerization of p53 effector Bax/Bak (multi‐domain BH3 proteins) to exert their intrinsic pro‐apoptotic activities. Subsequently, mitochondrial outer membranes become permeable, leading to cytochrome c release (from inter‐membrane spaces) and activating the intrinsic apoptotic pathway 21. Hitherto, accumulating data have shown that both caspase‐8 and caspase‐2 can act upstream of mitochondrial permeabilization by cleaving and activating Bid, leading to subsequent cytochrome c release, thus playing the key roles for inducing intrinsic apoptotic pathway 22 (Fig. 1b).
Other apoptotic modulators: cytochrome c, Apaf‐1 and IAPs
Cytochrome c, once released into the cytosol, can bind to apoptotic peptidase activating factor 1 (Apaf‐1) in the presence of dATP or ATP, thus promoting self‐oligomerization of Apaf‐1. It is well known that Apaf‐1 and caspase‐9 contain the protein interaction motif, caspase recruitment domain (CARD) 23. Apaf‐1 can bind caspase‐9 by CARD–CARD interactions that may result in formation of the apoptosome, and dimerization‐induced activation of caspase‐9. Once the apoptosome is formed and then caspase‐9 is activated, domains of Apaf‐1 can recruit downstream executioners such as caspases‐3, and ‐7 that target and cleave the key structural proteins for proteolysis, to trigger apoptosis. Subsequently, caspase‐9 processes these effector caspases, initiating their release from the complex. Also, the cysteine site in caspase‐9 and the cleavage aspartate site (D175) of caspase‐3 are required for efficient recruitment and activation of caspase‐3 24.
Caspase‐9 can be inactivated by some protein kinases including ERK1⁄2 and protein kinase A (PKA), that can be activated by extracellular survival signals. ERK1/2 can also inhibit apoptosis through phosphorylation of caspase‐9 during interphase of the cell cycle, while PKA can phosphorylate caspase‐9, which can block recruitment of caspase‐9 to the apoptosome by compromising Apaf‐1 oligomerization. PKA can directly phosphorylate Apaf‐1, suggesting it to be a regulator of caspase‐9 25. Survivin, which is often overexpressed in cancer cells, opposes caspase‐9 activity, thus having a role as a member of the anti‐apoptotic family. Like other anti‐apoptotic caspase antagonists such as cFLIPS and XIAP, survivin has a very short half‐life, predicting it to disappear from the cell over the course of G2/M, however, survivin plays a critical role during this time. During mitosis, survivin binds to and stabilizes the mitotic spindle. In the absence of survivin, cycling cells undergo mitotic collapse and caspase‐9‐mediated apoptosis 26.
Hitherto, eight human IAP family members have been identified, and X‐chromosome linked IAP (XIAP), containing three BIR domains, is the most efficient caspase inhibitor among them. XIAP can bind caspase‐9 through a groove in the BIR3 domain, associating it with the homodimerization domain of the enzyme and preventing its conformational change 27. The linker region to N‐terminus of the BIR2 domain binds the catalytic domain of caspase‐3 and blocks the active site of the enzyme by steric hindrance, a reverse orientation. In contrast to XIAPs, cellular IAPs (cIAP1 and cIAP2) can inhibit a JNK signalling pathway that promotes the activation of caspase‐8, and the cIAP2‐BIR2 domain directly binds and inhibits caspase‐2. This indicates that HIAP2 is a candidate inhibitor of the caspase‐2 containing complex 28. ML‐IAP/Livin though contains only a single BIR domain and inhibits both caspase‐3 and ‐9 in cancer cells 29.
Several mammalian IAPs, including XIAP, c‐IAP2 and c‐IAP1 possess a c‐terminal RING that may function as an E3 ubiquitin ligase, which can promote ubiquitination, and target caspase‐3 and ‐7 to the proteasome. It is suggested that caspase degradation may be a further mechanism by which IAPs suppress apoptosis 30. As a final apoptotic executioner, caspase‐3 can specifically activate the endonuclease CAD, which is complexed with its inhibitor, ICAD. Activated caspase‐3 can cleave ICAD to release CAD in apoptosis 31 and caspase‐3 can also cleave other substrates including PARP‐1, which may cause morphological and biochemical changes seen in apoptosis of cancer cells (Fig. 1c).
Caspase‐mediated autophagy in cancer
The intricate relationships between caspases and their apoptotic modulators (TNFRs, Bcl‐2 family, cytochrome c, Apaf‐1 and IAPs) have been elucidated at some levels; however, a new emerging correlation between caspases and Atgs has recently been explored in cancer. Beclin‐1 (mammalian homolog of Atg6) can enhance autophagic signalling pathways by combining with class III phosphatidyl inositol‐3‐kinase (PI3KCIII) at the initiating stage 32. In this process, Beclin‐1 and PI3KCIII are novel substrates that can be cleaved by caspases‐3, ‐7 and ‐8. Beclin‐1 and PI3KCIII are cleaved by caspases in both mitochondrial‐ and death receptor‐mediated apoptotic pathways in cancer cells 33. Under some circumstances (Bax over‐expression, nutrient deprivation and oxidative stress), Beclin‐1 can be cleaved by caspases 34. Apoptotic stimuli that activate death receptor‐ and mitochondria‐mediated signalling pathways may cause activation of autophagy, however, during sustained exposure to apoptotic stimuli, caspase‐mediated cleavage of Beclin‐1 produces fragments (Beclin‐1‐N and Beclin‐1‐C) that lose their abilities to induce autophagy, which might be explained by their mislocalization. To be specific, a Beclin‐1‐C fragment translocates to the mitochondrion, thus acquiring a new pro‐apoptotic function to sensitize cells to apoptotic signals by enhancing its local permeabilizing activity 33. Simultaneously Beclin‐1‐N, containing the BH3‐only domain, fails to promote cytoprotective autophagy when the Beclin‐1‐C fragment is stimulated 35 (Fig. 2).
Figure 2.
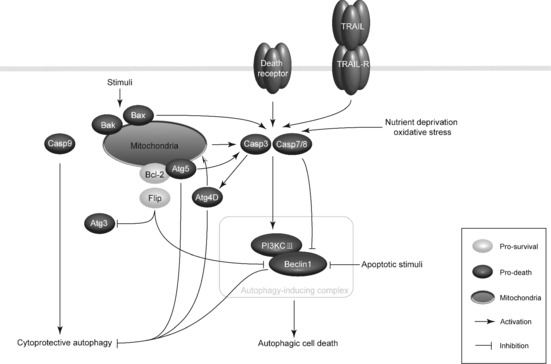
Caspase‐mediated autophagic pathways in cancer. Under some circumstances (for example nutrient deprivation and oxidative stress), Beclin‐1, key component of autophagy‐inducing complex, is cleaved by caspases‐3, ‐7 and ‐8. Fragments of Beclin‐1 may lose their ability to induce autophagy and acquire a new pro‐apoptotic function. Besides Beclin‐1, Atg5 can be cleaved, and generates a truncated protein that may interact with Bcl‐2 with apoptosis promotion, and it can also activate caspase‐3. Cleavage of Atg4D by caspase‐3 produces a fragment that increases autophagy activity and simultaneously amplifies the apoptotic response by the mitochondrial pathway. In particular conditions, Bcl‐2 and Flip, two anti‐apoptotic proteins, can form complexes with Beclin‐1 and Atg3 respectively, thus blocking caspase‐mediated autophagic pathways. Moreover, activation of caspase‐8 and caspase‐9 may act in pro‐apoptotic roles or enhance cytoprotective autophagy in the different biological contexts.
Furthermore, Beclin‐1, Atg5 (being required for the elongation of autophagosomes) and Atg4D (mediating proteolytic maturation of Atg8/LC3) can be cleaved in apoptosis. When apoptosis occurs, calpain‐mediated cleavage of Atg5 can generate a truncated protein that interacts with Bcl‐2 at the mitochondrion to facilitate apoptosis induction 36. Conversely, Atg5 may connect to several signalling pathways that may activate caspase‐3, of which the highest scoring one is that pathway that linking Atg5 to caspase‐3 by Rhophilin2 (RHPN2), cytokeratin 18 and DEDD. Although apoptosis‐associated cleavage of Beclin‐1 and Atg5 can inactivate autophagy, cleavage of Atg4D by caspase‐3 generates a fragment with increasing autophagic activity; this fragment has a cytotoxic effect that correlates with its recruitment to mitochondria 37, 38. Thus, apoptosis‐associated cleavage of Atg4D, similar to that of Beclin‐1 and Atg5, may amplify an apoptotic response via the mitochondrial pathway (Fig. 2).
A further link between autophagy and apoptosis has been explored, that FADD may play a key role in promoting caspase‐8 recruitment and survival via cytoprotective autophagic signalling, suggesting that caspase‐8 plays a part as autophagic regulator 39. In addition, caspase‐8 may have a pro‐apoptotic role while caspase‐9 can enhance cytoprotective autophagy in human breast MCF‐7 cells, which suggests that caspase‐9 can be considered to have connections between apoptosis and autophagy 9, 40. Bcl‐2 and Flip, two anti‐apoptotic proteins, can inhibit autophagy by forming complexes with Beclin‐1 and Atg3, respectively, suggesting another caspase‐mediated autophagic pathway in cancer (Fig. 2).
Targeted caspases and their substrates for drug discovery
Of note, TNF alpha and its family members of death ligands CD95L (Fas ligand) and TRAIL, all possess considerable potential in apoptosis induction in many types of cancer cells. However, systemic administration of TNF alpha and CD95L provoke severe adverse effects, thus their applicability as tumour cell‐specific agents remains to be clarified for cancer therapy. In contrast, TRAIL is a rather promising anti‐cancer cytokine as it has been shown to exert selectivity amongst various cancer cells. Even though TRAIL initiates apoptosis after binding to TRAIL receptors (TRAILR1/DR4, TRAILR2/DR5) in the death‐receptor (extrinsic) pathway, it can also recruit the mitochondrial pathway (intrinsic) for apoptotic response 6, 18.
Caspases are responsible for initiation and regulation of the apoptotic pathway, and thus play their important roles as tumour repressors. Currently, several anti‐cancer approaches (gene therapy and antisense strategies) have been developed to target some specific apoptotic regulators 11, 22. Thus, we focus here on presenting several known anti‐tumour agents that can target caspases and their substrates (for example PARP‐1 and Beclin‐1) in apoptotic and/or autophagic pathways in cancer cells (Fig. 3).
Figure 3.
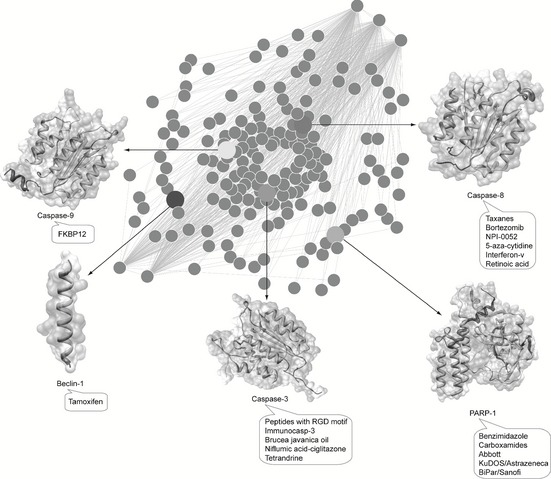
Anti‐tumour agents that target the caspase‐modulated programmed cell death network. Several anti‐tumour agents that may target caspase‐3 (peptides with the RGD motif, immunocasp‐3, Brucea javanica oil, niflumic acid–ciglitazone and tetrandrine), caspase‐8 (taxanes, Bortezomib, NPI‐0052, 5‐aza‐cytidine, interferon‐v and retinoic acid) and caspase‐9 (FKBP12), as well as their substrates PARP‐1 (benzimidazole, carboxamides, Abbott, KuDOS/Astrazeneca and BiPar/Sanofi) and Beclin‐1 (tamoxifen), have been utilized to modulate PCD networks for cancer therapy.
Caspase‐8, ‐9 and ‐3
Caspase‐8 is not only an apical caspase which triggers apoptosis following death receptor ligation, but also a highly versatile molecule that impacts on cellular signalling. It is worth observing that caspase‐8 is expressed heterogeneously amongst different types of cancer 41. Thus, it is possible to devise a range of anti‐neoplastic therapeutic strategies that can enhance death‐inducing functions of caspase‐8 by elevating its expression. Caspase‐8 regulation can be targeted by small molecules for potential applications. One classical type of these is the microtubule‐stabilizing agents, such as taxanes, that can amplify caspase‐8 activation via microtubule‐anchored DED ‘filaments’ to promote apoptosis 42. Caspase‐8‐mediated death may be linked to the status of cellular tubulin, as well as to production of reactive oxygen species (ROS) 43. Production of ROS from mitochondria may mobilize sequestered pools of existing caspase‐8. Interestingly, ROS (for example, NO) can lead to nitrosylation of NF‐κB, a nuclear transcription factor that modulates expression of many genes involved in apoptosis regulation in carcinogenesis, thereby acting as a molecular switch of apoptotic and non‐apoptotic functions of caspase‐8 44. Proteosomal inhibitors, such as Bortezomib and NPI‐0052, have been utilized against tumour cell populations that express caspase‐8. These small molecules may increase caspase‐8 expression presumably by blocking its degradation 45. Moreover, treatments with other agents such as 5‐azacytidine, interferon‐V and retinoic acid can up‐regulate transcription of caspase‐8, thus favouring activation of PCD pathways 46, 47, 48.
Caspase‐9 is the initiator caspase associated with the intrinsic or mitochondrial pathway of apoptosis. Once activated, caspase‐9 cleaves and activates downstream effector caspases‐3 and ‐7, thus resulting in apoptosis. FKBP12, a caspase‐9 fusion protein, is anti‐angiogenic in mouse models by induction of caspase‐9 dimerization 49.
Caspase‐3, the executioner caspase, is able to directly degrade multiple substrates including structural and regulatory proteins. Thus, therapeutic strategies designed to stimulate apoptosis by activating caspase‐3 may help in combating cancer caused by apoptosis deficiency. Some small molecules have been developed to be selective activators of caspase‐3, peptides containing the arginine‐glycine‐aspartate (RGD) motif, which promotes apoptosis by directly inducing auto‐processing of pro‐caspase‐3 50. A further reagent, immunocasp‐3, is a fusion protein comprised of a single‐chain anti‐erbB2/HER2 antibody, and an active caspase‐3 molecule. Significant tumour regression in mouse xenografts of HER2‐positive tumour cells has been observed upon over‐expression of chimaeric immunocasp‐3 gene, which can induce autoactivation of caspase‐3 51. Gene therapy approaches have been aimed at replacing defective caspases in cancer cells by their normal counterparts. For example, Ad‐G/iCasp3 is a replication‐incompetent adenoviral vector carrying the caspase‐3 gene, whose antitumor activity has been tested in mouse models of prostate cancer 52. There are a variety of anti‐cancer agents that target both caspase‐3 and ‐9 and promote the apoptotic process, including Brucea javanica 53 oil, niflumic acid–ciglitazone 54 and tetrandrine 55.
PARP‐1 and Beclin‐1
Several caspase substrates may be targeted in combined therapies of cancer. For instance, a typical substrate is Poly ADP‐ribose polymerase‐1 (PARP‐1), as the ‘guardian angel of DNA’. The PARP‐1‐mediated pathway is a major mechanism for DNA repair in cancer cells, thus leading to drug resistance and continued tumour growth 56, 57. Hence, PARP‐1 inhibitors or deletion of the PARP‐1 gene can act as potentiators in chemotherapeutics, damaging DNA repair mechanisms and resulting in genomic dysfunction and cell death. Inhibitors of PARP‐1, such as benzimidazole, carboxamides [Abbott, KuDOS/Astrazeneca] and BiPar/Sanofi 58, 59, 60, have been utilized for cancer therapy.
A new emerging caspase substrate is Beclin‐1, a component of the PI3KCIII complex required in autophagy. Beclin‐1 is a novel substrate of caspases‐3, ‐7 and ‐8, suggesting its crucial role in cross‐talk between apoptosis and autophagy. Interestingly, tamoxifen, an oestrogen receptor (ER) antagonist first targeted breast cancer therapeutic agent, can up‐regulate levels of Beclin‐1, thus inducing autophagy in breast cancer treatment 61, 62.
Concluding remarks
Cancer is a complex, multi‐step gene‐derived disease with astonishingly numerous links to PCD apoptosis and autophagy; nevertheless, cancer cells evolve a variety of strategies to circumvent apoptosis. This multiplicity of apoptosis‐evading mechanisms may reflect diversity of apoptosis‐inducing signalling pathways that cancer cell populations may encounter. The apoptotic machinery and strategies utilized by cancer cells to evade its actions have been widely appreciated, thus, any new therapeutic strategy should consider combination of apoptosis and autophagy in cancer treatment.
Currently, considerable effort is needed to further determine the molecular mechanisms of apoptosis and autophagy in cancer, to define how some apoptotic or autophagic modulators such as caspases (caspases‐3, ‐8 and ‐9) and their mediators (TNFRs, Bcl‐2 family, cytochrome c, Apaf‐1 and IAPs) can impact on cancer aetiology and pathology, specially focusing on distinguishing differences between physiological and pathological PCD, thus specifically targeting caspase‐modulated apoptotic and autophagic pathways as promising avenues for therapy. Thus, a new therapeutic strategy would be carried forward for drug discovery, if new PCD‐modulating agents could be designed to target not only caspases and their substrates (for example PARP‐1 and Beclin‐1), but also their aforementioned PCD modulators, for future cancer drug discovery.
Acknowledgements
We are grateful to Dr Jin‐ku Bao, Dr Yan Cheng, He‐jiao Bian, Qian Liu and Ming‐wei Min for their critical reviews on this manuscript. We also thank Huai‐long Xu, Lei‐lei Fu, Xu Zhao and Jia‐ying Yu for their valued suggestions. This work was supported in part by the grants from the Young Teacher's Fund of Sichuan University (No. 2010SCU11066), the Science Foundation for Post Doctorate Research of China (No. 20110491725) and the Major State Basic Research Development Program of China (973 Program) (No. 2010cb529900).
References
- 1. Hanahan D, Weinberg RA (2011) Hallmarks of cancer: the next generation. Cell 144, 646–674. [DOI] [PubMed] [Google Scholar]
- 2. Hengartner MO (2000) The biochemistry of apoptosis. Nature 407, 770–776. [DOI] [PubMed] [Google Scholar]
- 3. Kurokawa M, Kornbluth S (2009) Caspases and kinases in a death grip. Cell 138, 838–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Yuan J, Shaham S, Ledoux S, Ellis HM, Horvitz HR (1993) The C. elegans cell death gene ced‐3 encodes a protein similar to mammalian interleukin‐1 beta‐converting enzyme. Cell 75, 641–652. [DOI] [PubMed] [Google Scholar]
- 5. Degterev A, Boyce M, Yuan J (2003) A decade of caspases. Oncogene 22, 8543–8567. [DOI] [PubMed] [Google Scholar]
- 6. Ghobrial IM, Witzig TE, Adjei AA (2005) Targeting apoptosis pathways in cancer therapy. CA Cancer J. Clin. 55, 178–194. [DOI] [PubMed] [Google Scholar]
- 7. Li J, Yuan J (2008) Caspases in apoptosis and beyond. Oncogene 27, 6194–6206. [DOI] [PubMed] [Google Scholar]
- 8. Cho DH, Jo YK, Hwang JJ, Lee YM, Roh SA, Kim JC (2009) Caspase‐mediated cleavage of ATG6/Beclin‐1 links apoptosis in HeLa cells. Cancer Lett. 274, 95–100. [DOI] [PubMed] [Google Scholar]
- 9. Jeong HS, Choi HY, Lee ER, Kim JH, Jeon K, Lee HJ et al (2010) Involvement of caspase‐9 in autophagy‐mediated cell survival pathway. Biochim. Biophys. Acta 1813, 80–90. [DOI] [PubMed] [Google Scholar]
- 10. Li ZY, Yang Y, Ming M, Liu B (2011) Mitochondrial ROS generation for regulation of autophagic pathways in cancer. Biochem. Biophys. Res. Commun. 414, 5–8. [DOI] [PubMed] [Google Scholar]
- 11. Philchenkov A (2004) Caspases: potential targets for regulating cell death. J. Cell Mol. Med. 8, 432–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sprick MR, Weigand MA, Rieser E, Rauch CT, Juo P, Blenis J et al (2000) FADD/MORT1 and caspase‐8 are recruited to TRAIL Receptors 1 and 2 and are essential for apoptosis mediated by TRAIL Receptor 2. Immunity, 12, 599–609. [DOI] [PubMed] [Google Scholar]
- 13. Krammer PH (1999) CD95(APO‐1/Fas)‐mediated apoptosis: live and let die. Adv. Immunol. 71, 163–210. [DOI] [PubMed] [Google Scholar]
- 14. Kischkel FC, Hellbardt S, Behrmann I, Germer M, Pawlita M, Krammer PH et al (1995) Cytotoxicity‐dependent APO‐1 (Fas/CD95)‐associated proteins form a death‐inducing signaling complex (DISC) with the receptor. EMBO J. 14, 5579–5588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ivano A, Gerry M, Richard AK (2011) Cell death pathology: cross‐talk with autophagy and its clinical implications. Biochem. Biophys. Res. Commun. 414, 277–281. [DOI] [PubMed] [Google Scholar]
- 16. Deveraux QL, Roy N, Stennicke HR, Arsdale TV (1998) IAPs block apoptotic events induced by caspase‐8 and cytochrome c by direct inhibition of distinct caspases. EMBO J. 17, 2215–2223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Medema JP, Scaffidi C, Kischkel FC, Shevchenko A, Mann M, Krammer PH et al (1997) FLICE is activated by association with the CD95 death‐inducing signaling complex (DISC). EMBO J. 16, 2794–2804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sun S (2011) Understanding the Role of the Death Receptor 5/FADD/caspase‐8 Death Signaling in Cancer Metastasis. Mol. Cell. Pharmacol. 3, 31–34. [PMC free article] [PubMed] [Google Scholar]
- 19. Huang Z (2000) Bcl‐2 family proteins as targets for anticancer drug design. Oncogene 19, 6627–6631. [DOI] [PubMed] [Google Scholar]
- 20. Chipuk JE, Green DR (2008) How do BCL‐2 proteins induce mitochondrial outer membrane permeabilization. Trends Cell Biol. 18, 157–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Li P, Nijhawan D, Budihardjo I, Srinivasula SM, Ahmad M, Alnemri ES et al (1997) Cytochrome c and dATP‐dependent formation of Apaf‐1/caspase‐9 complex initiates an apoptotic protease cascade. Cell 91, 479–489. [DOI] [PubMed] [Google Scholar]
- 22. Ghavami S, Hashemi M, Ande SR, Yeganeh B, Xiao W, Eshraghi M et al (2009) Caspase genes apoptosis and cancer: mutations within caspase genes. J. Med. Genet. 46, 497–510. [DOI] [PubMed] [Google Scholar]
- 23. Kuida K, Haydar TF, Kuan CY et al (1998) Reduced apoptosis and cytochrome c‐mediated caspase activation in mice lacking caspase‐9. Cell 94, 325–337. [DOI] [PubMed] [Google Scholar]
- 24. Martin MC, Allan LA, Lickrish M, Sampson C, Morrice N, Clarke PR (2005) Protein kinase A regulates caspase‐9 activation by Apaf‐1 downstream of cytochrome c. J. Biol. Chem. 280, 15449–15455. [DOI] [PubMed] [Google Scholar]
- 25. Allan LA, Clarke PR (2009) Apoptosis and autophagy: regulation of caspase‐9 by phosphorylation. FEBS J. 276, 6063–6073. [DOI] [PubMed] [Google Scholar]
- 26. Shai WG, David TK, Christina VJ (2009) The role of protein synthesis in cell cycling and cancer. Mol. Oncol. 3, 402–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Srinivasula SM, Ashwell JD (2008) IAPs: what's in a name? Mol. Cell 30, 123–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Altieri DC (2010) Survivin and IAP proteins in cell‐death mechanisms. Biochem. J. 430, 199–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Shiozaki EN, Chai J, Rigotti DJ, Riedl SJ, Li P, Srinivasula SM et al (2003) Mechanism of XIAP‐mediated inhibition of caspase‐9. Mol. Cell 11, 519–527. [DOI] [PubMed] [Google Scholar]
- 30. Shi Y (2002) Mechanisms of caspase activation and inhibition during apoptosis. Mol. Cell 9, 459–470. [DOI] [PubMed] [Google Scholar]
- 31. Chen M, Wang J (2002) Initiator caspases in apoptosis signaling pathways. Apoptosis 7, 313–319. [DOI] [PubMed] [Google Scholar]
- 32. Liu B, Cheng Y, Liu Q, Bao JK, Yang JM (2010) Autophagic pathways as new targets for cancer drug development. Acta Pharmacol. Sin. 31, 1154–1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wirawan E, Vande Walle L, Kersse K, Cornelis S, Claerhout S, Vanoverberghe I et al (2010) Caspase‐mediated cleavage of Beclin‐1 inactivates Beclin‐1‐induced autophagy and enhances apoptosis by promoting the release of pro‐apoptotic factors from mitochondria. Cell Death Dis. 1, e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Luo S, Rubinsztein DC (2009) Apoptosis blocks Beclin‐1‐dependent autophagosome synthesis: an effect rescued by Bcl‐XL . Cell Death Differ. 17, 268–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Djavaheri‐Mergny M, Maiuri MC, Kroemer G (2010) Cross‐talk between apoptosis and autophagy by caspase‐mediated cleavage of Beclin‐1. Oncogene 29, 1717–1719. [DOI] [PubMed] [Google Scholar]
- 36. Yousefi S, Perozzo R, Schmid I, Ziemiecki A, Schaffner T, Scapozza L et al (2006) H‐U Simon, Calpain‐mediated cleavage of Atg5 switches autophagy to apoptosis. Nat. Cell Biol. 8, 1124–1132. [DOI] [PubMed] [Google Scholar]
- 37. Zalckvar E, Yosef N, Reef S, Ber Y, Rubinstein AD, Mor I et al (2010) A systems level strategy for analyzing the cell death network: implication in exploring the apoptosis/autophagy connection. Cell Death Differ. 17, 1244–1253. [DOI] [PubMed] [Google Scholar]
- 38. Betin VM, Lane JD (2009) Caspase cleavage of Atg4D stimulates GABARAP‐L1 processing and triggers mitochondrial targeting and apoptosis. J. Cell Sci. 122, 2554–2566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Bell BD, Leverrier S, Weist BM, Newton RH, Arechiga AF, Luhrs KA et al (2008) FADD and caspase‐8 control the outcome of autophagic signaling in proliferating T cells. Proc. Natl. Acad. Sci. USA. 105, 16677–16682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wang SY, Yu QJ, Zhang RD, Liu B (2011) Core signaling pathways of survival/death in autophagy‐related cancer networks. Int. J. Biochem. Cell Biol. 4, 1263–1266. [DOI] [PubMed] [Google Scholar]
- 41. Stupack DG (2010) Caspase‐8 as a therapeutic target in cancer. Cancer Lett. doi: 10.1016/j.canlet.2010.07.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Mielgo A, Torres VA, Clair K, Barbero S, Stupack DG (2009) Paclitaxel promotes a caspase 8‐mediated apoptosis through death effector domain association with microtubules. Oncogene 28, 3551–3562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Chen C, Liu Y, Zheng D (2009) An agonistic monoclonal antibody against DR5 induces ROS production, sustained JNK activation and endo G release in Jurkat leukemia cells. Cell Res. 19, 984–995. [DOI] [PubMed] [Google Scholar]
- 44. Kelleher ZT, Matsumoto A, Stamler JS, Marshall HE (2007) NOS2 regulation of NF‐kappaB by S‐nitrosylation of p65. J. Biol. Chem. 282, 30667–30672. [DOI] [PubMed] [Google Scholar]
- 45. Chauhan D, Singh A, Brahmandam M et al (2008) Combination of proteasome inhibitors bortezomib and NPI‐0052 trigger in vivo synergistic cytotoxicity in multiple myeloma. Blood 111, 1654–1664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Hopkins‐Donaldson S, Bodmer JL, Bourloud KB, Brognara CB, Tschopp J, Gross N (2000) Loss of caspase‐8 expression in highly malignant human neuroblastoma cells correlates with resistance to tumor necrosis factor‐related apoptosis‐inducing ligand‐induced apoptosis. Cancer Res. 60, 4315–4319. [PubMed] [Google Scholar]
- 47. Raguénez G, Mühlethaler‐Mottet A, Meier R, Duros C, Bénard J, Gross N (2009) Fenretinide‐induced caspase‐8 activation and apoptosis in an established model of metastatic neuroblastoma. BMC Cancer 9, 97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Kalli KR, Devine KE, Cabot MC, Arnt CR, Heldebrant MP, Svingen PA et al (2003) Heterogeneous role of caspase‐8 in fenretinide‐induced apoptosis in epithelial ovarian carcinoma cell lines. Mol. Pharmacol. 64, 1434–1443. [DOI] [PubMed] [Google Scholar]
- 49. Nör JE, Hu Y, Song W, Spencer DM, Núñez G (2002) Ablation of microvessels in vivo upon dimerization of iCaspase‐9. Gene Ther. 9, 444–451. [DOI] [PubMed] [Google Scholar]
- 50. Buckley CD, Pilling D, Henriquez NV, Parsonage G, Threlfall K, Scheel‐Toellner D et al (1999) RGD peptides induce apoptosis by direct caspase‐3 activation. Nature 397, 534–539. [DOI] [PubMed] [Google Scholar]
- 51. Liu JJ, Lin M, Yu JY, Liu B, Bao JK (2011) Targeting apoptotic and autophagic pathways for cancer therapeutics. Cancer Lett. 300, 105–114. [DOI] [PubMed] [Google Scholar]
- 52. Shariat SF, Desai S, Song W, Khan T, Zhao J, Nguyen C et al (2001) Adenovirus‐mediated transfer of inducible caspases: a novel ‘death switch’ gene therapeutic approach to prostate cancer. Cancer Res. 61, 2562–2571. [PubMed] [Google Scholar]
- 53. Lou GG, Yao HP, Xie LP (2010) Brucea javanica oil induces apoptosis in T24 bladder cancer cells via upregulation of caspase‐3, caspase‐9, and inhibition of NF‐kappa B and COX‐2 expressions. Am. J. Chin. Med. 38, 613–624. [DOI] [PubMed] [Google Scholar]
- 54. Kim BM, Maeng K, Lee KH, Hong SH (2011) Combined treatment with the Cox‐2 inhibitor niflumic acid and PPARγ ligand ciglitazone induces ER stress/caspase‐8‐mediated apoptosis in human lung cancer cells. Cancer Lett. 300, 134–144. [DOI] [PubMed] [Google Scholar]
- 55. Li X, Su B, Liu R, Wu D, He D (2011) Tetrandrine induces apoptosis and triggers caspase cascade in human bladder cancer cells. J. Surg. Res. 166, e45–e51. [DOI] [PubMed] [Google Scholar]
- 56. Ljungman M (2009) Targeting the DNA damage response in cancer. Chem. Rev. 109, 2929–2950. [DOI] [PubMed] [Google Scholar]
- 57. Miwa M, Masutani M (2007) Poly (ADP‐ribosyl) ation and cancer. Cancer 98, 1528–1535. [Google Scholar]
- 58. White AW, Almassy R, Calvert AH, Curtin NJ, Griffin RJ, Hostomsky Z et al (2000) Resistance‐modifying agents. 9. Synthesis and biological properties of benzimidazole inhibitors of the DNA repair enzyme poly(ADP‐ribose) polymerase. J. Med. Chem. 43, 4084–4097. [DOI] [PubMed] [Google Scholar]
- 59. Ferraris DV (2010) Evolution of poly (ADP‐ribose) polymerase‐1(PARP‐1) inhibitors. from concept to clinic. J. Med. Chem. 53, 4561–4584. [DOI] [PubMed] [Google Scholar]
- 60. Loh VM Jr, Cockcroft XL, Dillon KJ, Dixon L, Drzewiecki J, Eversley PJ et al (2005) Phthalazinones, Part 1: the design and synthesis of a series of novel potential inhibitors of poly (ADP‐ribose) polymerase. Bioorg. Med. Chem. Lett. 15, 2235–2238. [DOI] [PubMed] [Google Scholar]
- 61. Rubinsztein DC, Gestwicki JE, Murphy LO, Klionsky DJ (2007) Potential therapeutic applications of autophagy. Nat. Rev. Drug Discov. 6, 304–312. [DOI] [PubMed] [Google Scholar]
- 62. John S, Nayvelt I, Hsu HC et al (2008) Cancer cells regulation of estrogenic effects by Beclin 1 in breast. Cancer Res. 68, 7855–7863. [DOI] [PubMed] [Google Scholar]