

Summary
Aims
Compound MQA (1,5‐O‐dicaffeoyl‐3‐O‐[4‐malic acid methyl ester]‐quinic acid) is a natural derivative of caffeoylquinic acid isolated from Arctium lappa L. roots. However, we know little about the effects of MQA on the central nervous system. This study aims to investigate the neuroprotective effects and underlying mechanisms of MQA against the neurotoxicity of N‐methyl‐d‐aspartate (NMDA).
Methods and Results
Pretreatment with MQA attenuated the loss of cell viability after SH‐SY5Y cells treated with 1 mM NMDA for 30 min by MTT assay. Hoechst 33342 and Annexin V‐PI double staining showed that MQA inhibited NMDA‐induced apoptosis. In addition to preventing Ca2+ influx, the potential mechanisms are associated with increases in the Bcl‐2/Bax ratio, attenuation of cytochrome c release, caspase‐3, caspase‐9 activities, and expressions. Also, MQA inhibited NMDA‐induced phosphorylation of ERK1/2, p38, and JNK1/2. Furthermore, deactivation of CREB, AKT, and GSK‐3β, upregulation of GluN2B‐containing NMDA receptors (NMDARs), and downregulation of GluN2A‐containing NMDARs were significantly reversed by MQA treatment. Computational docking simulation indicates that MQA possesses a well affinity for NMDARs.
Conclusion
The protective effects of MQA against NMDA‐induced cell injury may be mediated by blocking NMDARs. The potential mechanisms are related with mitochondrial apoptosis, ERK‐CREB, AKT/GSK‐3β, p38, and JNK1/2 pathway.
Keywords: Apoptosis, Caffeoylquinic acid, Excitotoxicity, Neuroprotection, N‐methyl‐d‐aspartate (NMDA)
Introduction
Glutamate is a primary excitatory neurotransmitter in the central nervous system (CNS) 1. Excessive accumulation of glutamate and overactivation of its receptors are implicated in a number of CNS disorders, including hypoxia, cerebral ischemia, as well as Huntington's, Alzheimer's, and Parkinson's diseases 2, 3. Many studies have shown that the N‐methyl‐d‐aspartate (NMDA) subtype of glutamate receptors is the major mediator of glutamate excitotoxicity 4. Overactivation of NMDA receptors (NMDARs) can trigger high level of calcium influx, which activates many enzymes that damage cell structures leading to neuronal cell death 4.
NMDARs are heteromeric complexes, consisting of three types of subunits: GluN1, GluN2 (A, B, C, and D), and GluN3 (A and B) 5. The functional NMDARs are composed of two GluN1 subunits and two GluN2 subunits. It has been observed that GluN2A and GluN2B play different roles in pathological conditions, while excitotoxicity was thought to be triggered by the selective activation of GluN2B‐containing NMDARs 6. Further, great considerations have been given in the search for GluN2B antagonists because of the potential to cure a number of CNS diseases 7, 8.
1,5‐O‐dicaffeoyl‐3‐O‐(4‐malic acid methyl ester)‐quinic acid (MQA) is a natural caffeoylquinic acid derivative (Figure 1A) recently isolated from Arctium lappa L. roots by our group 9. Caffeoylquinic acid derivatives possess a broad range of biological properties, including antioxidant, antibacterial, anticancer, and anti‐α‐glucosidase activities 10, 11, 12. Previous study has demonstrated that caffeoylquinic acid derivatives inhibited Aβ42‐induced toxicity in SH‐SY5Y cells 13. Moreover, caffeoylquinic acid derivatives significantly improved spatial learning memory by increasing the mRNA expression level of phosphoglycerate kinase‐1 in vivo experiments 14. However, there are no comprehensive reports on the effects of MQA against the neurotoxicity of NMDA.
Figure 1.
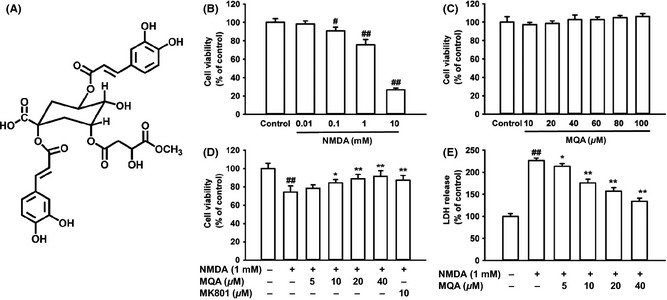
Effects of MQA on NMDA‐induced cytotoxicity in SH‐SY5Y cells. (A) Chemical structure of MQA. (B) Cytotoxic effects of NMDA on cell viability. (C) Effects of different concentrations of MQA on the cell viability. (D) Effects of MQA and MK801 on the cell viability in NMDA‐treated cells. (E) Effects of MQA on LDH leakage after exposure to NMDA. SH‐SY5Y cells were pretreated with different concentrations of MQA for 2 h prior to 1 mM NMDA for 30 min. Data were shown as means ± SD (n = 3). # P < 0.05 and ## P < 0.01 versus the control group. *P < 0.05 and **P < 0.01 versus the NMDA‐treated group.
Previous studies have indicated that the expression of NMDARs was detectable in SH‐SY5Y cells and this cell line is a useful model system for NMDARs studies in vitro 15, 16, 17. Therefore, SH‐SY5Y cells were applied to investigate the neuroprotective effects of MQA against NMDA‐induced cytotoxicity in this study. The finding that MQA inhibited NMDA‐induced apoptosis and calcium influx prompted us to explore the intracellular pathways. Further, computational molecular docking was performed to simulate the possible molecular interactions between MQA and NMDARs.
Materials and Methods
Materials
MQA (purity > 98%) was isolated from A. lappa L. roots by our group. MQA was dissolved in DMSO and the final concentration of DMSO was <0.1% (v/v). 3‐(4,5‐Dimethylthiazol‐2‐yl)‐2,5‐diphenyltetrazolium bromide (MTT), trypsin, and DMSO were purchased from Amresco (Solon, OH, USA). Fetal bovine serum and Dulbecco's modified Eagle's medium (DMEM) were obtained from Hyclone (Logan, UT, USA). The lactate dehydrogenase (LDH) assay kit was obtained from Nanjing Jiancheng Bioengineering Institute (Nanjing, China). Hoechst 33342, NMDA, and MK801 were purchased from Sigma‐Chemical (St. Louis, MO, USA). The Annexin V/propidium iodide (PI) apoptosis assay kit was obtained from BD Pharmingen (San Jose, CA, USA). The fluo‐3/AM, caspase‐3, and caspase‐9 activity kits were purchased from Beyotime (Haimen, China). The following antibodies were purchased from Cell Signaling Technology (Boston, MA, USA): phospho‐ERK1/2 (Thr202/Tyr204), ERK1/2, phospho‐JNK1/2 (Thr183/Tyr185), JNK1/2, phospho‐p38 (Thr180/Tyr182), p38, p‐CREB (Ser133), CREB, phospho‐GSK‐3β (Ser9), GSK‐3β, phospho‐AKT (Thr308), AKT, GluN2A, GluN2B. Antibodies against cytochrome c, Bcl‐2, Bax, caspase‐3, caspase‐9, β‐actin, and the secondary antibodies conjugated with horseradish peroxidase (HRP) were all purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). All other chemicals and reagents were of analytical grade.
Cell Culture and Treatment
Human neuroblastoma SH‐SY5Y cells were cultured in DMEM supplemented with 10% (v/v) fetal bovine serum. The cells were incubated in a humidified atmosphere of 5% CO2 at 37°C. The culture medium was routinely replaced every other day and subcultured once they reached 70–80% confluence.
SH‐SY5Y cells were seeded in different culture plates at an appropriate density. After grown in the incubator for 24 h, cells were treated with different concentrations of MQA (20, 40 μM/mL). After incubation for 2 h, the medium was changed to normal medium (control group) or with Mg2+‐free Lock's buffer (154 mM NaCl, 5.6 mM KCl, 3.6 mM NaHCO3, 2.3 mM CaCl2, 5.6 mM glucose, 5 mM HEPES, PH 7.4) supplemented with NMDA (1 mM) for 30 min. Then, cells were returned to the normal culture medium for another 12 h.
Cell Viability Assay
SH‐SY5Y cells were seeded into 96‐well culture plates at a density of 1 × 104 cells/well. The cells were grown for 24 h, and the medium was replaced with Mg2+‐free Lock's buffer that containing various concentrations of NMDA (final concentration, 0, 0.01, 0.1, 1, and 10 mM) for 30 min. For MQA‐mediated protection assay, the cells were pretreated with MQA (0, 5, 10, 20, and 40 μM) or MK801 (10 μM) for 2 h. Then, the medium was removed and replaced with Mg2+‐free Lock's buffer that containing 1 mM NMDA for 30 min. At the end of these treatments, the culture medium was changed to normal medium containing 0.5 mg/mL MTT. After incubation for 4 h at 37°C, the medium was replaced by 150 μL DMSO. Then, the optical density was read on a microplate reader (ELX 800; Bio‐tek, Winooski, VT, USA) at 490 nm. Cell viability was presented as a percentage of the absorbance of untreated cultures.
Measurement of LDH Release
SH‐SY5Y cells were seeded into 96‐well culture plates at a density of 1 × 104 cells/well. At the end of treatment, the supernatant was collected and used for the assay of LDH activity. The LDH activity was measured by an assay kit according to the manufacturer's instructions. The absorbance of the samples was determined at 450 nm using a microplate reader.
Hoechst 33342 Staining
SH‐SY5Y cells were cultured in 6‐well plates at a density of 2 × 105 cells/well. The treated cells were washed with PBS solution twice, stained with Hoechst 33342 dye (10 μg/mL) for 15 min 18. After the treatment, the cells were visualized using a fluorescence microscope (IX71; Olympus, Tokyo, Japan).
Flow Cytometry Analysis for Apoptosis
The apoptotic cells were measured by Annexin V‐FITC and propidium iodide (PI) apoptosis assay kit, according to the manufacturer's instructions. Briefly, SH‐SY5Y cells treated with NMDA and MQA were harvested, washed twice with cold PBS, and resuspended in binding buffer at the density of 1 × 106 cells/mL. Subsequently, 100 μL of the cells was incubated with 5 μL FITC‐Annexin V and 10 μL PI for 15 min in the dark 19. Then, 400 μL of binding buffer was added after incubation and samples were analyzed by flow cytometry (FACScan; Becton Dickinson, San Jose, CA, USA).
Measurement of Intracellular Calcium Concentration
The concentration of intracellular calcium was measured with Fluo‐3/AM 20. SH‐SY5Y cells were cultured in 3.5‐cm plates at a density of 1 × 105 cells/plate for 24 h, and then incubated with 40 μM MQA for 2 h. After the treatment, the supernatant was removed and cells were incubated with Fluo‐3/AM (5 μM) for 30 min. Then, cells were washed twice with Mg2+‐free extracellular solution (140 mM NaCl, 3 mM KCl, 2 mM CaCl2, 10 mM HEPES, 10 mM glucose, pH was adjusted to 7.2–7.3 with NaOH). The cells were measured for fluorescence using a confocal laser scanning microscope (LEICA). Prior to exposure to NMDA, the dye‐loaded cells were scanned for 1 min to obtain the basal level of intracellular Ca2+. Then, 1 mM NMDA was added to the cultures. Changes of Ca2+ concentration were measured by the fluorescence intensities of the fluo‐3/AM loaded cells for another 5 min. The results were expressed as the change ratio to the basal level.
Caspase‐3 and Caspase‐9 Activities Assay
The activities of caspase‐3 and caspase‐9 were determined by caspase‐3 and caspase‐9 fluorometric assay kit, respectively. After the treatment, the cells were collected and centrifuged at 2800 g for 10 min at 4°C. The pellet was washed twice with cold PBS and lysed with lysis buffer. After the lysate was centrifuged at 9500 g for 10 min at 4°C, 20 μL of the supernatant was added to 96‐well plates. Then, the 10 μL of peptide substrate for caspase‐3 (Ac‐DEVD‐pNA) or caspase‐9 (Ac‐LEHD‐pNA) and 70 μL of reaction buffer was added to start the reaction. After incubation at 37°C for 1 h, the production of yellow pNA was measured at 405 nm.
Western Blotting
After each treatment, SH‐SY5Y cells were collected and then suspended in lysis buffer. Protein concentration was determined using a BCA Protein Assay Kit (Beyotime, Haimen, China). Protein was separated by 8–12% SDS polyacrylamide gel and then transferred to a PVDF membrane (Millipore Corporation, Billerica, MA, USA). After blocking the membrane with 5% nonfat milk in TBST, target proteins were incubated overnight with primary antibodies including Bcl‐2 (1:600), Bax (1:600), caspase‐3 (1:400), caspase‐9 (1:800), cytochrome c (1:500), p‐ERK1/2 and ERK1/2 (1:1000), p‐JNK1/2 and JNK1/2 (1:1000), p‐p38 and p38 (1:1000), p‐AKT and AKT (1:800), p‐GSK‐3β and GSK‐3β (1:1000), p‐CREB and CREB (1:1000), GluN2A (1:800), GluN2B (1:1000), β‐actin (1:500). After three washed with TBST for 10 min, membranes were incubated with HRP‐conjugated secondary antibodies (1:5000) for 1 h at room temperature, followed by three TBST washes for 5 min. The blots were detected by an ECL detection kit and then analyzed by Image J program (NIMH, Bethesda, MD, USA).
Computer Molecular Docking
The crystal structures of NMDARs (PDB ID: 3QEM) 21 from RCSB Protein Data Bank (http://www.pdb.org/) were used in the docking studies. Preparation of the protein for docking was performed with the Biopolymer module of Sybyl X. Water and ligands were removed from the crystal structures of the receptor, and hydrogen atoms were then added. CHARMm force field was applied to the protein as well as MQA to minimize energy. The binding site was defined by the co‐complexed ligand (ifenprodil) in the crystal structure. Next, the docking process was performed using CDOCKER module of Discovery Studio 3.0 and the docking protocols were set up on the default setting. The conformation with the lowest CDOCKER interacting energy was selected as the most likely binding conformation. A 2D diagram was performed to show the interaction between MQA with the receptor using Analyze Complexes module.
Statistical Analysis
Data were expressed as means ± standard deviation (SD) of three independent experiments. Statistical significance was performed by analysis of variance (ANOVA) with the post hoc LSD test (SPSS 16.0, IBM, Armonk, NY, USA). P values <0.05 were used to denote as statistically significant.
Results
Effects of MQA on NMDA‐Induced Cytotoxicity in SH‐SY5Y Cells
To determine whether MQA display neuroprotective effects, we firstly evaluated the effects of MQA on SH‐SY5Y cells by MTT assay. Our results suggest that NMDA decreased cell viability in a dose‐dependent manner (Figure 1B). The cells that exposed to 1 mM NMDA for 1 h showed a significant decrease in cell viability (75.66 ± 5.61%, compared with the control group). Therefore, 1 mM NMDA was used in the subsequent experiments. The presence of MQA alone (at 10–100 μM) caused no significant changes in the cell viability compared with the control (Figure 1C). Pretreatment with MQA at 10, 20, 40 μM showed effective neuroprotection against NMDA‐induced cell injury. The cell viability was remarkably increased to 84.38 ± 3.7% (P < 0.05 vs. NMDA alone), 88.90 ± 4.7% (P < 0.01 vs. NMDA alone), and 91.46 ± 6.24% (P < 0.01 vs. NMDA alone), respectively (Figure 1D). NMDA antagonist MK801 also attenuated the toxicity induced by NMDA (Figure 1D).
To further investigate the protective effects of MQA against NMDA‐induced cytotoxicity, the LDH leakage was measured. Treatment with NMDA resulted in an increase of LDH leakage into the medium, 226.32 ± 5.75% of the control value. However, pretreatment with MQA at the different concentration (10, 20, 40 μM) markedly reduced LDH leakage to 175.58 ± 8.6%, 157.27 ± 7.67%, and 134.06 ± 7.25%, respectively (Figure 1E).
Effects of MQA on NMDA‐Induced Apoptosis in SH‐SY5Y Cells
Hoechst 33342 staining was applied to investigate the neuroprotective effects of MQA. As shown in Figure 2A, after NMDA treatment, nuclear fragmentation and DNA condensation were presented. However, pretreatment with MQA inhibited these typical apoptotic characteristics. To further justify antiapoptotic effects of MQA, Annexin V and PI double staining were performed on these cells. The untreated cells showed very little early apoptosis (5.54 ± 0.44%), whereas treatment of cells with 1 mM NMDA significantly increased the percentage of early apoptotic cells (25.63 ± 3.25%). Pretreatment with MQA at 20 and 40 μM significantly decreased the percentage of cells undergoing early apoptosis to 15.13 ± 2.03% and 9.57 ± 1.61%, respectively (Figure 2B,C).
Figure 2.
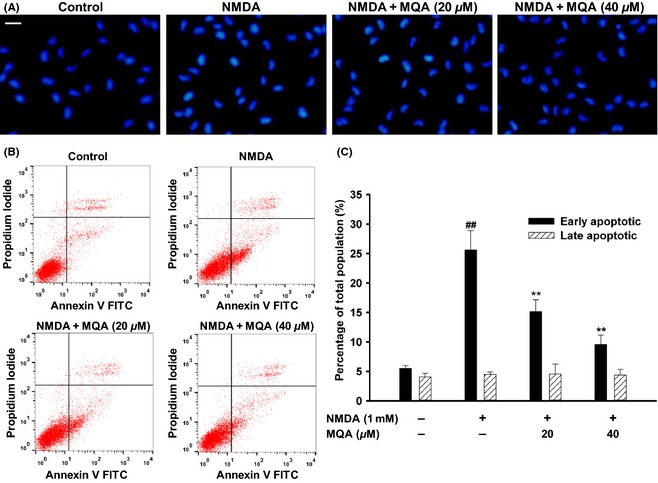
Effects of MQA on NMDA‐induced apoptosis in SH‐SY5Y cells. (A) Representative fluorescence images were obtained after Hoechst 33342 staining. Scale bar: 20 μm. (B) Effects of MQA on NMDA‐induced early and late apoptosis after AV‐PI double staining. (C) The quantitative analysis of early and late apoptotic cells. ## P < 0.01 versus the control group. **P < 0.01 versus the NMDA‐treated group.
Effects of MQA on NMDA‐Induced Ca2+ Overload
Considering that intracellular Ca2+ overload contributes to neuronal cells death following excessive activation of NMDARs, we focused on the effects of MQA on calcium influx. The fluorescence intensity represents cytoplasmic Ca2+ concentration. The Ca2+ concentration in control group was stable during our experiments (Figure 3A,B). One mM NMDA induced a fast elevation in Ca2+ concentration for the next 5 min (Figure 3A,C). However, the amplitude of Ca2+ concentration induced by NMDA exposure were significantly attenuated in the cells with pretreatment of MQA (Figure 3A,C).
Figure 3.
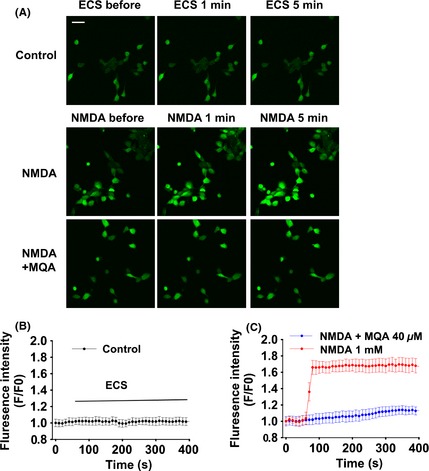
Effects of MQA on NMDA‐induced Ca2+ overload. (A) Fluorescence images were obtained under the laser scanning microscope. SH‐SY5Y cells were loaded with fluo‐3, followed by exposure to NMDA for 5 min. Scale bar: 20 μm. (B) Fluorescence intensity of the control group during detection time (n = 15). (C) Fluorescence intensity of cells in NMDA‐treated group (red), NMDA + MQA‐treated group (blue) (n = 15).
Effects of MQA on Apoptosis‐Related Proteins Expression
Western blot analysis showed that the ration of Bax/Bcl‐2 ratio was significantly increased to 744.93 ± 24.67% of the control group after exposure to NMDA. Meanwhile, cytochrome c release was markedly increased to 182.73 ± 14.17% of the control. However, these changes were remarkably reversed by treatment of MQA (Figure 4A,B).
Figure 4.
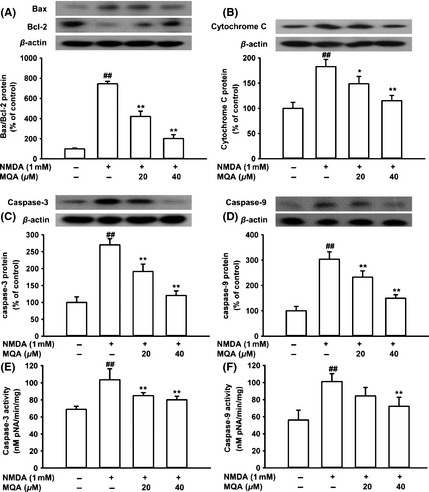
Effects of MQA on apoptosis‐related protein expressions, caspase‐3, and caspase‐9 activities in NMDA‐treated SH‐SY5Y cells. Western blot analysis for the expression of (A) Bax and Bcl‐2, (B) cytochrome c, (C) caspase‐3, (D) caspase‐9 proteins. (E) Caspase‐3 activity in SH‐SY5Y cell cultures. (F) Caspase‐9 activity in SH‐SY5Y cell cultures. Cells were pretreated with MQA (20 and 40 μM) for 2 h prior to 1 mM NMDA for 30 min. Data are shown as means ± SD (n = 3). ## P < 0.01 versus the control group. *P < 0.05 and **P < 0.01 versus the H2O2‐treated group.
Also, treatment with 1 mM NMDA markedly caused the upregulation of caspase‐3 to 270.12 ± 18.25% of the control value. In contrast, after cells were pretreated with 20 and 40 μM MQA, the upregulation of caspase‐3 was significantly decreased to 191.76 ± 21.6% and 120.89 ± 13.10%, respectively. Similar results were observed in the expression of caspase‐9 protein (Figure 4C,D). Further, after incubation of SH‐SY5Y cells with 1 mM NMDA, the activity of caspase‐3 was increased. On the other hand, MQA pretreatment significantly reduced the caspase‐3 activity (Figure 4E). The caspase‐9 activity showed the similar trend (Figure 4F). These data suggest that MQA inhibited NMDA‐induced cell death by downregulating activities and expressions of caspase‐3 and caspase‐9 protein.
Effects of MQA on the Expression of Phosphorylated ERK1/2, p38, JNK1/2, CREB, AKT, and GSK‐3β Proteins
ERK1/2, p38, and JNK1/2 are the main members of the MAPK family. We investigated the effects of MQA on the expression levels of phospho‐ERK1/2 (p‐ERK1/2), phospho‐p38 (p‐p38), and phospho‐JNK1/2 (p‐JNK1/2) in SH‐SY5Y cells in response to NMDA treatment. The time course results of p‐ERK1/2, p‐p38, and p‐JNK1/2 expressions are shown in Figure 5. The expression of p‐ERK1/2, p‐p38, and p‐JNK1/2 was increased, which peaked at 5, 10, 20 min, respectively. However, MQA pretreatment significantly decreased the phosphorylation of ERK1/2, p38, and JNK1/2.
Figure 5.
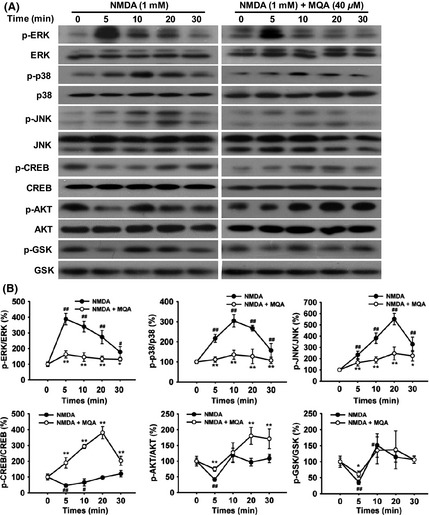
Effects of MQA on the expression of MAPKs, CREB, AKT, and GSK‐3β in NMDA‐treated SH‐SY5Y cells. (A) Time courses of phosphorylated ERK1/2, CREB, p38, JNK1/2, AKT, and GSK‐3β proteins. (B) Quantitative analysis of phosphorylated ERK1/2, CREB, p38, JNK1/2, AKT, and GSK‐3β levels. SH‐SY5Y cells were pretreated with 40 μM MQA for 2 h and then exposed to 1 mM NMDA for 5, 10, 20, and 30 min. # P < 0.05 and ## P < 0.01 versus the control group. *P < 0.05 and **P < 0.01 versus the NMDA‐treated group.
To further explore the underlying mechanisms of neuroprotection of MQA, phosphorylated CREB, AKT, and GSK‐3β were measured by Western blotting. As shown in Figure 5, NMDA led to significant reductions of p‐CREB, p‐AKT, and p‐GSK‐3β at 5 min. However, MQA restored phosphorylation of CREB, Akt, and GSK‐3β reduced by NMDA treatment.
Effects of MQA on the Expression of GluN2A‐ and GluN2B‐Containing NMDARs
It has been observed that GluN2A‐ and GluN2B‐containing NMDARs are involved in different intracellular cascades and play different roles in neuronal cells survival or death 22. Therefore, we examined effects of MQA on the expression of NMDAR subtypes. Exposure to NMDA induced decreases in the expression of GluN2A and increases in the expression of GluN2B. In contrast, MQA pretreatment attenuated these trends (Figure 6A,B). Thus, downregulation of GluN2B and upregulation of GluN2A subtype are suggested to be partly contributing to the neuroprotective effects of MQA against NMDA‐induced excitotoxicity.
Figure 6.
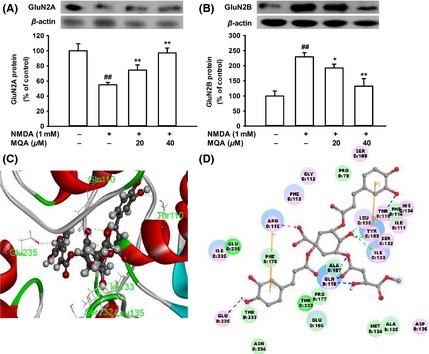
Effects of MQA on GluN2A and GluN2B expressions and molecular docking models of MQA with NMDARs. (A) Expression level of GluN2A protein. (B) Expression level of GluN2B protein. # P < 0.05 and ## P < 0.01 versus the control group. *P < 0.05 and **P < 0.01 versus the NMDA‐treated group. (C) Binding mode of MQA in the active site of NMDARs. The receptor was shown by ribbon, and MQA was shown by ball and stick style. (D) The two‐dimensional diagram of interactions between MQA and NMDARs. Hydrogen bond interactions (dashed line), pi–pi interactions (yellow line), and electrostatic interactions (magenta line) are presented.
Computational Molecular Docking Study
To further elucidate the molecular interaction mechanisms between MQA and NMDA receptor, we employed computational docking simulation. The most stable docking poses are shown in Figure 6C,D. In the MQA‐NMDARs complex, MQA served as hydrogen bond donor to form three hydrogen bonds with Gln110 of GluN2B. In addition, MQA formed hydrogen bonds with Glu235 of GluN2B, Thr110, Ser132, Ile133, and Leu135 of GluN1. This model indicates that MQA has the potential to interact with active sites of NMDARs.
Discussion
This study demonstrates that MQA attenuated the neuronal cell injury induced by NMDA, as suggested by the results from the MTT assay, flow cytometry analyses, and Western blot analysis. Our results indicate that the protective effects of MQA are associated with the suppression of apoptosis by inhibition of GluN2B‐containing NMDARs. The current study demonstrates that MQA exerted significantly neuroprotective effects against NMDA‐induced cytotoxicity and further influenced downstream of NMDARs.
In our experiment, the concentration of NMDA (1 mM) was consistent with Corasaniti's previous study 23. The high concentration of NMDA (1 mM) seems common in SH‐SY5Y cells. However, the EC50 for NMDA is usually in the range of micromolar in primary cultured neurons. The difference may result from different composition of NMDARs. Previous study suggested that NMDARs might be mainly constituted of triheteromeric GluN1/2A/2B receptors in SH‐SY5Y cells 17. The hippocampal NMDARs largely consist of diheteromeric GluN1/2B and triheteromeric GluN1/2A/2B subtypes. GluN2B subtype seems contribute more than GluN2A subtype to NMDA‐induced excitotoxicity 24. The lower ratio of GluN2B may partly explain the lower sensitivity to NMDA in SH‐SY5Y cells.
NMDAR‐mediated signals are critical for the neuronal development, survival, and synaptic plasticity 4. However, overactivation of NMDARs is a key step in glutamate‐induced excitotoxic injury in CNS, resulting in Ca2+ overload. Marked elevation in Ca2+ triggers several downstream reactions, including oxidative stress, mitochondrial dysfunction, and nitrosative stress, leading to the subsequent initiation of apoptosis 25. In this study, inhibition of Ca2+ influx induced by NMDA contributes to neuroprotection of MQA.
Mitochondria‐dependent apoptotic pathways are involved in NMDA‐induced cytotoxicity 26. Mitochondria take up substantial amounts of cytosolic Ca2+ as a very efficient Ca2+ buffer, leading to significant decreases in MMP 27. Disruption of MMP causes imbalances between Bcl‐2 and Bax. As a mitochondrial membrane‐associated protein, Bcl‐2 has been reported to inhibit Bax expression in mitochondria, and inhibit subsequent activation of the caspase‐3 28. The pro‐apoptotic protein Bax promotes cell apoptosis by translocating to the mitochondrial membrane and serves as one of the major causes of neurological disorders. Cell survival in the early phase of apoptosis depends mostly on the balance between the antiapoptotic protein Bcl‐2 and pro‐apoptotic protein Bax 29. Moreover, increased mitochondrial permeability promotes the release of cytochrome c from the mitochondria, resulting in activation of caspase‐9, and subsequent activation of caspase‐3. Caspase‐3 acts as one of the main apoptotic executors in the later phase of apoptosis, responsible for activating DNA fragmentation factor to cleave nuclear DNA, and eventually leads to cell death 30, 31. Furthermore, caspase‐3 and caspase‐9 activities were upregulated following exposure to NMDA. However, these effects were attenuated by MQA pretreatment. Therefore, our findings indicate that MQA rescued SH‐SY5Y cells from NMDA‐induced apoptosis possibly through inhibition of apoptosis‐related proteins activation.
Stimulation of NMDA receptors by different agonists triggers various signaling cascades that regulate a diverse of neuronal functions. The ERK MAP kinase is one of such target that plays a critical role in promoting neuronal cell death in neurodegenerative disorders. Phosphorylation of ERK1/2 was increased following stroke and treatment with inhibitors that blocked phosphorylated ERK1/2 led to a significant decrease in infarct volume 32, 33. Activated ERK1/2 was also observed in the damaged brain tissue from a mice traumatic brain model, while pretreatment with the ERK inhibitor U0126 resulted in a marked reduction in cortical lesion 34. In consistent with previous studies, phosphorylation of ERK1/2 was observed after exposure to NMDA. However, pretreatment with MQA attenuated this tendency in a dose‐dependent manner. To further explore neuroprotective effects of MQA, we focused on CREB, one downstream target of ERK1/2 signaling. CREB is an important component of the survival pathway, responsible for regulating transcription of potential neuroprotective genes, including Bcl‐2 and brain‐derived neurotrophic factor 35, 36, 37. Phosphorylation of CREB at serine 133 has been associated with cell survival and neuroprotection 36. Pretreatment of MQA increased phosphorylation of CREB as compared with NMDA treatment. These results indicate that ERK‐CREB signaling pathway is involved in the neuroprotection of MQA.
P38 and JNK1/2, another member of the MAPK family, are upregulated following NMDA stimulation 38, 39. Thus, we studied the effect of MQA on phosphorylation of p38 and JNK1/2. A three‐tiered kinase cascade is involved in regulating the activation of p38 and JNK1/2 40. The first member of this cascade is MAP kinase kinase kinase (MKKK family), which activates and phosphorylates MAP kinase kinase (MKK 3/6 for p38; MKK 4/7 for JNK1/2) that in turn activates and phosphorylates p38 and JNK1/2 MAPKs 40, 41. Activation of p38 and JNK1/2 lead to neuronal death and contribute to various neurological conditions such as neurodegenerative disorders and cerebral ischemia. Consisting with the previous reports 38, our study suggested that NMDA caused activation of p38 and JNK1/2. Furthermore, our data showed that MQA inhibited the phosphorylation of p38 and JNK1/2. These results indicate that the protective effect of MQA on NMDA‐induced cell injury was regulated via p38‐ and JNK1/2‐dependent pathway.
Exposure of SH‐SY5Y cells to NMDA led to a rapid deactivation of Akt kinase as monitored by Western blot. Although the mechanism of NMDA receptor deactivates Akt remains unclear, it is believed that calcium influx may be implicated. Activation of NMDARs increases the activity of protein phosphatases 1, which promotes de‐phosphorylation of Akt 42. Activated Akt promotes cell survival by phosphorylation of Ser473 and Thr308, whereas deactivated Akt participates in promoting cell death such as Bax, caspase‐9, and GSK‐3β 43, 44. In particular, Akt inactivates GSK‐3β by phosphorylation of Ser9 44. Moreover, a selective inhibitor of GSK‐3β has been reported to rescue SH‐SY5Y cells from NMDA‐induced cell injury 45. Consequently, the finding that MQA is able to attenuate deactivation of Akt kinase and GSK‐3β induced by NMDA suggests Akt/GSK‐3β signaling pathway involved in neuroprotective effects of MQA.
Western blot indicates that GluN2A‐ and GluN2B‐containing NMDARs participate in different roles in the neuroprotective effects of MQA. Activation of GluN2B‐containing NMDA receptors promotes neuronal damage, mediating NMDARs‐induced excitotoxicity 22, 24. In contrast, activation of either extra synaptic or synaptic GluN2A‐containing NMDARs promotes neuronal survival and exerts protective action against either NMDAR‐mediated or non‐NMDAR‐mediated neuronal injury 22. In the present study, the results showed that MQA reversed that upregulation of GluN2B and downregulation of GluN2A induced by NMDA, suggesting that the neuroprotection of MQA is likely to regulate a particular NMDAR subunit. However, we would further investigate subcellular localization of NMDAR subunits by immune‐cytological studies to sustain this hypothesis in subsequent experiments. Molecular docking was performed to further illustrate the possible combination mode of MQA‐NMDARs. Promisingly, the MQA moiety closely contacted with Gln110 and Glu235 of GluN2B to form four hydrogen bonds. In addition, there are at least other 25 active residues in the catalytic unit of NMDARs contributing to the stability of MQA‐NMDARs complex, such as Glu106, Arg115, and Phe176. These active residues will be confirmed in our future research.
Conclusion
In summary, to our knowledge, the present study is the first to show that MQA can provide protection against NMDA‐induced apoptotic cell death in SH‐SY5Y cells. The result of Ca2+ influx and molecular docking suggests that the protective effects of MQA may be mediated by blocking NMDARs. The potential mechanisms of its protective effects are associated with mitochondrial apoptosis, ERK‐CREB, AKT/GSK‐3β, P38, and JNK1/2 pathway. Therefore, MQA may be a potential neuroprotective compound for preventing and treating neurodegenerative disorders. Further works are required to elucidate explicit neuroprotective mechanisms in vitro and in vivo studies.
Conflict of Interest
The authors declare no conflict of interest.
Acknowledgments
This project was supported by Key National Science & Technology Specific Project of China (2014ZX09J14101‐05C).
References
- 1. Arundine M, Tymianski M. Molecular mechanisms of glutamate‐dependent neurodegeneration in ischemia and traumatic brain injury. Cell Mol Life Sci 2004;61:657–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Camins A, Pallas M, Silvestre JS. Apoptotic mechanisms involved in neurodegenerative diseases: experimental and therapeutic approaches. Methods Find Exp Clin Pharmacol 2008;30:43–65. [DOI] [PubMed] [Google Scholar]
- 3. Hovelso N, Sotty F, Montezinho LP, Pinheiro PS, Herrik KF, Mork A. Therapeutic potential of metabotropic glutamate receptor modulators. Curr Neuropharmacol 2012;10:12–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Paoletti P. Molecular basis of NMDA receptor functional diversity. Eur J Neurosci 2011;33:1351–1365. [DOI] [PubMed] [Google Scholar]
- 5. Paoletti P, Neyton J. NMDA receptor subunits: function and pharmacology. Curr Opin Pharmacol 2007;7:39–47. [DOI] [PubMed] [Google Scholar]
- 6. Loftis JM, Janowsky A. The N‐methyl‐d‐aspartate receptor subunit NR2B: localization, functional properties, regulation, and clinical implications. Pharmacol Ther 2003;97:55–85. [DOI] [PubMed] [Google Scholar]
- 7. Gogas KR. Glutamate‐based therapeutic approaches: NR2B receptor antagonists. Curr Opin Pharmacol 2006;6:68–74. [DOI] [PubMed] [Google Scholar]
- 8. Kim Y, Cho HY, Ahn YJ, Kim J, Yoon YW. Effect of NMDA NR2B antagonist on neuropathic pain in two spinal cord injury models. Pain 2012;153:1022–1029. [DOI] [PubMed] [Google Scholar]
- 9. Bai JP, Hu XL, Tian X, Zhao QC. Caffeic acids from roots of Arctium lappa and their neuroprotective activity. Chin Trad Herb Durg 2015;46:163–168. [Google Scholar]
- 10. Zhao JG, Yan QQ, Xue RY, Zhang J, Zhang YQ. Isolation and identification of colourless caffeoyl compounds in purple sweet potato by HPLC‐DAD‐ESI/MS and their antioxidant activities. Food Chem 2014;161:22–26. [DOI] [PubMed] [Google Scholar]
- 11. Fiamegos YC, Kastritis PL, Exarchou V, et al. Antimicrobial and efflux pump inhibitory activity of caffeoylquinic acids from Artemisia absinthium against gram‐positive pathogenic bacteria. PLoS One 2011;6:e18127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ooi KL, Muhammad TS, Tan ML, Sulaiman SF. Cytotoxic, apoptotic and anti‐alpha‐glucosidase activities of 3,4‐di‐O‐caffeoyl quinic acid, an antioxidant isolated from the polyphenolic‐rich extract of Elephantopus mollis Kunth. J Ethnopharmacol 2011;135:685–695. [DOI] [PubMed] [Google Scholar]
- 13. Miyamae Y, Kurisu M, Murakami K, et al. Protective effects of caffeoylquinic acids on the aggregation and neurotoxicity of the 42‐residue amyloid beta‐protein. Bioorg Med Chem 2012;20:5844–5849. [DOI] [PubMed] [Google Scholar]
- 14. Han J, Miyamae Y, Shigemori H, Isoda H. Neuroprotective effect of 3,5‐di‐O‐caffeoylquinic acid on SH‐SY5Y cells and senescence‐accelerated‐prone mice 8 through the up‐regulation of phosphoglycerate kinase‐1. Neuroscience 2010;169:1039–1045. [DOI] [PubMed] [Google Scholar]
- 15. Ndountse LT, Chan HM. Methylmercury increases N‐methyl‐d‐aspartate receptors on human SH‐SY 5Y neuroblastoma cells leading to neurotoxicity. Toxicology 2008;249:251–255. [DOI] [PubMed] [Google Scholar]
- 16. Petroni D, Tsai J, Mondal D, George W. Attenuation of low dose methylmercury and glutamate induced‐cytotoxicity and tau phosphorylation by an N‐methyl‐d‐aspartate antagonist in human neuroblastoma (SHSY5Y) cells. Environ Toxicol 2013;28:700–706. [DOI] [PubMed] [Google Scholar]
- 17. Zhou F, Xu Y, Hou XY. MLK3‐MKK3/6‐P38MAPK cascades following N‐methyl‐d‐aspartate receptor activation contributes to amyloid‐beta peptide‐induced apoptosis in SH‐SY5Y cells. J Neurosci Res 2014;92:808–817. [DOI] [PubMed] [Google Scholar]
- 18. Tan JW, Tham CL, Israf DA, Lee SH, Kim MK. Neuroprotective effects of biochanin A against glutamate‐induced cytotoxicity in PC12 cells via apoptosis inhibition. Neurochem Res 2013;38:512–518. [DOI] [PubMed] [Google Scholar]
- 19. Kwon SH, Hong SI, Kim JA, et al. The neuroprotective effects of Lonicera japonica THUNB against hydrogen peroxide‐induced apoptosis via phosphorylation of MAPKs and PI3K/Akt in SH‐SY5Y cells. Food Chem Toxicol 2011;49:1011–1019. [DOI] [PubMed] [Google Scholar]
- 20. Meng H, Li C, Feng L, et al. Effects of Ginkgolide B on 6‐OHDA‐induced apoptosis and calcium over load in cultured PC12. Int J Dev Neurosci 2007;25:509–514. [DOI] [PubMed] [Google Scholar]
- 21. Karakas E, Simorowski N, Furukawa H. Subunit arrangement and phenylethanolamine binding in GluN1/GluN2B NMDA receptors. Nature 2011;475:249–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Liu Y, Wong TP, Aarts M, et al. NMDA receptor subunits have differential roles in mediating excitotoxic neuronal death both in vitro and in vivo. J Neurosci 2007;27:2846–2857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Corasaniti MT, Maiuolo J, Maida S, et al. Cell signaling pathways in the mechanisms of neuroprotection afforded by bergamot essential oil against NMDA‐induced cell death in vitro. Br J Pharmacol 2007;151:518–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. von Engelhardt J, Coserea I, Pawlak V, et al. Excitotoxicity in vitro by NR2A‐ and NR2B‐containing NMDA receptors. Neuropharmacology 2007;53:10–17. [DOI] [PubMed] [Google Scholar]
- 25. Arundine M, Tymianski M. Molecular mechanisms of calcium‐dependent neurodegeneration in excitotoxicity. Cell Calcium 2003;34:325–337. [DOI] [PubMed] [Google Scholar]
- 26. Ghavami S, Shojaei S, Yeganeh B, et al. Autophagy and apoptosis dysfunction in neurodegenerative disorders. Prog Neurobiol 2014;112:24–49. [DOI] [PubMed] [Google Scholar]
- 27. McElnea EM, Quill B, Docherty NG, et al. Oxidative stress, mitochondrial dysfunction and calcium overload in human lamina cribrosa cells from glaucoma donors. Mol Vis 2011;17:1182–1191. [PMC free article] [PubMed] [Google Scholar]
- 28. Zuo W, Zhang W, Han N, Chen NH. Compound IMM‐H004, a novel coumarin derivative, protects against CA1 cell loss and spatial learning impairments resulting from transient global ischemia. CNS Neurosci Ther 2015;21:280–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kim S, Park SE, Sapkota K, Kim MK, Kim SJ. Leaf extract of Rhus verniciflua Stokes protects dopaminergic neuronal cells in a rotenone model of Parkinson's disease. J Pharm Pharmacol 2011;63:1358–1367. [DOI] [PubMed] [Google Scholar]
- 30. Le DA, Wu Y, Huang Z, et al. Caspase activation and neuroprotection in caspase‐3‐ deficient mice after in vivo cerebral ischemia and in vitro oxygen glucose deprivation. Proc Natl Acad Sci U S A 2002;99:15188–15193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Han BH, Xu D, Choi J, et al. Selective, reversible caspase‐3 inhibitor is neuroprotective and reveals distinct pathways of cell death after neonatal hypoxic‐ischemic brain injury. J Biol Chem 2002;277:30128–30136. [DOI] [PubMed] [Google Scholar]
- 32. Lu K, Liang CL, Liliang PC, et al. Inhibition of extracellular signal‐regulated kinases 1/2 provides neuroprotection in spinal cord ischemia/reperfusion injury in rats: relationship with the nuclear factor‐kappaB‐regulated anti‐apoptotic mechanisms. J Neurochem 2010;114:237–246. [DOI] [PubMed] [Google Scholar]
- 33. Wang ZQ, Wu DC, Huang FP, Yang GY. Inhibition of MEK/ERK 1/2 pathway reduces pro‐inflammatory cytokine interleukin‐1 expression in focal cerebral ischemia. Brain Res 2004;996:55–66. [DOI] [PubMed] [Google Scholar]
- 34. Clausen F, Lundqvist H, Ekmark S, Lewen A, Ebendal T, Hillered L. Oxygen free radical‐dependent activation of extracellular signal‐regulated kinase mediates apoptosis‐like cell death after traumatic brain injury. J Neurotrauma 2004;21:1168–1182. [DOI] [PubMed] [Google Scholar]
- 35. Meller R, Minami M, Cameron JA, et al. CREB‐mediated Bcl‐2 protein expression after ischemic preconditioning. J Cereb Blood Flow Metab 2005;25:234–246. [DOI] [PubMed] [Google Scholar]
- 36. Lee B, Butcher GQ, Hoyt KR, Impey S, Obrietan K. Activity‐dependent neuroprotection and cAMP response element‐binding protein (CREB): kinase coupling, stimulus intensity, and temporal regulation of CREB phosphorylation at serine 133. J Neurosci 2005;25:1137–1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Li XB, Yang ZX, Yang L, et al. Neuroprotective effects of flax lignan against NMDA‐induced neurotoxicity in vitro. CNS Neurosci Ther 2012;18:927–933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Fan J, Gladding CM, Wang L, et al. P38 MAPK is involved in enhanced NMDA receptor‐dependent excitotoxicity in YAC transgenic mouse model of Huntington disease. Neurobiol Dis 2012;45:999–1009. [DOI] [PubMed] [Google Scholar]
- 39. Waxman EA, Lynch DR. N‐methyl‐d‐aspartate receptor subtype mediated bidirectional control of p38 mitogen‐activated protein kinase. J Biol Chem 2005;280:29322–29333. [DOI] [PubMed] [Google Scholar]
- 40. Zarubin T, Han J. Activation and signaling of the p38 MAP kinase pathway. Cell Res 2005;15:11–18. [DOI] [PubMed] [Google Scholar]
- 41. Centeno C, Repici M, Chatton JY, et al. Role of the JNK pathway in NMDA‐mediated excitotoxicity of cortical neurons. Cell Death Differ 2007;14:240–253. [DOI] [PubMed] [Google Scholar]
- 42. Wang Y, Briz V, Chishti A, Bi X, Baudry M. Distinct roles for mu‐calpain and m‐calpain in synaptic NMDAR‐mediated neuroprotection and extrasynaptic NMDAR‐mediated neurodegeneration. J Neurosci 2013;33:18880–18892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Liang K, Ye Y, Wang Y, Zhang J, Li C. Formononetin mediates neuroprotection against cerebral ischemia/reperfusion in rats via downregulation of the Bax/Bcl‐2 ratio and upregulation PI3K/Akt signaling pathway. J Neurol Sci 2014;344:100–104. [DOI] [PubMed] [Google Scholar]
- 44. Sun B, Chen L, Wei X, Xiang Y, Liu X, Zhang X. The Akt/GSK‐3beta pathway mediates flurbiprofen‐induced neuroprotection against focal cerebral ischemia/reperfusion injury in rats. Biochem Biophys Res Commun 2011;409:808–813. [DOI] [PubMed] [Google Scholar]
- 45. Meijer L, Skaltsounis AL, Magiatis P, et al. GSK‐3‐selective inhibitors derived from Tyrian purple indirubins. Chem Biol 2003;10:1255–1266. [DOI] [PubMed] [Google Scholar]