

Summary
Aim
Poststroke depression (PSD) is one of the most common neuropsychiatric complications after stroke. TREK‐1, a two‐pore‐domain potassium channel, has been implicated in the pathogenesis of stroke and depression. The aim of this study was to investigate whether TREK‐1 plays a role in the therapeutic effects of the selective serotonin reuptake inhibitor (SSRI) escitalopram in a rat PSD model.
Methods
The whole‐cell patch‐clamp technique was performed to assess the effect of escitalopram on recombinant TREK‐1 currents in HEK293 cells. The expression of TREK‐1 mRNA and protein was measured in the hippocampus and prefrontal cortex (PFC), and neural stem cell (NSC) proliferation was detected in the hippocampal dentate gyrus (DG) in PSD rats after 3 weeks of escitalopram administration.
Results
Escitalopram reversibly inhibited TREK‐1 currents in a concentration‐dependent manner. Chronic treatment with escitalopram significantly reversed the reductions in weight gain, locomotor activity, and sucrose preference in PSD rats. The expressions of TREK‐1 mRNA and protein were significantly increased in hippocampal CA1, CA3, DG, and PFC in PSD rats, with the exception of TREK‐1 mRNA in hippocampal CA1. NSC proliferation was significantly decreased in hippocampal DG of PSD rats. Escitalopram significantly reversed the regional increases of TREK‐1 expression and the reduction of hippocampal NSC proliferation in PSD rats.
Conclusion
TREK‐1 plays an important role in the therapeutic effects of the SSRI escitalopram in PSD model, making TREK‐1 an attractive candidate molecule for further understanding the pathophysiology and treatment of PSD.
Keywords: Escitalopram; Poststroke depression; Therapeutic effects; TREK‐1 (TWIK‐related potassium channel, type 1)
Introduction
Stroke is a major cause of death and long‐existing disability in adult populations around the world 1, 2. Poststroke depression (PSD), with a prevalence of 30–50%, is one of the most common neuropsychiatric complications after stroke 3. PSD causes increased disability, more impaired cognitive function, reduced quality of life, and higher risk of suicide 4 and death 5. The majority of PSD studies have focused on the epidemiological aspects of this condition. Our current understanding of the neurobiological mechanisms of PSD is primarily based on older studies of ischemic lesions and depression, including the lesions of fronto‐subcortical circuit, the hyperactivation of hypothalamic–pitutary–adrenal (HPA) axis and neuroinflammation, and serotonergic dysfunction 6. Meta‐analyses have demonstrated the therapeutic effects of selective serotonin reuptake inhibitors (SSRIs) on PSD 7, which strengthen poststroke motor 8 and cognitive recovery 9 and reduce overall mortality 10. The mechanisms underlying the efficacy of SSRI antidepressants in PSD have not been clearly elucidated.
Recently, TREK‐1 (TWIK‐related potassium channel, type 1) has been implicated in the pathophysiological mechanisms of stroke and depression, resulting in it being an attractive candidate molecule for studying PSD. TREK‐1 is one of the six types of K2p (two‐pore domain background potassium) channels 11, and it is abundantly expressed in several brain regions involved in the emotional and cognitive aspects of depression, including the hippocampus, the prefrontal cortex (PFC), the amygdala, and the gyrus cinguli 11, 12, 13. The STAR*D study found an association between four SNP variants in the TREK‐1 gene and resistance to multiple antidepressants 14. Moreover, TREK‐1 knockout mice display a depression‐resistant phenotype that mimics treatment with antidepressants 15. The SSRI antidepressant fluoxetine and its metabolite norfluoxetine robustly inhibit TREK‐1 channels 16. These findings suggest that TREK‐1 may play an important role in the etiology and treatment of depression.
Increasing evidence has demonstrated the possible role of TREK‐1 in ischemic neuroprotection 17, 18, 19. TREK‐1 knockout mice are more sensitive to ischemia 20 and exhibit decreased vasodilation of the basilar artery in response to polyunsaturated fatty acids 21. The hippocampus and prefrontal cortex receive the serotoninergic neural projection from raphe nuclei and also have neural connections between each other, which are importantly implicated in the mood regulation and pathogenesis of depression. TREK‐1 expression had been found to be increased in the rat cortex and hippocampus 2 h after middle cerebral artery occlusion (MCAO) 18. The effect of increased TREK‐1 expression lasted for up to 30 days in the rat hippocampus following permanent bilateral carotid artery ligation 22. It is unclear whether TREK‐1 expression is altered in the brain regions implicated in PSD, a disease that has characteristics of stroke and depression. The suppression of TREK‐1 activity increases neuronal apoptosis 19 and inhibits the proliferation of astrocytes 17 in the cortex of rats exposed to hypoxia, suggesting that TREK‐1 might be involved in neuronal cell renewal and survival. Opposite trends have been consistently reported regarding hippocampal neurogenesis following cerebral ischemia and depression. Neurogenesis is elevated following brain ischemia 23 and decreased in depression 24. Our group recently reported that reduced hippocampal neurogenesis could be reversed by citalopram administration in a rat model of PSD 25. The question of whether TREK‐1 activity is related to changes in hippocampal neurogenesis following cerebral ischemia and PSD has not been studied. In this study, we conducted preliminary explorations into the role of TREK‐1 in the pathophysiological mechanisms of PSD and SSRI therapeutic efficacy. We assessed the effect of escitalopram (ESC), a widely prescribed SSRI antidepressant, on the recombinant TREK‐1 channel currents in human embryonic kidney cells (HEK293) by the whole‐cell patch‐clamp technique. Additionally, we investigated the effects of ESC on depressive behavior, hippocampal neurogenesis, and TREK‐1 expression in the hippocampus and prefrontal cortex (PFC) of PSD rats, a model that we previously established using MCAO followed by a 21‐day chronic unpredictable mild stress (CMS) paradigm combined with isolation rearing 26.
Methods
Electrophysiology
The electrophysiology experiments utilized HEK293 cells cultured at a density of 5 × 104 cells/35‐mm plate 48 h after transfection with TREK‐1‐pEGFP‐N1 plasmids. The HEK293 cells were cultured in Dulbecco's modified Eagle medium (DMEM) supplemented with 10% fetal calf serum (FBS) and antibiotics (ampicillin and streptomycin, 50 μg/mL each) at 37°C in a 5% CO2 incubator. TREK‐1‐pEGFP‐N1 plasmids were transfected at a concentration of 565 ng/μL according to the Polyjet Reagent protocol. The HEK293 cells were visualized by fluorescence microscopy 48 h after transfection.
The whole‐cell patch‐clamp data were recorded using a 200B patch‐clamp amplifier (Axon Instrument, Union City, CA, USA) and a 12‐bit data acquisition system (Axon CNS Molecular Devices, Singapore city, Singapore). The TREK‐1 currents were low‐pass‐filtered at 3 kHz and digitized by a 12‐bit converter at 10 kHz (Axon CNS Molecular Devices). The patch‐clamp pipettes were produced from borosilicate glass capillaries with a resistance of 3.0–4.0 MΩ. The patch‐clamp electrode solution contained 1 mM CaCl2, 2 mM MgCl2, 140 mM KCl, 10 mM HEPES, and 10 mM EGTA, and the pH was adjusted to 7.2 with KOH. The external medium contained 150 mM NaCl, 5 mM KCl, 1.2 mM MgCl2, 0.5 mM CaCl2, and 10 mM HEPES, and the pH was adjusted to 7.4 with NaOH. The cells were clamped at −80 mV, and the incremental voltage was set from −100 to 80 mV, with a rank of 20 mV and duration of 1 second. The seal resistance of the pipette was 1 GΩ in HEK293 cells. The TREK‐1 potassium current was recorded at room temperature (20–24°C) at escitalopram concentrations of 10, 30, 50, 100, 500 μM, and 1 mM (n = 6 at each concentration). Patch‐clamp software (pCLAMP 10.2) was used to analyze the TREK‐1 currents recorded by whole‐cell patch clamping, and the data were expressed as the mean ± SD.
Experimental Design and Drug Administration in Animal Experiments
Fifty‐two male Sprague‐Dawley rats (Shanghai Laboratory Animal Center, Shanghai, China) weighing 210–250 g each were used. The rats were housed four per cage with free access to food and water and were maintained with a 12‐h light/dark cycle (lights on at 7 am), temperature of 21 ± 2°C, and humidity of 55%. The experiments were compliant with the animal experiment guidelines developed by the National Institutes of Health (Publication No. 85–23, revised 1985) and approved by the Animal Study Committee of Southeast University, China (NO 2011ZDLL015.2).
After being screened with a sucrose preference test and an open field test (OFT), the animals were randomly divided into the sham surgery (SHAM) and MCAO groups. A left MCAO was performed in the ISCH rats, and neurological deficits were evaluated on a 5‐point scale 24 h after surgery, as previously described 27, 28. MCAO rats with a score ≥1 and <4 were further imaged with 7.0 T Magnetic Resonance Imaging (MRI) using a T2 protocol to confirm the area of cerebral infarction (Figure 1). The rate of infarction area of basal ganglia layer by T2, respectively, was analyzed to ensure the comparability of cerebral ischemia among the experimental groups (Supplementary Material). The overall mortality rate during the entire experiment was 35% in the rats undergoing MCAO and MRI scanning.
Figure 1.
Confirmation of permanent middle cerebral artery occlusion (MCAO). The rats in each group were scanned using 7.0‐T micro‐MRI according to the T2 protocol 24 h after surgery. MRI, magnetic resonance imaging; T2, transverse relaxation time.
The survivors of MCAO were further divided into the following four subgroups: ischemic stroke (ISCH), escitalopram‐treated ISCH (ISCH+escitalopram), PSD (ISCH+CMS), and escitalopram‐treated PSD (PSD+escitalopram). Cerebral infarctions in the ISCH rats were confirmed by visual inspection after the animals were sacrificed on day 21. The rats in the SHAM group underwent a similar MCAO procedure except that a nylon thread was not inserted. The CMS procedure was performed on the PSD and PSD+escitalopram groups for 21 consecutive days beginning 48 h after the MCAO. The CMS, non‐CMS, and control animals were kept in different rooms. The drug‐treated rats received daily intraperitoneal injection of escitalopram (Lundbeck, Copenhagen, Denmark) at a dose of 10 mg/kg and a volume of 1.0 mL/kg for 21 days. This dose was selected according to previous animal studies of depression 29, 30, 31. To label the proliferating cells, 5′‐bromodeoxyuridine (BrdU, 50 mg/kg, Sigma, Saint Louis, MO, USA) was delivered i.p. twice per day at 8‐h intervals for 3 days (days 19, 20, and 21 after ischemia). Sucrose preference tests and OFTs were performed at baseline and once per week over 3 consecutive weeks. The rats were sacrificed for sample collection 12 h after final drug administration.
CMS Regimen
The 21‐day CMS regimen included eight stressors: water deprivation for 18 h (followed by a 1‐h exposure to empty bottles), food and water deprivation for 20 h (followed by a sucrose preference test), 45° cage tilt for 17 h, overnight illumination (lights on for 36 h), swimming in 4°C water for 5 min, wet cage for 21 h (200 mL of water in 100 g of sawdust bedding), tail pinch for 1 min, and cage shaking for 2 h.
Body Weight
Body weight was recorded at 10:00 am 3 days before the end of the adaptive period (baseline) and then once per week for 3 consecutive weeks. The weight gain was calculated according to the following formula: weight gain (%) = [(body weight at measurement points (g)‐baseline body weight (g))/baseline body weight (g)] × 100%.
Open Field Test
To assess the general locomotor activity of the animals, OFT was performed using a plastic cube (75 cm on each side) with a black floor and white walls in a quiet and dusky room. Each rat was placed at the center of the floor, which was divided into 25 equal squares, including nine central squares and 16 peripheral squares. The rats were then allowed to move freely for 5 min, and their activity was subsequently recorded. Software (RD1112‐IOF‐R‐1 v2.2.0; Mobile Datum, Shanghai, China) was used to track the total distance that the rats moved in the box.
Sucrose Preference Test
A sucrose preference test was used to operationally measure anhedonia in the animals. Prior to the baseline test, the animals were freely exposed to a training procedure of water and a 1% sucrose solution for 24 h. In the formal test, the rats were exposed to water and to a 1% sucrose solution for 1 h after a 24‐h food and water deprivation period. The two bottles were randomly placed on the right/left side of the cage and switched after 30 min. Sucrose preference (SP %) was calculated using the following formula: [sucrose intake (g)/(sucrose intake (g)+water intake (g))] × 100%.
Tissue Processing
The animals were sacrificed by deep anesthesia using 10% chloral hydrate (30 mL/kg, i.p.) 12 h after the final escitalopram treatment. For immunostaining experiments, the rats were successively perfused with 0.1 M phosphate‐buffered saline (PBS, 300 mL) and 4% paraformaldehyde (PFA, 300 mL). The brains were removed and fixed overnight in 4% PFA followed by dehydration in 30% sucrose at 4°C overnight. A freezing cryostat was used to cut coronal frozen sections (30 μm) from the hippocampus (bregma −3.14 to −4.16 mm) and the PFC (bregma 3.70 to 2.70 mm), as determined by the rat brain atlas of Paxinos and Watson and then stored at −20°C in cryoprotectant. For real‐time PCR and Western blotting analyses, the rats were decapitated after deep anesthesia. The brains were quickly removed and stored in isopentane at −50°C. The brains were cut into 1‐mm‐thick coronal sections at −20°C using a freezing cryostat. Then, bilateral tissues containing the PFC and dentate gyrus (DG), the cornu ammonis 1 (CA1), and the cornu ammonis 3 (CA3) of the hippocampus were separated using a 16‐gauge needle. The tissues were immediately frozen in liquid nitrogen and stored at −80°C.
Immunofluorescence
BrdU and TREK‐1 were co‐labeled in the hippocampal sections. To detect the BrdU‐labeled nuclei, the DNA was denatured as follows: incubation for 2 h in 50% formamide and 2× SSC (0.3 M NaCl, 0.03 M sodium citrate) at 65°C, wash with 2× SSC for 5 min, wash with 2 N HCl for 30 min at 37°C, and incubation in 0.1 M boric acid, pH 8.5, for 10 min. The sections were blocked for 2 h in 3% normal goat serum/1% BSA/0.3% Triton X‐100 in PBS at room temperature. The sections were then incubated with a mouse monoclonal anti‐BrdU antibody (1:300; IVGN, B35128) and a TREK‐1 (C‐20) goat anti‐rat antibody (1:100, sc‐11557; Santa Cruz Biotechnology, Inc., Dallas, Texas, USA) for 24 h at 4°C. Subsequently, the sections were washed and incubated for 2 h with Alexa Fluor 488 donkey anti‐goat IgG (1:500; IVGN, A21206) and Alexa Fluor 546 donkey anti‐mouse IgG (1:500; IVGN, A10036) at room temperature. To label TREK‐1 in the PFC, the sections were incubated with antibodies as described above but were not subjected to the DNA denaturation steps. Images of the DG, CA1, CA3, and PFC were captured using an OLYMPUS FV1000 laser‐scanning microscope (Melville, NY, USA). The number of BrdU and TREK‐1 double‐labeled cells in three sections (300 μm apart) of each DG was analyzed by a designated blinded person using the manual counting option of the Image proplus software (version 4.1; Diagnostic, Inc., New York, NY, USA).
Real‐time PCR
Total RNA was isolated from micropunches of the DG, CA1, and CA3 tissues using the TRIzol reagent (TaKaRa, Dalian, Liaoning, China). RNA (1 μg) was converted to first‐strand cDNA using reverse transcriptase (TaKaRa), and the mRNA expression was determined using an ABI 7300 PCR instrument (Applied Biosystems, Oceanside, CA, USA). The 20‐μL total‐volume reactions contained the following: 10 μL SYBR Premix Ex Taq™ (TaKaRa), 1 μL cDNA, 0.5 μL each forward and reverse primer (0.25 μM each), and 8 μL ddH2O. The synthesized primer pairs were as follows: TREK‐1 sense, 5′‐TCA TCA CTC TGA CCA CCA TTG‐3′; TREK‐1 antisense, 5′‐ACC TCT TCC TTC GTC TTC TTA G‐3′; GAPDH sense, 5′‐ACA GCA ACA GGG TGG TGG AC‐3′; and GAPDH antisense, 5′‐TTT GAG GGT GCA GCG A AC TT‐3′. The PCR included an initial denaturation at 95°C for 5 min, followed by 40 cycles of denaturation at 95°C for 5 second and annealing/extension at 60°C for 1 min. The expression of the TREK‐1 mRNA was normalized to that of GAPDH based on the standard curve method.
Western Blotting Analysis
The DG, CA1, and CA3 micropunch tissues were homogenized in ice‐cold lysis buffer, and the protein concentration was detected by the BCA method. In total, 50 μg of proteins was separated by SDS‐PAGE using 10% polyacrylamide gels and then transferred to PVDF membrane (Millipore, Billerica, MA, USA). The membranes were blocked using 5% nonfat milk for 2 h at room temperature and then incubated overnight at 4°C with a TREK‐1 (C‐20) goat anti‐rat antibody (polyclonal, unconjugated; 1:200; Santa Cruz). The blots were incubated with donkey anti‐goat IgG‐HRP secondary antibody (1:5000; Santa Cruz) at room temperature for 2 h, and the protein bands were detected using the enhanced chemiluminescence (ECL) method. The density of each TREK‐1 band was normalized against a β‐actin control.
Statistical Analyses
All of the data were expressed as the mean ± SEM, except that the electrophysiology data were expressed as the means ± SD. The behavioral index and body weight data were all examined using repeated‐measures analysis of variance (ANOVA), with the different drug groupings and time as the independent variables. Subsequent group comparisons of the behavioral index, electrophysiology, protein, and mRNA data were performed using one‐way ANOVA and post hoc Bonferroni analyses. Statistical significance was set at P < 0.05.
Results
The Effect of Escitalopram on TREK‐1 Channel Currents
The whole‐cell patch‐clamp technique showed that the TREK‐1 background current was an outward rectification that reversed the K+ current in HEK293 cells transfected with the TREK‐1‐pEGFP‐N1 plasmids (Figure 2A). Bath application of escitalopram inhibited the TREK‐1 currents in a concentration‐dependent manner, and this could be clearly reversed following washing. The TREK‐1 current at 0 mV was decreased on average by 12.42 ± 0.47%, 25.21 ± 0.23%, 37.98 ± 0.22%, 54.02 ± 0.87%, 61.72 ± 0.93%, and 62.12 ± 0.92% with 10, 30, 50, 100, and 500 μM of escitalopram, respectively (Figure 2A). Escitalopram significantly inhibited the TREK‐1 currents at 0 mV at 50, 100, and 500 μM compared with vehicle treatment (n = 6 each, all P < 0.05) (Figure 2A,B). The concentration response curve indicated an IC50 value of 81.94 μM for escitalopram at 0 mV, with a Hill coefficient of 2.49 ± 0.31 (Figure 2C). Escitalopram (50 μM) voltage‐dependently inhibited the TREK‐1 current compared with a control ramp trace (Figure 2D). However, no significant change in the percent inhibition of the TREK‐1 current was found in the voltage range from −40 to +40 mV, indicating that blockage by escitalopram is not voltage dependent.
Figure 2.
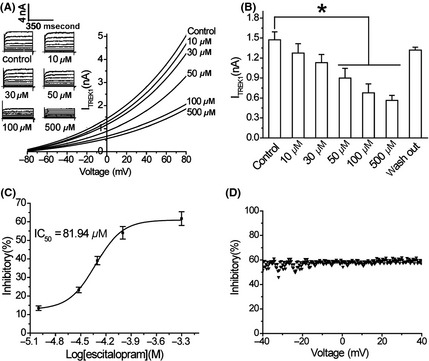
The inhibition of TREK‐1 channel currents by escitalopram. (A) The record of typical TREK‐1 background potassium currents by the whole‐cell patch‐clamp technique in HEK293 cells transfected with the TREK‐1‐pEGFP‐N1 plasmid. (B) The significant inhibition of the TREK‐1 current by escitalopram (at 0 mV with 10, 30, 50, 100, and 500 μM) as compared with the vehicle (n = 6 each, all *P < 0.05). (C) The concentration response curve for the escitalopram‐mediated inhibition of the TREK‐1 current. The IC50 value at 0 mV is 81.94 μM. (D) There is no significant change in the percentage inhibition of the TREK‐1 current within the voltage range from −40 to +40 mV. All of the data are represented as the mean ± SD.
The Effect of PSD and Escitalopram on Body Weight
Repeated‐measures ANOVA demonstrated a significant effect [F(5,227) = 74.70, P < 0.001] of ISCH, CMS, and escitalopram on body weight at the 3 weekly time points (Figure 3A). Bonferroni post hoc tests showed a significant reduction in body weight in the PSD group at day 7 (P < 0.05), day 14 (P < 0.05), and day 21 (P < 0.001) compared with the SHAM group. The body weight in the PSD rats was significantly decreased relative to the ISCH group on day 14 (P < 0.05) and day 21 (P < 0.001). Three weeks of escitalopram treatment significantly reversed the reduction of body weight of the PSD rats (P < 0.05).
Figure 3.
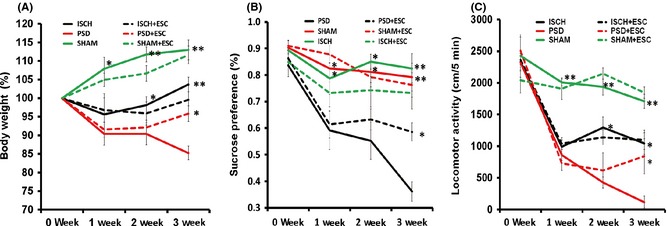
The effect of escitalopram administration on body weight (A), sucrose preference (B), and locomotor activity (C). *P < 0.05 and **P < 0.001 versus PSD group. Eight animals were included in the SHAM and ISCH groups, and 7 animals were included in the ISCH+ESC, PSD, and PSD+ESC groups. All of the data are shown as the mean ± SE. PSD, poststroke depression; SHAM, sham surgery; ISCH, ischemia; ESC, escitalopram.
The Effect of PSD and Escitalopram on Sucrose Preference
Repeated‐measures ANOVA demonstrated significant effects [F(5,227) = 97.52, P < 0.001] of ISCH, CMS, and escitalopram on the sucrose preference at the 3 weekly time points (Figure 3B). Bonferroni post hoc tests showed significant reductions in the sucrose preference at day 7, day 14, and day 21 in the PSD group relative to the SHAM group and the ISCH group (P < 0.05 and P < 0.001, respectively). The reduction of sucrose preference in the PSD group was significantly reversed following 3 weeks treatment by escitalopram (P < 0.05).
The Effect of PSD and Escitalopram on Locomotor Activity
Repeated‐measures ANOVA demonstrated robust effects [F(5,227) = 103.96, P < 0.001] of ISCH, CMS, and escitalopram on locomotor activity at the 3 weekly time points (Figure 3C). Post hoc tests showed a significant reduction of locomotor activity in the PSD group at day 7, day 14, and day 21 relative to the SHAM group (all P < 0.001). Locomotor activity was significantly decreased in the PSD group compared to the ISCH group at day 14 and day 21 (both P < 0.05). The reduction of locomotor activity was significantly reversed by escitalopram in the PSD group after 3‐week administration (P < 0.05).
The Effect of Escitalopram on TREK‐1 Expression in the PFC and Hippocampus of PSD Rats
Immunofluorescence staining revealed a broad distribution of TREK‐1‐positive cells in hippocampal DG, CA1, and CA3 and in the PFC (Figure 4A–D). No significant differences in TREK‐1 mRNA or protein expression were observed in the PFC, CA3, or DG regions between the ISCH and SHAM groups. The expression of TREK‐1 mRNA and protein was significantly higher in the PFC, CA3, and DG regions of the PSD group than in those of the ISCH and SHAM groups (all P < 0.05). The TREK‐1 protein level was also increased significantly in the CA1 of the ISCH (P < 0.05) and PSD (P < 0.05) groups relative to the SHAM group (Figure 4J). Escitalopram significantly reversed the increases of the expression of TREK‐1 mRNA and protein in these regions of the PSD rats after 3 weeks of administration (all P < 0.05) (Figure 4E–L, except 4F).
Figure 4.
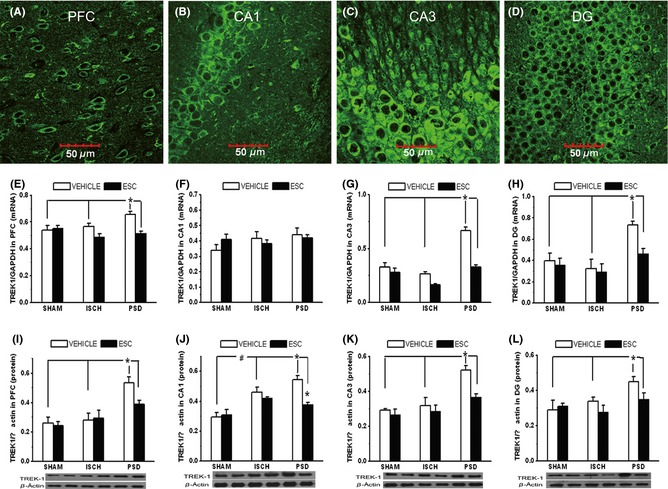
The distribution of TREK‐1 in the rat hippocampal CA1, CA3, DG, and PFC regions (A–D). The images shown as 60× magnification (E–H). The expression of TREK‐1 mRNA in the four brain regions (n = 8 each) (I–L). The expression of TREK‐1 protein in the four brain regions (n = 7 each). *P < 0.05 versus PSD group; #P < 0.05 versus ISCH group. PFC, prefrontal cortex; CA1, cornu ammonis 1; CA3, cornu ammonis 3; DG, dentate gyrus.
The Effect of Escitalopram on Cell Proliferation in the DG
Immunofluorescence staining showed that the TREK‐1 protein co‐localized with the BrdU staining in the cells of the DG (Figure 5A). The BrdU‐labeled cells frequently formed clusters and were nearly exclusively located near the subgranular zone (SGZ) (Figure 5B). The number of BrdU‐positive cells in the SGZ was significantly higher in the PSD and ISCH groups than in the SHAM group (Figure 5C). The number of BrdU‐positive cells in the SGZ was significantly lower in the PSD rats than in the ISCH rats. Escitalopram significantly increased the BrdU‐positive cells in the SGZ of the PSD rats (P < 0.05), but not in the ISCH or SHAM rats (Figure 5C), after 3 weeks of administration.
Figure 5.
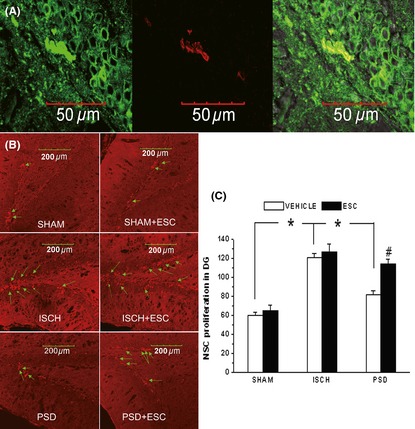
The double staining of TREK‐1 and newly proliferated cells in the subgranular zone (SGZ) of the hippocampal dentate gyrus (mean ± SEM). (A) TREK‐1 (green) co‐localized with BrdU‐labeled granule cells (red) in the SGZ of the dentate gyrus (60×). (B) A representative image of the newly proliferated cells in the SGZ of the dentate gyrus. (C) Group comparisons of the numbers of BrdU‐positive cells in the hippocampal dentate gyrus. *P < 0.05 versus ISCH group; #P < 0.05 versus PSD group. Eight animals were included in the SHAM and ISCH groups, and 7 animals were included in the ISCH+escitalopram, PSD, and PSD+escitalopram groups. PSD, poststroke depression; SHAM, sham surgery; ISCH, ischemia; ESC, escitalopram.
Discussion
To our knowledge, this is the first study to provide the evidence of the reversible inhibition of escitalopram on TREK‐1 currents in a concentration‐dependent manner, and the TREK‐1 elevation in the hippocampus and PFC in a rat model of PSD. The elevation of TREK‐1 levels was also associated with decreased cellular proliferation in the DG. The results further showed that chronic escitalopram administration could attenuate the upregulation of TREK‐1 expression and increase proliferation in the DG as well as improve weight gain, locomotor activity, and sucrose preference in PSD rats. Although the exact mechanisms by which TREK‐1 influences PSD pathogenesis and escitalopram regulates TREK‐1 expression remain unclear, these studies imply that TREK‐1 may be an important molecular mediator involved in the pathophysiology of PSD and the influence of SSRIs on PSD.
This study had performed a PSD‐like animal model that uses MCAO, CMS, and isolation rearing to mimic the complex clinical conditions of PSD patients by replicating stroke, stress, and psychosocial conditions 26. The results show a broad spectrum of behavioral alterations, including reduced sucrose preference, reduced motor activity, and reduced body weight, in PSD rats 1 week after MCAO surgery. These changes are similar to the core depression‐like symptoms. The chronic treatment by escitalopram for 3 weeks significantly reversed these behavioral changes in the PSD rats. These findings demonstrate that escitalopram has a significant therapeutic effect on depressive symptoms in PSD animals, which is consistent with the clinical efficacy of SSRI antidepressants, including escitalopram and tricyclic antidepressants 32, 33, 34, in PSD patients. Combined with our previous reports 35, this study provides evidence showing that this animal model is suitable for mimicking PSD in the clinical setting.
The presented study revealed dose‐dependent inhibition by escitalopram on the current mediated by recombinant TREK‐1 channels in HEK293 cells. The blockage by escitalopram was reversible by washing and was voltage independent, which is consistent with a previous report of the inhibition of the SSRI antidepressant fluoxetine on TREK‐1 currents 16. The mechanisms by which antidepressants inhibit TREK‐1 channels are poorly understood. It was previously suggested that fluoxetine does not bind to the pore region or the cytosolic C‐terminal domain of TREK‐1 channels 16. Recently, TREK‐1 has been importantly implicated in mood regulation, in which suppression of TREK‐1 channels may have an antidepressant efficacy. Both TREK‐1 knockout mice 15 and wild‐type mice treated with spadin, a peptide that blocks TREK‐1 channels 36, displayed depression‐resistant responses in five behavioral models. These responses were similar to the responses observed in antidepressant‐treated, wild‐type mice. It has been assumed that opening of the TREK‐1 channels leads to hyperpolarization of the cell, reduction of the firing rate, and a decrease in the release of serotonin from the dorsal raphe neurons (DRN) 37, 38. Consistently, TREK‐1 knockout mice and spadin treatment were found to increase the firing rate of the DRN 5‐HT neuron in vivo 36. The inhibition of TREK‐1 by the SSRI escitalopram that was shown in this study might increase DRN excitability and 5‐HT release, which could further contribute to its therapeutic efficacy for poststroke depression.
The results demonstrated a robust increase in the TREK‐1 protein in hippocampal CA1 after 21 days of left unilateral ISCH, which is partially consistent with prior findings indicating increased TREK‐1 expression in the cortex and hippocampal CA1 of MACO rats 3, 7, and 30 days after reperfusion 17. A previous in situ hybridization study reported that there were more TREK‐1‐positive neurons in the cortex and hippocampus at 3 and 30 days after bilateral carotid artery ligation 22. This increased TREK‐1 expression was expected to hyperpolarize the membrane potential, decrease neuronal excitability, and rescue homeostatic dysfunctions under ischemic conditions 17, 22, 39. The present study showed, for the first time, that TREK‐1 expression is significantly increased in hippocampal CA1 and CA3 and in the PFC of PSD rats, and this could be reversed by chronic escitalopram administration. Chronic, but not acute, SSRI administration has been found to inhibit the firing rate of pyramidal neurons in rat hippocampal CA1 and PFC regions, and this is paralleled by the amelioration of depressive symptoms 15, 40. However, the reduction in TREK‐1 expression following escitalopram treatment that was found in this study might seemingly increase neuronal excitability, which is opposite to the effects of chronic escitalopram on the regional firing rates in depressed animals. Interestingly, TREK‐1 channels could also preferentially pass an outward current at depolarized potentials to facilitate recovery and repetitive firing when C‐terminal serine S333 is phosphorylated by protein kinase A 41. Therefore, TREK‐1 theoretically possesses two distinct functional modes to fine‐tune neuronal excitability. The reduced TREK‐1 expression in the hippocampal CA1 and PFC regions might have a different role than that in the DRN and might decrease the firing rate of pyramidal neurons following chronic escitalopram treatment. The present results demonstrated that changes in TREK‐1 expression in the hippocampal CA1 and PFC regions were largely consistent with behavioral changes before and after escitalopram treatment. Therefore, these results support our hypothesis and emphasize the importance of future research into the mechanisms of the aforementioned effects.
This research was consistent with previous reports showing increased neural stem cell (NSC) proliferation in the SGZ of ischemic rats compared with SHAM rats 35, 42. Ischemia has been shown to promote neurogenesis in the germinative zones of the neostriatum and in the hippocampal DG, which contributes to brain recovery. These results present several potential therapeutic targets for PSD 43, 44. Our results showed a robust reduction of NSC proliferation in the SGZ of the PSD rats compared to the ischemic rats, and this could be ameliorated by chronic escitalopram treatment. Along with our previous report 35, the results of this study indicate a potential association between the deficits of adult hippocampal neurogenesis and the development of stroke‐induced depression, which are consistent with the well‐documented involvement of hippocampal neurogenesis in the etiology and therapy of endogenous depression 24, 45, 46, 47. This study is the first to find that TREK‐1 channels are implicated in the decreased hippocampal neurogenesis in PSD, showing that TREK‐1 expression in the hippocampal DG of PSD rats was significantly higher compared with that of ischemic rats, and this increased expression could be reversed by continual escitalopram administration. Interestingly, TREK‐1 blockage had been demonstrated by the antidepressant effects through hippocampal neurogenesis 36, 48. The TREK‐1 channel blocker spadin enhanced both hippocampal CREB phosphorylation and neurogenesis in mice, which have been regarded as key indicators of antidepressant action following continuous SSRI treatment 36. We previously demonstrated that the proliferation of embryonic NSCs was significantly inhibited by transfection of hTREK‐1 in vitro, and this proliferation was reversible following fluoxetine administration 49. Fluoxetine (3 mg/kg) robustly elevated NSC proliferation in the SGZ of TREK‐1 knockout mice compared to wild‐type mice 15. Therefore, TREK‐1 might be an important target in the regulation of antidepressants on hippocampal neurogenesis in PSD rats.
The following limitations should be particularly paid attention in the present study. Firstly, the Brdu‐labeled cells had not been double‐stained with a neural stem cell marker, which was further helpful to illuminate the effect of escitalopram on hippocampal neurogenesis and the relationship with TREK‐1 channel. A previous double staining study indicated that Brdu‐positive cells increased approximately sixfold in the subgranular zone of the hippocampal DG 7 days after MCAO, in which more than 80% of Brdu‐positive cells were the newly generated neural cells in the hippocampus 50. Secondly, we acknowledge that the present findings should be accepted with caution due to the difference between animal models and human syndrome. The current studies of PSD‐like rat model do not necessarily extrapolate to the PSD clinical symptoms in the human.
In summary, this study demonstrates that the SSRI escitalopram can reversibly inhibit the TREK‐1 current concentration‐dependently. Continuous escitalopram treatment decreased the TREK‐1 overexpression in the hippocampus and prefrontal cortex and elevated hippocampal NSC proliferation in PSD rats, which was accompanied by the amelioration of depressive symptoms. These data indicate that the K2p channel TREK‐1 plays an important role in the pathophysiology of PSD and might represent a potential therapeutic target for PSD.
Conflict of Interests
The authors declare no conflict of interest.
Supporting information
Table S1. The rate of infarction area of basal ganglia layer by T2 24 h after MCAO surgery.
Acknowledgments
This research was supported by the National Natural Science Foundation of China (No. 31371074 and No. 81420108012 to Zhijun Zhang), the National Key Science and Technology Program in the 12th Five Year Plan of China (The Project of New Drug Formulation, No. 2012ZX09506‐001‐009 to Zhijun Zhang), the Key Program for Clinical Medicine and Science and Technology: Jiangsu Provence Clinical Medical Research Center (No. BL2013025 to Zhijun Zhang), and the Jiangsu Province Natural Science Foundation of China (BK2012747 to Xiangrong Zhang). The authors thank H. Lundbeck A/S, Copenhagen, Denmark, for the generous gift of escitalopram.
The first two authors contributed equally to the work.
References
- 1. Arboix A. Stroke in the very old: Myths and realities. Med Clin (Barc) 2013;140:68–69. [DOI] [PubMed] [Google Scholar]
- 2. Adams HP, Froehler MT. Emergency management of acute ischemic stroke: The evolving roles of intravenous and endovascular therapies. JAMA Neurol 2013;70:828–830. [DOI] [PubMed] [Google Scholar]
- 3. Gaete JM, Bogousslavsky J. Post‐stroke depression. Expert Rev Neurother 2008;8:75–92. [DOI] [PubMed] [Google Scholar]
- 4. Carod‐Artal FJ. Post‐stroke depression (II): Its differential diagnosis, complications and treatment. Rev Neurol 2006;42:238–244. [PubMed] [Google Scholar]
- 5. Hama S, Yamashita H, Yamawaki S, Kurisu K. Post‐stroke depression and apathy: Interactions between functional recovery, lesion location, and emotional response. Psychogeriatrics 2011;11:68–76. [DOI] [PubMed] [Google Scholar]
- 6. Gothe F, Enache D, Wahlund LO, et al. Cerebro‐vascular diseases and depression: Epidemiology, mechanisms and treatment. Panminerva Med 2012;54:161–170. [PubMed] [Google Scholar]
- 7. Yi ZM, Liu F, Zhai SD. Fluoxetine for the prophylaxis of poststroke depression in patients with stroke: A meta‐analysis. Int J Clin Pract 2010;64:1310–1317. [DOI] [PubMed] [Google Scholar]
- 8. Chollet F, Tardy J, Albucher JF, et al. Fluoxetine for motor recovery after acute ischaemic stroke (flame): A randomised placebo‐controlled trial. Lancet Neurol 2011;10:123–130. [DOI] [PubMed] [Google Scholar]
- 9. Jorge RE, Acion L, Moser D, Adams HP Jr, Robinson RG. Escitalopram and enhancement of cognitive recovery following stroke. Arch Gen Psychiatry 2010;67:187–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ried LD, Jia H, Feng H, et al. Selective serotonin reuptake inhibitor treatment and depression are associated with poststroke mortality. Ann Pharmacother 2011;45:888–897. [DOI] [PubMed] [Google Scholar]
- 11. Fink M, Duprat F, Lesage F, et al. Cloning, functional expression and brain localization of a novel unconventional outward rectifier k+ channel. EMBO J 1996;15:6854–6862. [PMC free article] [PubMed] [Google Scholar]
- 12. Hervieu GJ, Cluderay JE, Gray CW, et al. Distribution and expression of trek‐1, a two‐pore‐domain potassium channel, in the adult rat cns. Neuroscience 2001;103:899–919. [DOI] [PubMed] [Google Scholar]
- 13. Medhurst AD, Rennie G, Chapman CG, et al. Distribution analysis of human two pore domain potassium channels in tissues of the central nervous system and periphery. Brain Res Mol Brain Res 2001;86:101–114. [DOI] [PubMed] [Google Scholar]
- 14. Perlis RH, Moorjani P, Fagerness J, et al. Pharmacogenetic analysis of genes implicated in rodent models of antidepressant response: Association of trek1 and treatment resistance in the star(*)d study. Neuropsychopharmacology 2008;33:2810–2819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Heurteaux C, Lucas G, Guy N, et al. Deletion of the back‐ground potassium channel trek‐1 results in a depression‐resistant phenotype. Nat Neurosci 2006;9:1134–1141. [DOI] [PubMed] [Google Scholar]
- 16. Kennard LE, Chumbley JR, Ranatunga KM, Armstrong SJ, Veale EL, Mathie A. Inhibition of the human two‐pore domain potassium channel, trek‐1, by fluoxetine and its metabolite norfluoxetine. Br J Pharmacol 2005;144:821–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wang M, Song J, Xiao W, et al. Changes in lipid‐sensitive two‐pore domain potassium channel trek‐1 expression and its involvement in astrogliosis following cerebral ischemia in rats. J Mol Neurosci 2012;46:384–392. [DOI] [PubMed] [Google Scholar]
- 18. Li ZB, Zhang HX, Li LL, Wang XL. Enhanced expressions of arachidonic acid‐sensitive tandem‐pore domain potassium channels in rat experimental acute cerebral ischemia. Biochem Biophys Res Commun 2005;327:1163–1169. [DOI] [PubMed] [Google Scholar]
- 19. Wu X, Liu Y, Chen X, et al. Involvement of trek‐1 activity in astrocyte function and neuroprotection under simulated ischemia conditions. J Mol Neurosci 2013;49:499–506. [DOI] [PubMed] [Google Scholar]
- 20. Heurteaux C, Guy N, Laigle C, et al. Trek‐1, a k+ channel involved in neuroprotection and general anesthesia. EMBO J 2004;23:2684–2695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Blondeau N, Petrault O, Manta S, et al. Polyunsaturated fatty acids are cerebral vasodilators via the trek‐1 potassium channel. Circ Res 2007;101:176–184. [DOI] [PubMed] [Google Scholar]
- 22. Xu X, Pan Y, Wang X. Alterations in the expression of lipid and mechano‐gated two‐pore domain potassium channel genes in rat brain following chronic cerebral ischemia. Brain Res Mol Brain Res 2004;120:205–209. [DOI] [PubMed] [Google Scholar]
- 23. Loubinoux I, Kronenberg G, Endres M, et al. Post‐stroke depression: Mechanisms, translation and therapy. J Cell Mol Med 2012;16:1961–1969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Santarelli L, Saxe M, Gross C, et al. Requirement of hippocampal neurogenesis for the behavioral effects of antidepressants. Science 2003;301:805–809. [DOI] [PubMed] [Google Scholar]
- 25. Wang SH, Zhang ZJ, Guo YJ, Sui YX, Sun Y. Involvement of serotonin neurotransmission in hippocampal neurogenesis and behavioral responses in a rat model of post‐stroke depression. Pharmacol Biochem Behav 2010;95:129–137. [DOI] [PubMed] [Google Scholar]
- 26. Wang SH, Zhang ZJ, Guo YJ, Zhou H, Teng GJ, Chen BA. Anhedonia and activity deficits in rats: Impact of post‐stroke depression. J Psychopharmacol 2009;23:295–304. [DOI] [PubMed] [Google Scholar]
- 27. Aspey BS, Cohen S, Patel Y, Terruli M, Harrison MJ. Middle cerebral artery occlusion in the rat: Consistent protocol for a model of stroke. Neuropathol Appl Neurobiol 1998;24:487–497. [DOI] [PubMed] [Google Scholar]
- 28. Longa EZ, Weinstein PR, Carlson S, Cummins R. Reversible middle cerebral artery occlusion without craniectomy in rats. Stroke 1989;20:84–91. [DOI] [PubMed] [Google Scholar]
- 29. Dremencov E, El Mansari M, Blier P. Noradrenergic augmentation of escitalopram response by risperidone: Electrophysiologic studies in the rat brain. Biol Psychiatry 2007;61:671–678. [DOI] [PubMed] [Google Scholar]
- 30. Jayatissa MN, Bisgaard C, Tingstrom A, Papp M, Wiborg O. Hippocampal cytogenesis correlates to escitalopram‐mediated recovery in a chronic mild stress rat model of depression. Neuropsychopharmacology 2006;31:2395–2404. [DOI] [PubMed] [Google Scholar]
- 31. Xi G, Hui J, Zhang Z, et al. Learning and memory alterations are associated with hippocampal N‐acetylaspartate in a rat model of depression as measured by 1H‐MRS. PLoS One 2011;6:e28686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lipsey JR, Robinson RG, Pearlson GD, et al. Nortriptyline treatment of post‐stroke depression: A double‐blind study. Lancet 1984;1:297–300. [DOI] [PubMed] [Google Scholar]
- 33. Lipsey JR, Robinson RG. Nortriptyline for post‐stroke depression. Lancet 1984;1:803. [DOI] [PubMed] [Google Scholar]
- 34. Robinson RG, Jorge RE, Moser DJ, et al. Escitalopram and problem‐solving therapy for prevention of poststroke depression: A randomized controlled trial. JAMA 2008;299:2391–2400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wang SH, Zhang ZJ, Guo YJ, Teng GJ, Chen BA. Hippocampal neurogenesis and behavioural studies on adult ischemic rat response to chronic mild stress. Behav Brain Res 2008;189:9–16. [DOI] [PubMed] [Google Scholar]
- 36. Mazella J, Petrault O, Lucas G, et al. Spadin, a sortilin‐derived peptide, targeting rodent TREK‐1 channels: A new concept in the antidepressant drug design. PLoS Biol 2010;8:e1000355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Gordon JA, Hen R. TREKing toward new antidepressants. Nat Neurosci 2006;9:1081–1083. [DOI] [PubMed] [Google Scholar]
- 38. Honore E. The neuronal background K2P channels: Focus on TREK1. Nat Rev Neurosci 2007;8:251–261. [DOI] [PubMed] [Google Scholar]
- 39. Mathie A, Veale EL. Therapeutic potential of neuronal two‐pore domain potassium‐channel modulators. Curr Opin Investig Drugs 2007;8:555–562. [PubMed] [Google Scholar]
- 40. Gronier BS, Rasmussen K. Electrophysiological effects of acute and chronic olanzapine and fluoxetine in the rat prefrontal cortex. Neurosci Lett 2003;349:196–200. [DOI] [PubMed] [Google Scholar]
- 41. Bockenhauer D, Zilberberg N, Goldstein SA. KCNK2: Reversible conversion of a hippocampal potassium leak into a voltage‐dependent channel. Nat Neurosci 2001;4:486–491. [DOI] [PubMed] [Google Scholar]
- 42. Darsalia V, Heldmann U, Lindvall O, Kokaia Z. Stroke‐induced neurogenesis in aged brain. Stroke 2005;36:1790–1795. [DOI] [PubMed] [Google Scholar]
- 43. Jin K, Wang X, Xie L, et al. Evidence for stroke‐induced neurogenesis in the human brain. Proc Natl Acad Sci U S A 2006;103:13198–13202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kuhn HG, Dickinson‐Anson H, Gage FH. Neurogenesis in the dentate gyrus of the adult rat: Age‐related decrease of neuronal progenitor proliferation. J Neurosci 1996;16:2027–2033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Sahay A, Hen R. Adult hippocampal neurogenesis in depression. Nat Neurosci 2007;10:1110–1115. [DOI] [PubMed] [Google Scholar]
- 46. Ming GL, Song H. Adult neurogenesis in the mammalian brain: Significant answers and significant questions. Neuron 2011;70:687–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Encinas JM, Vaahtokari A, Enikolopov G. Fluoxetine targets early progenitor cells in the adult brain. Proc Natl Acad Sci U S A 2006;103:8233–8238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Moha Ou Maati H, Veyssiere J, Labbal F, et al. Spadin as a new antidepressant: Absence of TREK‐1‐related side effects. Neuropharmacology 2012;62:278–288. [DOI] [PubMed] [Google Scholar]
- 49. Xi G, Zhang X, Zhang L, et al. Fluoxetine attenuates the inhibitory effect of glucocorticoid hormones on neurogenesis in vitro via a two‐pore domain potassium channel, TREK‐1. Psychopharmacology 2011;214:747–759. [DOI] [PubMed] [Google Scholar]
- 50. Takasawa K, Kitagawa K, Yagita Y, et al. Increased proliferation of neural progenitor cells but reduced survival of newborn cells in the contralateral hippocampus after focal cerebral ischemia in rats. J Cereb Blood Flow Metab 2002;22:299–307. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. The rate of infarction area of basal ganglia layer by T2 24 h after MCAO surgery.