

Summary
Aims
Compound IMM‐H004 (7‐hydroxy‐5‐methoxy‐4‐methyl‐3‐[4‐methylpiperazin‐1‐yl]‐2H‐chromen‐2‐one) is a new synthetic derivative of coumarin, and previous studies showed that it exhibited antioxidant and neuroprotective roles in focal cerebral ischemia. However, we know little about the compound's function in transient global ischemia. This study is to investigate whether compound IMM‐H004 can protect against transient global ischemic injury.
Methods
Four‐vessel occlusion (4VO) rat model was induced for a 20‐min occlusion and different times of reperfusion to mimic transient global cerebral ischemia. IMM‐H004 (3, 6, 9 mg/kg) or Edaravone (6 mg/kg) was administered after 30 min of reperfusion. Morris water maze tests were used to estimate the ability of spatial learning and memory. Nissl staining, TUNEL assay and Immunoblot for Bax/Bcl‐2 and activated caspase‐3 were used to detect hippocampal neuron injury. Immunoblot for PSD‐95 and synapsin 1, and electron microscopy were used to observe synaptic function.
Results
Compared with vehicle group, IMM‐H004 significantly improved the spatial learning performance and exhibited less CA1 neurons loss. The expressions of Bax/Bcl‐2 and activated caspase‐3 were decreased. IMM‐H004 also ameliorated synaptic structure, decreased PSD‐95 and increased synapsin 1 expression.
Conclusion
These findings suggested that IMM‐H004 exerted neuroprotective role in global ischemia by reducing apoptosis and maintaining the integrity of synaptic structure.
Keywords: Apoptosis, Global cerebral ischemia, Hippocampus, IMM‐H004, Neuroprotection
Introduction
Cerebral ischemia is one of the most major causes of death, only surpassed by heart disease and cancer 1. With the continuous improvement of living standards, the number of individuals with stroke will grow in the future 2. Tissue plasminogen activator, which can cause fast reperfusion, is the only approved treatment for acute ischemic stroke by FDA nowadays 3. However, various individual chemical cascades and major pathways during reperfusion are triggered, leading to neuronal apoptosis and necrosis 4, 5. In addition, because of the short therapeutic time window and high risk of bleeding, only few stroke patients are handled with tissue plasminogen activator 6. Therefore, it is imperative for us to find more effective and safer treatments for ischemic stroke.
Interruption of cerebral blood due to cardiac arrest is widespread in clinical that cause transient global cerebral ischemia. As we know, transient global cerebral ischemia results in delayed neuronal death (DND), which normally happens 3–7 days after the onset of ischemic injury 7 in certain areas of brain both in humans and in animals 8, especially in the region of hippocampal CA1 9. There was accumulating evidence that DNA fragmentation induced by ischemic injury, which is considered as a typical apoptosis process, is a key component of DND in the ischemic models 10, 11, 12. One of the possible reasons may attribute to the induction of apoptotic‐related factors, for instance, Bax, Bcl‐2 and caspase‐3 13, 14. Therefore, agents that inhibit the apoptosis pathway and promote the neurons destiny may protect against ischemic injury‐mediated DND. What is more, this neuronal death can be concurrent with severe cognitive impairments. Commonly, survivors of brain ischemia exhibit disorders in the task of learning and memory. Synaptic function and morphology are thought to be critical for information processing such as learning and memory in the brain. A variety of cellular signaling proteins underlie the damage of neurons and synaptic dysfunction induced by global ischemia. Among them, PSD‐95 is a synaptic density protein (PSD) involved in the interaction of the synaptic cytoskeletal structure and receptors, and can be modified in response to synaptic stimulation. Another important protein is synapsin 1 that plays a key role in regulation of synaptic vesicle exocytosis and neurotransmitter release.
Rat 4‐VO model can successfully imitate this pathological process of transient global cerebral ischemia including neuronal injury in hippocampus, and impairments in hippocampus‐dependent spatial learning and object recognition abilities. Thus, we could see that it is critical to promote the neuronal survival and improve the synaptic function after global ischemia.
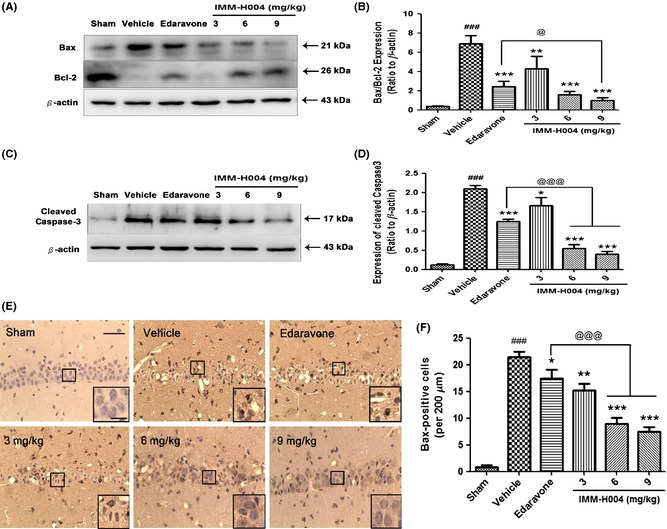
Recently, treatment options for ischemic stroke are not sufficient and a majority of clinical trials of antiischemic agents are disappointing 15, 16. However, neuroprotective agents are proved as a hopeful method to acute ischemic treatment in recent years. One plausible neuroprotection hypothesis suggested that ischemic injury can be prevented in some parts by blocking the core aspects of the ischemic cascade 17. Recent studies have demonstrated that coumarin compounds exhibited neuroprotective effects in vivo 18, 19 and in vitro 20, 21. Coumarins contain a group of phenolic compounds widely distributed in natural plants. It is well known that esculetin exerted a neuroprotective role in brain ischemic injury in mice and exerted antiapoptotic role through upregulating the expression of Bcl‐2 as well as downregulating the expression of Bax 22. Our previous work found that IMM‐H004 (Figure 1), a novel synthetic coumarin derivative, significantly ameliorated the PC12 survival subjected to OGD, Aβ 25–35 and rotenone injury in vitro as well as effectively protected rat brain subjected to middle cerebral artery occlusion (MCAO) injury 23, 24. It was worth to note that IMM‐H004 also showed a better effect in scavenging hydroxyl radicals than Edaravone, which was widely used as a neuroprotective agent in stroke treatment. Thus, we chose Edaravone as the positive control. Furthermore, compound IMM‐H004 not only retained the biological activity of nature coumarin derivatives, but could also be synthetic, solving the problem of complicated extraction process as well as low content of the active ingredient. However, there was no evidence indicating whether compound IMM‐H004 is also effective in global brain ischemia. Thus, we aim to investigate the role of compound IMM‐H004 in transient global cerebral ischemia.
Figure 1.
The chemical structure of compound IMM‐H004.
Materials and Methods
Experimental Animals
All experiments were performed on male Sprague–Dawley (SD) rats (weight, 280–300 g), which were obtained from Experimental Animal Center of Chinese Academy of Medical Sciences (Beijing China). The housing conditions were at 22 ± 2 °C, humidity 35–45% and 12 h light/dark. Animals were maintained for 1 week before drug treatment or surgery and had free access to water and food. Rats were treated according to the standard in the Guide for the Care and Use of Laboratory Animals published by the Institute of Laboratory Animals Resources of the National Research Council (United States), and the study was approved by the Animal Care and Use Committee of the Peking Union Medical College and the Chinese Academy of Medical Sciences. All efforts were made to reduce the suffering and the number of the animals.
Drug Administration and Experimental Group
Animals were divided randomly into (1) sham group, (2) vehicle + ischemia group, (3) Edaravone (6 mg/kg in 1 mL saline) + ischemia group, (4) compound IMM‐H004 (3, 6 and 9 mg/kg in 1 mL vehicle) + ischemia group. The compound was offered by Chinese Academy of Medical Science. The compound IMM‐H004 was dissolved in the vehicle, which contains 95% saline and 5% dehydrated alcohol. The solution was under agitation for about 20 min at room temperature and filtered before use. Drug or vehicle was injected into caudal veins of rats 30 min after I/R.
Transient Global Cerebral Ischemia
Global brain ischemia was carried out using 4VO model 25. On the first day, rats were anesthetized by 10% chloral hydrate (300 mg/kg, i.p.) and then fixed by stereotaxic ear bars. The head was placed at about 30° to the horizontal. Briefly, after disinfection using 75% alcoholic, both the vertebral arteries were occluded permanently by electrocauterization with a 0.5‐mm‐diameter needle through the foramen of the first cervical vertebra. Then, the two common carotid arteries (CCAs) were isolated gently and a nylon thread was loosely placed around both CCAs. After the surgery, each rat was injected penicillin to prevent wound infection. The rats were kept recovering for 24 h. The next day, animals were reanesthetized and both CCAs were occluded for 20 min using microvascular clips to induce cerebral ischemia. Within 10 seconds, rats became unresponsive, accompanied with absence of righting reflex, loss of pain and dilatation of the pupils. Reperfusion immediately started right after the both clips were softly removed. During the whole operation and in the following 6 h, a heating blanket was used to keep rectal temperature at 37 ± 0.5°C. Any rat returned to righting reflexes during the 20min occlusion or epileptic seizured after ischemia was excluded. Animals in sham group were subjected to similar treatment with ischemic group without occluding CCAs.
Morris Water Maze
The water maze (MWM) test was conducted as described previously 26. Briefly, MWM was divided into 4 quadrants. A hidden circular Plexiglas platform (40 cm height, 10 cm diameter) was placed in the middle of IV quadrant of the MWM (120 cm diameter, 80 cm deep), 1–2 cm lower than the water surface (24 ± 1°C) and remained there through all tests. The task contained two trails: spatial probe and orientation navigate. In the first trail, animals were released into the middle of II quadrant with the head up and toward the wall. If the rats cannot find it in a maximum time of 120 seconds, it was allowed to stay on it for 30 seconds. Each trail contained five tests, and data of the five tests were averaged. There was a 5‐min rest period between tests. Rats had been trained for 5 days consecutively from the 8th day after 4VO. The time used to find the platform called escape latency was recorded. After the last testing (13th day after 4VO), the platform got removed and the animals were released to swim in the pool for 120 seconds. The length of trajectory and the time passing through the place where the platform was previously located were recorded with a tracking system.
Nissl Staining
Nissl staining was performed with Cresyl Violet, which can selectively stain the Nissl body in survival neurons. Animals were sacrificed 3 days after ischemia and then immediately perfused by cold phosphate‐buffered saline (PBS, 0.1 M; PH 7.4) and 4% paraformaldehyde. Brain was removed and immersed in fixative. Then, tissues were embedded in paraffin, and 10 coronal sections (4 μm) were cut continuously at the dorsal hippocampus located between 3.3 and 4.0 mm posterior from bregma 27 for Nissl staining. 6 sections (randomly chosen) per rat were stained with 1% Cresyl Violet dissolved in 0.25% acetic acid for 15 min, washed, dehydrated and mounted by neutral balsam. The survived hippocampal neurons per 200 μm liner length of the medial CA1 cell layer were counted bilaterally in 6 sections per animal with microscope (400×). Cell counts from the left and right hippocampus on each of the six sections were average to provide a single value (number of neurons per 200 μm length) for each animal.
TUNEL Staining
TUNEL can selectively recognize fragmented DNA by binding to 3′‐OH ends. Thus, we adopted this method to detect apoptosis. TUNEL staining was performed using the Calbiochem® (MERCK) kit. Briefly, the sections were xylene‐ and ethanol‐treated for paraffin removal and for dehydration. Then, the sections were washed by TBS (PH 7.4) softly and excess liquid was removed using filter paper. Then, the sections were cultured at room temperature with protease K (20 μg/mL) for 20 min. And after raised in TBS, they were treated with reaction buffer at 37°C for 90 min. Then, the sections were incubated with TBS three times each for 1 min. Subsequently, they were mounted with mounting media. During the whole process, the samples should be kept moistening. The numbers of TUNEL‐positive cells were counted bilaterally in three sections per animal (400×).
Western Blot
Hippocampus was removed immediately after 72 h of reperfusion. The tissues were homogenized in ten volumes of ice‐cold RIPA lysis buffer (50 mmol/L Tris pH 7.4), 150 mmol/L NaCl, 0.1 mmol/L PMSF, 1% NP40, 0.5% sodium deoxycholate and 1% SDS). Then, the homogenized tissue were centrifuged at 12,000 g at 4°C for 30 min. Loading buffer was added into the supernatant to denature the protein and then the protein was stored at −70°C. Quantitative protein determination was carried out through bicinchoninic acid (BCA) method. Thirty micrograms of proteins from each tested group was separated on 10–15% gels by electrophoresis and then transferred to polyvinylidene difluoride (PVDF) membrane (Millipore, USA) in blotting buffer for an hour at 100 mA. The whole PVDF membrane was then blocked with 3% BSA for 2 h at room temperature. Primary antibodies used were anti‐Bcl‐2 (1:500, santa), anti‐Bax (1:500, santa), anticaspase‐3 (1:500, santa), anti‐PSD‐95 (1:500, santa), and antisynapsin 1 (1:500, santa). Following by horseradish peroxidase (HRP)‐conjugated secondary antibody (1:5000, Invitrogen), the expression of each protein was detected with enhanced chemiluminescence (ECL) plus detection system (Molecular Device, Lmax). The density of each band was quantified using image analysis software (Quantity one; Tokyo, Japan).
Immunohistological Staining
Immunohistological staining for Bax was performed. Brain samples were removed, embedded in paraffin, cut into 4‐μm‐thick slices. After deparaffinized, endogenous peroxidase was inactivated by 3% H2O2 for 15 min at 37°C. Then, the slices were permeabilized in 0.1% PBS for 5 min of three times at temperature. After blocked with 10% normal serum for 20 min, slices were then treated with primary antibody (anti‐Bax, sc‐493, diluted 1:100) overnight at 4°C. After washed three times in PBS, the slices were incubated with biotinylated goat anti‐rabbit Abs for 1 h, followed by avidin–biotin complex for 30 min. The expression of Bax was detected with 3, 3‐diaminobenzidin (DBA) as the substrate. At last, the slices were mounted by neutral balsam.
Electron Microscopy
Tissue sections were obtained after 13 days of reperfusion and processed for EM as described previously 28, 29. Briefly, dorsal hippocampus was cut into a thickness of 200 μm and prefixed in 3% glutaraldehyde overnight at 4°C. After washed with 0.1 M PBS, the slices were postfixed for 1 h with 1% osmium tetroxide, then rinsed in PBS and finally stained by 1% aqueous uranyl acetate. The samples were then dehydrated in ethanol, followed by dry acetone and at last embedded in Epon. For EM, sections were stained with uranyl acetate and lead citrate and then embedded in Durcupan ACM.
Statistics
All data were presented as mean ± SEM and were analyzed by one‐way anova followed by Newman–Keuls post hoc analysis on Graph pad Prism 5.0. Every sample was corresponded to one animal. P < 0.05 was considered as significant.
Results
IMM‐H004 Improved Spatial Memory in 4VO Rats
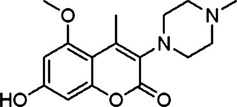
MWM task was to access spatial learning and memory. As shown in Figure 2, animals subjected to 4VO and treated with vehicle spent much more time to find the hidden platform (escape latency) when compared with the sham‐treated animals. In contrast, the escape latency was shortened by IMM‐H004 treatment significantly, especially the 9 mg/kg (Figure 2A, P < 0.01)‐treated group showing comparable effect with Edaravone (Figure 2A, P < 0.01). In model group, the time spending in the quadrant where the hidden platform was previously placed was significantly shorter than that in the sham group (Figure 2B, P < 0.01). However, the decreased time in 4VO‐treated group was significantly increased by IMM‐H004 administration, especially the dose of 6 mg/kg (P < 0.01) and 9 mg/kg (P < 0.001) showing comparable or better effect with Edaravone. However, the swimming speed showed no significantly difference between the groups (Figure 2C, P > 0.05). Figure 2D shows representative diagrams of search trajectories in MWM on day 4 of acquisition.
Figure 2.
Effects of IMM‐H004 on Morris water maze performance deficits induced by transient cerebral ischemia–reperfusion in rats. (A) Representative examples of search patterns in the circular water maze by members of each group on day 4 of acquisition. (B) Escape latency in the Morris water maze task in the acquisition trials. (C) Percentage of time spends in target quadrant within 120 seconds in the probe trial. (D) Swimming speed of each group. Data are expressed as means ± S.E.M. n = 8–12 in each group. ## P < 0.01, ### P < 0.001 vs. Sham, *P < 0.05, **P < 0.01,***P < 0.001 vs. Vehicle.
IMM‐H004 Improved CA1 Neuronal Survival
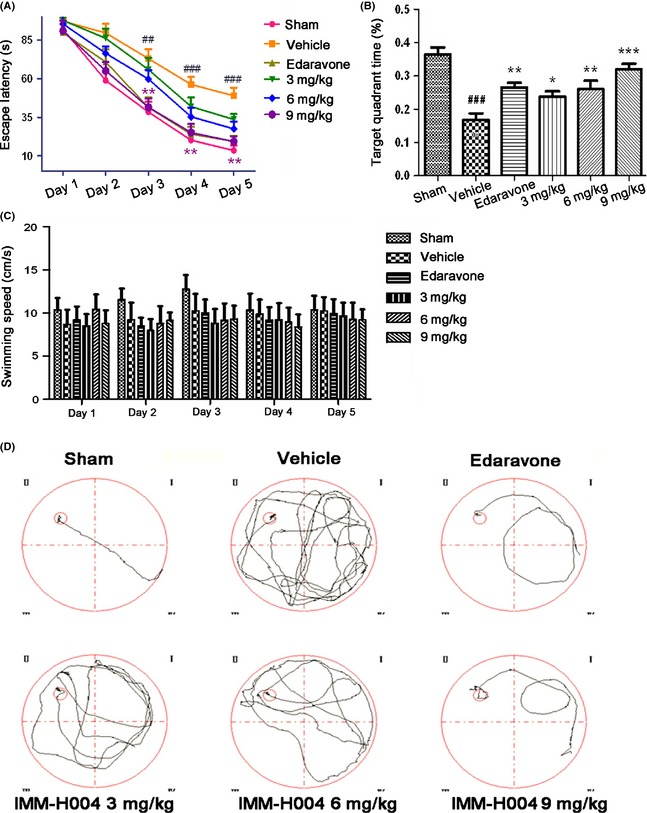
Transient global cerebral ischemia for 20 min caused selective loss of pyramidal neurons in hippocampal CA1. Nissl staining revealed that after I/R, the number of normal CA1 neurons of vehicle‐pretreated group sharply decreased, while the relict neurons showed karyopyknosis, anachromasis, nucleoli disappeared and vacuolization (Figure 3A). Only 9.1% of CA1 pyramidal cells were reserved in the model group. In contrast, compound IMM‐H004 at doses of 3, 6 and 9 mg/kg significantly increased the number of pyramidal cell by35.7. ± 4.1%, 53.7 ± 4.6% and 67.8 ± 4.4%, respectively (Figure 3B, P < 0.001). Increasing evidences have demonstrated that after cerebral ischemia injury there were different types of cell death including apoptosis, necrosis as well as necroptosis 30. Apoptosis was investigated with TUNEL staining that allowed in situ labeling of DNA fragmentation in nucleus. The results showed that apoptosis was appeared in the DND after 3 days of reperfusion in the hippocampus. Rarely TUNEL‐stained cells, which showed morphology of dark nuclei and shrunken cytoplasm, were detected in hippocampal CA1 of the sham‐operated group, whereas in the vehicle‐pretreated group, massive of TUNEL‐positive cells were observed with a strongly fluorescence (Figure 3C). After administered with compound IMM‐H004, ischemia‐induced DNA fragment in the CA1 region was significantly reduced compared to the vehicle‐treated group (Figure 3D, P < 0.001). Representative photomicrographs of the hippocampal CA1 region on Nissl‐stained brain slices suggested the degeneration of neurons induced by global cerebral ischemia and the potential neuroprotective effect of compound IMM‐H004.
Figure 3.
Effects of compound IMM‐H004 on morphologic changes and DNA fragmentation in hippocampal CA1 region that after 3 days of global ischemic reperfusion. (A) Representative photographs of Nissle‐stained hippocampal CA1 subfield from rats sacrificed 3 days after global ischemia (scale bar=50 μm). (B) Dose–response effect of compound IMM‐H004 treatment on CA1 neuronal destiny. For each experimental group, six randomly chosen brain sections form five rats were used for statistics. And cell counts from the left and right hippocampus on each of the six sections were average to provide a single value (number of neurons per 200 μm length) for each animal. (C) Representative photomicrographs displaying the CA1 substructure in DNA fragmentation assessed by TUNEL method after 3 days reperfusion (scale bar=37.5 μm). (D) The number of TUNEL‐positive cells per group after 3 days reperfusion. Data were expressed as mean±S.E.M. n = 5 per experimental group, ### P < 0.001 versus sham group, ***P < 0.001 versus vehicle group, @ P < 0.05, @@ P < 0.01 and @@@ P < 0.001 versus Edaravone treatment group.
IMM‐H004 Inhibited the Release of Proapoptosis Factors Induced by 4VO
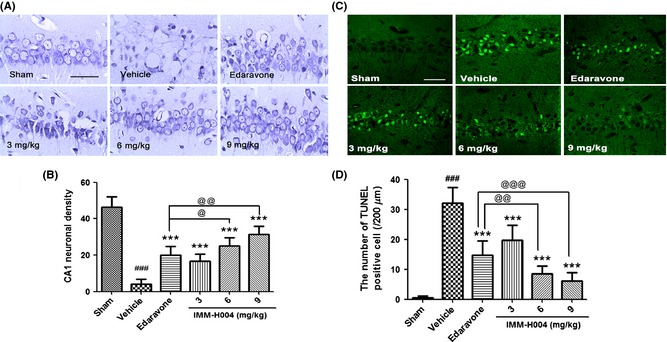
To investigate the antiapoptosis mechanism of compound IMM‐H004 in I/R injury, we detected the expression of apoptotic proteins including cleaved caspase‐3 and Bcl‐2/Bax. In sham group, the expression of Bax was low, and the expression of Bcl‐2 was fairly higher. Completely reversed data were found in the ischemic group. Treatment with compound IMM‐H004 significantly increased the expression of Bcl‐2 and decreased the expression of Bax in a dose‐dependent manner (Figure 4A,B, P < 0.01, P < 0.001). Bax immunostaining confirmed the results of Western blot (Figure 4E,F). In sham group, rare Bax‐positive cells were detected, but in vehicle group, numerous of Bax‐positive cells were found under microscope. However, treatment with compound IMM‐H004 decreased Bax‐positive cells. For cleaved caspase‐3 expression, there was nearly no expression of activated caspase‐3 in sham‐operated rats. In contrast, the cleaved caspase‐3 expression induced by transient global cerebral ischemia increased remarkably (Figure 4C,D, P < 0.001). After administered with compound IMM‐H004, the level of cleaved caspase‐3 expression decreased significantly in a dose‐dependent manner (Figure 4C,D, P < 0.05, P < 0.001).
Figure 4.
Effect of compound IMM‐H004 on Bax, Bcl‐2 and cleaved caspase‐3 expression induced by global cerebral ischemia after 3 days after reperfusion. (A) Representative Western blots of Bcl‐2, Bax and β‐actin. (B) Ratio of Bcl‐2/Bax proteins. (C) Representative Western blots of cleaved caspase‐3 and β‐actin. (D) Ratio of cleaved caspase‐3/β‐actin. (E) Representative photomicrographs of Bax expression in the CA1 substructure assessed by immunohistochemistry. (F) Histograms representing the number of Bax‐positive cells of each hippocampus (per 200 μm) (scale bar=100 μm and 10 μm). The lower right sections of the images showed the enlarged view of the black‐edged rectangle. Values are expressed as means ± S.E.M for three independent experiments. ### P < 0.001 versus sham group, *P < 0.05, **P < 0.01,***P < 0.001 versus vehicle group and @ P < 0.05, @@@ P < 0.001 versus Edaravone group.
IMM‐H004 Ameliorated Synaptic Injury Induced by 4VO
To study the structural and functional changes of the PSDs, we analyzed the expression of PSD‐95 that involved in the function of receptors and cytoskeletal at synapse and p‐synapsin 1 that is localized to synaptic vesicles and has essential functions in regulating synaptic vesicle exocytosis. Western blot results showed that the compound IMM‐H004, in a dose‐dependent manner, inhibited the overexpression of PSD‐95 and increased the expression of p‐synapsin 1 in the region of hippocampal CA1 at the early phase of global cerebral ischemia (Figure 5A–C, P < 0.05, P < 0.01).
Figure 5.
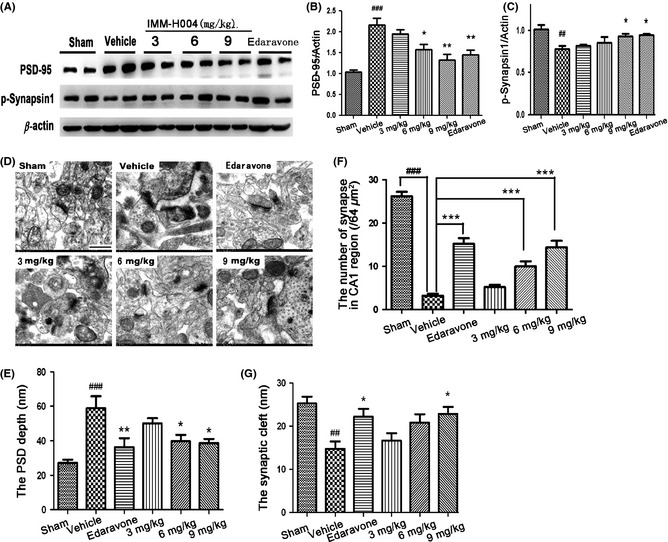
Effect of compound IMM‐H004 on synaptic function. (A) Representative Western blots of PSD‐95, p‐synapsin 1 and β‐actin. (B) Ratio of PSD‐95/β‐actin. (C) Ratio of p‐synapsin 1/β‐actin. (D) Representative EM images of structure of synapse and quantification of (E) PSD depth, (F) the number of synapse and (G) synaptic cleft in CA1 region. The number of synapse was counted in 8 × 8 μm electron microscopy photographs. Scale bar = 500 nm. Data were expressed as mean±S.E.M. ## P < 0.05 and ### P < 0.001 versus sham controls, *P < 0.05, **P < 0.01 and ***P < 0.01 versus vehicle group.
As biochemical investigations showed significant change of PSD protein expression, we took electron microscopy to observe the PSD ultrastructure in hippocampal CA1 region. Compared to control PSDs, postischemic PSDs were much thicker, less compact, more irregular and showing fluffier material extending from the postsynaptic region. Compound IMM‐H004 enhanced the number of synapses, reduced the thickness of PSD and increased the width of synaptic cleft (Figure 5D–G, P < 0.05, P < 0.0001) compared with the model group.
Discussion
Our studies, for the first time, demonstrated that IMM‐H004 exhibited a significantly neuroprotective effect in 4VO model such as improving spatial memory, decreasing the CA1 pyramidal cell death and preserving synaptic structure. The neuroprotective role of IMM‐H004 in global ischemic injury is better than Edaravone for its multibioactivities besides the ability of ROS scavenging. The mechanism may be related to inhibition of neuron apoptosis by inactivation of caspase‐3, decreasing the expression of Bax/Bcl‐2, and increasing the synaptic plasticity.
It is well known that a loss of cerebral blood flow develops a cascade of complex pathological signaling, including oxidative stress, cell death, intracellular calcium overload, inflammatory reaction and excitotoxic toxicity 31, which is badly related to miscellaneous of brain dysfunctions 32, 33, 34, 35. In this study, we observed that compound IMM‐H004 improved rat learning and memory performances measured by Morris test. Data obtained in these tests showed that compound IMM‐H004 improved the ability of objective recognition and spatial learning after global ischemia.
Global cerebral ischemia is usually triggered by a reduction of brain blood flow throughout the majority or overall regions of the brain as a consequence of sudden cardiac arrest 36 and eventually leading to brain cellular dysfunction and loss. Accumulating evidences have proposed that transient global cerebral ischemia in humans and rodents initiates a molecular cascade resulting in DND in CA1 pyramidal neurons selectively 8, 9, 11, 28. It is worth noting that the postischemic DNA fragmentation in rat cerebral ischemic models is a key component of DND, and stimulation of nucleosomal breaks of DNA is thought to be a characteristic exhibition of apoptosis, which appears simultaneously with neuronal death in vulnerable region of brain after ischemic injury 37, 38, 39, 40. Here, we demonstrated that DND in hippocampal CA1 area induced by 4VO was strongly reduced by treatment with compound IMM‐H004, for it conspicuously decreased TUNEL‐positive cells in CA1 compared to model group. Besides DNA fragmentation, Bcl‐2 and caspases family members seem to be extremely important regulators in complex apoptotic cell death pathways triggered by I/R. Bcl‐2 is considered as one of the major antiapoptotic member, while Bax is considered to be a marker of neurons experiencing apoptotic cell death 41. We found an increasing expression of Bax as well as a decreasing expression of Bcl‐2 in hippocampal of vehicle‐treated rats. Conversely, the expression of Bcl‐2 was increased while the expression of Bax was decreased in compound IMM‐H004‐treated groups. In Edaravone‐treated groups, the expression of Bax was also increased although the ration of Bax/Bcl‐2 was decreased. Among caspases family, caspase‐3 is considered as the main executioner of nucleus degradation in neurons subjected to ischemic injury, which leads to DNA fragmentation 29, 42, 43, 44. A large amount of evidences suggested that cerebral ischemia could cause activation and upregulation of caspase‐3 preceding cell death in experimental global cerebral ischemic models 45, 46, 47. What is more, inhibition of caspase‐3 also played a critical role in preventing DNA fragmentation 48. The effect of compound IMM‐H004 on caspase‐3 expression might contribute to its antiapoptotic activity.
Lots of theories have been proposed as possible stimulators and mechanisms for the above DND in transient ischemia 49, 50, 51, 52. Among them, excitotoxic hypothesis such as glutamate transmission which contributed to DND was first proposed since early 1980s. Physiological changes of synapses have been observed in the postischemic brain 53, 54, indicating there was a mechanism that regulates synaptic function. Serious of biochemical studies exhibited that the proteins related to the PSD (postsynaptic density) changed after ischemia. PSD is a cytoskeletal protein, especially predominance in glutamatergic synapse 55, 56. Among them, PSD‐95 functioned by binding to its receptors such as NMDA receptors. Researchers found that overactivation of PSD‐95 in hippocampal CA1 neurons could contribute to the excitatory synaptic maturation and increase the activities of glutamate receptors 57, 58. Studies also reported that overexpression of PSD‐95 could be induced by brain ischemic injury. Overactivation of PSD‐95 exaggerated the excitatory injury in ischemic brain by interacting with MLK3 and GluR6, while PSD‐95 antisense oligodeoxynucleotides reversed the harmful effect 59. Another key regulator of synaptic plasticity is synapsin 1, which is associated with neurotransmitter release and synaptic vesicle life cycle 60. One study proved that synapsin 1‐mutant mice displayed an increasing cognitive impairment associated with aging 61. Consistent to the above findings, our data demonstrated that IMM‐H004 reversed the overexpressed PSD‐95 and increased the level of synapsin 1, suggesting its role in improving synaptic plasticity. Changes in the structure of synapse were thought to involve in most types of synaptic plasticity as well. With EM, we observed that PSDs were thicker and less condensed in ischemic brain compared with control, while IMM‐H004 reversed these changes.
In conclusion, our findings demonstrate that treatment with compound IMM‐H004 after transient global ischemia protects against the loss of CA1 neurons as well as the impairment of spatial learning. Further research suggests that the neuroprotective properties are mediated by regulating the balance of Bcl‐2/Bax, inactivating caspase‐3 pathway and enhancing synaptic plasticity. These results contribute to a growing body of research literature investigating the effects of IMM‐H004 on neuronal and behavioral outcome after brain ischemic injury, and further contribute to the development of IMM‐H004‐based strategies for ischemia therapy.
Conflict of Interest
The authors declare no conflict of interest.
Acknowledgments
The authors thank the funding from the National Natural Science Foundation of China (Grant No. 81373997). Special Purpose for New Drug Development (2012ZX09103101‐006). Studies on Structure and Function of Bioactive Substances from Natural Medicines (IRT1007). Beijing Key Laboratory of New Drug Mechanisms and Pharmacological Evaluation Study (NO.BZ0150).
The first two authors contributed equally to this work.
References
- 1. Rosamond W, Flegal K, Friday G, et al. Heart disease and stroke statistics–2007 update: a report from the American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Circulation 2007;115:e69–e171. [DOI] [PubMed] [Google Scholar]
- 2. Lakhan SE, Kirchgessner A, Hofer M. Inflammatory mechanisms in ischemic stroke: therapeutic approaches. J Transl Med 2009;7:97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Sharma VK, Teoh HL, Wong LY, Su J, Ong BK, Chan BP. Recanalization therapies in acute ischemic stroke: pharmacological agents, devices, and combinations. Stroke Res Treat 2010;2010:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Sugawara T, Kinouchi H, Oda M, Shoji H, Omae T, Mizoi K. Candesartan reduces superoxide production after global cerebral ischemia. NeuroReport 2005;16:325–328. [DOI] [PubMed] [Google Scholar]
- 5. White BC, Sullivan JM, DeGracia DJ, et al. Brain ischemia and reperfusion: molecular mechanisms of neuronal injury. J Neurol Sci 2000;179:1–33. [DOI] [PubMed] [Google Scholar]
- 6. Medcalf RL. Plasminogen activation‐based thrombolysis for ischaemic stroke: the diversity of targets may demand new approaches. Curr Drug Targets 2011;12:1772–1781. [DOI] [PubMed] [Google Scholar]
- 7. Jung JE, Kim GS, Chen H, et al. Reperfusion and neurovascular dysfunction in stroke: from basic mechanisms to potential strategies for neuroprotection. Mol Neurobiol 2010;41:172–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kirino T. Delayed neuronal death in the gerbil hippocampus following ischemia. Brain Res 1982;239:57–69. [DOI] [PubMed] [Google Scholar]
- 9. Pulsinelli WA, Brierley JB, Plum F. Temporal profile of neuronal damage in a model of transient forebrain ischemia. Ann Neurol 1982;11:491–498. [DOI] [PubMed] [Google Scholar]
- 10. Heron A, Pollard H, Dessi F, et al. Regional variability in DNA fragmentation after global ischemia evidenced by combined histological and gel electrophoresis observations in the rat brain. J Neurochem 1993;61:1973–1976. [DOI] [PubMed] [Google Scholar]
- 11. Horn M, Schlote W. Delayed neuronal death and delayed neuronal recovery in the human brain following global ischemia. Acta Neuropathol 1992;85:79–87. [DOI] [PubMed] [Google Scholar]
- 12. Iwai T, Hara A, Niwa M, et al. Tmporal profile of nuclear DNA fragmentation in situ in gerbil hippocampus following transient forebrain ischemia. Brain Res 1995;671:305–308. [DOI] [PubMed] [Google Scholar]
- 13. Lockshin RA. Programmed cell death: history and future of a concept. J Soc Biol 2005;199:169–173. [DOI] [PubMed] [Google Scholar]
- 14. Yip KW, Reed JC. Bcl‐2 family proteins and cancer. Oncogene 2008;27:6398–6406. [DOI] [PubMed] [Google Scholar]
- 15. Chavez JC, Hurko O, Barone FC, Feuerstein GZ. Pharmacologic interventions for stroke: looking beyond the thrombolysis time window into the penumbra with biomarkers, not a stopwatch. Stroke 2009;40:e558–e563. [DOI] [PubMed] [Google Scholar]
- 16. Mantz J, Degos V, Laigle C. Recent advances in pharmacologic neuroprotection. Eur J Anaesthesiol 2010;27:6–10. [DOI] [PubMed] [Google Scholar]
- 17. Fisher M. Characterizing the target of acute stroke therapy. Stroke 1997;28:866–872. [DOI] [PubMed] [Google Scholar]
- 18. Kwon OS, Choi JS, Islam MN, Kim YS, Kim HP. Inhibition of 5‐lipoxygenase and skin inflammation by the aerial parts of Artemisia capillaris and its constituents. Arch Pharm Res 2011;34:1561–1569. [DOI] [PubMed] [Google Scholar]
- 19. Lee SJ, Lee US, Kim WJ, Moon SK. Inhibitory effect of esculetin on migration, invasion and matrix metalloproteinase‐9 expression in TNF‐alpha‐induced vascular smooth muscle cells. Mol Med Rep 2011;4:337–341. [DOI] [PubMed] [Google Scholar]
- 20. Lin TH, Lu FJ, Yin YF, Tseng TH. Enhancement of esculetin on arsenic trioxide‐provoked apoptosis in human leukemia U937 cells. Chem Biol Interact 2009;180:61–68. [DOI] [PubMed] [Google Scholar]
- 21. Park SS, Park SK, Lim JH, Choi YH, Kim WJ, Moon SK. Esculetin inhibits cell proliferation through the Ras/ERK1/2 pathway in human colon cancer cells. Oncol Rep 2011;25:223–230. [PubMed] [Google Scholar]
- 22. Wang C, Pei A, Chen J, et al. A natural coumarin derivative esculetin offers neuroprotection on cerebral ischemia/reperfusion injury in mice. J Neurochem 2012;121:1007–1013. [DOI] [PubMed] [Google Scholar]
- 23. Song XY, Hu JF, Sun MN, et al. IMM‐H007, A novel coumarin derivative compound, protects against amyloid beta‐induced neurotoxicity through a mitochondrial‐dependent pathway. Neuroscience 2013;242:28–38. [DOI] [PubMed] [Google Scholar]
- 24. Zuo W, Chen J, Zhang S, et al. IMM‐H004 prevents toxicity induced by delayed treatment of tPA in a rat model of focal cerebral ischemia involving PKA‐and PI3K‐dependent Akt activation. Eur J Neurosci 2014;39:2107–2118. [DOI] [PubMed] [Google Scholar]
- 25. Pulsinelli WA, Buchan AM. The four‐vessel occlusion rat model: method for complete occlusion of vertebral arteries and control of collateral circulation. Stroke 1988;19:913–914. [DOI] [PubMed] [Google Scholar]
- 26. Morris R. Developments of a water‐maze procedure for studying spatial learning in the rat. J Neurosci Methods 1984;11:47–60. [DOI] [PubMed] [Google Scholar]
- 27. Pulsinelli WA, Brierley JB. A new model of bilateral hemispheric ischemia in the unanesthetized rat. Stroke 1979;10:267–272. [DOI] [PubMed] [Google Scholar]
- 28. Bonnekoh P, Barbier A, Oschlies U, Hossmann KA. Selective vulnerability in the gerbil hippocampus: morphological changes after 5‐min ischemia and long survival times. Acta Neuropathol 1990;80:18–25. [DOI] [PubMed] [Google Scholar]
- 29. Broughton BR, Reutens DC, Sobey CG. Apoptotic mechanisms after cerebral ischemia. Stroke 2009;40:e331–e339. [DOI] [PubMed] [Google Scholar]
- 30. Meloni BP, Meade AJ, Kitikomolsuk D, Knuckey NW. Characterisation of neuronal cell death in acute and delayed in vitro ischemia (oxygen‐glucose deprivation) models. J Neurosci Methods 2011;195:67–74. [DOI] [PubMed] [Google Scholar]
- 31. Shi LL, Chen BN, Gao M, et al. The characteristics of therapeutic effect of pinocembrin in transient global brain ischemia/reperfusion rats. Life Sci 2011;88:521–528. [DOI] [PubMed] [Google Scholar]
- 32. Celsis P, Agniel A, Puel M, Rascol A, Marc‐Vergnes JP. Focal cerebral hypoperfusion and selective cognitive deficit in dementia of the Alzheimer type. J Neurol Neurosurg Psychiatry 1987;50:1602–1612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Heiss WD, Zeiler K, Havelec L, Reisner T, Bruck J. Long‐term prognosis in stroke related to cerebral blood flow. Arch Neurol 1977;34:671–676. [DOI] [PubMed] [Google Scholar]
- 34. Wardlaw JM, von Kummer R, Farrall AJ, Chappell FM, Hill M, Perry D. A large web‐based observer reliability study of early ischaemic signs on computed tomography. The Acute Cerebral CT Evaluation of Stroke Study (ACCESS). PLoS ONE 2010;5:e15757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Yoshida F, Sadoshima S, Fujii K, Iino K, Fujishima M. Regional cerebral blood flow in chronic stroke patients with dementia. Jpn J Med 1988;27:172–176. [DOI] [PubMed] [Google Scholar]
- 36. Traystman RJ. Animal models of focal and global cerebral ischemia. ILAR J 2003;44:85–95. [DOI] [PubMed] [Google Scholar]
- 37. Chen J, Jin K, Chen M, et al. Early detection of DNA strand breaks in the brain after transient focal ischemia: implications for the role of DNA damage in apoptosis and neuronal cell death. J Neurochem 1997;69:232–245. [DOI] [PubMed] [Google Scholar]
- 38. Kihara S, Shiraishi T, Nakagawa S, Toda K, Tabuchi K. Visualization of DNA double strand breaks in the gerbil hippocampal CA1 following transient ischemia. Neurosci Lett 1994;175:133–136. [DOI] [PubMed] [Google Scholar]
- 39. Linnik MD, Zobrist RH, Hatfield MD. Evidence supporting a role for programmed cell death in focal cerebral ischemia in rats. Stroke 1993;24:2002–2008. [DOI] [PubMed] [Google Scholar]
- 40. MacManus JP, Hill IE, Preston E, Rasquinha I, Walker T, Buchan AM. Differences in DNA fragmentation following transient cerebral or decapitation ischemia in rats. J Cereb Blood Flow Metab 1995;15:728–737. [DOI] [PubMed] [Google Scholar]
- 41. Chan PH. Mitochondria and neuronal death/survival signaling pathways in cerebral ischemia. Neurochem Res 2004;29:1943–1949. [DOI] [PubMed] [Google Scholar]
- 42. Chaudhry P, Singh M, Parent S, Asselin E. Prostate apoptosis response 4 (Par‐4), a novel substrate of caspase‐3 during apoptosis activation. Mol Cell Biol 2012;32:826–839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Enari M, Sakahira H, Yokoyama H, Okawa K, Iwamatsu A, Nagata S. A caspase‐activated DNase that degrades DNA during apoptosis, and its inhibitor ICAD. Nature 1998;391:43–50. [DOI] [PubMed] [Google Scholar]
- 44. Graham SH, Chen J. Programmed cell death in cerebral ischemia. J Cereb Blood Flow Metab 2001;21:99–109. [DOI] [PubMed] [Google Scholar]
- 45. Gillardon F, Kiprianova I, Sandkuhler J, Hossmann KA, Spranger M. Inhibition of caspases prevents cell death of hippocampal CA1 neurons, but not impairment of hippocampal long‐term potentiation following global ischemia. Neuroscience 1999;93:1219–1222. [DOI] [PubMed] [Google Scholar]
- 46. Ni B, Wu X, Su Y, et al. Transient global forebrain ischemia induces a prolonged expression of the caspase‐3 mRNA in rat hippocampal CA1 pyramidal neurons. J Cereb Blood Flow Metab 1998;18:248–256. [DOI] [PubMed] [Google Scholar]
- 47. Niwa M, Hara A, Iwai T, et al. Caspase activation as an apoptotic evidence in the gerbil hippocampal CA1 pyramidal cells following transient forebrain ischemia. Neurosci Lett 2001;300:103–106. [DOI] [PubMed] [Google Scholar]
- 48. Luo Y, Cao G, Pei W, O'Horo C, Graham SH, Chen J. Induction of caspase‐activated deoxyribonuclease activity after focal cerebral ischemia and reperfusion. J Cereb Blood Flow Metab 2002;22:15–20. [DOI] [PubMed] [Google Scholar]
- 49. Siesjo BK. Mechanisms of ischemic brain damage. Crit Care Med 1988;16:954–963. [DOI] [PubMed] [Google Scholar]
- 50. Kristian T, Siesjo BK. Calcium in ischemic cell death. Stroke 1998;29:705–718. [DOI] [PubMed] [Google Scholar]
- 51. Rothman SM, Olney JW. Excitotoxicity and the NMDA receptor—still lethal after eight years. Trends Neurosci 1995;18:57–58. [DOI] [PubMed] [Google Scholar]
- 52. Rosenblum WI. Histopathologic clues to the pathways of neuronal death following ischemia/hypoxia. J Neurotrauma 1997;14:313–326. [DOI] [PubMed] [Google Scholar]
- 53. Gao TM, Pulsinelli WA, Xu ZC. Prolonged enhancement and depression of synaptic transmission in CA1 pyramidal neurons induced by transient forebrain ischemia in vivo . Neuroscience 1998;87:371–383. [DOI] [PubMed] [Google Scholar]
- 54. Zhang L, Zhang Y, Tian GF, Wallace MC, Eubanks JH. Reversible attenuation of glutamatergic transmission in hippocampal CA1 neurons of rat brain slices following transient cerebral ischemia. Brain Res 1999;832:31–39. [DOI] [PubMed] [Google Scholar]
- 55. Hu BR, Kurihara J, Wieloch T. Persistent translocation and inhibition of Ca21/calmodulin‐dependent protein kinase II in the crudesynaptosomal fraction of the vulnerable hippocampus following hypoglycemia. J Neurochem 1995;64:1361–1369. [DOI] [PubMed] [Google Scholar]
- 56. Hu BR, Park M, Martone ME, Fischer WH, Ellisman MH, Zivin JA. Assembly of proteins to postsynaptic densities after transient cerebral ischemia. J Neurosci 1998;18:625–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. EL‐Husseini AE, Schnell E, Chetkovich DM. PSD‐95 involvement in maturation of excitatory synapses. Science 2000;290:1364–1368. [PubMed] [Google Scholar]
- 58. Takagi N, Logan R, Teves L. Altered interaction between PSD‐95 and the NMDA receptor following transient global ischemia. J Neurochem 2000;74:169–178. [DOI] [PubMed] [Google Scholar]
- 59. Pei DS, Sun YF, Guan QH, Hao ZB, Xu TL, Zhang GY. Postsynaptic density protein 95 antisense oligodeoxynucleotides inhibits the activation of MLK3 and JNK3 via the GluR6.PSD‐95.MLK3 signaling module after transient cerebral ischemia in rat hippocampus. Neurosci Lett 2004;367:71–75. [DOI] [PubMed] [Google Scholar]
- 60. Rosahl TW, Spillane D, Missler M, et al. Essential functions of synapsins I and II in synaptic vesicle regulation. Nature 1995;375:488–493. [DOI] [PubMed] [Google Scholar]
- 61. Corradi A, Zanardi A, Giacomini C, et al. Synapsin‐I‐ and synapsin‐II‐null mice display an increased age‐dependent cognitive impairment. J Cell Sci 2008;121:3042–3051. [DOI] [PubMed] [Google Scholar]