

Abstract
Abstract. Objectives: Recent studies show that measuring pharmacodynamic (PD) effects offers a unique possibility to predict immunosuppression. Thus, in this study we have monitored the PD properties of immunosuppressants on diverse T‐cell functions in heart transplant (HTx) recipients. Materials: PDs and blood concentrations (PK) of three different basis‐immunosuppressive drugs were studied: cyclosporin A (CsA); tacrolimus (TRL) and sirolimus (SRL). T‐cell function was analysed by expression of proliferating cell nuclear antigen (PCNA) labelling, expression of cytokines (IL‐2, IFN‐γ) and surface antigen (for example, CD25) by FACS analysis. Results: In group I, at time points C0 and C2, increased CsA‐PK significantly inhibited expression of IL‐2, IFN‐γ, PCNA and CD25 (P < 0.05). Correlations (r 2) at C2 between inhibition of T‐cell functions (PD) with PK and with drug doses were: CsA‐PK: 0.71–0.91 and CsA‐dose: 0.73–0.87. In group II, increased TRL‐PK over time did not further inhibit expression of CD25, but inhibited PCNA expression more on day 3, and IL‐2 and IFN‐γ expression was significantly higher on days 2 and 3 compared to PD effects of CsA (P < 0.05). Blood SRL concentrations in C0 group III, increased on day 1 and remained stable at days 3 and 4. Expression of PCNA was not altered in the SRL‐PK category, whereas expression of CD25 was higher and expression of cytokines was lower than PD effects of CsA. Conclusions: Our results show that PD effects on T‐cell function can be used to monitor immunosuppression bringing potential to increase the efficacy and safety of immunosuppressive therapy after HTx.
INTRODUCTION
Advances in immunosuppressive drug therapy have improved the outcome for heart transplant (HTx) recipients. New sampling strategies for treatment with cyclosporin (CsA), for example C2 monitoring, have been shown to enhance efficacy compared to standard C0 blood concentration measurements (Cantarovich et al. 1999). Furthermore, the introduction of new potent agents such as tacrolimus (TRL) and sirolimus (SRL) allow clinicians scope in immunosuppressive drug therapy, with better patient recovery from drug side effects (for example, renal dysfunction).
However, as a result of their small therapeutic windows, all these drugs require therapeutic monitoring, which is currently performed by routine measurements of blood‐drug concentration (pharmacokinetic, PK). This is to avoid the occurrence of toxic drug levels and to monitor recipients’ well‐being. However, failure of PK to measure pharmacodynamic (PD) drug effects has prevented preemptive immunosuppressive therapy from being administered at high levels to those recipients who require more intense immunosuppression; this leads to rejection and increased risk of late graft failure (Kahan 2002).
Genetic heterogeneity of the human immune system among transplant recipients means that even identical blood‐drug levels do not affect the immune cells of all recipients equally. Without the knowledge of PD, changes in the sensitivity of immune cells to immunosuppressive drug effects are empirical. Thus, studying PD variability by direct measurement of immunosuppressive drug effects on immune cell functions should increase the efficacy and safety of a class of drug (Dambrin et al. 2000). Over the last decade, an increasing number of methods aimed at studying the PD of immunosuppressants (mainly CsA) have been developed.
Methods described in these studies include cytokine production after mitogen stimulation either by enzyme‐linked immunosorbent assay (ELISA) quantification from serum, by intracellular cytometric analysis of whole blood or by detection at the messenger RNA (mRNA) level (van den Berg et al. 1998; Stein et al. 1999; Härtel et al. 2002; Hodge et al. 2005). Previous methods have included assays to measure activity of a drug target enzyme, lymphocyte proliferation or lymphocyte subsets (Chang et al. 1996; Budde et al. 2000; Weimer et al. 2000; Stalder et al. 2003; Grinyóet al. 2004; Hutchinson et al. 2004). All of these have one or more of the following limitations: (1) numbers of PD parameters measured; (2) low number of recipients; (3) analysis of PD effects at one time point only; (4) no correlation between PD and both PK and drug dose; and (5) labourious and time‐consuming methods to assess PD. Our group has developed various PD assays to evaluate T‐cell function, and an earlier study has assessed PD of immunosuppressive therapy in an experimental animal model. We determine the relationship between PD effects on different T‐cell functions and PK and drug doses and we have observed that changes of T‐cell function over time in immunosuppressed allograft recipients correlates closely with the severity of histopathological events within the allograft tissue. Furthermore, we have been able to detect new mechanisms of drug action and could distinguish between synergistic, additive or antagonistic drug interactions after combination therapy (Barten et al. 2002; Barten et al. 2004). In human whole blood, we measure drug potency and efficacy and determine interactions with a combination of drugs with various mechanisms of action in vitro, using T‐cell function assays (Barten et al. 2003).
We have designed the present study to evaluate potential benefits of our T‐cell function assays, which are based on the technologies of whole blood and flow cytometry, to assess PD effects of CsA, TRL or SRL‐based immunosuppression in stable HTx recipients at different time points, and also to define the correlation between PD and both PK and drug dose.
MATERIALS AND METHODS
Study design
Here we have monitored the PK and PD of three differently based immunosuppressive drugs in HTx recipients. Group I consisted of 45 HTx recipients taking CsA, which we monitored during their routine outpatient visits. The second group consisted of nine HTx recipients with gingival hypertrophy whose treatment was changed from CsA to TRL, and then they were monitored for 3 days. Group 3 consisted of 10 HTx patients with severe renal dysfunction who were changed from CsA to SRL, and were then monitored for 4 days. Patients from all groups received mycophenolate mofetil (MMF) as adjuvant therapy in doses 250, 500 or 1000 mg in the evening and of 250, 500, 1000 or 1500 mg in the morning.
In the first group, blood for measuring PK and PD values was taken from all recipients before the morning dose of CsA (trough value) and 2 h after dosing (2‐h value). Blood from patients of groups II and III was drawn to assess PK‐ and PD‐morning trough‐values of CsA‐therapy before their treatment was changed (day 0) and consecutively over 3 (group II) or 4 days (group III), respectively, before new‐drug dosing. For all groups, collected blood was stored at room temperature for analysis of PK and PD within 1 h after receipt.
Reagents
Culture medium (CM) was prepared using RPMI 1640 supplemented with 100 U/ml of penicillin and 100 µg/mg streptomycin, obtained from Sigma Chemical GmbH (Steinheim, Germany), and with 2 mm l‐glutamine (Biochrom, Berlin, Germany). Concanavalin A (Con A) was stored at −70 °C. Phorbol 12‐myristate 13‐acetate (PMA), ionomycin (IONO) and brefeldin (BFA) were purchased from Sigma and all were dissolved in dimethyl sulfoxide (DMSO, Sigma) as stock solution of 100 mg/ml (PMA), 500 mg/ml (IONO) and 5 mg/ml (BFA). All PMA, IONO and BFA were stored at −20 °C and working solutions were freshly prepared in phosphate buffered saline (PBS) for PMA and BFA and in CM for IONO, respectively. The IntraPrep® reagent kit for intracellular cytokine detection was purchased from Coulter, Marseille, France.
All fluorescein isothiocyanate (FITC), phycoerythrin (PE) and phycoerythrincyanin5 (Cy)‐mAbs (anti‐CD25, anti‐CD134, anti‐CD95, anti‐CD45, anti‐CD14, anti‐CD3, anti–INF‐γ, anti–TNF‐α, anti–IL‐2, anti–IL‐4) and the mouse isotype controls IgG2aκ, IgG2bκ and IgG1κ, were purchased from Becton Dickinson (Heidelberg, Germany). Anti‐PCNA (FTIC, Clone PC 10) was purchased from Dako Corporation (Hamburg, Germany). RNAse, propidium iodide (PI) and sodium azide were purchased from Sigma. PBS was made by dissolving 7.013 g NaCl, 0.2 g KCl, 1.513 g Na2HPO4 and 0.2 g KH2PO4, all purchased from Roth (Karlsruhe, Germany), in 1 l of distilled water, and by adjusting the pH to 7.4 by adding HCl (Riedel‐de‐Haen, Seelze, Germany) or 1 N NaOH (Merck, Darmstadt, Germany). Red blood cell (RBC) lysis buffer was made daily by dissolving 8.29 g NH4Cl, 1 g KHCO3 and 37.2 mg Na2‐ethylenediaminetetraacetic acid (EDTA), all obtained from Roth, in 1 l of deionized H2O (pH 7.2). Permeabilizing buffer contained 1% heat‐inactivated foetal calf serum (Sigma), 0.1% saponin (Roth) and 0.1% sodium azide in PBS. Formaldehyde solution and absolute methanol were purchased from Merck.
Pharmacokinetic analysis
At all time points, blood, CsA and TRL concentrations were detected using immunoassay EMIT® (Dade Behring, Newark, USA) and blood SRL concentrations were measured by liquid chromatography tandem mass‐spectroscopy (LC‐MS/MS; Bruker Daltonic, Bremen, Germany).
Pharmacodynamic analysis
For analysis of expression of cell proliferation markers and of surface antigens, aliquots of 200 µl of heparinized blood were diluted in 1791 µl of CM and were added to the wells of flat‐bottomed 24‐well tissue culture microtitre plates (TPP, Switzerland). To each well, 9 µl of Con A or 9 µl CM (unstimulated cultures) was added to provide the final volume of 2000 µl. Final dilution of blood in the wells was 1/10 and final concentration in 200 µl blood cultures for Con A was 2.5 µg/ml. Cultures were then incubated for 3 days at 37 °C in a humidified 5% CO2 air water‐jacketed incubator.
For detection of accumulated intracellular cytokines, 500 µl of undiluted heparinized blood was stimulated with 10 µl of each of PMA and IONO to reach a final concentration of PMA of 25 ng/ml and of IONO 750 ng/ml, respectively. Culture tubes were incubated upright in a humidified 37 °C, 7% CO2 incubator for a total time of 6 h, with the first hour of incubation in the absence of BFA to enable antigen processing by antigen‐presenting cells. All samples were analysed with a LSR II flow cytometer (Becton Dickinson, Heidelberg, Germany) equipped with an air‐cooled argon laser (488 nm) using FACSDIVATM software (Becton Dickinson). Unstimulated blood cultures were used as negative controls. Specific controls included replacement of primary mAbs with isotype mouse immunoglobulins. Analysed regions were set to achieve non‐specific binding of < 1% within the positive event region.
T‐cell proliferation
After incubation, the contents of each well were mixed and 800 µl was used for the detection of PCNA expression/DNA content, with the combination of FITC‐mAb and PI to identify the cells that were in S/G2M phases of the cell cycle. Eight millilitres of lysis buffer were mixed with 800 µl diluted blood, and RBCs were lysed for 10 min at room temperature. Leucocytes were pelleted (200 g, 10 min) and were again washed with 2 ml PBS. Leucocytes were fixed by adding 1 ml PBS containing 1% (v/v) formaldehyde, and were then kept on ice for 5 min. After washing (200 g, 5 min) with 2 ml PBS, the cells were re‐suspended with 2 ml of ice‐cold 100% methanol and stored for at least 10 min at 4 °C. Prior to staining, the cells were washed (200 g, 5 min) in 2 ml PBS to remove any remaining methanol. Cell pellet was then re‐suspended in a staining mixture containing 107 µl permeabilizing buffer, 10 µl RNAse A (stock solution 100 mg/ml in H2O), 5 ml PI (stock solution 1 mg/ml in H2O) and 2.5 µl anti‐PCNA FITC‐mAb. The cells were incubated in the staining mixture for 30 min at 37 °C. After washing with 2 ml PBS, leucocytes were pelleted (200 g, 5 min) and re‐suspended in 500 µl PBS containing 10 µg/ml PI. Total lymphocytes were collected using forward light scatter (FLS) and side light scatter (SLS) and were displayed as two‐colour PCNA/DNA dot plots as PCNA‐positive cells with S/G2M‐phase DNA content. Ten thousand events were analysed per sample.
T‐cell surface antigens
Surface antigens were detected post‐incubation time using the contents of 600 µl of diluted blood. A combination of FLS and SLS, with anti‐CD45/CD14‐analysis, was performed to distinguish leucocyte subpopulations. Lymphocyte gates were optimized by a three‐step sequential analysis of immunoflourescence and light scattering to include all normal and activated lymphocytes, according to methods described previously (Barten et al. 2003). A forward light 3 (FL3) histogram was created to confine the evaluation to CD3+ T cells, and FL2/FL3 staining characteristics (based on the contour plot) of each antibody were determined on FL1 (CD3)‐positive cells. To each 200 µl of diluted, incubated blood from pooled cultures, 50 µl of mAb (1/50 dilution in PBS containing 3% FCS and 0.1% NaN3) were added. After incubation for 30 min in the dark at room temperature (RT), samples were washed (200 g, 5 min) with 2 ml PBS. Four millilitres of lysis buffer was added to all tubes and RBCs were lysed at RT for 10 min. Leukocytes were washed (200 g, 5 min) with 2 ml PBS and then pelleted. Prior to analysis, leucocytes were re‐suspended in 250 µl PBS containing 1% (v/v) formaldehyde. Ten thousand events were analysed per sample.
T‐cell cytokines
Ten µl of secretion‐inhibitor BFA was added directly to the whole blood cultures to reach the BFA final concentration of 10 µg/ml after one hour of the incubation. After the incubation, 50 µl of blood was incubated with 5 µl of each mAb, anti‐CD3 PE‐Cy5, anti‐CD14 FITC and anti‐CD45 PE to distinguish between leucocyte subsets, and three times 50 µl was incubated with 5 µl of anti‐CD3 for further intracellular cytokine detection, for 15 min at RT. Subsequently, cells were fixed and permeabilized using a standard reagent kit and the manufacturer's immunofluorescence staining protocol (IntraPrep® Coulter). Cells were fixed by adding 100 µl of reagent I (containing 5.5% formaldehyde) to each sample and incubating at RT in the dark for 15 min. After washing (4 ml PBS, 200 g, 6 min), cells were permeabilized by adding 100 ml of reagent II (containing saponin) to each sample 5 min before adding 3 µl of each anticytokine mAb (INF‐γ FITC, TNF‐α PE, IL‐2 FITC, IL‐4 PE) and incubating the samples at RT in the dark for 20 min. After washing (4 ml PBS, 200 g, 6 min), 100 µl of PBS containing 1% formaldehyde(volume/volume) was added to each sample before flow cytometry analysis was performed. Ten thousand light scatter gated lymphocytes were analysed per sample.
Statistical analysis
Statistical analysis was performed using the spss software package (SPSS Inc., Chicago, USA). All data are expressed as median and statistical significance was analysed between two groups with equal variance, by one‐way anova followed by the Mann/Whitney (MW) rank sum test. Graphic data were presented as box plots with Sigmaplot (Systat, Erkrath, Germany). For study group I, PD effects before CsA (trough values, E0) were used as baseline values to calculate the percentage inhibition of PD effects 2 h after dosing (E2). Coefficients of correlation (r 2) between PD, CsA dose levels or PK were determined using the Pearson product rank order (anova on ranks). PD effects for each parameter in groups II and III of CsA‐therapy (day 0) were compared with the PD effects of TRL‐therapy and/or SRL‐therapy for all time points at days 1–3 or 4, respectively (anova on ranks). P < 0.05 was considered significant.
RESULTS
CsA‐based immunosuppression
At the time of the study, the mean age of our recipients was 62 years (range 46–70 years), 35 of whom were men. The mean existence after HTx was 70 months, with a range of 127–49 months. Figure 1 illustrates individual data of PCNA expression before (0 h) and after (2 h) CsA therapy for two HTx patients. Blood CsA concentrations at 0 h of 123 ng/ml (recipient 1) and of 128 ng/ml (recipient 2) produced PCNA expression levels of 15% and of 19%, respectively (Fig. 1a). A CsA dose of 75 mg increased blood concentrations to either 589 ng/ml or to 621 ng/ml for each recipient. Inhibition of PCNA expression between the recipients differed, however, 2 h after dosing (Fig. 1b).
Figure 1.
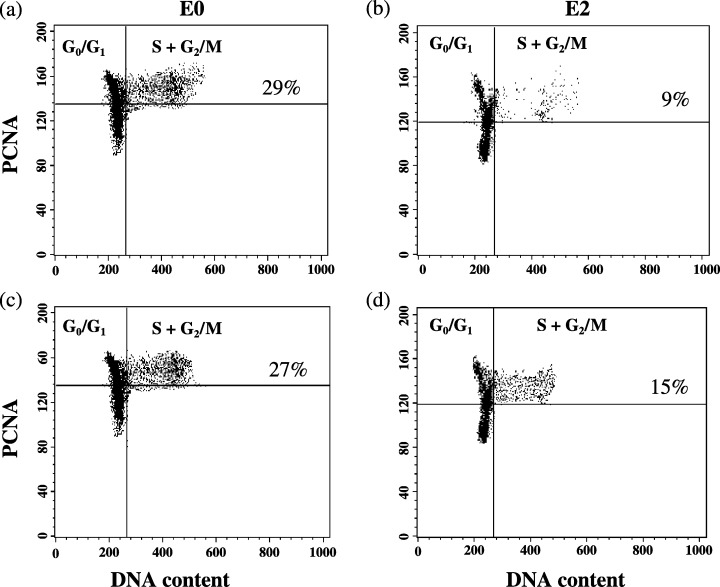
Examples of T‐cell proliferation of two HTx recipients before (EO, parts a and c) and two hours (E2, parts b and d) after dosing of cyclosporin. Two colour flow cytometric analysis of PCNA expression and DNA content of two HTx recipients before (E0, parts a and c) and two hours (E2, parts c and d), after intake of the morning dose of cyclosporin. Dot plots show Con A stimulated lymphocytes in whole blood after three days incubation.
For all 45 HTx patients, evening doses of CsA (25, 50, 75 or 100 mg) produced C0‐levels of 122 ng/ml and morning doses of CsA (50, 75 or 100 mg) produced C2‐levels of 408 ng/ml. These blood CsA concentrations significantly reduced expression of all T‐cell function parameters 2 h after dosing, in comparison to expression before dosing, with the exception of IL‐4 (Fig. 2 and b; P < 0.05, MW). Despite this high correlation of all PD parameters with PK, there was variation concerning dose correlation with PK among results with differing PD parameters (Table 1).
Figure 2.
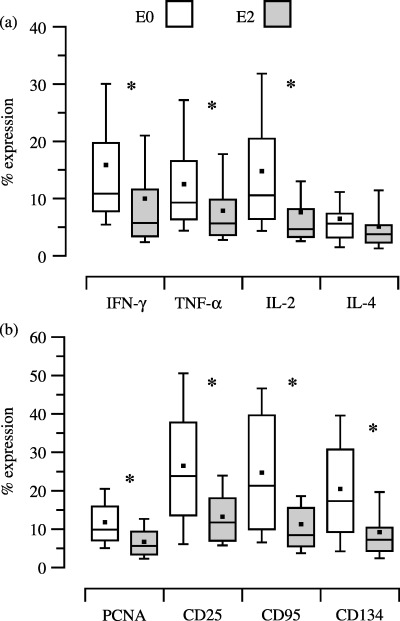
Pharmacodynamics of cyclosporin before (EO) and 2 h (E2) after dosing measured by expression of cytokines (part a), proliferation (PCNA) and surface antigens (part b). T‐cell function measured by levels of expression of intracellular cytokines (IFN‐γ, TNF‐α, IL‐2, IL‐4) and by expression of proliferation marker (PCNA) and surface antigens (CD25, CD95, CD134) of 45 HTx recipients before (E0) and 2 h (E2) after intake of the morning cyclosporin dose. Collected blood was either stimulated with PMA plus IONO for detection of intracellular cytokine expression, or with Con A for detection of PCNA and surface antigens. After an incubation period of 5 h for PMA, plus IONO assay or for 3 days for the Con A assay, harvested blood was analysed by multi‐colour flow cytometry. Statistically significant differences (MW rank sum test) between expression of intracellular cytokines, cell proliferation and surface antigen expression at E0 and E2 are indicated (*).
Table 1.
Correlation (r 2) between pharmacodynamic effects (PD) at E2, drug pharmacokinetics (PK) at C2 and with drug dose given at C0, of cyclosporin‐based immunosuppressive therapy in HTx recipients
| PD | PK | Dose |
|---|---|---|
| PCNA | 0.86 | 0.78 |
| CD25 | 0.84 | 0.79 |
| CD95 | 0.81 | 0.82 |
| CD134 | 0.89 | 0.91 |
| IL‐2 | 0.77 | 0.78 |
| TNF‐α | 0.67 | 0.70 |
| IL‐4 | 0.62 | 0.63 |
| IFN‐γ | 0.68 | 0.61 |
TRL‐based immunosuppression
In this study group, the mean age of the heart recipients was 52 years ranging between 32 and 64 years. Seven recipients were men and the mean time after HTx was 52 months with a range of 87–202 months. Figure 3 shows the PK values and PD effects of CsA therapy (day 0, time point 0 h) compared to PK values and PD effects of TRL therapy at day 1 to day 3. Dosing of 3 mg TRL b.i.d. increased blood TRL concentration (Fig. 3a). This increase in blood TRL concentration was reflected in the decrease in expression of the cytokines INF‐γ, IL‐2 and TNF‐α reaching significance on day 3 at time points 0 h and 12 h, compared to expression on day 0 at time point 0 h with CsA therapy (P < 0.05, MW; Fig. 3b). However, increased blood TRL concentration did not alter expression of either CD25 or CD95 (Fig. 3c). This was the opposite of the expression of PCNA and CD134, which were influenced by increased TRL concentrations, which lead to a decrease in expression of these, to a minimum on day 3 at 12 h (Fig. 3c). Compared to the effects of CsA therapy on day 0, TRL decreased expression of PCNA and CD134 on day 3 (P < 0.05, MW), although expression of CD25 and CD95 were comparably high before and after drug change (Fig. 3c).
Figure 3.
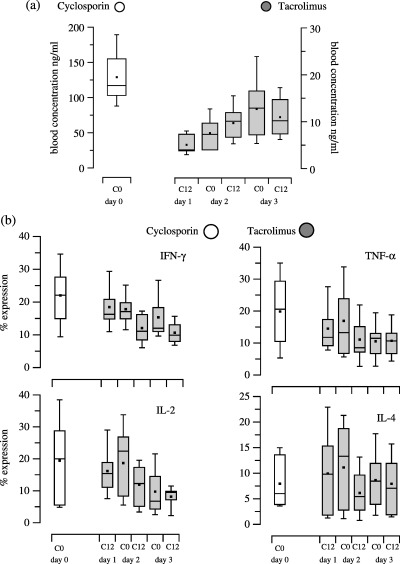
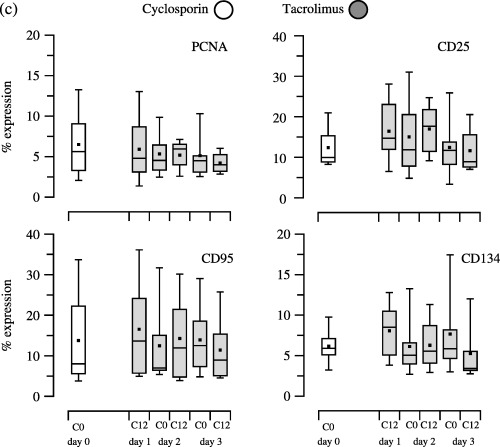
Morning through pharmacokinetic (part a) and pharmacodynamic (parts b and c) values of 9HTx recipients converted from cyclosporin treatment (day 0) to tacrolimus therapy (day 1 to day 3). Trough pharmacokinetic (part a) and pharmacodynamic (parts b and c) values of 9 HTx recipients with gingival hyperplasia, who were changed from cyclosporin treatment (day 0) to tacrolimus therapy (day 1 to day 3). Blood cyclosporin and tacrolimus concentrations were measured by EMIT®. Flow cytometric analysis of T‐cell function was measured by percentage expression of: intracellular cytokines (IFN‐γ, TNF‐α, IL‐2, IL‐4), cell proliferation (PCNA) and surface antigen expression (CD25, CD95, CD134).
SRL‐based immunosuppression
At the study time, the mean age of our recipients in this group was 59 years, ranging from 46 to 66 years. Seven of the recipients were men and the mean time after HTx was 46 months with a range of 8 to 70 months. Figure 4 shows individual PK and PD data of two recipients before and after changing their treatment from CsA to SRL. Dosing of 3 mg SRL q.i.d. on day 1 and of 1 mg SRL q.i.d. on days 2 and 3 increased blood SRL concentration for each recipient, with the maximum on day 3. On day 4, SRL concentrations dropped or remained stable for recipient 1 and 2, respectively (Fig. 4a,b). Despite the maximum of a SRL concentration on day 3, expression of all PD parameters increased in the blood of patient 1, but on day 4, expression of PD parameters decreased parallel with the decrease of SRL PK (Fig. 4b). Compared to CsA therapy, SRL reduced the expression of IFN‐γ and IL‐2 significantly (P < 0.05, MW), whereas expression of PCNA and CD25 were higher with SRL than with CsA in patient I (Fig. 4b). For patient 2, changes of expression of PD parameters were minimal over the study period regardless of the increased SRL PK, except for an increased expression of CD25 on day 4 (Fig. 4d). Besides the expression of CD25, the expression of PD parameters was comparably high in recipient 2 between CsA and SRL (Fig. 4d).
Figure 4.
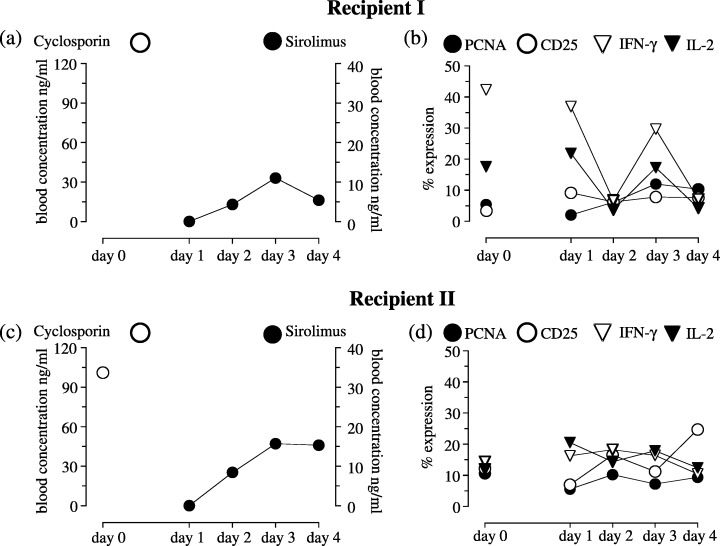
Pharmacokinetic and pharmacodynamic morning trough values of two HTx recipients converted from cyclosporin to sirolimus. Pharmacokinetic and pharmacodynamic morning trough values of two HTx recipients who were changed from cyclosporin to sirolimus‐based immunosuppressive therapy as a result of severe renal dysfunction. Blood cyclosporin and sirolimus concentrations were measured by EMIT® or LC‐MS/MS, respectively. Flow cytometric analysis of T cell function was measured by percentage expression of: intracellular cytokines (IFN‐γ and IL‐2), cell proliferation (PCNA) and surface antigen expression (CD25).
Table 2 shows the PK and PD values for all 10 HTx recipients. Dosing of 3 mg SRL q.i.d. at day 1 and of 1 mg q.i.d. at days 2 and 3 increased blood SRL concentrations over the study period.
Table 2.
Analysis of pharmacokinetics (PK) and pharmacodynamics (PD); morning trough values of 10 HTx recipients suffering from severe renal dysfunction, before and after treatment change from cyclosporin to sirolimus
| Cyclosporin | Sirolimus | ||||
|---|---|---|---|---|---|
| Day 0 | Day 1 | Day 2 | Day 3 | Day 4 | |
| PK | (ng/ml) | (µg/ml) | |||
| 98.0 | 0.0 | 9.0 | 9.8 | 9.3 | |
| PD | (% expression) | (% expression) | |||
| PCNA | 4.5 | 4.2 | 5.8 | 6.4 | 4.0 |
| CD25 | 12.4 | 13.1 | 13.4 | 15.7 | 19.4 |
| CD95 | 8.3 | 8.1 | 9.2 | 9.8 | 15.3 |
| CD134 | 8.0 | 7.0 | 8.2 | 7.1 | 12.3 |
| INF‐γ | 28.0 | 25.4 | 12.3 | 19.2 | 12.7 |
| TNF‐α | 16.4 | 15.8 | 13.6 | 14.5 | 13.5 |
| IL‐2 | 13.3 | 13.0 | 7.6 | 10.3 | 9.3 |
| IL‐4 | 5.7 | 5.6 | 7.6 | 5.5 | 6.4 |
Expression of PCNA and of all cytokines did not change after change to SRL treatment regardless of increasing blood SRL concentrations. However, expression of surface antigens increased on day 4 compared to that on day 3, despite similarly high SRL concentrations on both days (Table 2). Values of PD parameters on days 0 and 4 showed that SRL therapy caused a decrease in expression of IFN‐γ, TNF‐α and IL‐2, which was significantly different for IFN‐γ (P < 0.05, MW; Table 2). In contrast was that surface antigen expression was higher with SRL treatment compared to that with CsA; these differences were significant for CD25 and CD95 (P < 0.05, MW; Table 2).
DISCUSSION
Therapeutic drug monitoring based on measuring blood or PK of various components has been important in the clinical management of organ transplantation since its adoption for assessment of immunosuppressive drugs. Because rejection or infection occurs at irregular intervals, immunosuppressive drug therapy is often empirical and prophylactic in nature. In addition, occurrence of adverse effects may be insidious, and knowledge of an upper or lower drug concentration limit can be useful in avoiding drug treatment‐emergent adverse effects (Oellerich et al. 2006). Specifically, therapeutic drug monitoring requires application of reliable and effective methods in order to achieve safe and successful immunosuppressive therapy. Over the recent years, it has been realized that measuring biologically relevant events (PD) offer a unique possibility to predict drug efficacy in vivo (Barten et al. 2004).
In this study, we show for the first time the potential value of monitoring PD effects on T‐cell function with CsA‐, TRL‐ or SRL‐based immunosuppression therapy in peripheral blood, over time, to enhance PK‐based therapeutic drug monitoring in HTx patients. Previously, PD assays have been used to measure the activity of calcineurin or ionosin monophospathe dehydrogenase (IMPDH) (the target enzymes of CsA or MMF, respectively), in recipients of renal transplantation (NTx). These studies have shown that inhibition of calcineurin correlates with highest blood CsA concentration, which occurs 2 h after dosing (Grinyo et al. 2004), and that knowledge of pre‐transplant IMPDH activity may influence post‐transplant outcome (Budde et al. 2000). The disadvantages of measuring drug target enzyme activity alone lie in that these assays measure enzyme activity alone, not the numerous effects on immune cell function caused by enzyme inhibition. Furthermore, these assays cannot measure the net effect on the immune system caused by multiple immunosuppressive drugs with different mechanisms of action, such as for example, CsA and MMF.
Further approaches to monitoring PD effects have been performed, mainly for CsA, by studying inhibition of T‐cell cytokines such as IL‐2, IFN‐γ and TNF‐α. A study by Stein et al. demonstrated the close relationship between IL‐2 inhibition and peak blood CsA concentration in healthy human controls (Stein et al. 1999), whereas a second investigation showed (in NTx) that in contrast to IL‐2 levels, INF‐γ seemed to be a more specific marker in the PD of CsA (Grinyo et al. 2004). In contrast to these works, numerous studies have shown that using the ELISA technique for analysis, flow cytometric results can assess intracellular production of cytokines by T cells, after ex vivo mitogen stimulation of whole blood. Thus, in NTx recipients, the number of cytokine‐producing T cells is significantly reduced compared to levels in healthy controls; this is pronounced for IL‐2 compared to IFN‐γ and TNF‐α. However, CsA trough levels do not correlate with the decreased IL‐2 production (Stalder et al. 2003). This result has been confirmed in a study of lung transplanted (LTx) individuals where CsA and TRL trough levels did not correlate with the inhibition of cytokine expression (Hodge et al. 2005). However, our results have shown that among cytokines, the highest correlation is for IL‐2 between blood CsA concentration at C2 and a CsA dose given at C0 (Table 1). This confirms the results of a study in NTx patients with good correlation between CsA trough levels and inhibition of IL‐2 expression (van den Berg et al. 1998). In our change‐of‐drug studies, inhibition of cytokine expression correlated with trough blood TRL concentration on both study days (Fig. 2), whereas in the SRL group, increased blood SRL concentration correlated with distinct of cytokine inhibition only on day 4 (Table 2). The least effect on expression, regardless of which immunosuppressive‐based drug, in our study was reached with IL‐4 compared to inhibition of IL‐2, TNF‐α and IFN‐γ. Our observations have been confirmed in LTx recipients where a CsA and TRL based‐immunosuppression decreased IL‐2, TNF‐α and IFN‐γ expression, and increased IL‐4 expression in healthy controls (Hodge et al. 2005). An optional approach to cytokine analysis might include measurements at DNA transcriptional level, as observed in NTx recipients, where CsA therapy lead to a significant delay in IL‐2, IL‐4 and TNF‐α peaks in mRNA expression (Härtel et al. 2002).
Variations in correlation between PD effects and PK concentration seem to confirm that immunosuppressive drugs produce a variety of responses in patients even when drug level and the drug dosage are similar. In general, monitoring cytokine production is problematic as a result of restriction of events to certain cell cycle phases, different half‐lives for circulating of cytokines and changes in up‐ and down‐regulation of cytokine gene expression. Thus, technical diversions, for example time of collection and the assay procedure, can influence clinical results (Fitzpatrick & Kelso 1998).
However, in our laboratory we go beyond these approaches and assess not only T‐cell cytokine production, but we also simultaneously determine the expression of both T‐cell proliferation and T‐cell surface antigen expression, all of which potentially play roles in costimulation, adhesion and apoptosis of the immune cells (Barten et al. 2003). In the present study, increase in blood CsA concentration 2 h after dosing led to a significant decrease in T‐cell proliferation (PCNA measurement), as well as T‐cell surface antigen expression. We found a high correlation between PD effects after CsA and both blood CsA concentration and CsA dose. Furthermore, overall correlation was higher for T‐cell proliferation and surface antigen expression than compared to any correlation with T‐cell cytokines (Table 1). This may implicate the preferable use of PCNA and surface antigen expression as surrogate markers than cytokines alone for the combination therapy of CsA and MMF. Additionally, the early anti‐proliferative result to CsA treatment may be the result of additive effects in combination with the anti‐proliferative drug MMF, which was observed earlier by our group, in vitro and in vivo, using whole blood of humans and animals (Barten et al. 2003; Barten et al. 2005).
The potential of analysis of expression of T‐cell surface antigens has been shown in HTx patients, where increased expression of CD4+CD25+ by T cells correlated with grade of rejection, in contrast to expression of cytokines (Chang et al. 1996). In two other studies on NTx recipients there was a significant effect of CsA treatment on decreased expression of T‐cell surface antigens (for example, CD25 and CD95), and of proliferation at morning trough values compared to human healthy controls (Stalder et al. 2003; Hutchinson et al. 2004). These two investigations used either flow cytometry or the incorporation of radio‐nucleotides to measure drug effects on lymphocyte proliferation. We prefer the sensitivity, versatility and speed of flow cytometry compared to techniques that rely on radio‐nucleotide incorporation to measure inhibition of cell proliferation as an index of drug effect, as the technique of flow cytometry is more specific for the measurement of drug potencies and efficacies in vivo (Gummert et al. 1999).
On the other hand, all these studies measure drug effects at one single time point only, which could be misleading for interpretation of a drug's overall efficacy. Thus, it is suggested that the area of PD parameter expression over time may be more appropriate (Barten et al. 2002; Härtel et al. 2002). Yet, because of the difficulties in obtaining blood at several time points, especially in out‐patient clinic recipients, we propose to analyse the blood at least two time points (for example, C0 and C2 for CsA) to provide all information of drug efficacy and of knowledge of the relationship between PD and PK, to be able to predict outcome as previously (Brunet et al. 2003). Further work is required to determine whether a time point different from C0 would be the best predictor of the immunosuppressive effect (PD) of TRL and SRL, enhancing the value of PK monitoring for these two drugs.
An ongoing goal of our study is to monitor conditions of drug change over, using our PD assays of T‐cell function. Up to date, only one study has observed PD effects after the change from CsA to TRL, in NTx (Weimer et al. 2000). In that study, TRL further inhibited IL‐2 expression and expression of T‐cell surface antigens (CD154, CD28, CD54) compared to CsA therapy, which confirms our results. Thus, we observed that TRL inhibited expression of IL‐2, TNF‐α and IFN‐γ significantly more and expression of both PCNA and the surface antigen CD134 on day 3 compared to CsA therapy. However, no differences of inhibition were observed between TRL and CsA therapy for IL‐4, CD25 and CD95. Furthermore, increasing blood TRL concentrations did not further inhibit the expression of these latter PD parameters, showing the importance of measuring PD drug effects to complete therapeutic drug monitoring.
After conversion from CsA to SRL therapy, we determined that increased blood SRL concentrations over the study time had different effects on PD parameters. SRL had a significantly higher effect on the expression of the cytokines other than IL‐4 compared to CsA, but SRL less effectively inhibited expression of T‐cell surface antigens compared to CsA (Table 2). Our data suggest that SRL dose adjustment relying on blood SRL concentrations does not predict SRL effect on immune cells. This is in accordance with results of our previous experimental animal study where higher SRL doses lead to an increase of blood SRL concentration, but these concentrations did not further inhibit T‐cell proliferation or surface antigen expression (Barten et al. 2004). The individual data of our studies (1, 4) reflect the potential value of monitoring the PD drug effect to tailor immunosuppressive drug therapy individually to avoid toxicity and to enhance efficacy.
Despite these promising results, until now it is not known to what extent T‐cell function parameters, like IL‐2, PCNA or CD25 expression, may be altered by infection or by rejection of a transplanted organ. Furthermore, it is not clear whether baseline values for individual patients are required, nor is an optimal PD target range known. Based on the fact that our results were obtained from a small number of recipients, in the future, multicentre studies with an adequate number of transplant recipients would be needed to confirm our observations. Current research investigations in human solid organ transplants are focused on the field of pharmacogenetics to provide information concerning how genetic differences influence individual organ recipients’ responses to drug PK and PD, but prospective studies are needed to determine their relevance to clinical practice (van Gelder et al. 2004).
In conclusion, PD studies to determine drug efficacy in vivo could be useful prior to making clinical decisions to determine dose and type of immunosuppressive drug best suited to a patient. However, PD will not eliminate PK. Rather, the complementary technologies of PK and PD, defining the fate and the effect of a drug, respectively, could help improve therapeutic drug monitoring for a more effective and safe result. Furthermore, the recent rapid advances in our scientific understanding of both clinical chemistry and genomics will aid in developing diagnostic assays of biomarkers for PK and for PD; then pharmacogenetics should be the foundation for establishing personalized medical care in transplantation medicine in the future.
AUTHORS’ CONTRIBUTIONS
All authors have read and approved the final manuscript. M.J.B., A.T., H.B.B. and S.D. designed the study; M.J.B. wrote the manuscript; M.J.B. and A.T. carried out the flow cytometry assays; J.G. did the flow cytometry analysis; M.J.B., H.B.B., S.D. and F.W.M. critically discussed the manuscript; J.F.G. was the principal investigator of the study.
ACKNOWLEDGEMENTS
Part of this work was presented at the 16th Annual Conference of the German Society for Cytometry, DGfZ, October 12–18th, 2005, http://www.dgfz.org. We gratefully acknowledge the skillful technical assistance of Anja Sagner. Markus J. Barten and Jan F. Gummert were supported by Deutsche Forschungsgemeinschaft grant GU 472/2–1.
REFERENCES
- Barten MJ, Van Gelder T, Gummert JF, Boecke K, Shorthouse R, Billingham M, Morris RE (2002) Pharmacodynamics of mycophenolate mofetil after heart transplantation: new mechanisms of action and correlations with histologic severity of graft rejection. Am. J. Transplant. 2, 719–732. [DOI] [PubMed] [Google Scholar]
- Barten MJ, Dhein S, Chang H, Bittner HB, Tarnok A, Rahmel A, Mohr FW, Gummert JF (2003) Assessment of immunosuppressive drug interaction: inhibition of lymphocyte function in peripheral human blood. J. Immunol. Methods 283, 99–114. [DOI] [PubMed] [Google Scholar]
- Barten MJ, Shipkova M, Bartsch P, Dhein S, Streit F, Tarnok A, Armstrong VW, Mohr FW, Oellerich M, Gummert JF (2005) Mycophenolic acid interaction with cyclosporine and tacrolimus in vitro and in vivo. Evaluation of additive effects on rat blood lymphocyte function. Ther. Drug Monit. 27, 123–131. [DOI] [PubMed] [Google Scholar]
- Barten MJ, Streit F, Boeger M, Dhein S, Tarnok A, Shipkova M, Armstrong VW, Mohr FW, Oellerich M, Gummert JF (2004) Synergistic effects of sirolimus and tacrolimus: analysis of immunosuppression on lymphocyte proliferation and activation in whole blood. Transplantation 77, 1154–1162. [DOI] [PubMed] [Google Scholar]
- Van Den Berg AP, Twilhaar WN, Mesander G (1998) Quantitation of immunosuppression by flow cytometric measurement of the capacity of T cells for interleukin‐2 production. Transplantation 65, 1066–1071. [DOI] [PubMed] [Google Scholar]
- Brunet M, Campistol JM, Millan O, Vidal E, Esforzado N, Rojo I, Jimenez. O, Oppenheimer F, Corbella J, Martorell J (2003) Pharmacokinetic and pharmacodynamic correlations of cyclosporine therapy in stable renal transplant patients: evaluation of long‐term target C2. Int. Immunopharmacol. 3, 987–999. [DOI] [PubMed] [Google Scholar]
- Budde K, Glander P, Bauer S, Braun K, Waiser J, Fritsche L, Mai I, Roots I, Neumayer HH (2000) Pharmacodynamic monitoring of mycophenolate mofetil. Clin. Chem. Lab. Med. 38, 1213–1216. [DOI] [PubMed] [Google Scholar]
- Cantarovich M, Elstein E, De Varennes B, Barkun JS (1999) Clinical benefit of Neoral dose monitoring with cyclosporine 2‐hr post‐dose levels compared with trough levels in stable heart transplant patients. Transplantation 68, 1839–1842. [DOI] [PubMed] [Google Scholar]
- Chang DM, Ding YA, Kuo SY, Chang ML, Wei J (1996) Cytokines and cell surface markers in prediction of cardiac allograft rejection. Immunol. Invest. 25, 13–21. [DOI] [PubMed] [Google Scholar]
- Dambrin C, Klupp J, Morris RE (2000) Pharmacodynamics of immunosuppressive drugs. Curr. Opin. Immunol. 12, 557–562. [DOI] [PubMed] [Google Scholar]
- Fitzpatrick DR, Kelso A (1998) Independent regulation of cytokine genes in T cells. Transplantation 65, 1–5. [DOI] [PubMed] [Google Scholar]
- Van Gelder T, Hesselink DA, Van Hest RM, Mathot RAA, Van Schaik R (2004) Pharmacogenetics in immunosuppressive therapy: the best thing since TDM? Ther. Drug Monit. 26, 343–346. [DOI] [PubMed] [Google Scholar]
- Grinyó JM, Cruzado JM, Millán O, Caldes A, Sabate I, Gil‐Vernet S, Seron D, Brunet M, Campistol JM, Torras J, Martorell J (2004) Low‐dose cyclosporine with mycophenolate mofetil induces similar calcineurin activity and cytokine inhibition as does standard‐dose cyclosporine in stable renal allografts. Transplantation 78, 1400–1403. [DOI] [PubMed] [Google Scholar]
- Gummert JF, Barten MJ, Sherwood SW, Van Gelder T, Morris RE (1999) Pharmacodynamics of immunosuppression by mycophenolic acid: inhibition of both lymphocyte proliferation and activation correlates with pharmacokinetics. J. Pharmacol. Exp. Ther. 291, 1100–1112. [PubMed] [Google Scholar]
- Härtel C, Fricke L, Schuhmacher N, Kirchner H, Müller‐Steinhardt M (2002) Delayed cytokine mRNA expression kinetics after T‐lymphocyte costimulation: a quantitative measure of the efficacy of cyclosporin A‐based immunosuppression. Clin. Chem. 48, 2225–2231. [PubMed] [Google Scholar]
- Hodge G, Hodge S, Reynolds P, Holmes M (2005) Intracellular cytokines in blood T cells in lung transplant patients – a more relevant indicator of immunosuppression than drug levels. Clin. Exp. Immunol. 139, 159–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutchinson P, Matthew J, Atkins RC, Holdsworth SR (2004) Ex vivo lymphocyte proliferative function is severely inhibited in renal transplant patients on mycophenolate mofetil treatment. Transpl. Immunol. 13, 55–61. [DOI] [PubMed] [Google Scholar]
- Kahan BD (2002) Therapeutic drug monitoring of immunosuppressant drugs in clinical practice. Clin. Ther. 24, 330–350. [DOI] [PubMed] [Google Scholar]
- Oellerich M, Barten MJ, Armstrong VW (2006) Biomarkers: the link between therapeutic drug monitoring and pharmacodynamics. Ther. Drug Monit. 28, 35–38. [DOI] [PubMed] [Google Scholar]
- Stalder M, Brisan T, Holm B, Haririfar M, Scandling J, Morris RE (2003) Quantification of immunosuppression by flow cytometry in stable renal transplant recipients. Ther. Drug Monit. 25, 22–27. [DOI] [PubMed] [Google Scholar]
- Stein CM, Murray JJ, Wood AJJ (1999) Inhibition of stimulated interleukin‐2 production in whole blood: a practical measure of cyclosporine effect. Clin. Chem. 45, 1477–1484. [PubMed] [Google Scholar]
- Weimer R., Melk A, Daniel V, Friemann S, Padberg W, Opelz. G (2000) Switch from cyclosporine A to tacrolimus in renal transplant recipients: impact on Th1, Th2, and monokine responses. Hum. Immunol. 61, 884–897. [DOI] [PubMed] [Google Scholar]