

Abstract
Alteration of appropriate cell‐cycle progression and of closely related apoptotic process is a basic feature of tumour cells, and development of new tumour‐targeted agents focus on apoptosis, either during cell‐cycle arrest or following premature cell‐cycle checkpoint exit. Increasingly, epidemiological and experimental studies suggest that curcumin protects against cancer, not only because of its well‐known antioxidant properties, but also because it modulates intracellular signalling, which is related to cell proliferation and apoptosis. Cisplatin and oxaliplatin are first‐line drugs in treatment of many types of epithelial cancer and their combination with other cytostatics are under investigation to limit their side effects and resistance to them.
Objectives: The aim of this study was to evaluate effects of a combined treatment using curcumin with cisplatin or with oxaliplatin, in a human ovarian cancer cell line (2008) and in its cisplatin‐resistant variant (C13).
Results: Curcumin per se caused concentration‐dependent (0.1–100 µm) and time‐persistent (24–72 h) reduction in cell proliferation, as well as altered cell cycle parameters and induced apoptosis, in both cell lines. When carcinoma cells were simultaneously exposed to curcumin and to cisplatin or oxaliplatin (at concentrations lower than IC50) cell viability was reduced more than with single‐drug treatment. Moreover, dose and time related effects of curcumin, when combined with platinum drugs, were linked to consistent reduction in cell cycling and increased apoptosis, in comparison with single‐drug treatment. These effects were significant both in wild type and in cisplatin‐resistant cells, indicating that curcumin was also able to increase sensitivity of resistant ovarian cancer cells to cisplatin.
Conclusions: The data suggests that curcumin is an interesting natural compound capable of limiting cell proliferation and possibly increasing clinical impact of platinum drugs, in ovarian cancer patients.
Introduction
New strategies to improve clinical response and to reduce toxicity of cancer therapy focus on chemoprevention, which hopes to identify substances that can suppress transformation of initiated or precancerous cells to malignant ones (1). Chemoprevention includes using agents that affect cell‐cycle progression and apoptosis, signal transduction, oncogene activation, polyamine metabolism, angiogenesis, gap junctional intercellular communication, and more (1, 2). A large variety of natural compounds elicit pronounced chemopreventive effects, many of them have been used in traditional herbal medication and are present in our daily diet (3, 4). Increasingly, preclinical and clinical evidence supports curcumin's chemopreventive and antitumour progression properties against human malignancies (5). Curcumin (diferuloylmethane) a yellow pigment isolated from the rhizome of the turmeric plant has been consumed for centuries in Asian countries as a dietary spice and it is well known for its antioxidant, antibacterial, antiviral, antifungal, anti‐inflammatory, antispasmodic and hepato‐protective roles (6). In vitro, curcumin causes apoptosis and cell‐cycle arrest in many different tumour cells by affecting various molecular targets, such as up‐regulating Cdk inhibitors and p53 and down‐regulating cyclin D1, Cdk‐1, cdc2, NFκB (7, 8, 9, 10). In vivo, curcumin reduces incidence and multiplicity of both epithelial invasive and non‐invasive adenocarcinomas (11, 12, 13).
Cisplatin (cis‐diamminedichloroplatinum) is a first‐generation platinum drug that plays a central role in chemotherapy of bladder, testicular and ovarian cancers (14, 15). Oxaliplatin [1R, 2R‐diamnocycloexanoxalate‐platinum (II)] is a third‐generation platinum drug active against a broad range of tumours including cisplatin‐resistant ones, specially colon cancer (16, 17). Cisplatin's nephrotoxicity and oxaliplatin's neurotoxicity, however, as well as intrinsic or acquired tumour cell resistance, are side effects that still limit their clinical use (15, 18). To improve the platinum drugs’ pharmacological profile, combination with resistance modulators or newly molecularly targeted drugs are under preclinical and clinical investigation (19). The aim of the present work was to study the effect of a combination of curcumin with cisplatin or oxaliplatin on cell viability, cell‐cycle parameters and apoptosis in two human ovarian carcinoma cell lines, wild type (2008) and its cisplatin‐resistant variant (C13).
Materials and methods
Cells
Wild‐type (2008) cells derived from human ovary carcinoma and its parental cisplatin‐resistant variant (C13) cells were generated by in vitro selection in presence of increasing concentrations of cisplatin (20). C13 cells exhibit persistent 9‐ to 12‐fold resistance to cisplatin (21). Wild‐type (2008) and C13 cells were routinely grown in humidified condition at 5% CO2 and 37 °C, incubated with RPMI 1640 medium, 10% FBS, 2% glutamine and 1% pen‐strep. Cells were collected every 2 days using minimal amounts of 0.05% trypsin‐0.02% EDTA to wet the whole culture dish surface, and were seeded 1 × 106 in 100 mm dishes with RPMI 1640 medium, 10% FBS and 1% pen‐strep. All reagents were from Lonza (Basel, Switzerland). Growth curves were constructed after trypan blue exclusion assay and 24 h doubling time was measured both for 2008 and C13 cells.
Cell viability assay
Cell viability was determined using the 3‐(4,5‐dimethyl‐thiazole‐2‐yl)‐2,5‐diphenyltetrazolium bromide (MTT) assay. They were plated in 200 µl medium in 96‐well microtitre plates at a density of 2500/well. Following overnight incubation, cells were exposed to a range of different concentrations of test compounds, according to the experimental protocols. After 24 h, cells were washed and replaced in fresh medium. At the end of experimental protocol cells were added to 20 µl of MTT (5 mg/ml) (Sigma, St. Louis, MO, USA) and incubated for 4 h at 37 °C. They were then lysed by adding 200 µl of isopropanol acid. Absorbance at 570 nm was determined using a Victor2, multilabel counter, Wallac Instruments (Waltham, Massachusetts, USA).
Cell‐cycle analysis by flow cytometry
Cells were seeded at a density of 1.5 × 105 cells/6 wells. After 24 h they were treated according to the experimental protocol. At the end of treatment cells were collected using trypsin, were counted then fixed for 30 min on ice with 70% cold ethanol at a density of 1 × 106 cell/ml, washed and resuspended in a solution of 0.05 mg/ml propidium iodide (Molecular Probes, Invitrogen, Paisley, UK) and 0.2 mg/ml RNAase‐DNAase free (Sigma) in phosphate‐buffered saline for roughly 30 min, at room temperature in the dark. Approximately 3 × 104 cells/sample were analysed in an Epics XL‐flow cytometer Beckman Coulter (Fullerton, California, USA), with an Argon Laser λexc = 488 nm and a photomultiplier PMT2, at λem = 575 ± 20 nm. Percentages of cells in the different phases of cell‐cycle were calculated using Multicycle software provided by the manufacturer, considering diploid cycles and correcting for cell clusters.
Annexin V/propidium iodide staining for apoptosis
Cells were seeded at 1.5 × 105 cells/6 wells and incubated overnight. Cells were treated according to experimental protocols and were incubated for 24 h. They were harvested by quick trypsinization to minimize potentially high annexin V background levels in adherent cells. Cells were then washed and stained with Alexa 488/annexin V/propidium iodide (Molecular Probes, Invitrogen). Stained cells were placed on ice and were protected from light. They were then analysed in an Epics XL‐flow cytometer (Beckman Coulter), with laser excitation wavelength set at λ = 488 nm. The green signal from Alexa 488/annexin V was measured at λ = 525 nm and red signal from propidium iodide was measured at λ = 620 nm. Cells negative for both annexin V and propidium iodide are viable, cells annexin V+/propidium iodide− are in early apoptosis, and cells annexin V+/propidium iodide+ are necrotic or in late apoptosis.
Intracellular glutathione assay
Intracellular glutathione concentrations were measured according to Anderson (22), as described previously by Floreani et al. (23). Cells [≈ (5–6) × 106 for each sample] were washed with NaCl/Pi and treated with 6% metaphosphoric acid (1 ml/dish) at room temperature. After 10 min, the acid extract was collected, centrifuged for 5 min at 18 000 g at 4 °C, and processed. Cell debris remaining on the plate was solubilized with 0.5 m KOH and assayed for protein content as described by Lowry et al. (24). For total glutathione determination, the above acid extract was diluted (1 : 6) in 6% metaphosphoric acid; thereafter 0.1 ml supernatant, 0.75 ml of 0.1 m potassium phosphate/5 mm EDTA buffer (pH 7.4), 0.05 ml of 10 mm 5,5′‐dithiobis‐(2‐nitrobenzoic acid) (prepared in 0.1 m phosphate buffer), and 0.08 ml 5 mm NADPH were added. After a 3‐min equilibration period at 25 °C, the reaction was started by addition of 2 U glutathione reductase (type III, Sigma; from baker's yeast; diluted in 0.1 m phosphate/EDTA buffer). Product formation was recorded continuously at 412 nm (for 3 min at 25 °C) with a Shimadzu UV‐160 spectrophotometer (Kyoto, Japan). Total amount of glutathione in samples was determined from a standard curve obtained by plotting known amounts (0.05–0.4 µg/ml) of glutathione against rate of change in absorbance at 412 nm. Glutathione standards were prepared daily in 6% metaphosphoric acid and diluted in phosphate/EDTA buffer (pH 7.4).
Reactive oxygen species intracellular assay
Direct detection of intracellular steady‐state levels of reactive oxygen species (ROS) was carried out on living cells using 2′,7′‐dichlorofluorescin‐diacetate (Molecular Probes). Cells were incubated for 2 h mixed with 5 µm H2‐DCF‐DA and incubated for a further 30 min at 37 °C. At the end of incubation, cells were washed in phosphate‐buffered saline solution, detached from duplicate wells by 0.05% trypsin and 0.2% EDTA, centrifuged for 5 min at 800 g, and then suspended in medium. Sample fluorescence was analysed by Epics XL‐flow cytometer (Beckman Coulter, Miami). Dead cells were excluded by electronic gating data on the basis of forward vs. side scatter profiles; a minimum of 104 cells of interest were analysed further. Logarithmic detectors were used for the FL‐1 fluorescence channel necessary for 2′,7′‐dichlorofluorescin detection. Mean values were obtained by the analysis EXPO 32 software (Beckman Coulter).
Statistical analysis
Statistical comparison of differences among groups of data was carried out using a two‐tailed Student's t‐test. Significance was considered at p < 0.05.
Results
Effect of curcumin, cisplatin and oxaliplatin on cell viability
Cells were exposed for 24, 48 or 72 h to curcumin or to either cisplatin or oxaliplatin at (0.01 µm–100 µm) concentration range, and cell viability measured by MTT assay. All compounds caused concentration‐dependent inhibition of cell viability, which persisted after 72 h (Fig. 1). IC50 is reported in Table 1. This shows that curcumin was more potent in cisplatin‐resistant than in cisplatin‐sensitive ovarian cancer cells. Cisplatin, as expected, was more potent in sensitive cells than in resistant cells. Oxaliplatin was almost equipotent in wild type 2008 and C13 cells and less potent than in colon cancer cell lines (IC50 0.88 µm in HT29, human colon adenocarcinoma grade II cell line, and IC50 1.3 µm in LoVo human colon carcinoma cell line), results not shown. Isobologram analysis of cell viability assay with combinations of curcumin (0.1–1 µm) and cisplatin or oxaliplatin (0.1–5 µm), according to methods proposed by Luszczki & Czuczwar (25), indicated synergistic inhibitory effect on cell viability of curcumin with platinum drugs both in wild type 2008 and C13 cells (results not shown).
Figure 1.
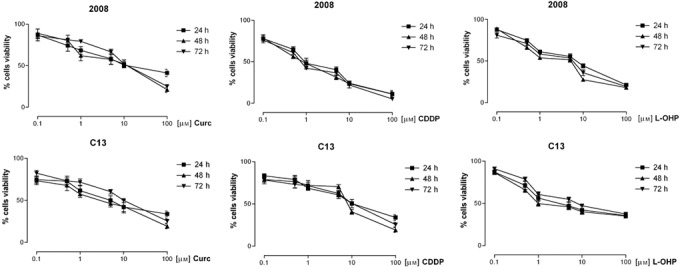
Effect of curcumin, CDDP and oxaliplatin on cell viability in 2008 and C13 cells. The cells were exposed to drugs at concentrations (0.1–100 µm) for 24, 48 and 72 hours and cell viability measured by MTT assay. Results are Mean ± ES from at least three independent experiments in quadruplicate.
Table 1.
| Drugs | 2008 | C13 | ||||
|---|---|---|---|---|---|---|
| IC50 µM % 95 CI | IC50 µM % 95 CI | |||||
| 24h | 48h | 72h | 24h | 48h | 72h | |
| Curcumin | 14.92 8.24 to 20.95 | 7.87 3.51 to 17.61 | 6.36 7.62 to 18.41 | 5.92 2.83 to 12.37 | 2.93 1.77 to 4.87 | 2.47 0.62 to 8.96 |
| Cisplatin | 1.35 0.79 to 2.29 | 0.96 0.74 to 1.24 | 0.94 0.51 to 1.73 | 13.52 8.50 to 21.50 | 7.97 2.15 to 29.53 | 9.00 4.42 to 20.59 |
| Oxaliplatin | 5.62 3.21 to 9.85 | 2.52 1.02 to 6.23 | 3.58 1.93 to 6.64 | 5.64 1.70 to 11.72 | 3.89 0.89 to 11.90 | 5.71 2.15 to 12.34 |
Alteration of ovarian cancer cell‐cycle parameters and apoptosis, induced by curcumin, cisplatin and oxaliplatin
Effects of curcumin (0.5, 1.0, 5.0 µm) on cell‐cycle progression were investigated in ovarian cancer cells cultured over different times (24, 48 and 72 h) by DNA content analysis, by flow cytometry. The cell‐cycle was not significantly different in untreated wild type 2008 and C13 cells. Curcumin in wild type 2008 cells significantly reduced the percentage of cells in G2/M phase in a concentration‐dependent fashion and caused apoptosis (sub‐G0), whereas in C13 cells it significantly increased numbers of cells in G0/G1 phase and reduced cells in S and G2/M, in comparison with untreated cells (Fig. 2). These effects were similar at 48 (Fig. 2) and 72 h (results not shown).
Figure 2.
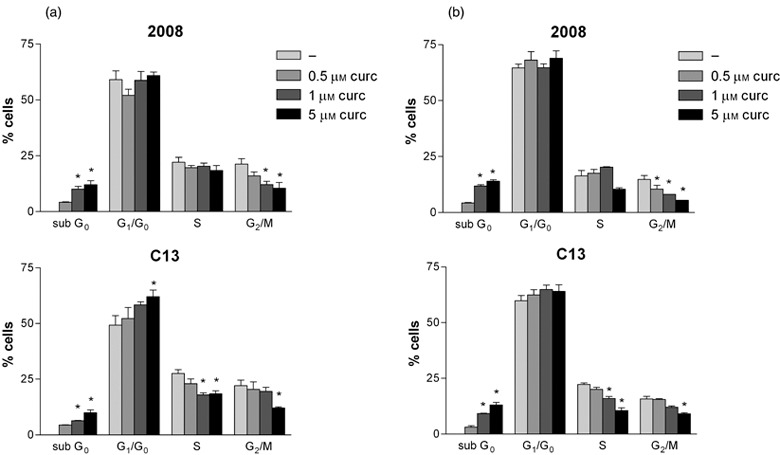
Effects of curcumin on the cell‐cycle progression in 2008 and C13 cells. The cells were treated with curcumin (0.5 µm) for 24 h and 48 h. Percentage of cells in G0/G1, S and G2/M phase were calculated using Multicycle software. Mean ± SD of at least three independent experiments. * p< 0.05 curcumin vs untreated cells.
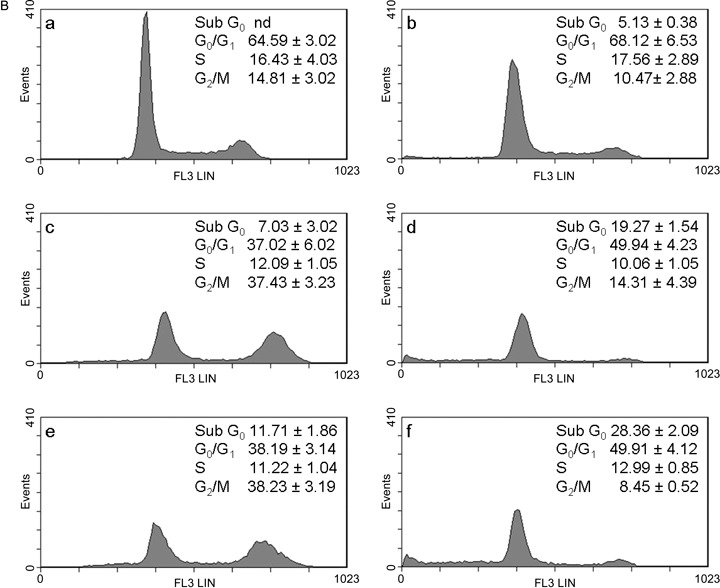
The aim of our study was to investigate effects of combination of curcumin with cisplatin and with oxaliplatin at concentrations lower than IC50 in order to increase efficacy and reduce toxicity and resistance to platinum drugs. Taking IC50 as a reference concentration (Table 1), we analysed the cell‐cycle in wild type 2008 and C13 cells exposed to 0.5 µm of curcumin and platinum drugs, which was the lowest concentration inhibiting cell viability (Fig. 1). Representative cytograms after 24 and 48 h are shown in 3, 3, 4, 4. In wild type 2008 cells (Fig. 3B) after 48 h treatment with curcumin and cisplatin, apoptosis (sub‐G0) increased (+175%) and also G0/G1 (+35%), and G2/M decreased (–60%) in comparison with cisplatin alone; after treatment with curcumin and oxaliplatin, apoptosis increased by 142% and G0/G1 by 30%, and cells in mitotic phase were reduced by 77%, as compared with oxaliplatin alone. In C13 cells (Fig. 4B), curcumin with cisplatin caused +39% apoptosis, +14% of G0/G1, –13% of S phase and –24% of G2/M phase; curcumin with oxaliplatin increased apoptosis almost 5‐fold, reduced G0/G1 by 16%, and G2/M phase by 9%.
Figure 3.
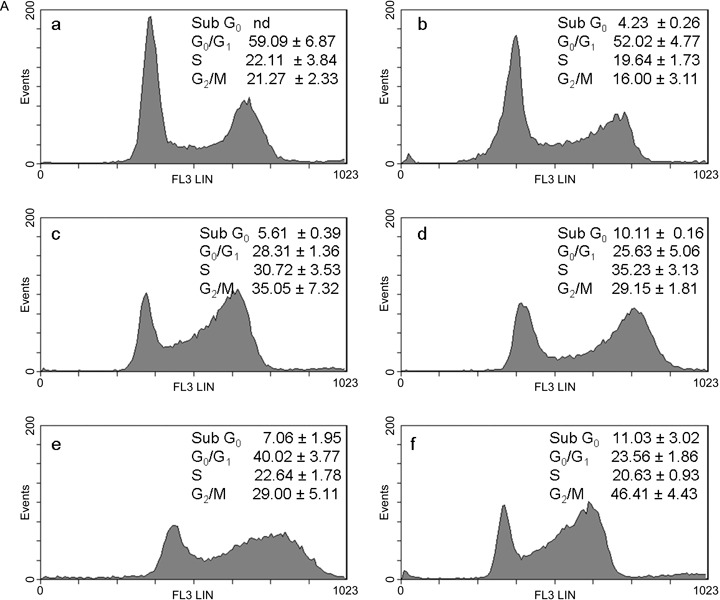
A. Effects of curcumin in combination with cisplatin or oxaliplatin on the cell‐cycle in 2008 cells. The cells were treated with drugs for 24 h: control (a); 0.5 µm curcumin (b); 0.5 µm CDDP (c); curc. + CDDP (d); 0.5 µm L‐OHP (e); curc. + L‐OHP (f). Plots of DNA content (PI fluorescence intensity) vs number of cell events. Percentage of cells in sub G0, G0/G1, S and G2/M phase were calculated using Multicycle software and are represented in the right side of the histograms. All pictures are typical of three independent experiments each performed under identical conditions.
Figure 3.
B. Effects of curcumin in combination with cisplatin or oxaliplatin on the cell‐cycle in 2008 cells. The cells were treated with drugs for 48 h: control (a); 0.5 µm curcumin (b); 0.5 µm CDDP (c); curc. + CDDP (d); 0.5 µm L‐OHP (e); curc. + L‐OHP (f). Plots of DNA content (PI fluorescence intensity) vs number of cell events. Percentage of cells in sub G0, G0/G1, S and G2/M phase were calculated using Multicycle software and are represented in the right side of the histograms. All pictures are typical of three independent experiments each performed under identical conditions.
Figure 4.
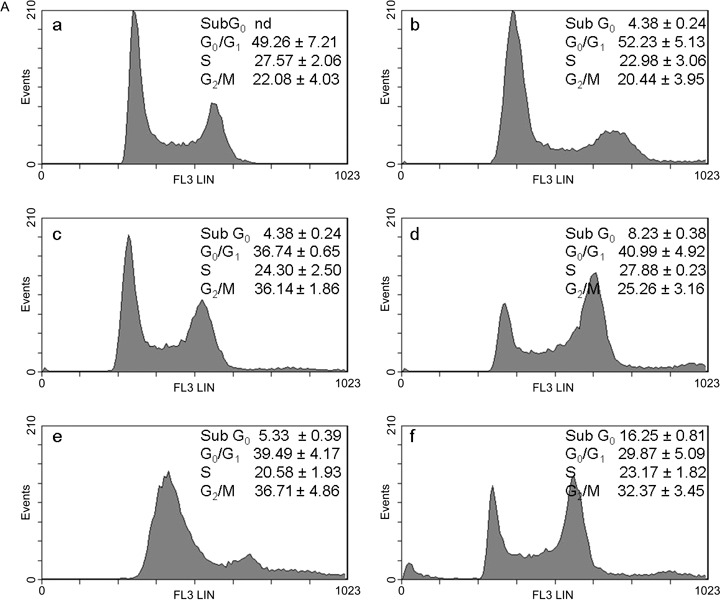
A. Effects of curcumin in combination with cisplatin or oxaliplatin on the cell‐cycle in C13 cells. The cells were treated with drugs for 24 h: control (a); 0.5 µm curcumin (b); 0.5 µm CDDP (c); curc. + CDDP (d); 0.5 µm L‐OHP (e); curc. + L‐OHP (f). Plots of DNA content (PI fluorescence intensity) vs number of cell events. Percentage of cells in G0/G1, S and G2/M phase were calculated using Multicycle software and are represented in the right side of the histograms. All pictures are typical of three independent experiments each performed under identical conditions.
Figure 4.
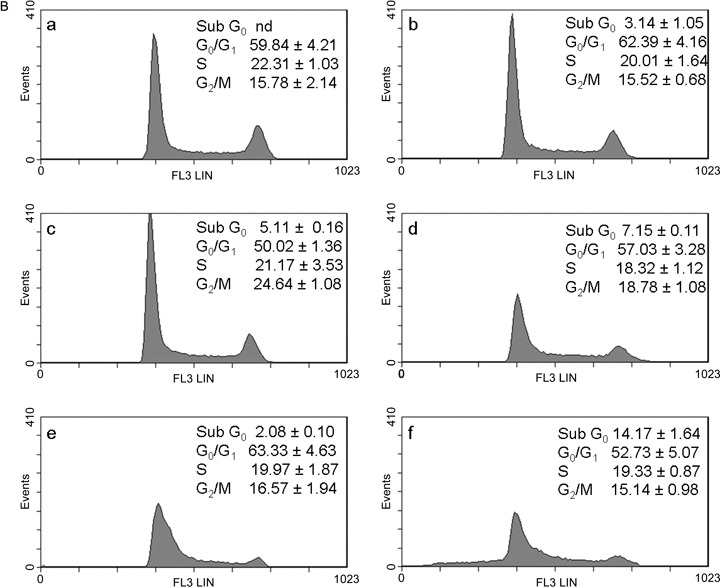
B. Effects of curcumin in combination with cisplatin or oxaliplatin on the cell‐cycle in C13 cells. The cells were treated with drugs for 48 h: control (a); 0.5 µm curcumin (b); 0.5 µm CDDP (c); curc. + CDDP (d); 0.5 µm L‐OHP (e); curc. + L‐OHP (f). Plots of DNA content (PI fluorescence intensity) vs number of cell events. Percentage of cells in G0/G1, S and G2/M phase were calculated using Multicycle software and are represented in the right side of the histograms. All pictures are typical of three independent experiments each performed under identical conditions.
Descriptive statistics derived from four different experiments at different curcumin concentrations (5, 6) confirmed the above‐described time and curcumin concentration‐dependent increased apoptosis and cell–cycle parameter alterations, both in absence and in presence of cisplatin or oxaliplatin.
Figure 5.
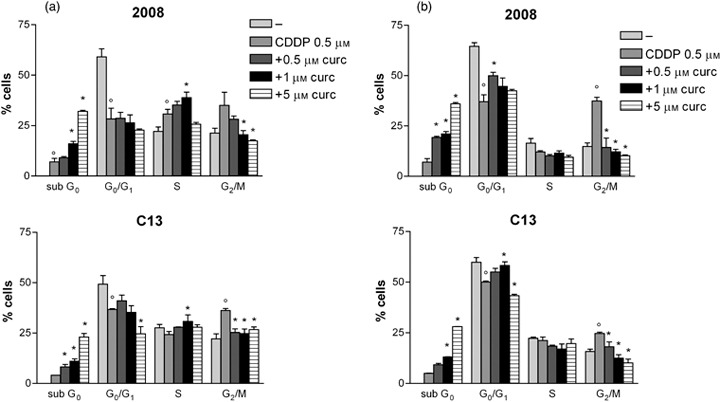
Descriptive statistic of the concentration and time dependent effect of curcumin plus CDDP on cell‐cycle in 2008 and C13 cells. The cells were treated with drugs for after 24 h (a) and 48 h (b). The results are expressed as percentage of cells in sub G0, G0/G1, S, G2/M phases. Mean ± SD of at least three independent experiments. p < 0.05 CDDP vs untreated cells *p < 0.05 cotreatment vs CDDP alone.
Figure 6.
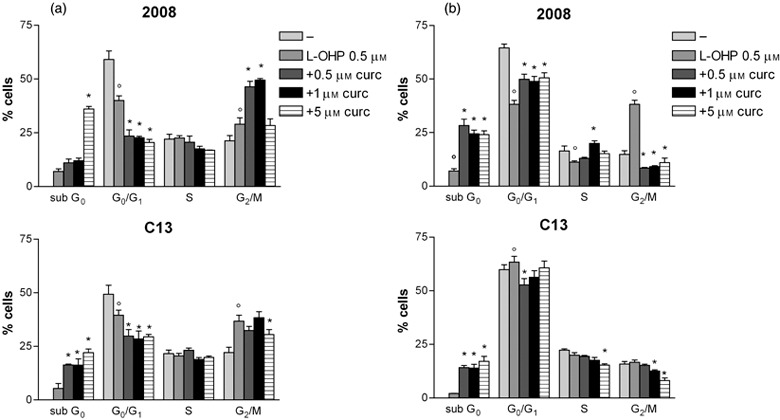
Descriptive statistic of the concentration and time dependent effect of curcumin plus L‐OHP on cell‐cycle in 2008 and C13 cells. The cells were treated with drugs for after 24 h (a) and 48 h (b).The results are expressed as percentage of cells in sub G0, G0/G1, S, G2/M phases. Mean ± SD of at least three independent experiments. p < 0.05 L‐OHP vs untreated cells *p < 0.05 cotreatment or L‐OHP alone.
By annexin V/propidium iodide assay, it is possible to detect early apoptosis. In wild type 2008 and C13 cells, exposure to 0.5 µm concentrations of curcumin and platinum drugs for 24 and 48 h caused highly significant increase of apoptotic fraction of cisplatin‐sensitive and cisplatin‐resistant cells (Fig. 7).
Figure 7.
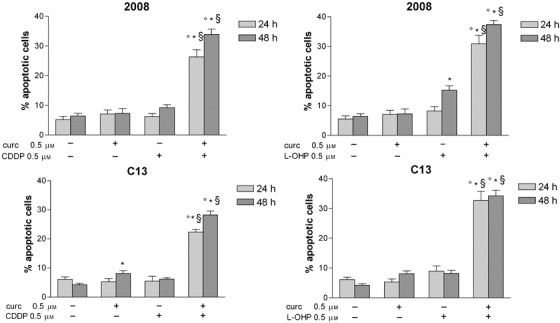
Early apoptosis in 2008 and C13 cells exposed to curcumin, cisplatin, oxaliplatin or drug combination. The cells were treated with (0.5 µm) curcumin, CDDP and L‐OHP for 24 h and 48 h and apoptosis was detected by flow citometry with annexin V/propidium iodide assay. Cells that are annexin V+/PI− are in early apoptosis. Data are Mean ± SD of at least three independent experiments. p < 0.05 vs untreated *p < 0.05 cotreatment vs CDDP alone or L‐OHP alone §p < 0.05 vs curcumin alone.
Effect of curcumin on ROS generation and glutathione levels
ROS generation was measured in wild type 2008 and C13 cells treated for 2 h with 0.5, 1.0 and 5 µm concentrations of curcumin. Basal ROS level was significantly lower in cisplatin‐resistant than in cisplatin‐sensitive cells, and curcumin induced concentration‐dependent increased ROS generation, both in wild type 2008 and C13 cells (Fig. 8).
Figure 8.
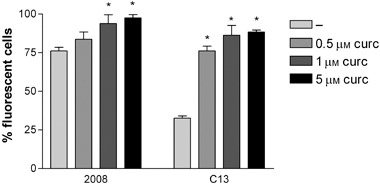
Effect of curcumin on ROS generation in 2008 and C13 cells. The cells were exposed for 2 hours to (0.5, 1.0 and 5.0 µm) curcumin and ROS detected by H2DCF staining and flow cytometry. Results are expressed as percentage of fluorescent cells. Each bar represents the mean ± SD from four replicates, and three independent experiments were carried out. *p < 0.01 compared to control.
Glutathione intracellular level was measured in wild type 2008 and C13 cells after 2 and 24 h of exposure to different curcumin concentrations (Fig. 9). Glutathione levels were higher in untreated cisplatin‐resistant cells than in cisplatin‐sensitive cells. Curcumin, in a concentration‐dependent fashion, caused reduction of glutathione after 2 h incubation, whereas it increased glutathione after 24 h. These effects of curcumin were similar both in wild type 2008 and C13 cells.
Figure 9.
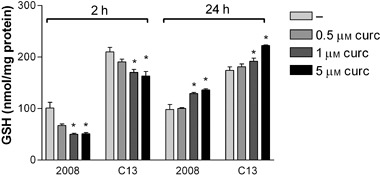
Effect of curcumin on glutathione (GSH) content in 2008 and C13 cells. Cells were exposed for 2 and 24 hours to (0.5, 1.0 and 5.0 µm) curcumin. Each bar represents the mean ± SD from three replicates, and three independent experiments were carried out. *p < 0.01 compared to control.
Discussion
There is still a high mortality rate of women with ovarian cancer, which has been attributed both to lack of early detection and to development of chemoresistance during treatment (14, 26). Cisplatin and oxaliplatin are first‐line chemotherapeutic drugs for ovarian, testicular and colon cancer, but because of increasing resistance to them, their combination with other cytostatic drugs or new molecular‐targeted drugs are currently under investigation (8, 27, 28, 29, 30, 31). As alteration in cell‐cycle progression and apoptotic processes are basic features of treated tumour cells (32, 33), new tumour‐targeted agents are needed to focus on substances that promote apoptosis, either during cell‐cycle arrest or following premature cell‐cycle checkpoint exit (34, 35). Experimental evidence increasingly demonstrates chemopreventive and anticarcinogenic properties of curcumin in vivo and its cell‐cycle and apoptosis‐related molecular mechanisms in vitro (for reviews, see 36, 37).
Here, we show curcumin's concentration‐ and time‐dependent inhibition of ovarian cancer cell viability and its synergism with cisplatin and oxaliplatin drugs. Furthermore, we demonstrate that compared to single‐drug treatment, curcumin combined with cisplatin or oxaliplatin, at concentrations lower than IC50, cause best dose‐ and time‐dependent increases in cell‐cycle arrest and apoptosis. Platinum drugs mainly indirectly cause cell‐cycle perturbation and apoptosis, by blocking DNA replication and transcription, after non‐enzymatic conversion to active derivatives, thus forming platinum‐DNA intra‐ and interstrand adducts, and DNA‐protein cross links (16, 38). Therefore our results suggest a crucial role for curcumin in alteration of cell‐cycle parameters, when combined with platinum drugs. Our data also clearly indicate that concurrent treatment of curcumin with platinum drugs makes cisplatin‐resistant ovarian carcinoma cells more susceptible to the action of the platinum drugs. Molecular mechanisms of cisplatin resistance consist of various altered pathways (27). Recent models describe platinum uptake by facilitated diffusion, perhaps involving a gated channel, which can be lost during selection of drug‐resistant variant(s) (39). p53 gene status is also implicated, mostly in cytotoxic cisplatin sensitivity (40). A further suggested mechanism for drug resistance is increase in intracellular thiols in the redox pathway, which may inactivate and remove platinum compounds (41, 42, 43). Here we demonstrate that in cisplatin‐resistant ovarian cancer cells, basal content of glutathione is higher than in cisplatin‐sensitive cells, as described in human ovarian cancer cell lines by Weir et al. (44). We also show that in cisplatin‐resistant cells (compared to cisplatin‐sensitive cells), higher glutathione content is parallel to lower ROS level, and that curcumin causes concentration‐dependent increase of ROS after 2 h incubation, but increase in glutathione after 24 h. Similarly, curcumin's early pro‐oxidative effect (that is, increased ROS and reduced glutathione) and late antioxidant effect (increased glutathione) have been shown by Sandur et al. (45) on human myeloid leukaemia cells, and by Weir et al. (44) on ovarian cancer cells. How curcumin mediates these effects is not fully understood, but we agree with Sandur et al. (45), who suggest that curcumin's anti‐inflammatory and apoptotic effects might be mediated through regulation of redox status of tumour cells. Our results also provide evidence that curcumin increased glutathion level both in cisplatin‐sensitive and cisplatin‐resistant ovarian cancer cells, suggesting that the increased susceptibility of ovary cancer cell to combined treatment of curcumin with platinum drugs is not related to a glutathion depletion.
In conclusion, our results consistently demonstrate that curcumin is cytotoxic to ovarian cancer cells. It also synergistically increases cisplatin's and oxaliplatin's cytotoxicity and resistant cells’ sensitivity to cisplatin. Our data warrant further investigation into chemopreventive and chemoprotective properties of curcumin, and its potential in improving clinical efficacy of platinum drugs for ovarian cancer patients.
Acknowledgements
The authors wish to thank Mr Enrico Secchi for his excellent technical assistance, Mr Stefano Lovison for software assistance, and Sara Eames, MD, for English language supervision. The Italian Ministry for University and Research supported this work (Grant no. 60A04‐8102/07).
References
- 1. Howells LM, Manson MM (2005) Prospects for plant‐derived chemopreventive agents exhibiting multiple mechanisms of action. Curr. Med. Chem. Anticancer Agents 5, 201–213. [DOI] [PubMed] [Google Scholar]
- 2. Aggarwal BB, Shishodia S (2006) Molecular targets of dietary agents for prevention and therapy of cancer. Biochem. Pharmacol. 71, 1397–1421. [DOI] [PubMed] [Google Scholar]
- 3. Hong WK, Sporn MB (1997) Recent advances in chemoprevention of cancer. Science 278, 1073–1077. [DOI] [PubMed] [Google Scholar]
- 4. Kelloff GJ, Hawk ET, Crowell JA, Boone CW, Nayfield SG, Perloff M, Steele VE, Lubet RA, Sigman CC (1996) Strategies for identification and clinical evaluation of promising chemopreventive agents. Oncology 10, 1471–1480. [PubMed] [Google Scholar]
- 5. Aggarwal BB, Kumar A, Bharti AC (2003) Anticancer potential of curcumin: preclinical and clinical studies. Anticancer Res. 23, 363–398. [PubMed] [Google Scholar]
- 6. Ammon HP, Wahl MA (1991) Pharmacology of Curcuma longa. Planta Med. 57, 1–7. [DOI] [PubMed] [Google Scholar]
- 7. Aggarwal S, Ichikawa H, Takada Y, Sandur SK, Shishodia S, Aggarwal BB (2006) Curcumin (diferuloylmethane) down‐regulates expression of cell proliferation and antiapoptotic and metastatic gene products trough suppression of IB kinase and Akt activation. Mol. Pharmacol. 69, 1195–1206. [DOI] [PubMed] [Google Scholar]
- 8. Park MJ, Kim EH, Park C, Lee HC, Woo SH, Lee JY (2002) Curcumin inhibits cell cycle progression of immortalized human umbilical vein endothelial (ECV304) cells by up‐regulating cyclin‐dependent kinase inhibitor, p21WAF1/CIP1, p27KIP1 and p53. Int. J. Oncol. 21, 379–383. [PubMed] [Google Scholar]
- 9. Kuttan G, Kumar KB, Guruvayoorappan C, Kuttan R (2007) Antitumor, anti‐invasion, and antimetastatic effects of curcumin. Adv. Exp. Med. Biol. 595, 173–184. [DOI] [PubMed] [Google Scholar]
- 10. Liu E, Wu J, Cao W, Zhang J, Liu W, Jiang X, Zhang X (2007) Curcumin induces G2/M cell cycle arrest in a p53‐dependent manner and upregulates ING4 expression in human glioma. J. Neurooncol. 85, 263–270. [DOI] [PubMed] [Google Scholar]
- 11. Cheng AL, Hsu CH, Lin JK (2001) Phase I clinical trial of curcumin, a chemopreventive agent, in patients with high‐risk or pre‐malignant lesions. Anticancer Res. 21, 2895–2900. [PubMed] [Google Scholar]
- 12. Sharma RA, McLelland HR, Hill KA (2001) Pharmacodynamic and pharmacokinetic study of oral Curcuma extract in patients with colorectal cancer. Clin. Cancer Res. 7, 1894–1900. [PubMed] [Google Scholar]
- 13. Sharma RA, Euden SA, Platton S (2004) Phase I clinical trial of oral curcumin: biomarkers of systemic activity and compliance. Clin. Cancer Res. 10, 6847–6854. [DOI] [PubMed] [Google Scholar]
- 14. Kelland L (2007) The resurgence of platinum‐based cancer chemotherapy. Nat. Rev. Cancer 7, 573–584. [DOI] [PubMed] [Google Scholar]
- 15. Jung Y, Lippard SJ (2007) Direct cellular responses to platinum‐induced DNA damage. Chem. Rev. 107, 1387–1407. [DOI] [PubMed] [Google Scholar]
- 16. Woynarowski JM, Faivre S, Herzig MCS, Arnett B, Chapman WG, Trevino AV, Raymond E, Chaney SG, Vaisman A, Varchenko M, Juniewicz PE (2000) Oxaliplatin‐induced damage of cellular DNA. Mol. Pharmacol. 58, 920–927. [DOI] [PubMed] [Google Scholar]
- 17. Raymond E, Faivre S, Chaney S, Woynarowski J, Cvitkovic E (2002) Cellular and molecular pharmacology of oxaliplatin. Mol. Cancer Ther. 3, 227–235. [PubMed] [Google Scholar]
- 18. Wong E, Giandomenico CM (1999) Current status of platinum‐based antitumor drugs. Chem. Rev. 99, 2451–2466. [DOI] [PubMed] [Google Scholar]
- 19. Pietras RJ, Fendly BM, Chazin VR, Pegram MD, Howell SB, Slamon DJ (1994) Antibody to HER‐2/neu receptor blocks DNA repair after cisplatin in human breast and ovarian cancer cells. Oncogene 9, 1829–1838. [PubMed] [Google Scholar]
- 20. Andrews PA, Albright KD (1992) Mitochondrial defects in cis‐diamminedichloroplatinum (II)‐resistant human ovarian carcinoma cells. Cancer Res. 52, 1895–1901. [PubMed] [Google Scholar]
- 21. Zinkewich‐Peotti K, Andrews PA (1992) Loss of cis‐ diamminedichloroplatinum (II) resistance in human ovarian carcinoma cells selected for rhodamine 123 resistance. Cancer Res. 52, 1902–1906. [PubMed] [Google Scholar]
- 22. Anderson ME (1985) Determination of glutathione and glutathione disulfide in biological samples. Methods Enzymol. 113, 548–555. [DOI] [PubMed] [Google Scholar]
- 23. Floreani M, Skaper SD, Facci L, Lipartiti M, Giusti P (1997) Melatonin maintains glutathione homeostasis in kainic acid‐exposed rat brain tissue. FASEB J. 11, 1309–1315. [DOI] [PubMed] [Google Scholar]
- 24. Lowry OH, Rosebrough NJ, Farr AL, Randall RJ (1951) Protein measurement with the Folin phenol reagent. J. Biol. Chem. 193, 265–275. [PubMed] [Google Scholar]
- 25. Luszczki JJ, Czuczwar SJ (2004) Three‐dimensional isobolographic analysis of interactions between lamotrigine and clonazepam in maximal electroshock‐induced seizures in mice. Naunyn Schmiedebergs Arch. Pharmacol. 370, 369–380. [DOI] [PubMed] [Google Scholar]
- 26. Smith LH, Morris CR, Yasmeen S, Parikh‐Patel A, Cress RD, Romano PS (2005) Ovarian cancer: Can we make the clinical diagnosis earlier? Cancer. 104, 1398–1407. [DOI] [PubMed] [Google Scholar]
- 27. Brabec V, Kasparkova J (2002) Molecular aspects of resistance to antitumor platinum drugs. Drug Resist. Update 5, 147–161. [DOI] [PubMed] [Google Scholar]
- 28. Lévi F, Misset JL, Brienza S, Adam R, Metzger G, Itzakhi M, Caussanel JP, Kunstlinger F, Lecouturier S, Descorps‐Declère A (1992) A chronopharmacologic phase II clinical trial with 5‐fluorouracil, folinic acid, and oxaliplatin using an ambulatory multichannel programmable pump. High antitumour effectiveness against metastatic colorectal cancer. Cancer 69, 893–900. [DOI] [PubMed] [Google Scholar]
- 29. Machover D, Diaz‐Rubio E, De Gramont A, Schilf A, Gastiaburu JJ, Brienza S, Itzhaki M, Metzger G, N'Daw D, Vignoud J, Abad A, Francois E, Gamelin E, Marty M, Sastre J, Seitz JF, Ychou M (1996) Two consecutive phase II studies of oxaliplatin (L‐OHP) for treatment of patients with advanced colorectal carcinoma who were resistant to previous treatment with fluoropyrimidines. Ann. Oncol. 7, 95–98. [DOI] [PubMed] [Google Scholar]
- 30. Giacchetti S, Perpoint B, Zidani R, Le Bail N, Faggiuolo R, Focan C, Chollet P, Llory JF, Letourneau Y, Coudert B, Bertheaut‐Cvitkovic F, Larregain‐Fournier D, Le Rol A, Walter S, Adam R, Misset JL, Lévi F (2000) Phase III multicenter randomized trial of oxaliplatin added to chronomodulated fluorouracil‐leucovorin as first‐line treatment of metastatic colorectal cancer. J. Clin. Oncol. 18, 136–147. [DOI] [PubMed] [Google Scholar]
- 31. Goldberg RM, Gill S (2004) Recent phase III trials of fluorouracil, irinotecan, and oxaliplatin as chemotherapy for metastatic colorectal cancer. Cancer Chemother. Pharmacol. 54, 57–64. [DOI] [PubMed] [Google Scholar]
- 32. Vermeulen K, Van Bockstaele DR, Berneman ZN (2003) The cell cycle: a review of regulation, deregulation and therapeutic targets in cancer. Cell Prolif. 36, 131–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Malumbres M, Carnero A (2003) Cell cycle deregulation: a common motif in cancer. Prog. Cell Cycle Res. 5, 5–18. [PubMed] [Google Scholar]
- 34. Ruetz S, Fabbro D, Zimmermann J, Meyer T, Gray N (2003) Chemical and biological profile of dual Cdk1 and Cdk2 inhibitors. Curr. Med. Chem. Anticancer Agents 3, 1–14. [DOI] [PubMed] [Google Scholar]
- 35. Schwartz GK, Shah MA (2005) Targeting the cell cycle: a new approach to cancer therapy. J. Clin. Oncol. 23, 9408–9421. [DOI] [PubMed] [Google Scholar]
- 36. Aggarwal BB, Sethi G, Baladandayuthapani V, Krishnan S, Shishodia S (2007) Targeting cell signaling pathways for drug discovery: an old lock needs a new key. J. Cell. Biochem. 102, 580–592. [DOI] [PubMed] [Google Scholar]
- 37. Sharma RA, Gescher AJ, Steward WP (2005) Curcumin: the story so far. Eur. J. Cancer 41, 1955–1968. [DOI] [PubMed] [Google Scholar]
- 38. Kelland LR (2005) Emerging drugs for ovarian cancer. Expert. Opin. Emerg. Drugs 10, 413–424. [DOI] [PubMed] [Google Scholar]
- 39. Martelli L, Di Mario F, Ragazzi E, Apostoli P, Leone R, Perego P, Fumagalli G (2006) Different accumulation of cisplatin, oxaliplatin and JM216 in sensitive and cisplatin‐resistant human cervical tumour cells. Biochem. Pharmacol. 72, 693–700. [DOI] [PubMed] [Google Scholar]
- 40. Martelli L, Di Mario F, Botti P, Ragazzi E, Martelli M, Kelland L (2007) Accumulation, platinum‐DNA adduct formation and cytotoxicity of cisplatin, oxaliplatin and satraplatin in sensitive and resistant human osteosarcoma cell lines, characterized by p53 wild‐type status. Biochem. Pharmacol. 74, 20–27. [DOI] [PubMed] [Google Scholar]
- 41. Kelley SL, Basu A, Teicher BA, Hacker MP, Hamer DH, Lazo JS (1988) Overexpression of metallothionein confers resistance to anticancer drugs. Science 241, 1813–1815. [DOI] [PubMed] [Google Scholar]
- 42. Mistry P, Kelland LR, Abel G, Sidhar S, Harrap KR (1991) The relationships between glutathione, glutathione‐S‐transferase and cytotoxicity of platinum drugs and melphalan in eight human ovarian carcinoma cell lines. Br. J. Cancer 64, 215–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Jansen BA, Brouwer J, Reedijk J (2002) Glutathione induces cellular resistance against cationic dinuclear platinum anticancer drugs. J. Inorg. Biochem. 89, 197–202. [DOI] [PubMed] [Google Scholar]
- 44. Weir NM, Selvendiran K, Kutala VK, Tong L, Vishwanath S, Rajaram M, Tridandapani S, Anant S, Kuppusamy P (2007) Curcumin induces G2/M arrest and apoptosis in cisplatin‐resistant human ovarian cancer cells by modulating Akt and p38 MAPK. Cancer Biol. Ther. 6, 178–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Sandur SK, Ichikawa H, Pandey MK, Kunnumakkara AB, Sung B, Sethi G, Aggarwal BB (2007) Role of pro‐oxidants and antioxidants in the anti‐inflammatory and apoptotic effects of curcumin (diferuloylmethane). Free Radic Biol. Med. 43, 568–580. [DOI] [PMC free article] [PubMed] [Google Scholar]