

Abstract.
Changes in intracellular Ca2+ correlate with specific events in the cell cycle. Here we investigated the role of Ca2+ in the G1 phase. HEK 293 cells were arrested in mitosis and subjected to short‐term treatments that alter Ca2+ homeostasis prior to their release into G1. Treatment with thapsigargin (TG), an irreversible inhibitor of the sarco‐endoplasmic reticulum Ca2+ ATPase (SERCA) lengthened the G1 phase. Moreover, TG treatment also resulted in a dramatic alteration in cellular morphology and attachment and in the reduction of MAPK activity and lower levels of cyclin D1 and cyclin E proteins. Treatments with reagents that transiently increase or decrease cytosolic Ca2+ or that temporarily inactivate SERCA did not alter any of the above parameters. Cells expressing a TG‐resistant form of SERCA progressed normally through the G1/S transition after TG treatment. These results suggest that long‐term SERCA inactivation affects cell cycle‐dependent events and compromises progression through G1/S.
Introduction
The role of calcium in the progression through the cell division cycle has been studied in a number of systems (Poenie et al. 1985; Ratan, Maxfield & Shelanski 1988; Tombes & Borisy 1989; Berridge 1995). The advent of intracellular calcium indicator dyes in the mid to late 1980s made it possible to measure calcium fluctuations directly during specific stages of the cell cycle (Grynkiewicz, Poenie & Tsien 1985). Studies in sea urchin eggs revealed a correlation between intracellular Ca2+ increases and mitotic events following fertilization, including pronuclear migration, nuclear envelope breakdown, metaphase/anaphase transition and cell–cell cleavage (Poenie et al. 1985). In cultured mammalian cells, rapid Ca2+ fluctuations and a continuous Ca2+ elevation during mitosis can be temporally correlated with anaphase onset and cytokinesis (Ratan et al. 1988; Tombes & Borisy 1989).
The first indication that Ca2+ might play a role in G1 came from studies where cell growth was reversibly blocked by lowering the level of extracellular Ca2+ (Whitfield et al. 1980). Interestingly, SV40 transformed Madin Darby Canine Kidney (MDCK) cells are able to grow in low Ca2+ levels, which inhibit normal cell growth, suggesting that transformation is associated with the constitutive activation of a Ca2+‐sensitive process. These transformed cells display spontaneous long‐lasting Ca2+ oscillations (Wojnowski et al. 1994). These observations suggest a role for Ca2+ during G1 when coupled with observations which show that: (1) mitogenic stimuli, such as hormones or growth factors are known to elicit cytoplasmic Ca2+ increases (Peres & Giovannardi 1990b; Peres et al. 1990a; Cheyette & Gross 1991) and (2) incubation of human fibroblasts in low Ca2+ media during early G1 results in a reduced number of cells that progress to S phase (Wahl & Gruenstein 1993). Some of the Ca2+ effects during late G1 which lead to the G1/S transition are thought to be mediated by calmodulin (CaM), since inactivation of CaM kinase II blocks cells in S phase (Hiroyouki, Inoue & Hiroyoushi 1994). The molecular nature of the Ca2+‐dependent events in early G1 remains elusive however.
Ca2+ has been implicated in multiple cellular processes such as gene expression, secretion, contraction, cellular metabolism, as well as the transduction of extracellular stimuli leading to cell proliferation and/or cell death (Jiang et al. 1994; Berridge 1995). These diverse effects are possible through the tight regulation of cytosolic free Ca2+ levels via release from compartmentalized intracellular Ca2+ stores or influx from the extracellular environment. Increases in cytosolic free Ca2+ are spatio‐temporally regulated and can elicit highly localized short‐term responses or propagate into rapidly spreading waves which are often longer lasting and regulate more global events (Bootman, berridge & lipp 1997). A major regulator of intracellular Ca2+ dynamics is inositol 1,4,5‐triphosphate (InsP3), which directly controls the release of stored Ca2+, an event that in turn stimulates the capacitative re‐entry of Ca2+ from the extracellular environment. InsP3 sensitizes the InsP3 receptor to Ca2+, which then promotes the release of Ca2+ from the ER, setting up a regenerative process of Ca2+ induced Ca2+ release. In muscle and nonmuscle cells, cytosolic Ca2+ homeostasis is maintained largely by the sarco/endoplasmic reticulum Ca2+‐ATPase (SERCA) family of proteins. SERCA proteins catalyse the ATP‐dependent translocation of Ca2+ from the cytoplasm into the ER lumen, thus restoring resting cytoplasmic Ca2+ concentrations (Carafoli 1987; Penner, Fasolato & Hoth 1993; Martonosi 1996; MacLennan, Rice & Green 1997; Parekh & Penner 1997). It is likely that Ca2+ oscillations are directly translated into biochemical signals by a diverse number of Ca2+‐binding and/or Ca2+‐dependent enzymes with various affinities for the cation.
Here we investigated the role of Ca2+ in G1 progression and in the G1/S transition. We used various pharmacological reagents that alter intracellular Ca2+ homeostasis and noted the effect of this manipulation on cellular ability to finish mitosis and progress through G1/S and complete S phase. Human epithelial kidney (HEK 293) cells were arrested in mitosis using the thymidine/nocodazole double block. Synchronized cells were either left untreated or subjected to short‐term treatments with the reagents that alter intracellular Ca2+ levels, and subsequently released from the mitotic block. The ability of cells to attach and spread after release was evaluated by light microscopy and their ability to finish mitosis, progress through G1 and enter S phase was evaluated using flow cytometric analysis of DNA content. Previous work on control of G1/S progression has focused on serum‐induced return to growth of serum starved, quiescent cells (G0). Here we have used conditions that attempt to mimic the M‐G1 transition which is characteristic of cycling cells, under conditions in which serum is present throughout the entire length of the experiment.
Two essential requirements for G1/S transition are growth factor activation of transmembrane receptors and integrin activation via cell attachment to the extracellular matrix. Both of these pathways contribute to the long‐term activation of mitogen‐activated protein kinases (MAPKs) (Leof et al. 1983; Straus 1984; Zhu & Assoian 1995; Bottazzi & Assoian 1997; Bottazzi et al. 1999). Sustained MAPK activation leads to the increased expression of cyclin D1 and reduced expression of cyclin dependent kinase inhibitors (CKIs), such as p21 and p27 (Lavoie et al. 1996; Weber et al. 1997). Cyclin D1 is an essential regulator of G1 phase progression: its expression levels determine its degree of association with cyclin‐dependent kinase (CDK) 4 and 6. Once activated, CDK4/6‐Cyclin D1 complexes phosphorylate several essential targets which lead to cyclin E expression (Bates et al. 1994; Horton, Qian & Templeton 1995). Cyclin E association with CDK2 and activation of this complex leads to expression of cyclin A, which is essential for S phase onset (Girard et al. 1991; Ohtsubo et al. 1995; Resnitzky & Reed 1995). We find that treatments that result in the long term depletion of intracellular Ca2+ stores lead to a 12‐hour delay in G1/S progression and the reduced activity and levels of important cell cycle regulators such as MAPK, cyclin D1 and cyclin E.
MATERIALS AND METHODS
Cell culture, growth and synchronization
HEK 293 cells were grown in DMEM containing 10% FBS, L‐glutamine and non‐essential amino acids. A 70% confluent culture was synchronized using a thymidine/nocodazole double block. Briefly, cells were incubated in the presence of 2 m m thymidine (Sigma, St Louis, MO, USA) for 16 h. Cells were washed once and incubated in fresh media containing 0.1 mg/ml nocodazole (Sigma) for 12–16 h. Cells were left untreated or were treated with a combination of 1 µm Thapsigargin (TG) and 5 m m EGTA, 10 µm cyclopiazonic acid (CPA), 30 µm 2,5‐di‐(tert‐butyl)‐1,4‐benzohydroquinone (BHQ), or 10 µm[1,2‐bis(o‐Aminophenoxy)ethane‐N,N,N′,N′‐tetraacetic acid tetra(acetomymethyl) ester] (BAPTA‐AM) (all from Calbiochem, La Jolla, CA, USA) for 30 min while still in nocodazole. Cells were treated with 1 µm ionomycin (Sigma) for 2 min prior removal of nocodazole. The cultures were released from mitotic arrest by washing once with PBS, and placed into fresh DMEM with 10% FBS (t = 0).
Flow cytometry
At indicated times after release from nocodazole arrest, an aliquot of control and drug treated cells was removed and processed for flow cytometric analysis. Cells were washed 1x in PBS, and fixed for 1 h in cold 80% ethanol. Fixed cells were centrifuged at 3000 r.p.m. for 5 min at 4 °C and washed once with PBS and once with PIB buffer (PBS, 0.12% Triton‐X, 0.12 m m EDTA). Fixed cells were treated with 10 µg/mL DNAse‐free RNAse for 45 min at 37 °C. DNA was stained using 5 µg/mL propidium iodide (Sigma) for 30 min at room temperature.
Samples were analysed on a Becton Dickinson FACSCalibur (Mountain View, CA, USA) and data analysed using Becton Dickinson Cellquest software. Alignment was checked using Immunocheck beads (Coulter Electronics, Fullerton, CA, USA) and the instrument was calibrated using Calibrite beads (Becton Dickinson, St Louis, MO, USA) and Autocomp software. Forward and side scatter were used to gate on live cells.
Cell lysis and western blot analysis
At appropriate times after treatment and release from synchronization, cell samples were lysed in MLB buffer (25 m m HEPES, pH 7.5; 150 m m NaCl; 1% NP‐40; 0.25% sodium deoxycholate; 10% glycerol; 1 m m EDTA; 10 m m MgCl2) supplemented with phosphatase inhibitors (25 m m NaF; 40 m m sodium pyrophosphate; 40 m mβ‐glycerol phosphate; 1 m m sodium‐orthovanadate) and protease inhibitors (10 mg/ml leupeptin; 10 µg/ml aprotinin; 0.1 m m AEBSF). Total protein concentration of cell lysates was determined using the Coomassie Plus Protein Assay Reagent (Pierce, Rockford, IL, USA). Twenty milligrams of lysate protein was assayed per sample and subjected to SDS‐PAGE electrophoresis and western blotting.
The relative protein levels of cyclin D1 and Cyclin E at various times after release from mitotic arrest and drug treatment were determined using polyclonal antibodies H295 and C‐19 (Santa Cruz Biotech, Santa Cruz, CA, USA), respectively. Antibodies were used as described by the manufacturers. The levels of phosphorylated MAPK were determined by Western blot analysis of control and TG‐treated lysates. Commercially available (New England Biolabs, Beverly, MA, USA) antibodies were used to measure the phosphorylation status of p44/42 MAP kinases (Erk1 and Erk2). This antibody detects phosphorylated threonine 202 and tyrosine 204 of catalytically activated p42 and p44.
Viability assays
An aliquot of untreated or treated cells was stained with 0.05% trypan blue. Cell viability was determined by counting the number of trypan blue unstained and stained cells at 12, 24 and 48 h after release from mitotic arrest.
An in situ cell death detection kit was used to determine the percentage of apoptotic cells in untreated and treated cell samples (Boehringer Mannheim, Indianapolis, IN, USA). This assay utilizes the TUNEL method (TdT‐mediated dUTP‐X nick end labelling) to label the free 3′‐OH termini of cleaved DNA with fluorescein dUTP. Synchronized control or drug treated cells (~5 × 105 cells) were washed and attached to polylysine coated coverslips (12 mm circle, Fisher Scientific, Napean, ON, Canada) in fresh media. At various times after release from mitotic arrest, cells were fixed with 2% paraformaldehyde at room temperature for 30 min and with 80% cold ethanol for 60 min at 4 °C. Cells were then permeabilized for 10 min, using 0.1% Triton X‐100 in 0.1% sodium citrate. Coverslips were then incubated with 25 µL of TUNEL reaction mixture (1 : 10 mixture of Reagent 1 and Reagent 2) for 60 min in a dark, humid environment. Finally cells were treated with RNAse (10 µg/µl) and counterstained with Propidium iodide (1 µg/µl). Coverslips were mounted on microscope slides using immuno floure mounting medium (ICN, Costa Mesa, CA, USA), and analysed using an Axiovert 100 Zeiss (Zeiss, North York, ON, Canada) inverted microscope.
Calcium imaging
The Photon Technologies Inc. (PTI, Rockwall, TX, USA) microfluorimetry system was used to measure the effect of various pharmacological agents on intracellular Ca2+ levels in HEK 293 cells. Briefly, cells were synchronized as described above. Ca2+ photometry or imaging were begun on intact cells prior to their release from nocodazole. Mitotically arrested cells were resuspended in carbon dioxide‐independent media (Gibco‐BRL, Rockville, MD, USA) containing nocodazole (0.1 µg/ml) and placed on a polylysine coated coverslip (25 mm circle, Fisher Scientific). This media was supplemented with Fura‐2 AM (molecular probes, 1 µm for photometry and 2.5 µm for imaging) and 0.02% pluronic F‐127 and cells were incubated for 30 min at 37 °C. After Fura‐2 AM loading, the coverslip was placed in a holder on the heated (37 °C) stage of an inverted Diaphot microscope (Zeiss Canada). The samples were excited alternately at 340 nm and 380 nm using a dual monochromator. The emitted fluorescence was filtered through a 510‐nm filter, before feeding into the photomultiplier tube. Averaged light intensities over excitation periods of 0.05 s at each of the two wavelengths were used to calculate the 340/380 nm ratio using Felix (PTI) software. Solutions containing the test pharmacological agents were added at one side of the chamber and aspirated on the opposite side.
Preparation of stable cell lines expressing thapsigargin resistant serca1
The SERCA 1 cDNA bearing the thapsigargin‐resistant F256V mutation and a C‐terminal myc‐tag (Yu et al. 1999) was subcloned into the pCDNA3 (Invitrogen, Carlsbad, CA, USA) vector. The EcoRI‐XhoI fragment (~3.2 Kb) was inserted into the pCDNA3 multiple cloning site. The resulting vector was transfected into HEK 293 cells using the calcium phosphate transfection method (Stone, Moran & Pawson 1991). Stable clones expressing the mutant SERCA protein were selected for neomycin resistance and screened by Western analysis using the 9E‐10 antimyc monoclonal antibody. One clone (G17) expressing low‐medium levels of myc‐tagged SERCA was selected for further study.
RESULTS
Manipulation of intracellular calcium levels
We altered intracellular Ca2+ levels of mitotically synchronized HEK 293 cells on a short and long‐term basis. The effect of each of these reagents on Ca2+ dynamics was monitored by Ca2+ imaging of Fura‐2 loaded cells (Grynkiewicz et al. 1985; see MATERIALS AND METHODS).
Firstly, we used thapsigargin (TG), a sesquiterpene lactone that selectively and irreversibly binds to and inhibits the SERCA family of proteins, including the SERCA‐2b isomer found in HEK 293 cells (Irvine 1989; Thastrup et al. 1990; Lytton, Westlin & Hanley 1991; Anger et al. 1994). This blocks the formation of ER Ca2+ stores and inhibits the response of cells to InsP3, thereby abolishing cellular ability to produce regenerative Ca2+ oscillations. As shown in Fig. 1a, TG treatment resulted in a rapid 5‐fold increase in cytoplasmic Ca2+ levels of approximately 100–150 s in duration. As Ca2+ is pumped out of the cytoplasm via plasma membrane pumps, intracellular Ca2+ levels dropped to a level slightly higher than basal (Fig. 1a). The presence of 5 m m EGTA in the extracellular media during TG treatment blocks Ca2+ re‐entry into cells and therefore results in lower than basal intracellular Ca2+ levels for prolonged periods of time (Fig. 1b).
Figure 1.
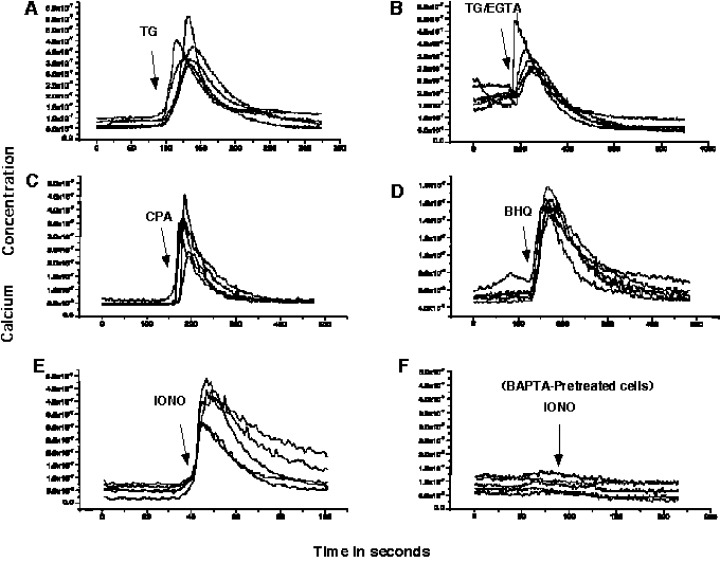
Effect of Specific Reagents on Intracellular Calcium Levels. HEK 293 were synchronized as described in ‘Materials and Methods’. Approximately 1 × 105 cells were resuspended in carbon dioxide‐independent medium and 10% FBS, placed on polylysine coated coverslips and loaded with Fura‐2‐AM for 30 min at 37 °C. Coverslips were then transferred to a growth chamber in fresh medium and calcium imaging was initiated. (a) 100 s recording was followed by change of medium containing 1 µm thapsigargin (TG). (b) 180 s recording followed by treatment with 1 µm thapsigargin in medium containing 5 m m EGTA (TG/EGTA). (c) 160 s recording followed by addition of 10 µm cyclopiazonic acid (CPA) in fresh medium. (d) 120 s recording followed by addition of 2,5‐di‐(ter‐butyl)‐1,4‐benzohydroquinone (BHQ). (e) 40 s recording followed by addition of fresh media containing 1 µm ionomycin (IONO). (f) cells were preloaded with 10 µm BAPTA‐AM for 30 min at 37 °C, 75 s recording followed by addition of fresh medium containing 1 µm ionomycin. Representative measurements of 5 cells are shown for all conditions.
Transient inactivation of SERCA using cyclopiazonic acid (CPA) and 2,5‐Di(t‐butyl)‐1,4‐hydroquinone (BHQ), resulted in the reversible, short‐term depletion of intracellular stores by mobilization of the InsP3‐sensitive Ca2+ pools (Mason, Garcia‐Rodriguez & Grinstein 1991; Foskett & Wong 1992). A transient 5–8‐fold increase in intracellular free Ca2+, of approximately 100 s duration was measured upon treatment with 10 µm CPA (Fig. 1c). Treatment of cells with 30 µm BHQ resulted in a 2–3‐fold increase in cytoplasmic Ca2+ levels, which had a duration of about 150 s (Fig. 1d). The levels of cytosolic Ca2+ returned to basal within seconds after CPA or BHQ treatment.
Intracellular Ca2+ levels were also altered by allowing extracellular influx of Ca2+ ions using ionomycin, a Ca2+ ionophore (Liu & Hermann 1978; Kauffman, Taylor & Pfeiffer 1980). Ionomycin increased intracellular Ca2+ levels 2–5‐fold for extended periods ranging from 100 to 300 s (Fig. 1e). Ionomycin has a detrimental effect on cellular integrity due to its time‐dependent and nonspecific effect on cellular membranes. Therefore, we chose to expose cells to ionomycin for shorter periods of time (2 min) in subsequent experiments.
BAPTA‐AM, a membrane permeable form of the BAPTA Ca2+ chelator, was used to reduce levels cytoplasmic free Ca2+ (Billman 1993). Chelation of intracellular Ca2+ by BAPTA resulted in diminished cellular response to ionomycin treatment (Fig. 1f). Similarly, BAPTA prevented the cytoplasmic Ca2+ increase caused by TG treatment (data not shown). The effect BAPTA as a cytosolic Ca2+ chelator is directly related to its presence in the cytosol. Under our experimental conditions (incubation at 37 °C), BAPTA remained within the cytosol for approximately 2–3 h, after which it is either metabolized or it accumulated within intracellular organelles (V.S. unpublished observations).
Cell recovery and morphological changes after release from mitotic arrest
Mitotically arrested cells have a rounded morphology and are loosely attached to the surface of the tissue culture dish. We determined the ability of synchronized HEK 293 cells to re‐attach and spread after release from mitotic arrest and evaluated their subsequent morphology by light microscopy, after treatment with various calcium altering reagents. Cell viability was evaluated by staining the cells with trypan blue. Treatment of cells with hydrogen peroxide, which induces rapid cell death, was used as a control for viability in these experiments.
Figure 2a depicts the time‐dependent behaviour of untreated or TG‐treated cells after release from the mitotic block. Upon removal of nocodazole, rounded untreated cells reattached to the surface of the tissue culture plate and were fully spread by 3 h, remaining attached for the rest of the experiment. Cells treated with TG or with a combination of TG/EGTA for 30 min prior to release from mitotic arrest, were able to reattach and spread in the tissue culture plates in a manner that is indistinguishable from untreated cells (Fig. 2a; 3 h and 6 h). Around 9–12 h after release from mitotic arrest, TG‐treated cells begin to lose their attachment to the extracellular matrix and lifted off the surface of the plates such that by 24 h almost 100% were rounded up and floating (Fig. 2a; 9 h, 12 h and 24 h).
Figure 2.
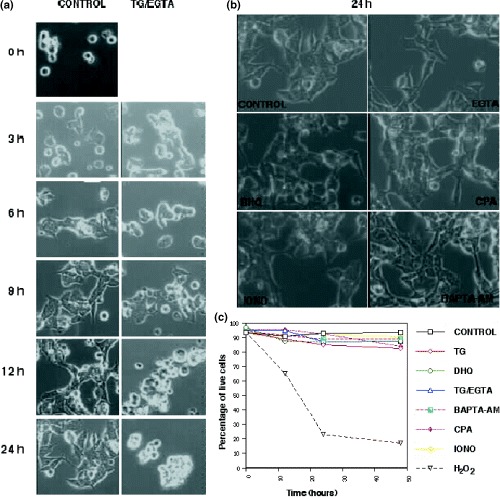
Cellular Behaviour After Release From Nocodazole Arrest. (a) Synchronized HEK 293 cells were either left untreated (CONTROL) or treated with TG/EGTA for 30 min prior to release from nocodazole arrest. Upon release from mitotic arrest and removal of TG/EGTA, a different group of cells from each population were photographed at (3, 6, 9,12 and 24 h) after removal of the drug. (b) Synchronized HEK 293 cells were treated with various reagents for 30 min at 37 °C prior to their release from mitotic arrest. Reagents were then removed and cells were replated in fresh DMEM containing 10% FBS. Pictures were taken 24 h after replating. CONTROL, EGTA (5 m m), BHQ (30 µm), CPA (10 µm), IONO (1 µm) and BAPTA‐AM (10 µm). (c) Cell viability of the culture was determined using the trypan blue exclusion assay at 0, 12, 24 and 48 h after removal of reagents and release from arrest. Hydrogen peroxide (H2O2) was used to induce cell death as a positive control.
The change in morphology observed in TG and TG/EGTA treated cells 9–12 h after removal of the drug raised the possibility that TG may trigger an apoptotic pathway. Since a hallmark of apoptosis is DNA fragmentation, we used a commercially available Tunel (TdT‐mediated dUTP‐X nick end labelling) assay kit. This assay labels fragmented DNA ends with fluorescein‐dUTP, thus labelling only apoptotic cells. We find that the percentage of Tunel‐positive cells is very low (<5%) in untreated and TG‐treated cells at 12 h and 24 h after removal of TG (data not shown). In contrast, a control population of cells treated with H2O2 was 95–100% Tunel‐positive within one hour after treatment. Therefore, the changes in cellular morphology and cellular attachment that are observed in TG/EGTA treated cells are not due to the induction of an apoptotic pathway.
Synchronized cells treated with EGTA alone, CPA or BHQ, for 30 min or with ionomycin for 2 min prior release from mitotic arrest, displayed no detectable difference in attachment, spreading or morphology when compared to untreated cells even 24 h after treatment (Fig. 2b). In a similar manner, cells preloaded with BAPTA‐AM before their release from mitotic arrest behaved indistinguishably from untreated cells (Fig. 2b). Trypan blue exclusion suggests that cell viability remains high for at least 48 h after treatment in cells treated with any of the above reagents (Fig. 2c).
Calcium dynamics and G1/S progression
Synchronized HEK 293 cells were exposed to a single, short‐term treatment with each of the reagents described in the previous section. The ability of these cells to complete mitosis, progress through G1 and enter S phase was evaluated by measuring the DNA content of cells at various times after removal from mitotic arrest. Treated and untreated HEK 293 cell samples were processed for flow cytometry and stained with propidium iodide.
Approximately 70–80% of cells have a 4N DNA content at the time prior release from mitotic arrest (Fig. 3a; C, 0 h). Untreated control cells finished mitosis and cytokinesis within 2 and 6 h after release from mitotic arrest, as measured by the decrease in the percentage of cells with a 4N DNA content and the increase in percentage of cells with a 2N DNA content (Fig. 3a; C, 6 h). Twelve hours after release from mitotic arrest, approximately 60% of control cells appeared to have begun replicating their DNA, as evidenced by the increasing number of cells having a DNA content between 2N and 4N (Fig. 3a; C, 12 h). By 24 h after release from mitotic arrest, untreated cells have finished cell division and although synchrony is diminished, some cells appeared to have begun a new round of DNA replication by 30 h after release (Fig. 3a; C, 24 h and 30 h).
Figure 3.
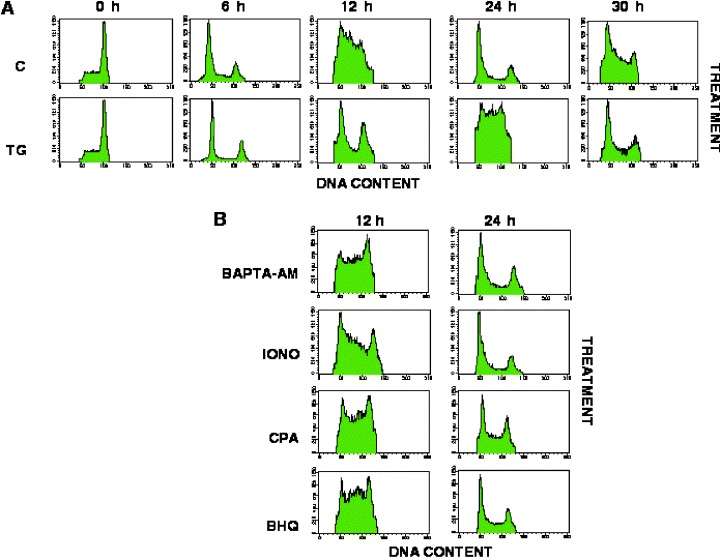
Thapsigargin Delays G1/S‐Phase Progression. (a) Synchronized cells were treated with reagents as described in Figure 2a; Control, TG/EGTA. At the times indicated, aliquots of the culture were fixed using 80% ethanol and stained using propidium iodide as described in ‘Materials and Methods’. DNA content was analysed using standard flow cytometric techniques. (b) Cells were treated with BAPTA‐AM (10 µm), ionomycin (IONO; 1 µm), CPA (10 µm) and BHQ (30 µm) as described in ‘Materials and Methods.’ Flow cytometry profiles of treated cells, fixed at 12 h and 24 h, are shown.
Synchronized cells, which had been treated with TG/EGTA for 30 min prior to their release from mitotic arrest, finished mitosis with the same dynamics as untreated cells (Fig. 3a; TG/EGTA, 6 h). Yet they failed to begin DNA replication until 24 h after release from mitotic arrest (Fig. 3a; TG/EGTA, 24 h). Thus, treatment of cells with a combination of TG/EGTA resulted in a 12‐h lengthening of the G1 phase. DNA replication appeared to be complete by 30 h after TG/EGTA treatment and release however, suggesting that this represents a delay in G1/S rather than a block in G1.
Treatments which result in the temporary increase of Ca2+ in the cytoplasm (ionomycin, BHQ or CPA) or chelation of cytosolic Ca2+ (BAPTA‐AM), did not alter the dynamics of cell cycle progression when compared to untreated cells (Fig. 3b). Moreover, chelation of the initial TG‐induced Ca2+ increase with BAPTA, failed to rescue timely progression through G1 and S phase entry in TG‐treated cells (data not shown). It is therefore unlikely that the delay in G1 observed in TG‐treated cells was due to the initial Ca2+ rise induced by this reagent. Instead, the delay in progression through G1/S could be due to: (a) an indirect effect caused by TG on an unidentified target; or (b) SERCA inactivation, which leads to the depletion of intracellular stores and the inability of cells to regenerate Ca2+; or (c) SERCA inhibition, but unrelated to intracellular Ca2+.
Delay in G1 is due to the specific long‐term inactivation of SERCA
In an attempt to determine whether the effect of TG on progression through G1 was due to inactivation of SERCA rather than to its effect on a yet unidentified cellular protein, we examined cell cycle progression in cells resistant to TG. Previous studies have shown that a single amino acid substitution (F256V) in the chicken SERCA‐1 protein could render this pump up to 40‐fold less sensitive to inhibition by TG (Yu et al. 1999). The SERCA‐1 protein bearing the F256V substitution and a C‐terminal myc‐tag was stably expressed in HEK 293 cells under the control of the CMV promoter. A characteristic clone, G17, was selected as neomycin resistant and for mid to high expression of SERCA. SERCA expression was confirmed using the 9E‐10 antimyc monoclonal antibody (Fig. 4a).
Figure 4.
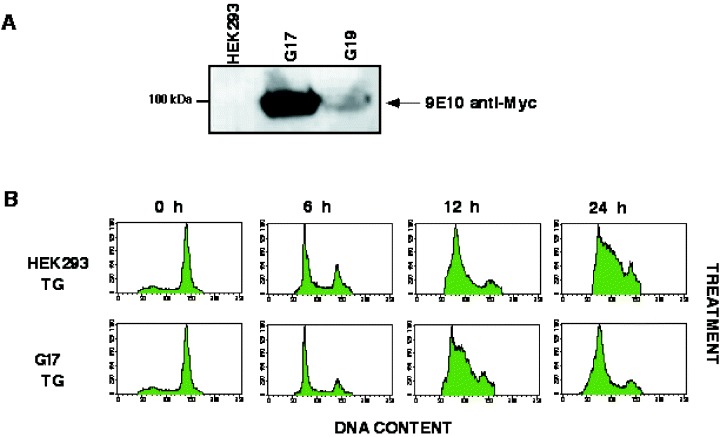
The Thapsigargin Effect on HEK 293 Cells is due to Inactivation of SERCA. (a) A stable cell line bearing the myc‐F256V‐SERCA substitution (clone G17) was prepared as described in ‘Materials and Methods’. Untransfected HEK 293 cells and two different clones G17 and G19 cells were lysed and subjected to SDS‐PAGE and Western blotting (30 µg of protein per sample). Blots were probed with the anti‐myc mouse monoclonal 9E10 antibody. (b) HEK 293 cells and cells from clone G17 were synchronized and treated with thapsigargin as described in Figure 2(A). At the times indicated, aliquots of the culture were fixed and processed for flow cytometry as described in Figure 3.
We find that a 30‐min treatment of mitotically synchronized G17 cells with TG/EGTA did not affect cell morphology, attachment or the timely progression through G1/S phase (Fig. 4b). This finding, combined with the observation that temporary inhibition of SERCA (BHQ and CPA) fail to reproduce the TG effect, suggest that it is the irreversible inactivation of the SERCA pump which results in the delay in cell cycle progression.
Cell cycle progression is sensitive to thapsigargin treatment early during G1
In an attempt to determine the sensitivity of the G1 phase to depletion of intracellular Ca2+ stores, cells were exposed to TG for 30 min intervals at various times after release from mitotic arrest. Cells were treated with TG/EGTA at 2, 4, 6, 8, 10 and 12 h for 30 min, and following media replacement, cells were allowed to progress through the rest of the cycle undisturbed. We found that cells treated within the first 4–6 h after release from mitotic arrest were delayed in G1/S progression, as determined by the low percentage of cells with a DNA content between 2N and 4N by 12 h (Fig. 5, TG2–12 h, TG4–12 h and TG6–12 h) when compared to control cells. In contrast, increasing number of cells with a DNA content greater than 2N are found in cells treated with TG/EGTA at times later than 6 h (Fig. 5; TG6–12 h – TG12–12 h). This experiment defines a period of TG sensitivity during early G1 phase.
Figure 5.
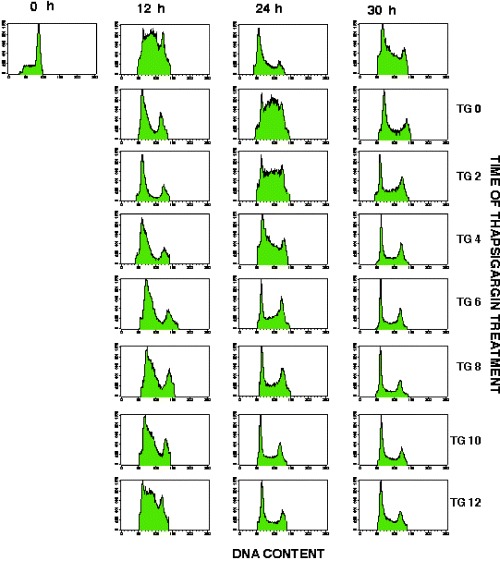
HEK 293 Cells are Sensitive to Thapsigargin Early During G1. Cells were released from synchronization and plated on fresh DMEM with 10% FBS, as described in ‘Materials and Methods’. Cells were treated with 1 µm TG and 5 m m EGTA for 30 min at the times indicated. The media was then replaced and cells were allowed to progress through the cell cycle. Treated and untreated cells were fixed and processed for flow cytometry at 12, 24 and 30 h after release from mitotic arrest.
Calcium store depletion alters the activity and expression of G1 phase cell cycle regulators
To determine if TG/EGTA treatment affected any of the major signalling pathways required for G1 progression, we examined the level of expression of cyclin D1 and cyclin E proteins and the level of activity of MAPKs. Activation of Erk1 and Erk2 leads to their phosphorylation on key residues (threonine 202 and tyrosine 204); the level of phosphorylation is therefore indicative of activity. In control cells the levels of Erk1 and Erk2 phosphorylation remained high during G1 and S phases, with a consistent slight reduction around 12 h after release from mitotic arrest. In contrast, Erk1 phosphorylation levels were significantly reduced by 9 h after release from mitotic arrest in TG/EGTA treated cells (Fig. 6). Erk2 phosphorylation levels are also reduced in TG/EGTA treated cells, but to a lesser extent than Erk1. Treatment of cells with the other reagents that alter cytosolic Ca2+ levels or treatment of G17 (TG resistant cells) with TG/EGTA had no effect on MAPK phosphorylation (data not shown).
Figure 6.
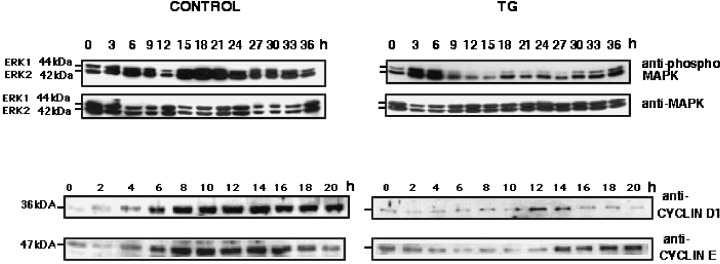
Thapsigargin Treatment Affects The Activity and Expression of G1/S Phase Molecular Regulators. HEK 293 cells were lysed at indicated times after release from nocodazole arrest, as described in ‘Materials and Methods’. Samples were then subjected to SDS‐PAGE and Western blotting. (20 µg of lysate/per lane). Blots were probed with the following antibodies: antiphospho‐MAPK, anti‐MAPK, anti‐cyclin D1, anti‐cyclin E.
Cyclin D1 levels increased steadily from 4 to 14 h after release from mitotic arrest in untreated cells. In contrast, the levels of cyclin D1 were significantly reduced in TG/EGTA treated cells, where some increase in protein levels is observed only around 12–14 h (Fig. 6). In control cells, cyclin E protein levels increased four‐fold from 6 to 16 h after release from mitotic arrest. TG/EGTA treatment, however, resulted in significantly lower levels of cyclin E protein, which only increased two‐fold between 12 and 14 h after release from mitotic arrest (Fig. 6).
The loss of MAPK activity and the lower levels of cyclin D1 and cyclin E proteins can be temporally correlated with the loss of cellular attachment in TG‐treated cells. These results suggests that SERCA inactivation by TG has a deleterious effect on essential G1 phase regulators.
DISCUSSION
Elementary and localized Ca2+ signals affect processes such as cell cycle progression, protein traffic, potassium channel opening, mitochondrial metabolism and apoptosis (Burgoyne & Morgan 1998; Krebs 1998; Lemaster et al. 1998; Vergara et al. 1998). Global Ca2+ signals, or waves, affect cellular processes such as muscle contraction, fertilization, gene transcription and cell proliferation. Ca2+ waves can also spread within tissues and affect events such as wound healing, ciliary beating and insulin secretion, among others (Beckingham, lu & andruss 1998; Borkakoti 1998; Brainman, Zagoory & Priel 1998; Hardingham & Bading 1998; Maechler & Wollheim 1998).
Since Ca2+ signalling has been implicated in controlling cell cycle events, it is essential to determine how changes in intracellular Ca2+ dynamics affect cell proliferation. Specific pharmacological agents, such as Ca2+ ionophores, Ca2+ chelators, and inhibitors of intracellular and plasma membrane Ca2+ pumps have been utilized to determine the effects of altering the tight intracellular regulation of Ca2+ levels on cell cycle progression. In view of the fact that the ER Ca2+ pools play a major role in the generation of intracellular Ca2+ fluxes, the major ER Ca2+ pump, SERCA, has been the focus of several studies which aim to eliminate Ca2+ stores using specific SERCA inhibitors (For review see: Treiman, Caspersen & Christensen 1998; Misquitta, Mack & Grover 1999).
We used various agents, which alter intracellular Ca2+ homeostasis to treat HEK 293 cells arrested in mitosis prior to their release into G1. The purpose was to evaluate the effect of short‐term treatments, which lead to predictable and finite changes in intracellular Ca2+ levels, on cellular ability to finish mitosis, progress through G1 and transit into S phase. Since previous studies were performed after addition of serum to serum‐deprived cells, which focuses on the G0/G1 transition (Gosh et al. 1991; Short et al. 1993; Jiang et al. 1994), we wanted to evaluate G1 entry in the continuous presence of serum, under conditions in which only the intracellular Ca2+ homeostasis had been altered.
Short‐term treatments which result in temporary Ca2+ increases, Ca2+ chelation or reversible and temporary inactivation of SERCA, do not affect progression through M, G1 or S phases. In contrast, a short (30 min) treatment with TG, which irreversibly blocks the SERCA Ca2+ pump activity, delays DNA replication for approximately 12 h. This delay is not due to the inability of cells to finish mitosis, since TG‐treated cells complete mitosis with approximately the same kinetics as untreated cells. DNA content analysis of TG‐treated HEK 293 cells indicated that DNA synthesis is not inhibited but merely delayed.
The TG‐induced delay in G1/S transition observed in HEK 293 cells could be explained in a number of different ways. Long‐term inactivation of the SERCA pumps abolishes cellular ability to generate Ca2+ oscillations or transients. Since Ca2+ oscillations have been implicated in the regulation of events during other stages of the cell cycle, it is possible that they may also be essential during G1. The loss of Ca2+ oscillations may activate a Ca2+‐dependent cell cycle checkpoint required for DNA synthesis. Such a checkpoint could involve the inability to assemble the DNA replication machinery, or inability to activate Ca2+ dependent enzymes or transcription factors. By treating cells with TG at various times after the release from mitotic arrest we have established that the delay in S‐phase entry can be directly correlated with the time of TG treatment. The longest delay of the G1/S transition occurs when cells are treated during the first 0–6 h after release from mitotic arrest. Therefore, we have defined a window of time during early G1, in which Ca2+ oscillations and/or SERCA activity are absolutely necessary for progression into S phase. If this speculation is correct, the nature of this requirement deserves further investigation.
These findings are reminiscent of a serum‐dependent early G1 period described by Zetterberg & Larsson (1985). In this early report, the authors described a 3–4 h post mitotic serum dependent period in Swiss 3T3 which was essential for timely progression through G1. Platelet‐derived growth factor (PDGF), but not epidermal growth factor or insulin, was able to rescue normal progression through G1 in serum‐deprived cells. It is possible that serum factors or PDGF stimulate InsP3‐dependent Ca2+ oscillations which are required for the direct activation of Ca2+ dependent processes.
In addition, irreversible inactivation of the SERCA pump with TG in DDT1MF‐2 smooth muscle cells results in exit from the cell cycle and entry into a G0‐like growth state (Gosh et al. 1991). Growth arrested, TG‐treated DDT1MF‐2 cells remain intact, viable and maintain a normal cellular morphology for several days after treatment. High serum (20%) treatment of TG‐treated cells induces recovery of functional Ca2+ pools, due to synthesis of new pump protein, and reentry into the cell cycle (Short et al. 1993). We find that TG‐treated HEK 293 cells are also unable to progress through G1, but instead of arresting in G0, they display a delay at the G1/S transition. Under our experimental conditions HEK 293 cells are exposed to relatively high serum levels (10% FBS) throughout the experiment, we cannot rule out the possibility that the delay corresponds to the time required for new SERCA synthesis.
A possibility, which we cannot determine from these experiments, is that TG binding to SERCA inactivates a Ca2+‐independent function of the pump, which is necessary for cell cycle progression. For example, SERCA structure may be compromised by TG binding in such a way that it compromises the ability of the pump to interact with additional factors necessary for cellular stability and/or cell cycle progression.
We have also examined the possibility that TG may have an effect on a target different from SERCA. Cells expressing a TG‐resistant form of SERCA are not delayed in the G1/S transition after TG‐treatment. Therefore, we conclude that the G1/S delay is not due to the inactivation of a target different from SERCA. Moreover, since CPA and BHQ, which inactivate SERCA temporarily, do not cause a delay in G1/S progression we propose that only a long‐term inactivation of SERCA can cause this effect. The long‐term inactivation of SERCA and depletion of intracellular Ca2+ stores could also result in the failure of ER processing functions, leading to impaired processing of essential for cell function.
The delay in G1/S progression can be correlated with a complete loss of cellular attachment to the extracellular matrix several hours after treatment with TG. These morphological changes, suggest a deleterious effect on the cytoskeleton and/or integrin activity and signalling. The loss of cellular attachment could certainly result in the loss of integrin mediated signalling, which has previously been linked to the cellular ability to progress through the cell cycle (Han et al. 1993; Bottazzi & Assoian 1997; Bottazzi et al. 1999) since it contributes to activation of MAPKs. We find that levels of phosphorylated ERK1 are significantly lower 6 h after TG treatment, whereas the levels of ERK2 are also lower but to a lesser extent after this period. The timing of these events suggests that the loss of MAPK activity occurs prior to cellular rounding and detachment. It remains to be determined whether these events are directly related. This suggests that SERCA activity is necessary for the maintenance of high levels of active MAPK during G1.
Previous studies have shown that sustained levels of active MAPK are necessary for expression of cyclin D1 (Lavoie et al. 1996). Consistent with the lower activity of MAPK, we find reduced levels of cyclin D1 and cyclin E in TG‐treated cells. Since active cyclin D1/CDK4/6 complexes are required for cyclin E expression, it is likely that the lower levels of cyclin D1 can directly result in lower cyclin E protein accumulation. Since we also find decreased levels of cyclin E, one could speculate that the G1/S delay observed in these experiments is due to the decreased levels of both cyclins and decreased activity of these cyclin‐dependent complexes. Therefore, one interpretation of our findings is that the time delay in G1 is directly related to the inability of cells to accumulate sufficient levels of cyclin D1. Previous findings have established that the length of G1 is directly related to the expression levels of D1 and E cyclins, since overexpression of these proteins in cycling Rat‐1 fibroblasts results in a decrease in the length of the G1 phase (Resnitzky et al. 1994). The specific effect of Ca2+ and/or SERCA on expression or activation of these molecules is currently being studied.
These studies point to the potential existence of a organelle‐integrity checkpoint, which may be essential for the successful completion of cell division. Our current efforts are focused on elucidating the exact molecular pathways affected by TG treatment and SERCA inactivation.
Acknowledgements
This work was supported by the National Cancer Institute of Canada and the Canadian Cancer Society. M.F.M. is a Medical Research Council of Canada Scientist, and V.R.S. an MRC Fellow. We would like to thank Dr A. Hussain, who kindly provided us with the TG‐resistant form of the SERCA1 cDNA. We thank H. Chen for technical assistance, J. Tong for assistance with calcium imaging equipment and C. Smith for assistance with flow cytometry. We would also like to thank Dr D. MacLennan for critical comments on this manuscript.
References
- Anger M, Samuel JL, Marotte F, Wuytack F, Rappaport L, Lompre AM (1994) In situ mRNA distribution of sarco (endo) plasmic reticulum Ca(2+)‐ATPase isoforms during ontogeny in the rat. J. Mol. Cell Cardiol. 26, 539. [DOI] [PubMed] [Google Scholar]
- Bates S, Bonetta L, MacAllan D et al. (1994) CDK6 (PLSTIRE) and CDK4 (PSK‐J3) are a distinct subset of the cyclin‐dependent kinases that associate with cyclin D1. Oncogene 9, 71. [PubMed] [Google Scholar]
- Beckingham K, Lu AQ, Andruss BF (1998) Calcium‐binding proteins and development. Biometals 11, 359. [DOI] [PubMed] [Google Scholar]
- Berridge MJ (1995) Calcium signalling and cell proliferation. Bioessays 17, 491. [DOI] [PubMed] [Google Scholar]
- Billman GE (1993) Intracellular calcium chelator, BAPTA‐AM, prevents cocaine‐induced ventricular fibrillation. Am. J. Physiol. 265, H1529. [DOI] [PubMed] [Google Scholar]
- Bootman MD, Berridge MJ, Lipp P (1997) Cooking with calcium: the recipes for composing global signals from elementary events. Cell 91, 367. [DOI] [PubMed] [Google Scholar]
- Borkakoti N (1998) Matrix metalloproteases: variations on a theme. Prog. Biophys. Mol. Biol. 70, 73. [DOI] [PubMed] [Google Scholar]
- Bottazzi ME & Assoian RK (1997) The extracellular matrix and mitogenic growth factors control G1 phase cyclins and cyclin dependent kinase inhibitors. Trends Cell Biol. 7, 348. [DOI] [PubMed] [Google Scholar]
- Bottazzi ME, Zhu X, Bohmer RM, Assoian RK (1999) Regulation of p21 (cip1) expression by growth factors and the extracellular matrix reveals a role for transient ERK activity in G1 phase. J. Cell Biol. 146, 1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brainman A, Zagoory O, Priel Z (1998) PKA induces Ca2+ release and enhances ciliary beat frequency in a Ca2+‐dependent and ‐independent manner. Am. J. Physiol. 275, c790. [DOI] [PubMed]
- Burgoyne RD & Morgan A (1998) Calcium sensors in regulated exocytosis. Cell Calcium 24, 367. [DOI] [PubMed] [Google Scholar]
- Carafoli E (1987) Intracellular calcium homeostasis. Ann. Rev. Biochem. 56, 395. [DOI] [PubMed] [Google Scholar]
- Cheyette TE & Gross DJ (1991) Epidermal growth factor‐stimulated calcium ion transients in individual A431 cells: initiation kinetics and ligand concentration dependence. Cell Regul. 2, 827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foskett JK & Wong D (1992) Calcium oscillations in parotid acinar cells induced by microsomal Ca2+‐ATPase inhibition. Am. J. Physiol. 262, C656. [DOI] [PubMed] [Google Scholar]
- Girard F, Strausfeld U, Fernandez A, Lamb NJ (1991) Cyclin A is required for the onset of DNA replication in mammalian fibroblasts. Cell 67, 1169. [DOI] [PubMed] [Google Scholar]
- Gosh TK, Bian J, Short AD, Rybak SL, Gill DL (1991) Persistent intracellular calcium pool depletion by thapsigargin and its influence on cell growth. J. Biol. Chem. 266, 24690. [PubMed] [Google Scholar]
- Grynkiewicz G, Poenie M, Tsien RY (1985) A new generation of Ca2+ indicators with greatly improved fluorescence properties. J. Biol. Chem. 260, 3440. [PubMed] [Google Scholar]
- Han EK, Guadagno TM, Dalton SL, Assoian RK (1993) A cell cycle and mutational analysis of anchorage‐independent growth: cell adhesion and TGF‐beta 1 control G1/S transit specifically. J. Cell Biol. 122, 461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardingham GE & Bading H (1998) Nuclear calcium: a key regulator of gene expression. Biometals 11, 345. [DOI] [PubMed] [Google Scholar]
- Hiroyouki M, Inoue S, Hiroyoushi H (1994) The effect of KN‐62, Ca2+/calmodulin dependent protein kinase II inhibitor on cell cycle. Biochem. Biophys. Res. Comm. 199, 241. [DOI] [PubMed] [Google Scholar]
- Horton LE, Qian Y, Templeton DJ (1995) G1 cyclins control the retinoblastoma gene product growth regulation activity via upstream mechanisms. Cell Growth Differ. 6, 395. [PubMed] [Google Scholar]
- Irvine RF (1989) How do inositol 1,4,5‐trisphosphate and inositol 1,3,4,5‐tetrakisphosphate regulate intracellular Ca2+? Biochem. Soc. Trans. 17, 6. [DOI] [PubMed] [Google Scholar]
- Jiang S, Chow SC, Nicotera P, Orrenius S (1994) Intracellular Ca2+ signals activate apoptosis in thymocytes: studies using the Ca2+‐ATPase inhibitor thapsigargin. Exp. Cell Res. 212, 84. [DOI] [PubMed] [Google Scholar]
- Kauffman RF, Taylor RW, Pfeiffer DR (1980) Cation transport and specificity of ionomycin. Comparison with ionophore A23187 in rat liver mitochondria. J. Biol. Chem. 255, 2735. [PubMed] [Google Scholar]
- Krebs J (1998) The role of calcium in apoptosis. Biometals 11, 375. [DOI] [PubMed] [Google Scholar]
- Lavoie JN, L'Allemain G, Brunet A, Muller R, Pouyssegur J (1996) Cyclin D1 expression is regulated positively by the p42/p44 MAPK and negatively by the p38/HOGMAPK pathway. J. Biol. Chem. 271, 20608. [DOI] [PubMed] [Google Scholar]
- Lemasters JJ, Nieminen AL, Qian T et al. (1998) The mitochondrial permeability transition in cell death: a common mechanism in necrosis, apoptosis and autophagy. Biochim. Biophys. Acta 1366, 177. [DOI] [PubMed] [Google Scholar]
- Leof EB, Van Wyk JJ, O'Keefe EJ, Pledger WJ (1983) Epidermal growth factor (EGF) is required only during the traverse of early G1 in PDGF stimulated density‐arrested BALB/c‐3T3 cells. Exp. Cell Res. 147, 202. [DOI] [PubMed] [Google Scholar]
- Liu C & Hermann TE (1978) Characterization of ionomycin as a calcium ionophore. J. Biol. Chem. 253, 5892. [PubMed] [Google Scholar]
- Lytton J, Westlin M, Hanley MR (1991) Thapsigargin inhibits the sarcoplasmic or endoplasmic reticulum Ca2+‐ATPase family of calcium pumps. J. Biol. Chem. 266, 17067. [PubMed] [Google Scholar]
- MacLennan DH, Rice WJ, Green MN (1997) The mechanism of Ca2+ transport by sarco (Endo) plasmic reticulum Ca2+‐ATPases. J. Biol. Chem. 272, 28815. [DOI] [PubMed] [Google Scholar]
- Maechler P & Wollheim CB (1998) Role of mitochondria in metabolism‐secretion coupling of insulin release in the pancreatic Beta‐cell. Biofactors 8, 255. [DOI] [PubMed] [Google Scholar]
- Martonosi AN (1996) Structure‐function relationships in the Ca2+‐ATPase of sarcoplasmic reticulum: facts, speculations and questions for the future. Biochim. Biophys. Acta 1275, 111. [DOI] [PubMed] [Google Scholar]
- Mason MJ, Garcia‐Rodriguez C, Grinstein S (1991) Coupling between intracellular Ca2+ stores and the Ca2+ permeability of the plasma membrane. Comparison of the effects of thapsigargin, 2,5‐di‐(tert‐butyl)‐1,4‐hydroquinone, and cyclopiazonic acid in rat thymic lymphocytes. J. Biol. Chem. 266, 20856. [PubMed] [Google Scholar]
- Misquitta CM, Mack DP, Grover AK (1999) Sarco/endoplasmic reticulum Ca2+ (SERCA)‐pumps: link to heart beats and calcium waves. Cell Calcium 25, 277. [DOI] [PubMed] [Google Scholar]
- Ohtsubo M, Theodoras AM, Schumacher J, Roberts JM, Pagano M (1995) Human cyclin E, a nuclear protein essential for the G1‐to‐S phase transition. Mol. Cell Biol. 15, 2612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parekh AB & Penner R (1997) Store depletion and calcium influx. Physiol. Rev. 77, 901. [DOI] [PubMed] [Google Scholar]
- Penner R, Fasolato C, Hoth M (1993) Calcium influx and its control by calcium release. Curr. Opin. Neurobiol. 3, 368. [DOI] [PubMed] [Google Scholar]
- Peres A & Giovannardi S (1990b) Mitogen‐induced oscillations of membrane potential and Ca2+ in human fibroblasts. FEBS Lett. 261, 35. [DOI] [PubMed] [Google Scholar]
- Peres A, Racca C, Zippel R, Sturani E (1990a) Cytosolic calcium and membrane conductance in response to platelet‐ derived growth factor and bradykinin stimulation in single human fibroblasts. Eur J. Cell Biol. 53, 290. [PubMed] [Google Scholar]
- Poenie M, Alderton J, Tsien RY, Steinhardt RA (1985) Changes of free calcium levels with stages of the cell division cycle. Nature 315, 147. [DOI] [PubMed] [Google Scholar]
- Ratan RR, Maxfield FR, Shelanski ML (1988) Long‐lasting and rapid calcium changes during mitosis. J. Cell Biol. 107, 993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Resnitzky D, Gossen M, Hermann B, Reed SI (1994) Acceleration of the G1/S phase transition by expression of cyclins D1 and E with an inducible system. Mol. Cell. Biol. 14, 1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Resnitzky D & Reed SI (1995) Different roles for cyclins D1 and E in regulation of the G1‐to‐S transition. Mol. Cell. Biol. 15, 3463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Short AD, Bian J, Ghosh TK, Waldron RT, Rybak SL, Gill DL (1993) Intracellular Ca2+ pool content is linked to control of cell growth. Proc. Natl. Acad. Sci. USA 90, 4986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stone JC, Moran MF, Pawson T (1991) Construction and Expression of Linker Insertion and Site‐Directed Mutants of v‐fps Protein‐Tyrosine Kinase. Meth Enzymol. 200, 673. [DOI] [PubMed] [Google Scholar]
- Straus DS (1984) Growth‐stimulatory actions of insulin in vitro and in vivo. Endocr. Rev. 5, 356. [DOI] [PubMed] [Google Scholar]
- Thastrup O, Cullen PJ, Drobak BK, Hanley MR, Dawson AP (1990) Thapsigargin, a tumor promoter, discharges intracellular Ca2+ stores by specific inhibition of the endoplasmic reticulum calcium ATPase. Proc. Natl. Acad. Sci. USA 87, 2466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tombes RM & Borisy GG (1989) Intracellular free calcium and mitosis in mammalian cells: anaphase onset is calcium modulated, but is not triggered by a brief transient. J. Cell Biol. 109, 627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treiman M, Caspersen C, Christensen SB (1998) A tool coming of age: thapsigargin as an inhibitor of sarco‐endoplasmic reticulum Ca(2+)‐ATPases. Trends Pharmacol. Sci. 19, 131. [DOI] [PubMed] [Google Scholar]
- Vergara C, Latorre R, Marrion NV, Adelman JP (1998) Calcium‐activated potassium channels. Curr. Op. Neurobio. 8, 321. [DOI] [PubMed] [Google Scholar]
- Wahl M & Gruenstein E (1993) Intracellular free Ca2+ in the cell cycle in human fibroblasts: transitions between G1 and G0 and progression into S phase. Mol. Biol. Cell 4, 293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber JD, Raben DM, Phillips PJ, Baldassare JJ (1997) Sustained activation of extracellular‐signal‐regulated kinase 1 (ERK1) is required for the continued expression of cyclin D1 in G1 phase. Biochem. J. 326, 61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitfield JF, Boynton AL, MacManus JP et al. (1980) The roles of calcium and cyclic AMP in cell proliferation. Ann. N. Y. Acad. Sci. 339, 216. [DOI] [PubMed] [Google Scholar]
- Wojnowski L, Hoyland J, Mason WT, Schwab A, Westphale HJ, Oberleithner H (1994) Cell transformation induces a cytoplasmic Ca2+ oscillator in Madin‐Darby canine kidney cells. Pflugers Arch. 426, 89. [DOI] [PubMed] [Google Scholar]
- Yu M, Lin J, Khadeer M, Yeh Y, Inesi G, Hussain A (1999) Effects of various amino acid 256 mutations on sarcoplasmic/endoplasmic reticulum Ca2+ ATPase function and their role in the cellular adaptive response to thapsigargin. Arch. Biochem. Biophys. 362, 225. [DOI] [PubMed] [Google Scholar]
- Zetterberg A & Larsson O (1985) Kinetic analysis of regulatory events in G1 leading to proliferation or quiescence of Swiss 3T3 cells. Proc. Natl. Acad. Sci. USA 82, 5365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu X & Assoian RK (1995) Integrin‐dependent activation of MAP kinase: a link to shape‐dependent cell proliferation [published erratum appears in Mol. Biol. Cell. 1996 Jun; 7, 1001]. Mol. Biol. Cell 6, 273. [DOI] [PMC free article] [PubMed] [Google Scholar]