

Abstract
Objectives
Up to now it has been unclear whether stromal/epithelial interaction affects progression of colon cancer. This study was designed to examine effects of tumour necrosis factor alpha (TNFα)‐activated stromal cyclooxygenase‐2 (COX‐2) signalling on proliferation and invasiveness of colon cancer epithelial cells.
Materials and methods
Cyclooxygenase‐2 mRNA and protein were determined by real‐time PCR and western blotting and prostaglandin E2 (PGE2) was assayed by radioimmunoassay. Cell proliferation and invasiveness were determined by transwell chamber assays and protein kinase C (PKC) was assayed by Biotrak™ PKC Assay System.
Results
Our results indicated that TNFα, a powerful inflammatory cytokine, strongly promoted COX‐2 expression and PGE 2 production in colon cancer‐associated fibroblasts. Using in vitro assays for estimating proliferative and invasive potential, we discovered that activation of stromal COX‐2 signalling significantly promoted proliferation and invasiveness of colon cancer epithelial cells. In addition, selective COX‐2 inhibitor N‐[2‐(Cyclohexyloxy)‐4‐nitrophenyl]methanesulfonamide, blocked such proliferative and invasive effects on the cancer epithelial cells. In this process, PKC was involved in activation of COX‐2 signalling in the fibroblasts.
Conclusion
We conclude that activation of stromal COX‐2 signalling by TNFα played a major role in promoting proliferation and invasiveness of colon cancer epithelial cells.
Introduction
Colon cancer ranks second in all malignant fatal diseases in the United States 1 and abnormal expression of cyclooxygenase‐2 (COX‐2) and overproduction of its product, prostaglandin E2 (PGE2), have been noted in most of colon cancer tissues 2. Nevertheless, which cell compartments play major roles in regulation of COX‐2 expression and PGE2 production are still controversial. In addition, how activation of COX‐2 signalling affects behaviour of colon cancer at the cellular lever remains unclear.
Traditionally, genetic alterations in the epithelial compartment have been considered to be the major cause of colon cancer 3. However, recent evidence has suggested that fibroblasts in stromal cell or tissue compartments were involved in COX‐2 signalling 4, 5, 6, 7, 8 and tumorigenesis 4, 9, 10, 11.
Stroma provides the connective tissue framework compartment, largely consisting of fibroblasts. Previously, one report has revealed that fibroblasts from the stromal compartment play a critical role in development and progression of carcinomas 12. Recently, an investigation has demonstrated that cancer‐associated fibroblasts (CAFs) were associated with initiation and development of epithelial ovarian cancer 13. We have shown that colonic CAFs are a potent source of COX‐2 expression and PGE2 synthesis 5, 6, 7 and inflammatory factors such as interleukin 1β and deoxycholic acid may induce prolonged COX‐2 expression and PGE2 production in CAFs 5, 6, 7; such activation of stromal COX‐2 signalling promotes proliferative and invasive responses of colon cancer epithelial cells 8, 14, 15. It is unclear whether inflammatory cytokines such as TNFα can activate CAFs, and if so, how, thus promoting proliferative and invasive responses of colon cancer epithelial cells.
A previous report has noted that TNFα can activate COX‐2 expression via a variety of types of extranuclear signalling, such as by protein kinase C (PKC), mitogen‐activated protein kinase (MAP‐kinase), phospholipase C‐γ2, tyrosine kinases and Rho proteins and NFκB 15, 16, 17, 18, 19 in endothelial cells 16, 18, epithelial cells 20 and fibroblasts 15. However, it is unclear which signalling controls TNFα‐mediated events such as proliferation and invasiveness of the fibroblasts.
The aim of our study described here was to investigate whether activation of COX‐2 signalling in CAFs by TNFα could modulate behaviour of colon cancer epithelial cells, including their proliferation and invasiveness, and signalling that may be responsible for such changes in behaviour of colon cancer epithelial cells.
Materials and methods
Materials
Human colon cancer cell lines (HT29, Caco2, HCA7 and HCT116) were obtained from American Type Culture Collection (Manassas, VA, USA) and cell culture reagents, TNFα, NS398 and bisindoylmalemide I (BIM) were purchased from Sigma (St. Louis, MO, USA). Real‐time PCR primer probes were purchased from Applied Biosystems (Foster City, CA, USA) and COX‐2 antibody was obtained from Cayman Chemical (Cayman, Ann Arbor, MI, USA). PGE2 radioimmunoassay kits were obtained from Amersham (Arlington Heights, IL, USA). Primary normal and CAFs cultures were initiated from pinch biopsies obtained from normal and cancer patients, with approval of Institutional Review Board of the Buffalo Veterans Affairs Medical Center, USA; colonoscopies were performed in the course of routine clinical care.
Real‐time PCR analysis of COX‐2 mRNA
Total RNA was extracted using RNeasy Mini Kit (Qiagen, Valencia, CA, USA) and was reverse‐transcribed using High Capacity Reverse Transcription Kit (Applied Biosystems, Foster City, CA, USA). cDNA of each cell sample was amplified by real‐time RT‐PCR using specific primer‐probe mixtures and DNA polymerase in 7000 Real‐time PCR System (Applied Biosystems). Real‐time RT‐PCR profile consisted of 10 min initial activation at 95 °C, followed by 40 cycles 15 s denaturation at 95 °C and 1 min annealing and extension at 60 °C. Genuine identity of each PCR product was confirmed by size determination using 2% agarose gels followed by ethidium bromide staining, together with PCR marker in EC3 Imaging System (BioImaging System, Upland, CA, USA). Each experiment was repeated 4 times.
Western blotting analysis of COX‐2 proteins
Cyclooxygenase‐2 protein level was determined as described below. Confluent cultured fibroblasts were treated with test compounds and lysates were electrophoresed on polyacrylamide gels in the presence of sodium dodecyl sulphate (SDS‐PAGE) and electroblotted to an Immobilon‐P membrane (Millipore Corporation, Bedford, MA, USA). The membrane was incubated for 4 h in PBS containing 10% non‐fat milk and 0.1% Tween 20 at pH 7.4. This was followed by 2 h incubation at room temperature with anti‐human COX‐2 antibody (Cayman, Ann Arbor, MI, USA) diluted 1:1000 in PBS containing 10% non‐fat milk and 0.1% Tween 20 at pH 7.4. The membrane was washed three times in PBS containing 0.1% Tween 20 at pH 7.4, then incubated for 1 h with horseradish peroxidase‐conjugated goat anti‐rabbit IgG (Sigma) for detection of COX‐2 protein. The membrane was washed four times in PBS containing 0.1% Tween 20 at pH 7.4. Bound COX‐2 antibody was detected by chemiluminescence using reagents purchased from KPL (Gaithersburg, MD, USA) and resulting bands were analysed densitometrically; protein loading difference was corrected by density of β‐actin. Each experiment was repeated 4 times.
PGE2 assay
Fibroblasts were seeded in 24‐well plastic culture plates at 5 × 105 cells/cm2 and were cultured for 1 week in DMEM supplemented with 10% FBS. It is well recognized that serum is a potent inducer of COX‐2 and, thus, PGE2. Therefore, 24 h before administering cytokine or other treatments to fibroblast cultures, medium was replaced with DMEM supplemented with 1% FBS. To determine effects of TNFα, selected cultures were incubated in medium supplemented with TNFα (10 ng/ml). For the final 30 min before harvesting cultures, medium was replaced with PBS supplemented with 1% gelatin. Test compounds were present during this final incubation period. PGE2 levels in harvested PBS‐gelatin were determined using radioimmunoassay kits (Amersham) according to the manufacturer's instructions. Protein concentration in the harvest was determined by the Bradford method. All determinations of PGE2 levels were derived from experiments in four separate fibroblast cultures.
Co‐culture and proliferation assay
Fibroblasts (1 × 107/well, grown to 90% confluence) were first prepared in the lower compartment and were treated with PBS, TNFα (10 ng/ml) for 24 h. Colon cancer epithelial cells (1 × 106/well) were later cultured in the upper compartment of 12 mm transwell chambers (0.4 μm; Corning Costar Co., Cambridge, MA, USA) for 24 h. Four hours prior to termination of the experiment, 3[H]thymidine (0.8 μCi/apical transwell compartment) was added. Proliferation was determined from incorporation of 3[H]thymidine into cell DNA of the cancer epithelial cells. Cultures without fibroblasts were used as controls. Each experiment was repeated 4 times.
Co‐culture and invasion assay
Colon cancer epithelial cells (1 × 105/well) were grown 8 μm pore size filters of chambers coated in Matrigel (Biocoat chambers; Becton Dickinson, Bedford, MA, USA) for 24 h, in the upper compartment, with 90% confluent fibroblasts in the lower, pre‐treated with PBS or TNFα (10 ng/ml) for 24 h. Cultures without fibroblasts were used as controls. Cells on upper surfaces of the filters were removed and cells adhering to undersurfaces of filters were counted. Each experiment was repeated 4 times.
COX‐2 inhibitor assay
A highly selective COX‐2 inhibitor (NS 398, 10 μm) was combined with TNFα in selected cultures. Each experiment was repeated 4 times.
PKC and PKC inhibitor assay
Involvement of PKC signalling was addressed by measurement of PKC activity. Normal fibroblast (NFs) and CAFs were seeded in 60‐mm dishes at 5 × 105/well and cultured to 90% confluence. Selected cultures were then treated with TNFα for periods of 1–10 min. Treated and control cultures were washed in PBS and homogenized in buffer containing 50 mm Tris/HCl at pH 7.5, 0.3% β‐mercaptoethanol, 50 μg/ml phenylmethylsulphonyl fluoride and 10 mm benzamidine. The homogenate was centrifuged at 100 000 ×g and pellet re‐suspended in the same buffer supplemented with 0.2% Triton X‐100 and mixed on ice for 60 min, before re‐centrifugation at 100 000 ×g. Solubilized particulate fractions were assayed for PKC activity using the Biotrak™ PKC Assay System (Amersham Pharmacia Biotech, Piscataway, NJ, USA). The system was based on PKC‐catalysed transfer of [γ‐32P]ATP to a PKC‐specific peptide and activity was expressed as pmol/min/mg protein (n = 4). In a further experiment, a selective PKC inhibitor, BIM (10 μm), was combined with TNFα.
Cell viability
Cell viability was determined by trypan blue exclusion 5. Cells were simultaneously seeded from a single parent culture and cultures were incubated without (control) or with TNFα. Treated and control cultures were harvested at the same time, after careful washing to remove cells that had detached during incubation. Harvested cells were incubated with trypan blue and counted using a haemacytometer. From each culture, four fields of triplicate preparations were counted for percentage of cells that excluded the dye. Cell viability was >95% after all treatment periods (data not shown).
Statistical analysis
All summary data were reported as means ± SE calculated for each group and compared using analysis of variance test and Student's unpaired t‐test by MicroSoft Excel™ (MicroSoft, Redmont, WA, USA). Test results were reported as two‐tailed P values, where P < 0.05 was considered statistically significant.
Results
TNFα elicited significantly higher expression of COX‐2 mRNA, protein and PGE2 in CAFs than in NFs
Resting CAFs expressed 6‐fold higher COX‐2 mRNA than resting NFs, indicating that COX‐2 was already activated in resting CAFs. Stimulation by TNFα elicited 9‐fold upregulation of COX‐2 mRNA in NFs; in contrast, stimulation by TNFα induced 44‐fold upregulation of COX‐2 mRNA in CAFs, compared to control of NFs, 7‐fold upregulation of COX‐2 transcripts compared to control of CAFs (Fig. 1a), suggesting that COX‐2 signalling could be activated by TNFα in both NFs and CAFs. However, activation of COX‐2 mRNA expression by TNFα was far greater in CAFs. Correspondingly, resting CAFs expressed 3‐fold more COX‐2 protein than that of resting NFs, indicating that COX‐2 protein was already overexpressed in resting CAFs. Stimulation by TNFα elicited 3‐fold upregulation of COX‐2 protein in NFs, but 6‐fold upregulation of COX‐2 protein in CAFs compared to control of NFs, 2‐fold upregulation of COX‐2 protein in CAFs compared to control of CAFs (Fig. 1b), suggesting that TNFα induced far greater expression of COX‐2 protein in CAFs than that in NFs. As expected, stimulation by TNFα elicited 13‐fold upregulation of PGE2 synthesis in NFs, 50‐fold upregulation of PGE2 synthesis in CAFs compared to control of NFs, 6‐fold upregulation of PGE2 synthesis in CAFs compared to control of CAFs (Fig. 1c), indicating that higher production of PGE2 was induced by TNFα in CAFs than in NFs.
Figure 1.

(a) Tumour necrosis factor alpha (TNFα) stimulated cyclooxygenase‐2 (COX‐2) mRNA in Normal fibroblast (NFs) and cancer‐associated fibroblasts (CAFs). NFs or CAFs were cultured in medium with or without TNFα (10 ng/ml). Stimulation by TNFα elicited 10‐fold upregulation of COX‐2 mRNA in NFs, compared to 7‐fold upregulation of COX‐2 mRNA in CAFs (n = 4, *P < 0.05 and **P < 0.01, respectively). Level of COX‐2 in resting CAFs was 6‐fold higher than that in resting NFs (n = 4, #P < 0.05), while level of COX‐2 in TNFα‐stimulated CAFs was 4‐fold higher than that in TNFα‐stimulated NFs (n = 4, #P < 0.05). (b) TNF α induced COX ‐2 protein expression in NF s and CAF s. NFs or CAFs were cultured in medium with or without TNFα (10 ng/ml). Stimulation by TNFα elicited 3‐fold upregulation of COX‐2 protein in NFs, but 2‐fold upregulation of COX‐2 protein in CAFs (n = 4, *P < 0.05). Level of COX‐2 protein in resting CAFs was 3‐fold greater that in resting NFs (n = 4, #P < 0.05), while level of COX‐2 protein in TNFα‐stimulated CAFs was 2‐fold greater than that in TNFα‐stimulated NFs (n = 4, #P < 0.05). (c) TNF α increased synthesis of PGE 2 in NF s and CAF s. NFs or CAFs were cultured in medium with or without TNFα (10 ng/ml). Stimulation by TNFα elicited 13‐fold upregulation of PGE 2 synthesis in NFs compared to 6‐fold upregulation of synthesis in CAFs (n = 4, *P < 0.05 and **P < 0.01 respectively). Level of COX‐2 protein in resting CAFs was 7‐fold greater than that in resting NFs (n = 4, #P < 0.05), while level of COX‐2 protein in TNFα‐stimulated CAFs was 4‐fold greater than that in TNFα‐stimulated NFs (n = 4, #P < 0.05).
CAFs activated by TNFα promoted greater proliferative and invasive responses of colon cancer epithelial cells than corresponding NFs
Baseline observation showed that NFs and CAFs promoted proliferation of cancer epithelial cells 2‐ and 3‐fold (Fig. 2a,b), respectively. CAFs activated by TNFα promoted a greater proliferative response of cancer epithelial cells than corresponding NFs (5‐fold and 3‐fold increase respectively compared to fibroblast controls, Fig. 2a,b), indicating that activation of COX‐2 signalling by TNFα in CAFs promoted expansion of cancer epithelial cells. In parallel, baseline NFs and CAFs promoted invasive potential by increasing invasive cell numbers by 40/view and 75/view, respectively (Fig. 2c,d). CAFs activated by TNFα promoted quantitatively greater invasive responses (increased by 90 invasive cells/view) than corresponding NFs (increased by 50 invasive cells/view), compared to corresponding controls, suggesting that activation of COX‐2 signalling in CAFs by TNFα promoted greater invasiveness in colon cancer epithelial cells than in NFs (Fig. 2c,d).
Figure 2.
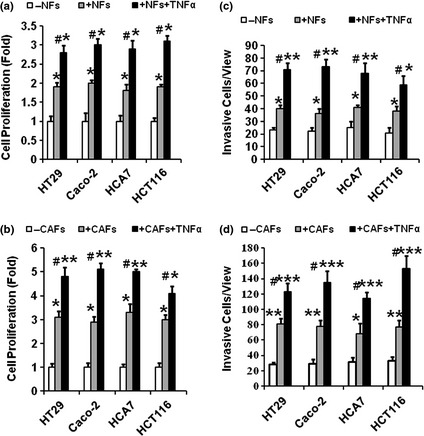
(a and b) Activation of cyclooxygenase‐2 ( COX ‐2) signalling by Tumour necrosis factor alpha ( TNF α) in Normal fibroblast ( NF s) and cancer‐associated fibroblasts ( CAF s) promoted proliferation of colon cancer epithelial cells. Colon cancer cell lines (HT29, and the others) and NFs or CAFs, respectively, were cultured in upper and lower compartments of Snapwell co‐culture chambers (0.4 μm pores). In selected experiments, some fibroblast cultures were stimulated with TNFα (10 ng/ml) for 24 h before co‐culture. Proliferation of colon cancer epithelial cells was measured by incorporation of 3[H]thymidine. Activation of COX‐2 signalling in CAFs (b), with or without treatment of TNFα, elicited quantitatively greater proliferative response in colon cancer epithelial cells than in NFs (a) (*P < 0.05 and **P < 0.01 compared to their corresponding −NFs control, n = 4; #P < 0.05 when compared to their corresponding +NFs group, n = 4). As a result, activation of COX‐2 signalling by TNFα in CAFs elicited quantitatively greater proliferative response in colon cancer epithelial cells when co‐cultured with CAFs than when co‐cultured with NFs (5‐ and 3‐fold increase in proliferation respectively). (c and d) Activation of COX ‐2 signalling by TNF α in NF s and CAF s promoted invasiveness of colon cancer epithelial cells. NFs or CAFs and the cancer epithelial cells were cultured in separate compartments of Matrigel chambers (Becton Dickinson, Bedford, MA, USA), separated by a filter with 0.8 μm pore size. Upper surface of the filter, on which epithelial cells were cultured, was coated with Matrigel; lower compartment contained no cells (control), or fibroblast cultures in unsupplemented medium or medium supplemented with TNFα (10 ng/ml) for 24 h before co‐culture. After co‐culture for 24 h, cells on the upper surface of filters were removed. Cells on the lower surface, which had invaded through the Matrigel layer and the filter pores, were counted (*P < 0.05, **P < 0.01 and ***P < 0.001, respectively when compared to the −NFs control, n = 4; #P < 0.05 when compared to the +NFs control, n = 4). As a result, activation of COX‐2 signalling by TNFα in CAFs elicited quantitatively greater invasiveness response in colon cancer epithelial cells when co‐cultured with CAFs than when co‐cultured with NFs (~120 versus 70 invasive cells per view).
COX‐2 inhibitor NS398 blocked proliferative and invasive effects of TNFα‐activated NFs and CAFs on colon cancer epithelial cells
Our results indicate that NS 398, a specific COX‐2 inhibitor, almost completely blocked paracrine effects of NFs and CAFs activated by TNFα, on proliferative responses in colon cancer epithelial cells (Fig. 3a,b), suggesting that PGE2 elicited by the fibroblasts was mostly responsible for proliferative responses. In parallel, NS 398 also attenuated paracrine effects of NFs and CAFs activated by TNFα on invasive responses of the cancer epithelial cells (Fig. 3c,d), suggesting that PGE2 elicited by the fibroblasts was largely responsible.
Figure 3.

(a) Inhibition of cyclooxygenase‐2 ( COX ‐2) signalling by NS 398 in Normal fibroblast ( NF s) and cancer‐associated fibroblasts ( CAF s) blocked proliferation of colon cancer epithelial cells. NS 398 (10 μm) was added with or without TNFα (10 ng/ml). NS 398, a specific COX‐2 inhibitor, blocked most paracrine effects induced by TNFα from NFs (a) and CAFs (b) on proliferation of colon cancer epithelial cells (*P < 0.05 and **P < 0.01 compared to their corresponding controls, n = 4). (B) Inhibition of COX ‐2 signalling by NS 398 in NF s and CAF s attenuated invasiveness of colon cancer epithelial cells. NS 398 (10 μm) was added with or without TNFα (10 ng/ml). As a result, NS 398 also blocked most effects of NFs and CAFs stimulated by TNFα on invasiveness of colon cancer epithelial cells (n = 4, *P < 0.05, **P < 0.01, ***P < 0.001 when compared to their corresponding controls, n = 4).
TNFα‐stimulated PGE2 synthesis was mediated by PKC
Previously, TNFα has been reported to activate COX‐2 expression via extranuclear PKC signalling in endothelial cells 16, 18, epithelial cells 20 and fibroblasts 15. However, whether activation of COX‐2 signalling by TNFα in NFs and CAFs would be associated with proliferation and invasiveness of colon cancer epithelial cells had been unknown.
To clarify this issue, we performed PKC assay with samples of NFs and CAFs challenged with TNFα. Our results indicated that activation of PKC by TNFα peaked at 3 min in both NFs and CAFs (Fig. 4a). Immediate addition of PKC inhibitor BIM to cultures treated with TNFα for 24 h completely blocked activation of PKC (data not shown), attenuated TNFα‐stimulated synthesis of PGE2 in both NFs and CAFs (Fig. 4b) and blocked their proliferative and invasive effect on colon cancer epithelial cells (data not shown), suggesting that TNFα‐stimulated COX‐2 gene expression in the fibroblasts was PKC‐dependent and was associated with their proliferative and invasive effect on colon cancer epithelial cells.
Figure 4.
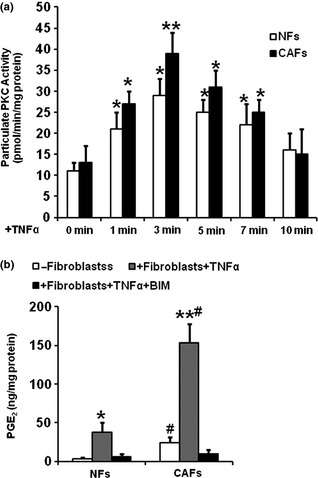
(a) Tumour necrosis factor alpha activated protein kinase C ( PKC ) activity in Normal fibroblast ( NF s) and cancer‐associated fibroblasts ( CAF s). NFs (a) and CAFs (b) were exposed to TNFα 10 ng/ml) for 1–10 min. Cell homogenates were pelleted, and particulate fractions were resolubilized. PKC activity of these samples was determined from transfer of [−32]ATP to a PKC‐specific peptide and were expressed as pmol/min/mg protein. Bars represent mean of results from 4 separated cultures ± SE. *P < 0.05 compared to their respective controls n = 4. (b) Effect of PKC on synthesis of PGE 2 . NFs and CAFs were co‐incubated with TNFα 10 ng/ml) and PKC inhibitor, bisindoylmalemide (BIM, 10 μm) for 24 h. PGE 2 levels in harvested media were determined by RIA. Bars represent mean of results from 4 separated cultures ± SE. *P < 0.05, **P < 0.01 compared to their respective controls, #P < 0.05 compared between NFs and CAFs.
Discussion
An earlier study has attributed the cause of colorectal cancer largely to genetic alterations within the epithelial compartment 3. However, more recent research evidence suggests that COX‐2 is expressed in stromal cells, but not in epithelial cells of intestinal microadenomas in genetically modified mice 10 and human colorectal adenomas 21. Here, we demonstrate that even without stimulation of TNFα, COX‐2 expression was already upregulated in CAFs (Fig. 1). In contrast, TNFα elicited significant upregulation of COX‐2 mRNA in NFs and robust upregulation of COX‐2 mRNA in CAFs, indicating that stromal fibroblasts, especially CAFs, are major sources of COX‐2 (Fig. 1a). Correspondingly, TNFα elicited significant upregulation of COX‐2 protein in NFs, but profound upregulation of COX‐2 protein in CAFs (Fig. 1b), confirming that CAFs are important sources of COX‐2, which might be regulated by TNFα. Those results were similar to our previous reports that interleukin 1β and deoxycholic acid might induce COX‐2 expression in colonic fibroblasts 5, 6, 7. In addition, TNFα also elicited higher production of PGE2 in CAFs than in NFs (Fig. 1c), suggesting that stromal fibroblasts, especially CAFs, were the source of PGE2, which might be regulated by TNFα. We believe that TNFα can specifically activate COX‐2 expression in both NFs and CAFs, but activated stromal COX‐2 in NFs physiologically activated stromal COX‐2 in CAFs pathologically, to a dangerous level associated with colon cancer.
To determine whether activation of COX‐2 signalling in CAFs played an important role in carcinogenesis of colon cancer in vitro, we investigated whether activation of COX‐2 signalling by TNFα in CAFs promoted proliferation and invasiveness of colon cancer epithelial cells. Interestingly, activation of COX‐2 signalling in CAFs promoted far greater proliferative response of the cancer epithelial cells than in NFs (Fig. 2a,b), suggesting that activation of COX‐2 signalling promoted population growth of the cancer epithelial cells. Similarly, activation of COX‐2 signalling in CAFs elicited quantitatively greater invasive responses in colon cancer epithelial cells than in NFs (Fig. 2c,d), demonstrating that activation of COX‐2 signalling in CAFs also promoted invasiveness of the cancer epithelial cells. As suggested by previous reports that PGE2 promoted malignant transformation and progression in ovarian cancer 22 and in pancreatic cancer 23, we speculate that enhanced proliferation and invasiveness in colon cancer epithelial cells might be due to the increased production of PGE2 in CAFs.
To confirm that activation of COX‐2 signalling and overproduction of PGE2 in CAFs were major factors promoting proliferation and invasiveness of the cancer epithelial cells, we used COX‐2 inhibitor, NS 398 to block COX‐2 expression and PGE2 production from the fibroblasts. Our results indicated that without addition of TNFα, NS 398 blocked COX‐2 expression and PGE2 production from NFs and CAFs and their proliferative and invasive effect on the cancer epithelial cells (data not shown). The effect of CAFs activated by TNFα on proliferation and invasiveness of the cancer epithelial cells was mostly attenuated by selective COX‐2 inhibitor, NS398 (Fig. 3), suggesting that PGE2 elicited by the fibroblasts was largely responsible for proliferative and invasive responses in colon cancer epithelial cells. This conclusion was also confirmed by treatment of COX‐2 siRNA, as results from COX‐2 siRNA experiments were similar to those treated with NS398 (data not shown). Our conclusion was in line with previous reports that NS‐398 significantly reduced proliferation of Barrett's oesophageal epithelial cells by inhibition of PGE2 production 24, suppressed proliferation and invasiveness and delayed liver metastasis in colon cancer 25.
We recognize that fibroblasts may retain significant ability to induce epithelial cell proliferation and invasiveness by non‐COX‐2‐related paracrine mechanisms. For example, transforming growth factor β2 and hepatocyte growth factor, enhance epithelial cell proliferation and both might be synthesized by fibroblasts 26. However, we believe that those factors might only play a minor role in proliferative and invasive effects elicited by colonic fibroblasts activated by TNFα as specific COX‐2 inhibitor, NS398, or COX‐2 siRNA inhibited most proliferative and invasive effects of CAFs activated by TNFα on the cancer epithelial cells.
To further investigate mechanisms of action, we performed a time‐course study of PKC activities on the fibroblasts. Our results demonstrate that activation of PKC signalling by TNFα peaked at 3 min in both NFs and CAFs (Fig. 4a). When NS‐398 was only added to fibroblast cultures without TNFα, synthesis of COX‐2 and PGE2 in NFs and CAFs was completely blocked and their proliferative and invasive effects on the cancer epithelial cells was also blocked (data not shown). Immediate addition of PKC inhibitor BIM (10 μm) to cultures treated with TNFα for 24 h blocked TNFα‐stimulated COX‐2 expression (data not shown), production of PGE2 in both NFs and CAFs (Fig. 4b), and thus their proliferative and invasive effects on the cancer epithelial cells (data not shown), suggesting that TNFα‐stimulated COX‐2 gene expression; thus proliferative and invasive effects of NFs and CAFs on the cancer epithelial cells were PKC‐dependent, similar to our previous reports that lypopolysaccharide and deoxycholate‐activated COX‐2 gene expression was PKC‐dependent 5, 6, 7, 8. When PKC activities were inhibited by PKC inhibitor BIM in the fibroblasts especially CAFs, their proliferative and invasive effect on the cancer epithelial cells was also completely inhibited (data not shown), indicating that activation of PKC in fibroblasts by TNFα played a major role in promoting proliferation and invasiveness of the cancer epithelial cells.
In summary, scientific relevance here can be summarized as follows: stromal fibroblasts from neoplastic colorectal tissue are the potent source of COX‐2 expression, which may be regulated by TNFα; PGE2 elicited from CAFs activated by TNFα is responsible for change in behaviour of human colon cancer epithelial cells.
Acknowledgements
This work was supported in part by Arizona Cancer Center (NIH Institutional Research Grant # 7400128) and a small research grant from the University of Arizona. No potential conflict of interest is declared.
References
- 1. Jemal A, Siegel R, Xu J, Ward E (2010) Cancer statistics, 2010. CA Cancer J. Clin. 60, 277–300. [DOI] [PubMed] [Google Scholar]
- 2. Kargman SL, O'Neill GP, Vickers PJ, Evans JF, Mancini JA, Jothy S (1995) Expression of prostaglandin G/H synthase‐1 and ‐2 protein in human colon cancer. Cancer Res. 55, 2556–2559. [PubMed] [Google Scholar]
- 3. Fodde R (2002) The APC gene in colorectal cancer. Eur. J. Cancer 38, 867–871. [DOI] [PubMed] [Google Scholar]
- 4. Mueller MM, Fusenig NE (2004) Friends or foes – bipolar effects of the tumour stroma in cancer. Nat. Rev. Cancer 4, 839–849. [DOI] [PubMed] [Google Scholar]
- 5. Zhu Y, Hua P, Lance P (2003) Cyclooxygenase‐2 expression and prostanoid biogenesis reflect clinical phenotype in human colorectal fibroblast strains. Cancer Res. 63, 522–526. [PubMed] [Google Scholar]
- 6. Zhu Y, Hua P, Rafiq S, Waffner EJ, Duffey ME, Lance P (2002) Ca2+‐ and PKC‐dependent stimulation of PGE2 synthesis by deoxycholic acid in human colonic fibroblasts. Am. J. Physiol. Gastrointest. Liver Physiol. 283, G503–G510. [DOI] [PubMed] [Google Scholar]
- 7. Kim EC, Zhu Y, Andersen V, Sciaky D, Cao HJ, Meekins H et al (1998) Cytokine‐mediated PGE2 expression in human colonic fibroblasts. Am. J. Physiol. 275, C988–C994. [DOI] [PubMed] [Google Scholar]
- 8. Zhu Y, Zhu M, Lance P (2012) iNOS signaling interacts with COX‐2 pathway in colonic fibroblasts. Exp. Cell Res. 318, 2116–2127. [DOI] [PubMed] [Google Scholar]
- 9. Bissell MJ, Radisky D (2001) Putting tumours in context. Nat. Rev. Cancer 1, 46–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kenny PA, Bissell MJ (2003) Tumor reversion: correction of malignant behavior by microenvironmental cues. Int. J. Cancer 107, 688–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kuperwasser C, Chavarria T, Wu M, Magrane G, Gray JW, Carey L et al (2004) Reconstruction of functionally normal and malignant human breast tissues in mice. Proc. Natl. Acad. Sci. USA 101, 4966–4971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bhowmick NA, Neilson EG, Moses HL (2004) Stromal fibroblasts in cancer initiation and progression. Nature 432, 332–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Schauer IG, Sood AK, Mok S, Liu J (2011) Cancer‐associated fibroblasts and their putative role in potentiating the initiation and development of epithelial ovarian cancer. Neoplasia 13, 393–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zhu Y, Zhu M, Lance P (2012) Stromal COX‐2 signaling activated by deoxycholic acid mediates proliferation and invasiveness of colorectal epithelial cancer cells. Biochem. Biophys. Res. Commun. 425, 607–612. [DOI] [PubMed] [Google Scholar]
- 15. Zhu Y, Zhu M, Lance P (2012) IL1beta‐mediated Stromal COX‐2 signaling mediates proliferation and invasiveness of colonic epithelial cancer cells. Exp. Cell Res. 318, 2520–2530. [DOI] [PubMed] [Google Scholar]
- 16. Schmeck B, Brunsch M, Seybold J, Krull M, Eichel‐Streiber C, Suttorp N et al (2003) Rho protein inhibition blocks cyclooxygenase‐2 expression by proinflammatory mediators in endothelial cells. Inflammation 27, 89–95. [DOI] [PubMed] [Google Scholar]
- 17. Caughey GE, Cleland LG, Penglis PS, Gamble JR, James MJ (2001) Roles of cyclooxygenase (COX)‐1 and COX‐2 in prostanoid production by human endothelial cells: selective up‐regulation of prostacyclin synthesis by COX‐2. J. Immunol. 167, 2831–2838. [DOI] [PubMed] [Google Scholar]
- 18. Blanco A, Habib A, Levy‐Toledano S, Maclouf J (1995) Involvement of tyrosine kinases in the induction of cyclo‐oxygenase‐2 in human endothelial cells. Biochem. J. 312(Pt 2), 419–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hirai K, Ezumi Y, Nishida E, Uchiyama T, Takayama H (1999) Comparative study of vanadate‐ and phorbol ester‐induced cyclo‐oxygenase‐2 expression in human endothelial cells. Thromb. Haemost. 82, 1545–1552. [PubMed] [Google Scholar]
- 20. Chen CC, Sun YT, Chen JJ, Chiu KT (2000) TNF‐alpha‐induced cyclooxygenase‐2 expression in human lung epithelial cells: involvement of the phospholipase C‐gamma 2, protein kinase C‐alpha, tyrosine kinase, NF‐kappa B‐inducing kinase, and I‐kappa B kinase 1/2 pathway. J. Immunol. 165, 2719–2728. [DOI] [PubMed] [Google Scholar]
- 21. Cao Y, Prescott SM (2002) Many actions of cyclooxygenase‐2 in cellular dynamics and in cancer. J. Cell. Physiol. 190, 279–286. [DOI] [PubMed] [Google Scholar]
- 22. Rask K, Zhu Y, Wang W, Hedin L, Sundfeldt K (2006) Ovarian epithelial cancer: a role for PGE2‐synthesis and signalling in malignant transformation and progression. Mol. Cancer 5, 62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ito H, Duxbury M, Benoit E, Clancy TE, Zinner MJ, Ashley SW et al (2004) Prostaglandin E2 enhances pancreatic cancer invasiveness through an Ets‐1‐dependent induction of matrix metalloproteinase‐2. Cancer Res. 64, 7439–7446. [DOI] [PubMed] [Google Scholar]
- 24. Buttar NS, Wang KK, Anderson MA, Dierkhising RA, Pacifico RJ, Krishnadath KK et al (2002) The effect of selective cyclooxygenase‐2 inhibition in Barrett's esophagus epithelium: an in vitro study. J. Natl. Cancer Inst. 94, 422–429. [DOI] [PubMed] [Google Scholar]
- 25. Yao M, Lam EC, Kelly CR, Zhou W, Wolfe MM (2004) Cyclooxygenase‐2 selective inhibition with NS‐398 suppresses proliferation and invasiveness and delays liver metastasis in colorectal cancer. Br. J. Cancer 90, 712–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Nakagawa H, Liyanarachchi S, Davuluri RV, Auer H, Martin EW Jr, de la CA et al (2004) Role of cancer‐associated stromal fibroblasts in metastatic colon cancer to the liver and their expression profiles. Oncogene 23, 7366–7377. [DOI] [PubMed] [Google Scholar]