

Abstract
Object
The purpose of this study was to explore whether melatonin could protect mesenchymal stem cells (MSCs) against ischaemic injury, by inhibiting endoplasmic reticulum (ER) stress and autophagy both in vivo and in vitro.
Materials and Methods
To confirm the protective effect of melatonin against ER stress in MSCs, markers of cell viability, apoptosis and autophagy were analysed. To further investigate the regenerative effect of melatonin‐treated MSCs in ischaemic tissues, a murine hindlimb ischaemic model was established.
Results
Under oxidative stress conditions, treatment with melatonin suppressed the activation of ER stress–associated proteins and autophagy‐associated proteins acting through upregulation of cellular prion protein (PrPC) expression. Consequently, inhibition of apoptotic cell death occurred. Melatonin also promoted the activation of MnSOD and catalase activities in MSCs. In a murine hindlimb ischaemia model, melatonin‐treated MSCs also enhanced the functional limb recovery as well as neovascularization. These beneficial effects of melatonin were all blocked by knock‐down of PrPC expression.
Conclusion
Melatonin protects against ER stress/autophagy‐induced apoptotic cell death by augmenting PrPC expression. Thus, melatonin‐treated MSCs could be a potential cell‐based therapeutic agent for ER stress–induced ischaemic diseases, and melatonin‐induced PrPC might be a key molecule in ameliorating ER stress and autophagy.
Keywords: autophagy, cellular prion protein, endoplasmic reticulum stress, melatonin, mesenchymal stem cell
1. INTRODUCTION
Mesenchymal stem cells (MSCs) represent one type of stem/progenitor cell‐based therapies used to treat ischaemic diseases, such as stroke, cardiovascular disease, peripheral arterial disease, and ischaemic kidney disease.1 Since these cells have a self‐renewal ability, multi‐differentiation potential, and an immunomodulatory effect, various studies have investigated the potential benefit of MSC‐based therapies in pre‐clinical and clinical trials.1, 2 Unfortunately, transplanted MSCs have shown a low survival rate in ischaemic‐damaged tissue because a sudden or prolonged blockage of blood flow induces hypoxia, oxidative stress, endoplasmic reticulum (ER) stress and nutrient deprivation.3, 4 To improve the therapeutic efficacy of MSC‐based therapies in pathophysiological conditions, a number of different approaches have been explored, including preconditioning, genetic modification, co‐transplantation of MSCs with other supporting cells, administration of MSCs along with medication and application of the MSCs with biomaterials.5 Although these strategies have improved the functionalities of MSCs, it is important to understand the underlying mechanisms behind these strategies in order to safely use MSC‐based therapies in ischaemic diseases. The ER is an important organelle for protein folding, calcium homoeostasis, and lipid and carbohydrate metabolism.6 Endoplasmic reticulum stress occurs in response to a range of cellular stresses, such as ischaemic damage, redox imbalance, oxygen and glucose deprivation and perturbations in calcium homoeostasis.6, 7 To maintain cellular homoeostasis against ER stress, cells have well‐orchestrated processes, including ER‐assisted degradation, autophagy, the hypoxia response and mitochondrial biogenesis.6, 8 Among these processes, autophagy plays a pivotal role in cell survival, cell growth, energy metabolism and lifespan in several cellular stresses under physiological conditions.6, 8 However, under pathophysiological conditions, severe stress induces excessive autophagy, causing cell death.9 Therefore, in order to create a successful cell‐based therapy for ischaemic diseases, the key is to inhibit the excessive autophagy without eliminating the basal autophagy level.
The normal cellular prion protein (PrPC) is encoded by the Prnp gene, which plays a physiological role in various cells.10 Accumulating evidence indicates roles for PrPC in a wide range of cellular processes, such as stress protection, oxidative stress‐induced apoptosis, the ER‐stress response, cellular differentiation, the immune system, mitochondrial homoeostasis, and metal ion homoeostasis.11 Knockout of the Prnp gene induces a dysfunction in synaptic transmission and plasticity, memory formation, calcium homoeostasis, neurite outgrowth and metabolism.10 In stem cell biology, PrPC is essential for cell proliferation, differentiation and self‐renewal.12, 13, 14 In addition, our previous studies have shown that PrPC is up‐regulated by hypoxia preconditioning and melatonin treatment, and that transplanted MSCs facilitate neovascularization in a murine hindlimb ischaemia model.15, 16 Moreover, augmentation of PrPC expression by treatment of MSCs with tauroursodeoxycholic acid inhibited ER stress–mediated cell death.17 Therefore, an understanding of the role of PrPC may provide insights into the development of stem/progenitor cell‐based therapies.
Melatonin, N‐acetyl‐5‐methoxytryptamine, is an indoleamine containing hormone secreted by several tissues including the bone marrow, ovary, testes, gut, placenta and liver, as well as the pineal gland.18, 19 Melatonin has a variety of functions, including the regulation of sleep and circadian rhythms, as well as having anti‐inflammatory, antioxidant and anti‐cancer effects.20, 21, 22, 23 Numerous studies have shown that melatonin‐treated MSCs can facilitate a therapeutically functional recovery in myocardial infarction, skin wounds, lung ischaemia‐reperfusion injury and sepsis‐induced kidney injury.24, 25, 26, 27 In addition, our previous study has shown that melatonin‐treated MSCs enhance neovascularization in hindlimb ischaemia through PrPC expression.16 This study, as an extension of this research, focuses on the effect of melatonin on ER stress in ischaemic disease. We show here that melatonin protects against autophagy‐mediated apoptosis both in vitro and in vivo following ischaemic‐induced ER stress and this occurs through the upregulation of PrPC expression. In addition, we demonstrate the therapeutic effect of melatonin‐treated MSCs on neovascularization in a murine hindlimb ischaemia model.
2. MATERIAL AND METHODS
2.1. Cell culture
Human adipose‐derived MSCs were obtained from the American Type Culture Collection (ATCC, Manassas, VA, USA). The supplier certified that the MSCs expressed the cell surface markers (CD73 and CD105) and could undergo adipogenic or osteogenic differentiation when cultured with the appropriate differentiation medium. MSCs were maintained in alpha‐minimum essential medium (Hyclone, Logan, UT, USA) supplemented with 10% (v/v) foetal bovine serum (Hyclone) and 100 U/mL penicillin/streptomycin (Thermo Fisher Scientific, Waltham, MA, USA). MSCs were incubated in a humidified incubator at 37°C and 5% CO2.
2.2. Chemical treatment of MSCs
Mesenchymal stem cells were washed twice with phosphate‐buffered saline (PBS), and suspended in fresh α‐MEM supplemented with 10% FBS. To detect the activation of the apoptotic signalling pathway, MSCs were pretreated with melatonin (1 µM) at 37°C for 30 min and then treated with H2O2 (200 µM) for the indicated durations (0, 2, 4, or 8 hours).
2.3. Animal care procedures and experiments
The Institutional Animal Care and Use Committee of Soonchunhyang University approved all surgical interventions and post‐operative animal care (IACIC2013‐5), and these were performed in accordance with the National Research Council Guidelines for the Care and Use of Laboratory Animals. Animal experiments were performed on 8‐week‐old male Balb/C nude mice (Biogenomics, Seoul, Korea), which were maintained in a pathogen‐free facility under a 12‐hours light/dark cycle at 25°C with free access to water and regular laboratory chow in accordance with the regulations of Soonchunhyang University, Seoul Hospital.
2.4. Murine hindlimb ischaemia model
Experiments using a murine hindlimb ischaemia model were performed as previously reported with minor modifications.15, 28 Ischaemia was induced by the ligation and excision of the proximal femoral artery and the boundary vessels of the mice. No later than 6 hours after surgery, PBS, untreated MSCs, or melatonin‐treated MSCs transfected with PRNP siRNA or a scrambled siRNA were injected intramuscularly into the ischaemic thigh area (5 × 105 cells/100 μL PBS per mouse; five mice per treatment group). Each mouse was injected with cells (or PBS) at five ischaemic sites. Thereafter, melatonin (20 mg/kg/day) was administered intraperitoneally daily for 28 days. The melatonin stock solution was diluted in water, so that the final ethanol concentration was below 1%. Blood perfusion was assessed by measuring the ratio of the blood flow in the ischaemic (left) limb to that in the non‐ischaemic (right) limb on post‐operative days 0 and 28 using laser Doppler perfusion imaging (LDPI; Moor Instruments, Wilmington, DE, USA).
2.5. Statistical analysis
All data are expressed as the mean ± SE of the mean (SEM) and all of the experiments were analysed using a one‐way analysis of variance (ANOVA). Some comparisons of ≥3 groups were made using the Bonferroni‐Dunn test. A P value <0.05 was considered statistically significant. These data were statistically analysed using SigmaPlot (Systat Software, San Jose, CA, USA).
Detailed some methods are provided in Appendix [Link].
3. RESULTS
3.1. Ischaemia‐mediated ROS induces apoptosis through ER stress in MSCs and in hindlimb ischaemic‐injured tissues
ROS is one of the inducers of ER stress which is known to be involved in cell life and death.29 Our previous study demonstrated that ischaemia‐induced ROS‐mediated ER stress–triggered apoptosis in ischaemic tissues and led to a deterioration in the quality and efficacy of the transplanted MSCs used as a cell‐based therapy for ischaemic diseases.17 To further investigate the effect of ROS on ER stress in MSCs and ischaemic tissues, the activation and expression of ER stress–associated proteins was assessed in MSCs treated with H2O2 (Figure 1), as well as in ischaemic‐injured tissues in a murine hindlimb ischaemia model. In MSCs, H2O2, as an ER‐stress inducer, increased the activation and expression of these ER stress–associated proteins (Figure 1A–D). In addition, the activation and expression of these proteins were also increased in ischaemic‐injured tissues (Figure 1E–H). These data indicate that ischaemia‐induced ROS induces the activation and expression of ER stress–mediated proteins. Furthermore, we assessed the level of apoptosis‐associated proteins in MSCs and in ischaemic tissues under these same ROS‐mediated ER‐stress conditions (Figure 1I–L). After treatment of MSCs with H2O2, the level of the anti‐apoptotic protein, BCL2 decreased and the expression of pro‐apoptotic proteins, including BAX, cleaved caspase‐3 and cleaved PARP‐1 increased. In ischaemic tissues, similar apoptosis expression level with treatment MSCs with H2O2. These results indicate that ischaemia‐mediated ROS leads to apoptosis via ER stress in MSCs and in hindlimb ischaemic‐injured tissues.
Figure 1.
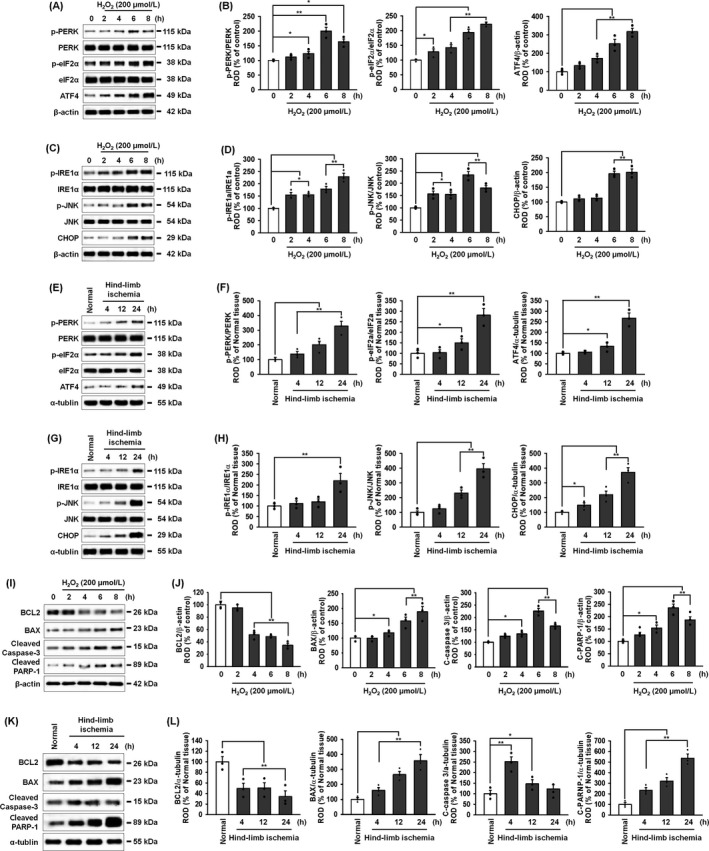
Endoplasmic reticulum stress–mediated apoptotic cell death in MSCs and ischaemic tissue. A, Western blot of p‐PERK, PERK, p‐eIF2α, eIF2α and ATF4 after treatment of MSCs with hydrogen peroxide (200 μmol/L) for 0, 2, 4, 6 and 8 hours. B, The levels of p‐PERK, p‐eIF2α and ATF4 were normalized to those of PERK, eIF2α and β‐actin, respectively. Values represent the mean ± SEM. *P < 0.05; **P < 0.01 vs untreated MSCs. C, Western blot of p‐IRE1α, IRE1α, p‐JNK, JNK and CHOP after treatment of MSCs with hydrogen peroxide (200 μmol/L) for 0, 2, 4, 6 and 8 hours. D, The levels of p‐IRE1α, p‐JNK and CHOP were normalized to those of IRE1α, JNK and β‐actin, respectively. Values represent the mean ± SEM. *P < 0.05; **P < 0.01 vs untreated MSCs. E, Western blot of p‐PERK, PERK, p‐eIF2α, eIF2α and ATF4 in ischaemic‐injured tissues from the murine hindlimb ischaemia model, 4, 12 and 24 hours after surgery. F, The expression of p‐PERK, p‐eIF2α and ATF4 were normalized to those of PERK, eIF2α and α‐tubulin, respectively. Values represent the mean ± SEM. *P < 0.05; **P < 0.01 vs normal tissue. G, Western blot of p‐IRE1α, IRE1α, p‐JNK, JNK and CHOP in ischaemic‐injured tissues from the murine hindlimb ischaemia model, 4, 12 and 24 hours after surgery. H, The expression of p‐IRE1α, p‐JNK and CHOP were normalized to those of IRE1α, JNK and α‐tubulin, respectively. Values represent the mean ± SEM. *P < 0.05; **P < 0.01 vs normal tissue. I, Western blot of BCL2, BAX, cleaved caspase‐3, and cleaved PARP‐1 after treatment of MSCs with hydrogen peroxide (200 μmol/L) for 0, 2, 4, 6 and 8 hours. J, All protein expression levels were normalized to that of β‐actin. Values represent the mean ± SEM. *P < 0.05; **P < 0.01 vs untreated MSCs. K, Western blot of BCL2, BAX, cleaved caspase‐3 and cleaved PARP‐1 in ischaemic‐injured tissues from the murine hindlimb ischaemia model, 4, 12 and 24 hours after surgery. L, All protein expression levels were normalized to that of α‐tubulin. Values represent the mean ± SEM. *P < 0.05; **P < 0.01 vs normal tissue
3.2. ROS‐mediated ER stress activates autophagy in both MSCs and hindlimb ischaemic‐injured tissues
Under physiological conditions, ER stress induces autophagy in order to remove misfolded proteins, resulting in the inhibition of cell death; however, under pathophysiological conditions, severe autophagy leads to cell death.30 To evaluate the effect of ROS‐mediated ER stress on autophagy in MSCs, the expression of autophagy marker proteins was examined after treatment of the MSCs with H2O2. Treatment with H2O2 increased activation of the ER‐stress proteins, PERK and IRE1α, but the autophagy inhibitor 3‐MA (500 μmol/L) did not significantly decrease this H2O2‐induced activation of these proteins (Figure 2A,B). ROS‐mediated ER stress also increased the expression of the autophagy markers, LC3B and Beclin‐1, and 4‐PBA (10 μmol/L) an ER‐stress inhibitor, suppressed this ROS‐induced augmentation of autophagy marker expression (Figure 2A,B). In addition, ROS‐mediated ER stress increased the expression of another autophagy marker, ATG7, as well as LC3B and Beclin‐1, and decreased the level of p62, which is known to be decreased following autophagy31 (Figure 2C,D). These findings indicate that autophagy is activated following ROS‐mediated ER stress. To further examine the effect of ischaemic‐induced ER stress on autophagy in ischaemic‐injured tissues, the expression of LC3B, Beclin‐1, and ATG7 were increased and the level of p62 was decreased (Figure 2E,F). In addition, immunofluorescence staining for LC3 showed that the level of LC3 was significantly increased in ischaemic tissue, compared with that in normal tissue (Figure 2G,H). Taken together, these results suggest that ischaemia‐induced ER stress activates the autophagy process.
Figure 2.
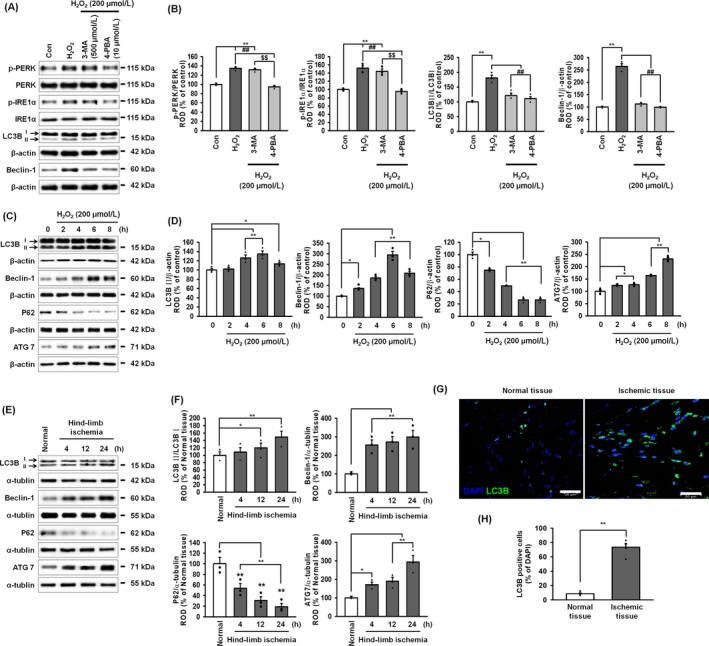
ER stress–mediated autophagy in MSCs and ischaemic tissue. A, Western blot of p‐PERK, PERK, p‐IRE1α, IRE1α, LC3B and Beclin‐1 after pre‐treatment of MSCs with 3‐MA (500 μmol/L; an autophagy inhibitor) or 4‐PBA (10 μmol/L; an ER‐stress inhibitor) followed by treatment with hydrogen peroxide (200 μmol/L). B, The levels of p‐PERK, p‐eIF2α, LC3BII and Beclin‐1 were normalized to those of PERK, eIF2α, LC3BI and β‐actin, respectively. Values represent the mean ± SEM. **P < 0.01 vs untreated MSCs, ## P < 0.01 vs MSCs treated with H2O2, and $ P < 0.05; $$ P < 0.01 vs 3‐MA‐pretreated MSCs treated with H2O2. C, Western blot of LC3B, Beclin‐1, p62 and ATG7 in cells after treatment of MSCs with hydrogen peroxide (200 μmol/L) for 0, 2, 4, 6 and 8 hours. D, The levels of LC3BII were normalized to LC3B1 whereas the levels of Beclin‐1, p62, and ATG7 were normalized to β‐actin. Values represent the mean ± SEM. *P < 0.05; **P < 0.01 vs untreated MSCs. E, Western blot of LC3B, Beclin‐1, p62, and ATG7 in ischaemic‐injured tissues from the murine hindlimb ischaemia model, 4, 12 and 24 hours after surgery. F, The levels of LC3BII were normalized to LC3B1 whereas the levels of Beclin‐1, p62 and ATG7 were normalized to α‐tubulin. Values represent the mean ± SEM. *P < 0.05; **P < 0.01 vs normal tissue. G, Immunofluorescent staining for DAPI (blue) and LC3B (green) in normal and ischaemic tissues. Scale bar = 30 μm. H, Autophagic cells were quantified as the percentage of LC3B‐positive cells per total DAPI‐stained cells. Values represent the mean ± SEM. **P < 0.01 vs normal tissue
3.3. Melatonin protects cells from oxidative stress through the melatonin‐MT2‐PrPC axis
Our previous study demonstrated that melatonin enhances MSC bioactivities, including proliferation, anti‐oxidative effects and immunomodulatory effects, through the upregulation of PrPC expression.16 Expression of the melatonin receptor 2 (MT2) gene is higher than that of MT1 in MSCs, and melatonin significantly upregulates MT2.16 To further investigate whether melatonin regulates the activity of ROS scavenger enzymes via the MT2‐mediated expression of PrPC, we first confirmed the anti‐oxidative effect of melatonin in MSCs. Treatment of MSCs with H2O2 significantly increased PrPC levels, whereas treatment with luzindole, a melatonin receptor antagonist that preferentially targets MT2 over MT1, blocked the increase in PrPC (Figure 3A,B). Under oxidative stress, melatonin increased cell viability, but inhibition of the melatonin receptor significantly decreased the survival of melatonin‐treated MSCs (Figure 3C). Furthermore, melatonin significantly inhibited the expression of cleaved caspase‐3 and cleaved PARP‐1 under oxidative stress, whereas this protective effect was blocked by luzindole (Figure 3D,E). These results indicate that melatonin protects MSCs from oxidative stress through the melatonin receptor.
Figure 3.
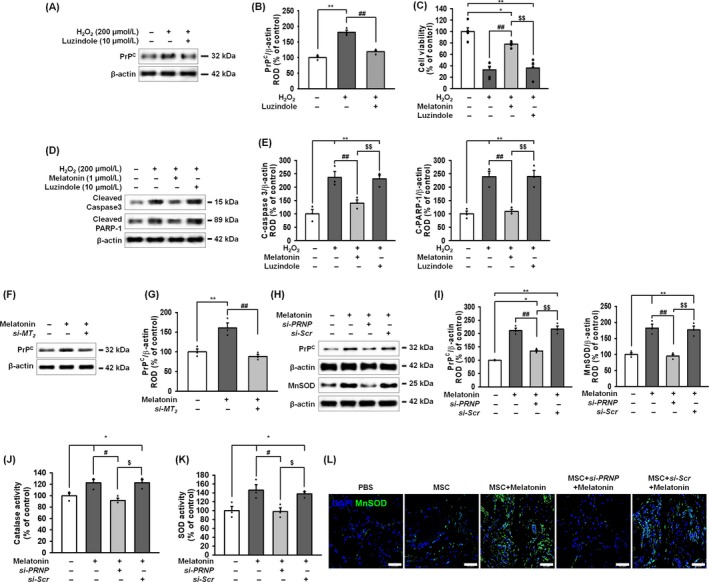
The anti‐oxidative effect of melatonin on MnSOD and catalase reduced cell viability through the melatonin‐MT2‐PrPC axis under oxidative stress. A, Expression of PrPC after treatment of MSCs with melatonin and a combination of melatonin and luzindole (10 μmol/L; an MT1 and MT2 antagonist). B, PrPC was quantified by densitometry and normalized to that of β‐actin. Values represent the mean ± SEM. **P < 0.01 vs untreated MSCs and ## P < 0.01 vs MSCs treated with melatonin. C, Viability of melatonin‐treated MSCs after treatment with H2O2 with/without luzindole. Values represent the mean ± SEM. *P < 0.05; **P < 0.01 vs untreated MSCs and ## P < 0.01 vs MSCs +H2O2. $$ P < 0.01 vs MSCs + melatonin + H2O2. D, Expression of cleaved caspase‐3 and cleaved PARP‐1 after treatment of melatonin‐pretreated MSCs with H2O2 (200 μmol/L). E, The levels of these proteins were normalized to that of β‐actin. Values represent the mean ± SEM. **P < 0.01 vs untreated MSCs, ## P < 0.01 vs MSCs + H2O2, and $$ P < 0.01 vs MSCs + melatonin + H2O2. F, Western blot of PrPC after treatment of MSCs with melatonin and a combination of melatonin and MT2 siRNA (si‐MT2). G, The level of PrPC was quantified by densitometry and normalized relative to the β‐actin expression level. Values represent the mean ± SEM. **P < 0.01 vs untreated MSCs and ## P < 0.01 vs MSCs treated with melatonin. H, Western blot of PrPC and MnSOD after treatment of MSCs with melatonin, a combination of melatonin and PRNP siRNA (si‐PRNP), or a combination of melatonin and scrambled siRNA (si‐Scr). I, The expression of PrPC and MnSOD were quantified by densitometry and normalized relative to the β‐actin expression level. Values represent the mean ± SEM. **P < 0.01 vs untreated MSCs, ## P < 0.01 vs MSCs treated with melatonin, and $$ P < 0.01 vs MSCs treated with melatonin and si‐PRNP. J, Catalase activity after treatment of MSCs with melatonin, a combination of melatonin and si‐PRNP, or a combination of melatonin and si‐Scr. Values represent the mean ± SEM. *P < 0.05 vs untreated MSCs, # P < 0.05 vs MSCs treated with melatonin, and $ P < 0.05 vs MSCs treated with melatonin and si‐PRNP. K, SOD activity after treatment of MSCs with melatonin, a combination of melatonin and si‐PRNP, or a combination of melatonin and si‐Scr. Values represent the mean ± SEM. *P < 0.05 vs untreated MSCs, # P < 0.05 vs MSCs treated with melatonin, and $ P < 0.05 vs MSCs treated with melatonin and si‐PRNP. L, Immunofluorescent staining for DAPI (blue) and MnSOD (green) in ischaemic‐injured tissues injected with PBS, MSCs, melatonin‐treated MSCs, melatonin‐treated MSCs plus si‐PRNP or melatonin‐treated MSCs plus si‐Scr. Scale bar = 50 μm
To further investigate the protective effect of melatonin on oxidative stress via the MT2‐PrPC axis, we knocked‐down MT2 in MSCs and analysed the anti‐oxidative effect of melatonin. Treatment of MSCs with melatonin significantly increased PrPC expression and knock‐down of MT2 blocked this melatonin‐induced increase in PrPC expression (Figure 3F,G). Melatonin also significantly augmented the expression of MnSOD in MSCs (Figure 3H,I). In addition, melatonin increased both catalase and MnSOD activities confirming that melatonin can enhance antioxidant activity (Figure 3J,K). Furthermore, in a murine hindlimb ischaemia model, immunofluorescent staining for MnSOD showed that the expression of MnSOD was increased in transplanted MSCs treated with melatonin (Figure 3L). Importantly, silencing of PrPC in MSCs blocked these anti‐oxidant effects of melatonin (Figure 3H–L and Figure 2). These data suggest that melatonin enhances ROS scavenger enzyme activity via the MT2‐mediated upregulation of PrPC.
3.4. Melatonin attenuated ER stress under ischaemic conditions by regulation of PrPC
To elucidate whether melatonin regulates ER stress through PrPC expression, the activation and expression of ER stress–associated proteins were analysed after treatment of MSCs with H2O2 and melatonin. Melatonin significantly decreased the activation and expression ER stress–associated protein (Figure 4A–D). To further investigate the protective effect of melatonin in vivo, the activation and/or expression levels of these ER stress–associated proteins were also significantly decreased following transplantation with MSCs treated with melatonin (Figure 4E‐H). However, knock‐down of PrPC in the MSCs abolished these inhibitory effects on ER stress–associated proteins. These results suggest that melatonin attenuated ER stress via upregulation of PrPC both in vitro and in vivo.
Figure 4.
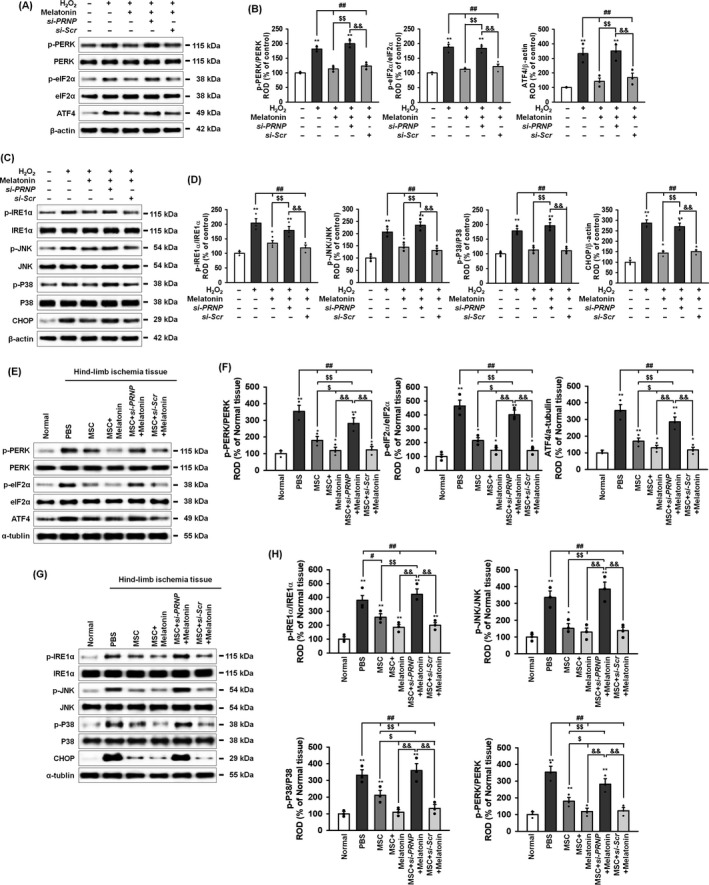
Melatonin protects against oxidative ER stress through the upregulation of PrPC. A, Western blot of p‐PERK, PERK, p‐eIF2α, eIF2α and ATF4 after treatment of melatonin‐pretreated MSCs with hydrogen peroxide (200 μmol/L). B, The levels of p‐PERK, p‐eIF2α and ATF4 were normalized to those of PERK, eIF2α and β‐actin, respectively. Values represent the mean ± SEM. *P < 0.05; **P < 0.01 vs untreated MSCs, ## P < 0.01 vs MSCs treated with H2O2, $$ P < 0.01 vs MSCs treated with melatonin, and && P < 0.01 vs MSCs treated with melatonin and si‐PRNP. C, Western blot of p‐IRE1α, IRE1α, p‐JNK, JNK, p‐p38, p38 and CHOP after treatment of melatonin‐pretreated MSCs with hydrogen peroxide (200 μmol/L). D, The levels of p‐IRE1α, p‐JNK, p‐p38 and CHOP were normalized to those of IRE1α, JNK, p38 and β‐actin, respectively. Values represent the mean ± SEM. *P < 0.05; **P < 0.01 vs untreated MSCs, ## P < 0.01 vs MSCs treated with H2O2, $$ P < 0.01 vs MSCs treated with melatonin, and && P < 0.01 vs MSCs treated with melatonin and si‐PRNP. E, Western blot of p‐PERK, PERK, p‐eIF2α, eIF2α and ATF4 in ischaemic‐injured tissues 1 days after injection with PBS, MSCs (MSC), MSCs treated with melatonin (MSC + Melatonin), MSCs treated with melatonin and PRNP siRNA (MSC + si‐PRNP + Melatonin), or MSCs treated with melatonin and scrambled siRNA (MSC + si‐Scr + Melatonin) in a murine hindlimb ischaemia model. F, The expressions of p‐PERK, p‐eIF2α and ATF4 were normalized to those of PERK, eIF2α, and α‐tubulin, respectively. Values represent the mean ± SEM. *P < 0.05; **P < 0.01 vs normal, ## P < 0.01 vs PBS, $ P < 0.05; $$ P < 0.01 vs MSC, and && P < 0.01 vs MSC + si‐PRNP + Melatonin. G, Western blot of p‐IRE1α, IRE1α, p‐JNK, JNK, p‐p38, p38 and CHOP in the same tissues as in E. H, The expression levels of p‐IRE1α, p‐JNK, p‐p38 and CHOP were normalized to those of IRE1α, JNK, p38 and α‐tubulin, respectively. Values represent the mean ± SEM. *P < 0.05; **P < 0.01 vs normal, # P < 0.05; ## P < 0.01 vs PBS, $ P < 0.05; $$ P < 0.01 vs MSC, and && P < 0.01 vs MSC + si‐PRNP + Melatonin
3.5. Melatonin‐induced PrPC protects cells from apoptotic cell death under conditions of ER stress through the regulation of autophagy
To explore whether melatonin protects ER stress–mediated autophagy in MSCs and ischaemic tissues through the regulation of PrPC, we analysed the expression of autophagy markers in the MSCs and ischaemic tissues transplanted with MSCs. Autophagy is regulated through the mTOR and AMPK signalling pathway.32 In MSCs, treated with H2O2 to induce ROS‐mediated ER stress, melatonin activated mTOR and inhibited AMPK phosphorylation (Figure 5A,B). Silencing of PrPC in the MSCs inhibited these melatonin‐mediated effects on the upstream autophagy signalling pathway. Under this same oxidative stress condition, in MSCs, melatonin suppressed the H2O2‐mediated induction of the autophagy inducing proteins, LC3B, Beclin‐1 and ATG7, and in parallel increased the expression of the autophagy repressor, p62 (Figure 5C,D). Moreover, in ischaemic‐injured tissues from the murine hindlimb ischaemia model, the levels of autophagy inducer proteins (LC3B, Beclin‐1, and ATG7) were decreased and the level of the autophagy repressor protein p62 was increased following transplantation with MSCs treated with melatonin (Figure 5E,F). These protective effects of melatonin on autophagy were all significantly ablated by silencing of PrPC, both in vitro and in vivo. These findings suggest that melatonin‐induced PrPC protected autophagy under oxidative ER‐stress condition through regulation of the mTOR‐AMPK axis.
Figure 5.
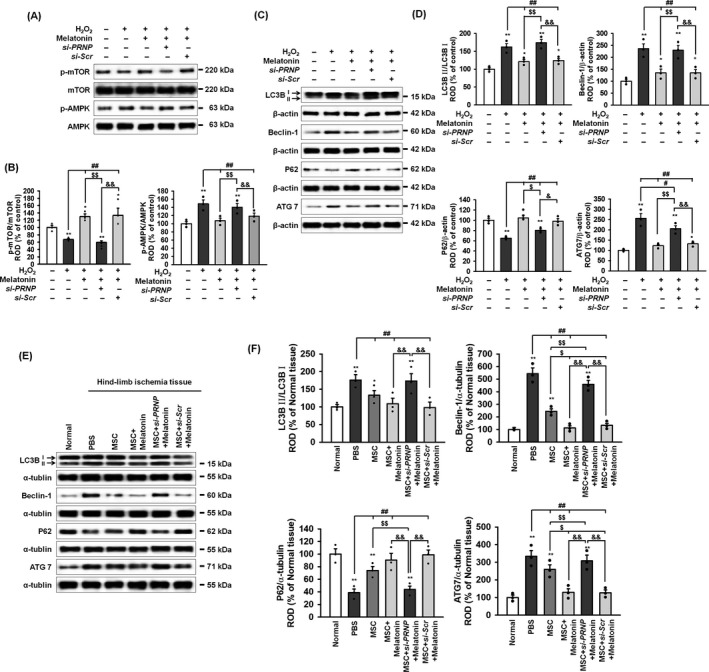
Melatonin inhibits oxidative ER stress–mediated autophagy via PrPC expression. A, Western blot of p‐mTOR, mTOR, p‐AMPK and AMPK after treatment of melatonin‐pretreated MSCs with hydrogen peroxide (200 μmol/L). B, The levels of p‐mTOR and p‐AMPK were normalized to those of mTOR and AMPK, respectively. Values represent the mean ± SEM. **P < 0.05; **P < 0.01 vs untreated MSCs, ## P < 0.01 vs MSCs treated with H2O2, $$ P < 0.01 vs MSCs treated with melatonin, and && P < 0.01 vs MSCs treated with melatonin and si‐PRNP. C, Western blot of LC3B, Beclin‐1, p62 and ATG7 after treatment of melatonin‐pretreated MSCs with hydrogen peroxide (200 μmol/L). D, The levels of LC3BII were normalized to LC3B1 whereas the levels of Beclin‐1, p62 and ATG7 were normalized to β‐actin. Values represent the mean ± SEM. *P < 0.05; **P < 0.01 vs untreated MSCs, # P < 0.05; ## P < 0.01 vs MSCs treated with H2O2, $ P < 0.05; $$ P < 0.01 vs MSCs treated with melatonin, and & P < 0.05; && P < 0.01 vs MSCs treated with melatonin and si‐PRNP. E, Western blot of LC3B, Beclin‐1, p62 and ATG7 in ischaemic‐injured tissues 1 days after injection with PBS, MSCs (MSC), MSCs treated with melatonin (MSC + Melatonin), MSCs treated with melatonin and PRNP siRNA (MSC + si‐PRNP + Melatonin), or MSCs treated with melatonin and scrambled siRNA (MSC + si‐Scr + Melatonin) in a murine hindlimb ischaemia model. F, The levels of LC3BII were normalized to LC3B1, whereas the levels of Beclin‐1, p62, and ATG7 were normalized to α‐tubulin. Values represent the mean ± SEM. *P < 0.05; **P < 0.01 vs normal, ## P < 0.01 vs PBS, $ P < 0.05; $$ P < 0.01 vs MSC, and && P < 0.01 vs MSC + si‐PRNP + Melatonin
To assess whether melatonin protects against apoptotic cell death under conditions of ER stress, through the upregulation of PrPC expression, the level of apoptosis‐associated proteins and cell death were assessed in MSCs and in ischaemic tissues transplanted with MSCs. In MSCs treated with H2O2 to induce ROS‐mediated ER stress, melatonin reduced apoptosis pathway (Figure 6A,B). Moreover, a PI‐Annexin V staining assay of MSCs showed that melatonin protected against ROS‐mediated apoptotic cell death (Figure 6C,D). In the hindlimb ischaemia, transplantation with MSCs treated with melatonin reduced expression of apoptosis protein (Figure 6E‐H). In addition, a TUNEL assay of the ischaemically injured tissues showed that ischaemia‐induced, ER stress–mediated, cellular apoptosis was significantly decreased following transplantation of MSCs treated with melatonin, compared with other MSC‐treated groups (Figure 6I,J). These protective effects of melatonin on apoptotic cell death under oxidative ER‐stress conditions, both in vitro and in vivo, were reversed by silencing of PrPC in the MSCs. Our findings suggest that the melatonin‐induced upregulation of PrPC protects against apoptotic cell death under oxidative ER‐stress conditions through regulation of the autophagy process.
Figure 6.
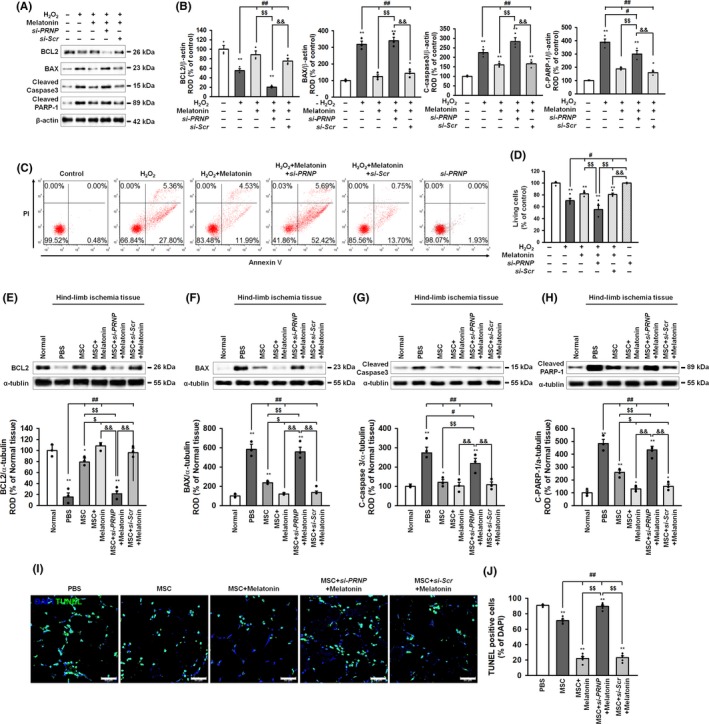
Melatonin protects against oxidative ER stress–mediated cell death via upregulation of PrPC. A, Western blot of BCL2, BAX, cleaved caspase‐3 and cleaved PARP after treatment of melatonin‐pretreated MSCs with hydrogen peroxide (200 μmol/L). B, The expressions of these proteins were normalized to that of β‐actin. Values represent the mean ± SEM. *P < 0.05; **P < 0.01 vs untreated MSCs, ## P < 0.01 vs MSCs treated with H2O2, $$ P < 0.01 vs MSCs treated with melatonin, and && P < 0.01 vs MSCs treated with melatonin and si‐PRNP. C, Flow cytometry analysis of PI and Annexin V staining after treatment of melatonin‐pretreated MSCs with H2O2. D, The number of surviving cells was quantified as the percentage of PI and Annexin V negative cells. Values represent the mean ± SEM. **P < 0.01 vs untreated MSCs, # P < 0.05 vs MSCs treated with H2O2, and $$ P < 0.01 vs MSCs treated with melatonin and si‐PRNP, and && P < 0.01 vs MSCs treated with si‐PRNP. (E‐H) Western blot of BCL2, BAX, cleaved caspase‐3 and cleaved PARP ischaemic‐injured tissues 1 days after injection with PBS, MSCs (MSC), MSCs treated with melatonin (MSC + Melatonin), MSCs treated with melatonin and PRNP siRNA (MSC + si‐PRNP + Melatonin), or MSCs treated with melatonin and scrambled siRNA (MSC + si‐Scr + Melatonin) in a murine hindlimb ischaemia model. The expression levels of BCL2, BAX, cleaved caspase‐3 and cleaved PARP were normalized to that of α‐tubulin. Values represent the mean ± SEM. *P < 0.05; **P < 0.01 vs normal, # P < 0.05; ## P < 0.01 vs PBS, $ P < 0.05; $$ P < 0.01 vs MSC, and && P < 0.01 vs MSC + si‐PRNP + Melatonin. I, TUNEL assay in ischaemic‐injured tissues from the groups in E‐H. Scale bar = 50 μm. J, Apoptotic cells were quantified as the percentage of TUNEL (green)‐positive cells per total DAPI‐stained cells. Values represent the mean ± SEM. *P < 0.05; **P < 0.01 vs PBS, # P < 0.05 vs MSC, $ P < 0.05; $$ P < 0.01 vs MSC + Melatonin
3.6. Melatonin‐treated MSCs enhance neovascularization in a murine hindlimb ischaemia model
To examine whether melatonin‐treated MSCs could promote the formation of neovessels in a murine hindlimb ischaemia model, blood perfusion and tissue recovery were assessed following transplantation of MSCs into the ischaemic‐injured tissue. To stimulate the transplanted MSCs over the long‐term, an intraperitoneal injection of melatonin was performed every day for 28 days following surgery. The blood perfusion ratio was analysed using LDPI at post‐operative day 0, 3, 7, 14, 21 and 28. The blood perfusion ratio was significantly higher in limbs transplanted and maintained with the melatonin‐treated MSCs than with any other group (Figure 7A,B). In addition, the percentage of limb salvaged was significantly higher in limbs transplanted and maintained with the melatonin‐treated MSCs than with any other group (Figure 7C). In ischaemic‐injured tissues, the secretion of the angiogenic cytokines, such as human VEGF, HGF and FGF, was significantly higher in mice transplanted and maintained with the melatonin‐treated MSCs than with any other group (Figure 7D). Immunofluorescent staining for CD31 (capillary) and α‐SMA (arterioles) revealed that the densities of capillaries and arterioles were significantly increased in mice transplanted and maintained with the melatonin‐treated MSCs than with any other group (Figure 7E‐H). Inhibition of PrPC expression significantly prevented these beneficial effects of melatonin on neovascularization. However, the densities of capillaries and arterioles at the sites injected with melatonin‐treated MSCs transfected with PRNP siRNA were significantly lower than those at the sites injected with MSCs alone. This is the reason for the significant increase in the level of pro‐apoptotic proteins, including BAX, cleaved caspase‐3 and cleaved PARP‐1, at sites injected with melatonin‐treated MSCs transfected with PRNP siRNA compared with that at the sites injected with MSCs alone (see, Figure 6F‐H); this effect also led to the increased apoptosis in ischaemic‐injured tissues (see, Figure 6I,J). In addition, oxidative stress increased the expression PrPC (see, Figure 3A). Therefore, PRNP siRNA might suppress the physiological increase in PrPC, thereby reducing the functional recovery at sites injected with melatonin‐treated MSCs transfected with PRNP siRNA compared with that at the sites injected with MSCs alone. These results indicate that transplantation with MSCs stimulated with melatonin improves neovascularization and functional recovery through the expression of PrPC.
Figure 7.
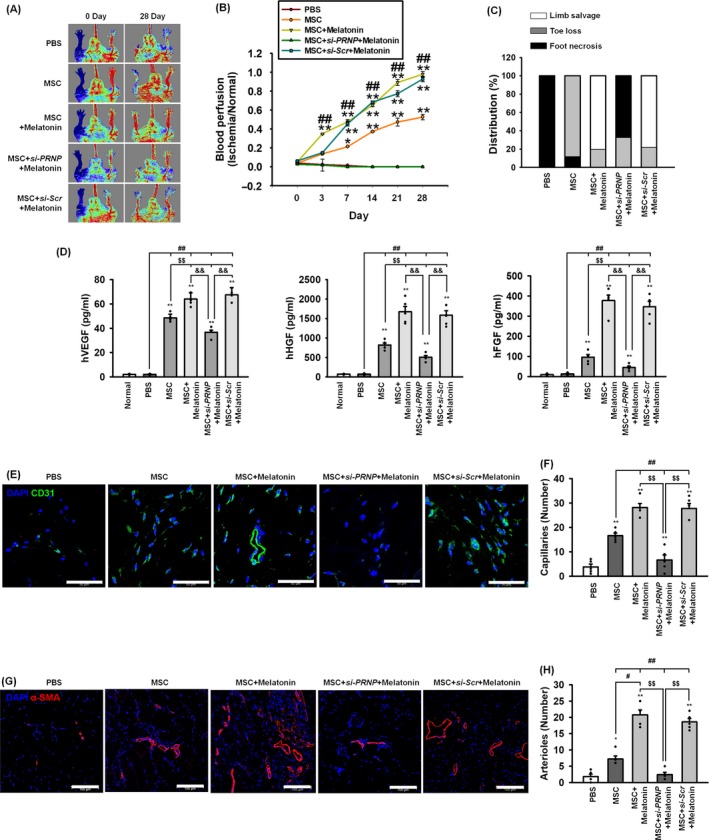
Assessment of functional recovery and neovascularization in a murine hindlimb ischaemia model. A, Blood perfusion was assessed using laser Doppler perfusion imaging analysis in the ischaemic limbs of mice injected with PBS, MSCs (MSC), melatonin‐treated MSCs (MSC + Melatonin), MSCs treated with melatonin and PRNP siRNA (MSC + si‐PRNP + Melatonin), or MSCs treated with melatonin and scrambled siRNA (MSC + Scr‐siRNA + Melatonin). B, The blood perfusion ratio was measured by LPDI analysis (blood flow in the left ischaemic limb/blood flow in the right non‐ischaemic limb). Values represent the mean ± SEM. *P < 0.05; **P < 0.01 vs PBS, and ## P < 0.01 vs MSC. C, Distribution of the different outcomes (limb salvage, toe loss and foot necrosis) in each of the treatment groups at post‐operative day 28. D, Secretion of human VEGF, HGF and FGF from the ischaemic‐injured tissues in each group was assessed by ELISA. Values represent the mean ± SEM. **P < 0.01 vs normal, ## P < 0.01 vs PBS, $$ P < 0.01 vs MSC, and && P < 0.01 vs MSC + si‐PRNP + Melatonin. E‐G At post‐operative day 28, the formation of capillaries and arterioles were analysed using immunofluorescent staining for CD31 (green; scale bar = 50 μm; E) and α‐SMA (red; scale bar = 100 μm; G). The capillary (F) and arteriole (H) densities were quantified as the number of CD31‐positive cells and α‐SMA‐positive cells, respectively. Values represent the mean ± SEM. *P < 0.05; **P < 0.01 vs PBS, # P < 0.05; ## P < 0.01 vs MSC, $$ P < 0.01 vs MSC+si‐PRNP + Melatonin
4. DISCUSSION
Ischaemic‐injured sites have harsh tissue conditions, including low oxygen levels, restricted nutrients, and high levels of ROS, with the result that effectiveness of the transplanted stem/progenitor cells used for regenerative medicine to treat ischaemic diseases is decreased. Among the pathophysiological conditions, ischaemia/ROS‐induced ER stress is known to induce apoptotic cell death in host tissues as well as in the transplanted stem/progenitor cells.6, 7, 17, 29 To solve this issue, several studies have suggested that melatonin could be used to protect against ER stress in neurotoxic states, liver diseases, immunotoxic states, pulmonary fibrosis and diabetes.33 Our previous study revealed that melatonin could enhance MSC functionality for the treatment of ischaemic disease by upregulating the express of PrPC.16 To further examine whether melatonin protects against ER stress through the upregulation of PrPC, we have confirmed the protective effect of melatonin on autophagy‐mediated cell death in MSCs with ischaemia‐induced ER stress. Our results showed that melatonin‐induced PrPC expression decreased ER stress/autophagy‐mediated apoptosis both in vitro and in a murine hindlimb ischaemia model.
Various reports have indicated that PrPC, which is expressed in many cells beyond the central nerve system,34 contributes to the regulation of several cellular process, including differentiation, proliferation, stress protection, myelin maintenance, mitochondrial homoeostasis and interaction with signal transduction pathways.11, 35 In particular, PrPC has been shown to have a protective effect against ROS‐mediated oxidative stress.11 PrPC suppresses ROS production by regulating the activities of superoxide dismutase and glutathione peroxidase.36, 37 In fact, our previous study revealed that TUDCA‐induced PrPC increased MnSOD expression via phosphorylation of Akt and activation of its signalling pathway.17 In keeping with the concept of PrPC regulating the activity of enzymes that control ROS levels it has been shown in a PrP‐null mouse, that total SOD activity was significantly decreased through modulation of mitochondrial complex I.38 Since ischaemic‐injured tissues represent a ROS‐rich oxidative stress environment, reductions in ROS levels are important for the successful transplantation of stem/progenitor cells in cell‐based therapies. One study has shown that PrPC enhances MSC engraftment to bone marrow.39 Hypoxia‐induced PrPC expression has also been shown to facilitate the survival of transplanted MSCs at ischaemic‐damaged sites in hindlimb model of ischaemia.15 Our data indicated that melatonin increased the activation of MnSOD and catalase activity in MSCs under ROS‐mediated oxidative stress through the upregulation of PrPC. Moreover, the expression of MnSOD in transplanted MSCs treated with melatonin was augmented in the ischaemic‐injured sites, resulting in the suppression of apoptosis.
Melatonin is known to be a powerful ROS scavenger through this regulation of antioxidant enzymes.40, 41 Several studies have investigated the protective effect of melatonin in MSCs with ischaemic injury in order to increase their efficacy in transplantation. In small bowel ischaemia‐reperfusion injury, injection with melatonin, along with adipose‐derived MSCs, decreased ischaemic damage through the regulation of antioxidant enzymes, notably NAD(P)H dehydrogenase, glutathione reductase and glutathione peroxidase.42 In addition, melatonin blocked oxidative stress‐induced premature senescence and promoted the therapeutic efficacy of MSCs in a murine myocardial infarction model via a sirtuin‐1‐dependent mechanism.24, 43 Our results therefore confirmed that melatonin stimulation of MSCs enhances the functional recovery and neovascularization in a murine hindlimb ischaemia model via PrPC expression. Consistent with these results, our previous studies have shown that PrPC is a key molecule in regulating the functionality and therapeutic potential of MSCs in ischaemic diseases.15, 16, 17 Taken together, our data show that melatonin protects against ROS‐mediated oxidative stress by regulating PrPC‐dependent antioxidant enzyme activity.
This study also showed that ROS/ischaemia triggered the activation of ER stress–associated proteins in both MSCs in vitro, and in ischaemic‐injured tissues in vivo. Prolonged activation of PERK and IRE1α induce apoptotic cell death under pathophysiological conditions.44 In addition to activation of the unfolded protein response ER stress stimulates autophagy. Autophagy is cellular process that is essential for cell survival, development, differentiation and cellular homoeostasis.45 Under physiological conditions, the basal level of autophagy plays an important role in cellular adaptation to nutrient starvation, endo‐lysosomal degradation, the removal of misfolded and long‐lived proteins, and other cellular stresses caused by detrimental cellular substances and invading pathogens.8 However, excessive autophagy, initiated by long‐term stress, triggers cellular damage, resulting in the induction of apoptotic cell death.8, 46 However, some studies have suggested that inhibition of autophagy protects against ischaemic‐induced injury, whereas other studies indicate that activation of autophagy inhibits apoptosis.33, 46, 47 Our data indicated that melatonin protected against ischaemia‐induced ER stress in both MSCs and in a murine hindlimb ischaemia model. Treatment of MSCs with melatonin increased antioxidant effects, thereby increasing the survival of transplanted cells. In addition, melatonin‐treated MSCs increased the secretion of angiogenic cytokines, such as VEGF, HGF and FGF, in ischaemic‐injured tissues, thereby increasing neovascularization. Of particular interest is that these protective effects were regulated by melatonin‐mediated PrPC upregulation. Several other studies have documented that melatonin can inhibit ischaemia‐induced ER stress. For example, treatment melatonin restored hypoxic‐ischaemic encephalopathy and ischaemia‐reperfusion injury in liver through reduced ER stress activity.48, 49 In addition, melatonin treatment has been reported to attenuate ischaemia/reperfusion‐induced ER stress by regulating the PERK and IRE1 pathways.46 This study showed melatonin protected against ER stress/autophagy‐mediated apoptotic cell death by appropriately regulating the expression of pro‐ and anti‐apoptotic proteins. Of particular importance, these beneficial effects were blocked by silencing of PrPC. These results are in good agreement with our previous findings showing that melatonin regulated apoptosis‐associated proteins under oxidative stress conditions by upregulating PrPC.16 In the liver of leptin‐deficient mice, melatonin has also been shown to inhibit oxidative damage through a reduction in ER stress and autophagy.50
This study therefore suggests that melatonin‐stimulated PrPC protects against ischaemic injury and apoptotic cell death by inhibiting the ER stress‐autophagy axis both in vitro and in vivo. Of particular note, we demonstrated for the first time that PrPC is involved in melatonin‐mediated ER stress and autophagy inhibition both in MSCs and in a hindlimb ischaemia model (Figure 8). Our findings therefore indicate that melatonin is a key regulator of ER stress and autophagy. In addition, our results suggest that melatonin‐induced PrPC is a crucial protective molecule for cell survival against apoptosis induced by ER stress–mediated autophagy. These observations provide important insights into other ischaemia‐induced cell death mechanisms that are inhibited by melatonin‐induced PrPC expression.
Figure 8.
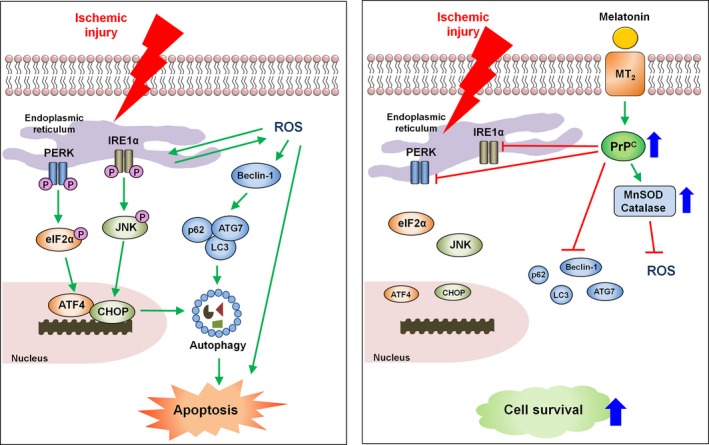
Schematic representation of the proposed mechanisms by which melatonin protects against ER stress/autophagy‐induced cell death through upregulation of the cellular prion protein (PrPC) levels. Treatment of MSCs with melatonin inhibits ER stress, autophagy and apoptosis via PrPC upregulation. Moreover, melatonin increases the activation of MnSOD and catalase activity. Transplantation of melatonin‐treated MSCs improves the functional recovery and vessel formation in ischaemic diseases through this melatonin‐mediated PrPC expression
CONFLICT OF INTEREST
The authors declare no conflict of interests.
AUTHOR CONTRIBUTIONS
JHL: study concept and design, acquisition of data, analysis and interpretation of data, drafting of the manuscript. YMY, YSH and SKJ: acquisition of data, analysis and interpretation of data, statistical analysis. SHL: study concept and design, acquisition of data, analysis and interpretation of data, drafting of the manuscript, procurement of funding, study supervision.
Supporting information
ACKNOWLEDGEMENTS
This work was supported by the National Research Foundation grant funded by the Korean government (NRF‐2017M3A9B4032528). The funders had no role in the study design, data collection or analysis, decision to publish, or preparation of the manuscript.
Lee JH, Yoon YM, Han Y‐S, Jung SK, Lee SH. Melatonin protects mesenchymal stem cells from autophagy‐mediated death under ischaemic ER‐stress conditions by increasing prion protein expression. Cell Prolif. 2019;52:e12545 10.1111/cpr.12545
REFERENCES
- 1. Caplan AI, Correa D. The MSC: an injury drugstore. Cell Stem Cell. 2011;9(1):11‐15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Liew A, O'Brien T. Therapeutic potential for mesenchymal stem cell transplantation in critical limb ischemia. Stem Cell Research & Therapy. 2012;3(4):28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Zhu XY, Lerman A, Lerman LO. Concise review: mesenchymal stem cell treatment for ischemic kidney disease. Stem cells. 2013;31(9):1731‐1736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Monsel A, Zhu YG, Gennai S, Hao Q, Liu J, Lee JW. Cell‐based therapy for acute organ injury: preclinical evidence and ongoing clinical trials using mesenchymal stem cells. Anesthesiology. 2014;121(5):1099‐1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Li L, Chen X, Wang WE, Zeng C. How to improve the survival of transplanted mesenchymal stem cell in ischemic heart? Stem Cells Int. 2016;2016:9682757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Senft D, Ronai ZA. UPR, autophagy, and mitochondria crosstalk underlies the ER stress response. Trends Biochem Sci. 2015;40(3):141‐148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hoyer‐Hansen M, Jaattela M. Connecting endoplasmic reticulum stress to autophagy by unfolded protein response and calcium. Cell Death Differ. 2007;14(9):1576‐1582. [DOI] [PubMed] [Google Scholar]
- 8. Rashid HO, Yadav RK, Kim HR, Chae HJ. ER stress: Autophagy induction, inhibition and selection. Autophagy. 2015;11(11):1956‐1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Liu Y, Levine B. Autosis and autophagic cell death: the dark side of autophagy. Cell Death Differ. 2015;22(3):367‐376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wulf MA, Senatore A, Aguzzi A. The biological function of the cellular prion protein: an update. BMC Biol. 2017;15(1):34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Castle AR, Gill AC. Physiological functions of the cellular prion protein. Front Mol Biosci. 2017;4:19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lopes MH, Santos TG. Prion potency in stem cells biology. Prion. 2012;6(2):142‐146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Martin‐Lanneree S, Hirsch TZ, Hernandez‐Rapp J, et al. PrP(C) from stem cells to cancer. Front Cell Dev Biol. 2014;2:55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Martin‐Lanneree S, Halliez S, Hirsch TZ, et al. The cellular prion protein controls notch signaling in neural stem/progenitor cells. Stem Cells. 2017;35(3):754‐765. [DOI] [PubMed] [Google Scholar]
- 15. Han YS, Lee JH, Yoon YM, Yun CW, Noh H, Lee SH. Hypoxia‐induced expression of cellular prion protein improves the therapeutic potential of mesenchymal stem cells. Cell Death Dis. 2016;7(10):e2395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lee JH, Han YS, Lee SH. Potentiation of biological effects of mesenchymal stem cells in ischemic conditions by melatonin via upregulation of cellular prion protein expression. J Pineal Res. 2017;62(2):e12385. [DOI] [PubMed] [Google Scholar]
- 17. Yoon YM, Lee JH, Yun SP, et al. Tauroursodeoxycholic acid reduces ER stress by regulating of Akt‐dependent cellular prion protein. Scientific reports. 2016;6:39838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Reiter RJ. Pineal melatonin: cell biology of its synthesis and of its physiological interactions. Endocr Rev. 1991;12(2):151‐180. [DOI] [PubMed] [Google Scholar]
- 19. Garcia JJ, Lopez‐Pingarron L, Almeida‐Souza P, et al. Protective effects of melatonin in reducing oxidative stress and in preserving the fluidity of biological membranes: a review. J Pineal Res. 2014;56(3):225‐237. [DOI] [PubMed] [Google Scholar]
- 20. Jan JE, Reiter RJ, Wasdell MB, Bax M. The role of the thalamus in sleep, pineal melatonin production, and circadian rhythm sleep disorders. J Pineal Res. 2009;46(1):1‐7. [DOI] [PubMed] [Google Scholar]
- 21. Mauriz JL, Collado PS, Veneroso C, Reiter RJ, Gonzalez‐Gallego J. A review of the molecular aspects of melatonin's anti‐inflammatory actions: recent insights and new perspectives. J Pineal Res. 2013;54(1):1‐14. [DOI] [PubMed] [Google Scholar]
- 22. Reiter RJ, Mayo JC, Tan DX, Sainz RM, Alatorre‐Jimenez M, Qin L. Melatonin as an antioxidant: under promises but over delivers. J Pineal Res. 2016;61(3):253‐278. [DOI] [PubMed] [Google Scholar]
- 23. Su SC, Hsieh MJ, Yang WE, Chung WH, Reiter RJ, Yang SF. Cancer metastasis: mechanisms of inhibition by melatonin. J Pineal Res. 2017;62(1):e12370. [DOI] [PubMed] [Google Scholar]
- 24. Han D, Huang W, Li X, et al. Melatonin facilitates adipose‐derived mesenchymal stem cells to repair the murine infarcted heart via the SIRT1 signaling pathway. J Pineal Res. 2016;60(2):178‐192. [DOI] [PubMed] [Google Scholar]
- 25. Lee SJ, Jung YH, Oh SY, Yun SP, Han HJ. Melatonin enhances the human mesenchymal stem cells motility via melatonin receptor 2 coupling with Galphaq in skin wound healing. J Pineal Res. 2014;57(4):393‐407. [DOI] [PubMed] [Google Scholar]
- 26. Yip HK, Chang YC, Wallace CG, et al. Melatonin treatment improves adipose‐derived mesenchymal stem cell therapy for acute lung ischemia‐reperfusion injury. J Pineal Res. 2013;54(2):207‐221. [DOI] [PubMed] [Google Scholar]
- 27. Chen HH, Lin KC, Wallace CG, et al. Additional benefit of combined therapy with melatonin and apoptotic adipose‐derived mesenchymal stem cell against sepsis‐induced kidney injury. J Pineal Res. 2014;57(1):16‐32. [DOI] [PubMed] [Google Scholar]
- 28. Limbourg A, Korff T, Napp LC, Schaper W, Drexler H, Limbourg FP. Evaluation of postnatal arteriogenesis and angiogenesis in a mouse model of hind‐limb ischemia. Nat Protoc. 2009;4(12):1737‐1746. [DOI] [PubMed] [Google Scholar]
- 29. Xu C, Bailly‐Maitre B, Reed JC. Endoplasmic reticulum stress: cell life and death decisions. J Clin Investig. 2005;115(10):2656‐2664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kim I, Xu W, Reed JC. Cell death and endoplasmic reticulum stress: disease relevance and therapeutic opportunities. Nat Rev Drug Discov. 2008;7(12):1013‐1030. [DOI] [PubMed] [Google Scholar]
- 31. Moscat J, Diaz‐Meco MT. Feedback on fat: p62‐mTORC1‐autophagy connections. Cell. 2011;147(4):724‐727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Fleming A, Noda T, Yoshimori T, Rubinsztein DC. Chemical modulators of autophagy as biological probes and potential therapeutics. Nature chemical biology. 2011;7(1):9‐17. [DOI] [PubMed] [Google Scholar]
- 33. Fernandez A, Ordonez R, Reiter RJ, Gonzalez‐Gallego J, Mauriz JL. Melatonin and endoplasmic reticulum stress: relation to autophagy and apoptosis. J Pineal Res. 2015;59(3):292‐307. [DOI] [PubMed] [Google Scholar]
- 34. Su AI, Wiltshire T, Batalov S, et al. A gene atlas of the mouse and human protein‐encoding transcriptomes. Proc Natl Acad Sci USA. 2004;101(16):6062‐6067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Linden R, Martins VR, Prado MA, Cammarota M, Izquierdo I, Brentani RR. Physiology of the prion protein. Physiological reviews. 2008;88(2):673‐728. [DOI] [PubMed] [Google Scholar]
- 36. Rachidi W, Vilette D, Guiraud P, et al. Expression of prion protein increases cellular copper binding and antioxidant enzyme activities but not copper delivery. J Biol Chem. 2003;278(11):9064‐9072. [DOI] [PubMed] [Google Scholar]
- 37. Miele G, Jeffrey M, Turnbull D, Manson J, Clinton M. Ablation of cellular prion protein expression affects mitochondrial numbers and morphology. Biochem Biophys Res Commun. 2002;291(2):372‐377. [DOI] [PubMed] [Google Scholar]
- 38. Paterson AW, Curtis JC, Macleod NK. Complex I specific increase in superoxide formation and respiration rate by PrP‐null mouse brain mitochondria. J Neurochem. 2008;105(1):177‐191. [DOI] [PubMed] [Google Scholar]
- 39. Mohanty ST, Cairney CJ, Chantry AD, et al. A small molecule modulator of prion protein increases human mesenchymal stem cell lifespan, ex vivo expansion, and engraftment to bone marrow in NOD/SCID mice. Stem Cells. 2012;30(6):1134‐1143. [DOI] [PubMed] [Google Scholar]
- 40. Rodriguez C, Mayo JC, Sainz RM, et al. Regulation of antioxidant enzymes: a significant role for melatonin. J Pineal Res. 2004;36(1):1‐9. [DOI] [PubMed] [Google Scholar]
- 41. Galano A, Tan DX, Reiter RJ. On the free radical scavenging activities of melatonin's metabolites, AFMK and AMK. J Pineal Res. 2013;54(3):245‐257. [DOI] [PubMed] [Google Scholar]
- 42. Chang CL, Sung PH, Sun CK, et al. Protective effect of melatonin‐supported adipose‐derived mesenchymal stem cells against small bowel ischemia‐reperfusion injury in rat. J Pineal Res. 2015;59(2):206‐220. [DOI] [PubMed] [Google Scholar]
- 43. Zhou L, Chen X, Liu T, et al. Melatonin reverses H2 O2 ‐induced premature senescence in mesenchymal stem cells via the SIRT1‐dependent pathway. J Pineal Res. 2015;59(2):190‐205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Tabas I, Ron D. Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nat Cell Biol. 2011;13(3):184‐190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kourtis N, Tavernarakis N. Autophagy and cell death in model organisms. Cell Death Differ. 2009;16(1):21‐30. [DOI] [PubMed] [Google Scholar]
- 46. Feng D, Wang B, Wang L, et al. Pre‐ischemia melatonin treatment alleviated acute neuronal injury after ischemic stroke by inhibiting endoplasmic reticulum stress‐dependent autophagy via PERK and IRE1 signalings. J Pineal Res. 2017;62(3):e12395. [DOI] [PubMed] [Google Scholar]
- 47. Chen J, Wang L, Wu C, et al. Melatonin‐enhanced autophagy protects against neural apoptosis via a mitochondrial pathway in early brain injury following a subarachnoid hemorrhage. J Pineal Res. 2014;56(1):12‐19. [DOI] [PubMed] [Google Scholar]
- 48. Carloni S, Albertini MC, Galluzzi L, Buonocore G, Proietti F, Balduini W. Melatonin reduces endoplasmic reticulum stress and preserves sirtuin 1 expression in neuronal cells of newborn rats after hypoxia‐ischemia. J Pineal Res. 2014;57(2):192‐199. [DOI] [PubMed] [Google Scholar]
- 49. Zaouali MA, Boncompagni E, Reiter RJ, et al. AMPK involvement in endoplasmic reticulum stress and autophagy modulation after fatty liver graft preservation: a role for melatonin and trimetazidine cocktail. J Pineal Res. 2013;55(1):65‐78. [DOI] [PubMed] [Google Scholar]
- 50. de Luxan‐Delgado B, Potes Y, Rubio‐Gonzalez A, et al. Melatonin reduces endoplasmic reticulum stress and autophagy in liver of leptin‐deficient mice. J Pineal Res. 2016;61(1):108‐123. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials