

Summary
Aims
μ‐opioid receptor (OPRM1) exerts many functions such as antinociception, neuroprotection, and hippocampal plasticity. A body of evidence has shown that OPRM1 activation could stimulate downstream effectors of mechanistic/mammalian target of rapamycin (mTOR). However, it is not clear whether OPRM1 protects neurons against β‐amyloid peptide (Aβ) neurotoxicity through mTOR signaling.
Methods
The effects of OPRM1 activation on Aβ oligomers‐induced neurotoxicity were assessed by cell viability and neurite outgrowth assay in primary cultured cortical neurons. The activities of mTOR, protein kinase B (Akt) and p70 ribosomal S6 kinase (p70 S6k) upon OPRM1 activation by morphine were measured by immunoblotting their phosphorylation status.
Results
Morphine dose‐dependently attenuated Aβ oligomers‐induced neurotoxicity. Aβ oligomers downregulated mTOR signaling. Morphine significantly rescued mTOR signaling by reversal of Aβ oligomers’ effect on mTOR and its upstream signaling molecule Akt, as well as its downstream molecule p70 S6k. Moreover, the neuroprotective effect of morphine could be reversed by OPRM1 selective antagonist and phosphatidylinositol 3‐kinases (PI3K), Akt and mTOR inhibitors. Furthermore, endogenous opioids–enkaphalins also attenuated Aβ oligomers‐induced neurotoxicity.
Conclusions
Our findings demonstrated OPRM1 activation attenuated Aβ oligomers‐induced neurotoxicity through mTOR signaling. It may provide new insight into the pathological process and useful strategy for therapeutic interventions against Aβ neurotoxicity.
Keywords: Alzheimer's disease, Mechanistic/mammalian target of rapamycin, β‐Amyloid peptide, μ‐Opioid receptor
Introduction
μ‐opioid receptor (OPRM1), one of the rhodopsin class of G protein‐coupled receptors (GPCRs), is widely expressed in sensory neurons and has a broad range of biological effects 1. Besides its critical role in antinociception, many other functions have been described such as neuroprotection, hippocampal plasticity, gastrointestinal motility, and modulation of the immune response 2, 3, 4. It has been demonstrated that OPRM1 activation by morphine promotes cell proliferation and also affects neuronal differentiation both in vitro and in vivo 5, 6, 7. Several studies also demonstrate that morphine protects neurons against microglia‐mediated neuroinflammation and oxidative stress 8, 9. Preconditioning with morphine induces neuroprotective effects against hypoxia/hypoglycemia 10, 11. Such neuroprotection is shown to be OPRM1 dependent 9, 11.
Mechanistic/mammalian target of rapamycin (mTOR), which is widely distributed throughout all eukaryotic cells, belongs to the phosphatidylinositol kinase‐related kinase (PIKK) family. PIKK family members contain serine/threonine kinase domain in carboxyl terminus that resemble the catalytic domain of phosphatidylinositol 3‐kinases (PI3Ks) and PI4Ks 12. PI3K activation concomitantly leads to the protein kinase B (Akt) activation, which further activates mTOR by phosphorylating and inactivating the tuberous sclerosis complex. mTOR primarily controls cell growth in response to favorable nutrients and other growth stimuli, whereas in the adult with relatively limited growth, mTOR influences aging and other aspects of nutrient‐related physiology such as protein synthesis, ribosome biogenesis and cell proliferation 13. In central nervous system (CNS), mTOR plays a critical role in neuronal plasticity and learning and memory and has been shown to associate with many neurodevelopmental and neuropsychiatric disorders 14. Brain‐derived neurotrophic factor (BDNF)‐dependent survival of primary hippocampal neurons requires mTOR activation 15. Conditional knockout of intrinsic negative regulators of mTOR such as phosphatase and tensin homolog (PTEN) or tuberous sclerosis complex 1 promotes axon regeneration after CNS injury 16. Moreover, mTOR activation promotes changes of dendritic morphology and formation of synaptic contacts 17. mTOR‐mediated translational control is required for late‐long‐term potentiation (L‐LTP) and memory reconsolidation 18.
Alzheimer's disease (AD) is a neurodegenerative disease leading to a progressive decline of the cognitive functions. Extracellular accumulation of β‐amyloid peptide (Aβ) has been reported to be a major etiological cause of AD 19, 20. Intensive research has been carried out to clarify the molecular mechanisms of Aβ in mediating the neuronal death. Several studies demonstrate that Aβ exposure induces a rapid but sustained reduction of the phosphorylation of mTOR and its downstream effector p70 ribosomal S6 kinase (p70 S6k) in murine neuroblastoma cells and hippocampal slices 21, 22, 23. Furthermore, reduced mTOR phosphorylation is manifested in the cortex or hippocampus of transgenic AD mouse models and in lymphocytes of AD patients 22, 24, 25, 26. Glycogen synthase kinase 3 (GSK3) inhibitor ameliorates Aβ pathology and reactivates mTOR 26. Moreover, mTOR activation has been shown to play a key role in cytoprotective effect of erythropoietin in AD 27. Thus, mTOR activation could slow or reverse the pathological process of AD.
OPRM1 activation has been shown to stimulate p70 S6k and other downstream effectors of mTOR, eukaryotic initiation factor 4E‐binding (eIF4E‐binding) proteins (4E‐BP1 and 4E‐BP2) 28. Furthermore, the activation of these downstream effectors of mTOR could be blocked by rapamycin 28. Therefore, whether such effect is involved in neuroprotection mediated by OPRM1 needs to be illuminated. In current study, we demonstrated that OPRM1 activation could activate mTOR and protect neurons against Aβ‐induced toxicity. Besides activating PI3K/Akt, MAP kinase kinase (MEK1/2)‐extracellular signal regulated kinases (ERK1/2) signaling cooperates with the PI3K/Akt axis to modulate mTOR activity 29. Moreover, protein kinase C (PKC) which is similar to Akt can be modulated by mTOR. Expression of PKC has been suggested to play a role in the preservation of cell survival and the formation and consolidation of memory 30, 31, 32. Increased activity of PKC in neurons is required in neuroprotection against several exogenous insults 33. The phosphorylation of Akt has been reported with OPRM1 activation by [D‐Ala2, N‐Me‐Phe4, Gly5‐ol]‐enkephalin (DAMGO) 28, 34. Moreover, PKC and ERK1/2 activation is observed after OPRM1 activation 35. Thus, the signaling pathway leading to mTOR activation by OPRM1 was further investigated.
Materials and Methods
Materials
Morphine and naloxone were supplied by the National Institute on Drug Abuse. Rapamycin, H‐D‐Phe‐Cys‐Tyr‐D‐Trp‐Arg‐Thr‐Pen‐Thr‐NH2 (CTAP), mouse monoclonal anti‐β‐actin antibody and hexafluoroisopropanol were purchased from Sigma Chemical Co (St. Louis, MO, USA). All the other antibodies were purchased from Cell Signaling Technology (Beverly, MA, USA). Aβ‐(1‐40) was purchased from Biosource International (Camarillo, CA, USA). Cell counting kit‐8 (CCK‐8) kits were purchased from Dojindo laboratories (Kumamoto, Japan). Cellomics neurite outgrowth assay kit was purchased from Thermo Scientific (Pittsburgh, PA, USA). All the cell culture reagents were purchased from GIBCO (Grand Island, NY, USA).
Primary Rat Cerebral Cortical Neuron Culture
Primary rat cerebral cortical neurons were prepared as previously described 36. Neurons were prepared from embryonic day 17 (E17) rat embryos (Sprague‐Dawley, Slac Laboratories, Chinese Academy of Sciences, Shanghai, China). The embryos were collected, and the cortices were dissected out in ice‐cold Hank's‐buffered salt solution without Ca2+ and Mg2+. Dissected pieces of cortical tissue were pooled together and transferred to an enzymatic dissociation medium containing 0.05% trypsin‐EDTA. Then, enzymatic dissociation medium was aspirated, and the tissue was triturated with a sterile Pasteur pipette in MEM with 10% fetal bovine serum (v/v), 5% horse serum (v/v), 2 mM L‐glutamine, 100 units/mL penicillin, and 100 μg/mL streptomycin plus 2000 IU/mL DNase. After centrifuging at 800 × g for 8 min, the pellets were resuspended in MEM medium and plated on poly‐L‐lysine precoated 96‐well or 24‐well plates. After culturing in a humidified atmosphere of 95% air and 5% CO2 at 37°C for 2 h, the medium was replaced with neurobasal medium plus 2% B27 and 2 mM GlutaMAX™‐I supplements.
Cell Viability
Cell viability was measured as previously described 36. Briefly, cells were plated in 96‐well plate at the density of 1–2 × 104 cells per well. On day 7 after culturing, half of the medium was aspirated, and 1 μM morphine was added for 2 h before the addition of 1 μM Aβ‐(1–40) oligomers prepared as previously described 36. 10 μM naloxone, 5 μM CTAP or 1 μM rapamycin were added 10 min before morphine. Then, the cells were incubated for 24 h, and cell viability was measured with CCK‐8 kit according to manufacturer's instructions. The absorbance of the samples was read by Varioskan Flash plate reader (Thermo Scientific) at 450 nm and 650 nm. Nuclear size and cell permeability were assessed using Cellomics HCS multiparameter cytotoxicity 2 kits according to the manufacturers’ instructions, and cells were then scanned and analyzed using the ArrayScan HCS Reader with appropriate image‐processing BioApplication software module.
Neurite Outgrowth
The neurite outgrowth assay was performed using Cellomics neurite outgrowth assay kit according to the manufacturer's instructions. In brief, primary cultured cerebral cortical neurons were treated with Aβ‐(1‐40) oligomers and morphine and then stained with neurite outgrowth primary antibody plus Dylight™ 488‐conjugated secondary antibody and Hoechst33342. The stained cells were scanned by Cellomics KineticScan reader (Thermo Scientific).
Western Blot
After treatment, cortical neurons were extracted with lysis buffer (20 mM Tris‐HCl, pH 7.5, 150 mM NaCl, 1 mM Na2EDTA, 1 mM EGTA, 1% Triton X‐100, 2.5 mM sodium pyrophosphate, 1 mM β‐glycerophosphate, 1 mM Na3VO4, 1 μg/mL leupeptin, and 1 × protease inhibitor cocktail (Sigma, Shanghai, China). After centrifuging at 12 000 × g for 5 min, sample loading buffer was added to the supernatants and boiled for 5 min. Approximately 30–40 μg of protein was subjected to SDS‐PAGE and transferred to polyvinylidene difluoride membranes (Millipore, Bedford, MA, USA). The membranes were blocked with 5% milk and then incubated with primary antibodies against mTOR phosphorylated at Ser2448 (1:1000), mTOR (1:1000), Akt phosphorylated at Thr308 (1:1000), Akt (1:1000), p70 S6k phosphorylated at Thr389 (1:1000), p70 S6k (1:1000), and β‐actin (1:5000) overnight at 4°C. Membranes were then incubated with goat anti‐rabbit/mouse HRP‐conjugated secondary antibody (1:10,000) for 1 h at room temperature, visualized with SuperSignal West Pico Chemiluminescent Substrate (Pierce Chemical, Rockford, IL, USA) and analyzed by Image J (National Institutes of Health) software.
Statistical Analysis
Data were presented as mean ± standard error of the mean (SEM). All experiments were carried out at least three times independently with triplicate repeats in cell viability assay. The results were analyzed by analysis of variances (ANOVA) followed by Dunnett's test using GraphPad Prism 5.0 statistical analysis software (GraphPad Software, La Jolla, CA, USA), with P < 0.05 taken to indicate significant difference.
Results
OPRM1 Activation by Morphine Protected Aβ Oligomers‐Induced Neurotoxicity
To evaluate whether OPRM1 activation could protect the neurons against the injury induced by exposure to Aβ, 1 μM Aβ‐(1‐40) oligomers and OPRM1 agonist morphine were administered to primary cultured cerebral cortical neurons. As shown in Figure 1A, 24 h exposure of cortical neurons to Aβ‐(1‐40) oligomers decreased the cell viability to 46.5 ± 4.1% of control, while co‐incubation of Aβ‐(1‐40) oligomers with 0.01, 0.1, 1 μM morphine increased the cell viability to 48.4 ± 3.9%, 67.9 ± 2.9% (P < 0.01 vs. Aβ‐(1‐40) oligomers treatment), 77.9 ± 2.6% (P < 0.001 vs. Aβ‐(1‐40) oligomers treatment).
Figure 1.
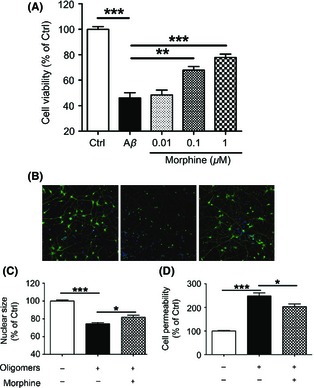
Morphine attenuated Aβ oligomers‐induced neurotoxicity in primary cultured cortical neurons. (A) Primary cultured rat cerebral cortical neurons were incubated with 0, 0.01, 0.1, and 1 μM morphine for 2 h before 1 μM Aβ‐(1‐40) oligomers was added. Cells were then cultured for 24 h, and cell viability was measured by CCK‐8 assay. (B) Primary cultured rat cerebral cortical neurons were incubated with 1 μM morphine for 2 h before 1 μM Aβ‐(1‐40) oligomers was added for another 24 h. Then, the neurons were stained with Hochest 33342 for nuclei and neurite primary antibody for cell bodies and neurites. Representative images were taken by Cellomics KineticScan HCS Reader. Magnification was 512 × 512 pixels (10 × for parameter settings, one pixel represents 0.625 μM). (C–D) Primary cultured rat cerebral cortical neurons were incubated with 1 μM morphine for 2 h before 1 μM Aβ‐(1‐40) oligomers was added for another 24 h. Nuclear size (C) and cell permeability (D) were measured and calculated using multiparameter HCS cytotoxicity kit and Cellomics BioApplication Modules. *P < 0.05; **P < 0.01; ***P < 0.001.
Synaptic changes reflected by monitoring neurite outgrowth provides subtler indication of neuronal function. As shown in Figure 1B left panel, normal neuronal cells had distinguishable cell bodies and neurites with corresponding nucleus. The dendrites and axon were clearly defined and neurons communicated with each other as indicated by numerous branches and cross‐points. The neurons treated with Aβ oligomers demonstrated ambiguous outlines of cell bodies and retraction of preformed neurites (Figure 1B, middle panel). The volumes of neurons decreased, and staining of cytoplasm and neurites was dimmer than that of normal neurons. Furthermore, karyopyknosis and nuclear accumulation were observed. 1 μM morphine administration significantly rescued the morphology damage as indicated by the recovery of normal‐sized nucleus and integral neurites (Figure 1B, right panel).
Toxic insults, such as Aβ oligomers, often cause cells to undergo necrosis or apoptosis, which can be manifested by changes in nuclear size and/or morphology 37, 38. Meanwhile, cell permeability can be increased by the impaired cell membrane integrity 39. To further evaluate the neurotoxicity of Aβ oligomers and the protective role of morphine on in vitro cultured neurons more vividly, we applied multiparameter HCS neurotoxicity kits to assess the change of nuclear size and cell permeability. As shown in Figure 1C, 24 h exposure of cortical neurons to Aβ‐(1‐40) oligomers decreased the nuclear size to 74.4 ± 1.5% of control (P < 0.001 vs. control), while co‐incubation of Aβ‐(1‐40) oligomers with 1 μM morphine significantly increased the nuclear size to 81.8 ± 2.3% of control (P < 0.05 vs. Aβ‐(1‐40) oligomers treatment). In line with nuclear abnormal morphology, cell membrane permeability increased to 248.4 ± 12.8% of control (P < 0.001 vs. control) after incubation with Aβ‐(1‐40) oligomers, while co‐incubation of Aβ‐(1‐40) oligomers with 1 μM morphine significantly decreased the cell permeability to 203.5 ± 11.4% of control (P < 0.05 vs. Aβ‐(1‐40) oligomers treatment). These results collectively demonstrated the apparent neuroprotective properties of morphine.
To further confirm that the neuroprotective effect of morphine is through OPRM1 activation, opioid receptor antagonist naloxone or OPRM1 selective antagonist CTAP was applied 10 min prior to morphine and Aβ oligomers administration. 10 μM naloxone reduced the cell viability to 46.5 ± 4.9% of control, and 10 μM CTAP reduced the cell viability to 56.9 ± 1.5% of control (Figure 2A). These data indicated that the neuroprotective effect of morphine was dependent on OPRM1 activation.
Figure 2.
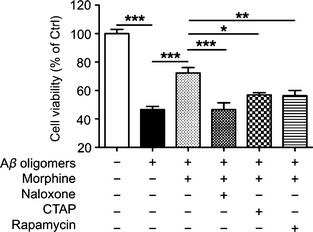
Morphine attenuated Aβ oligomers‐induced neurotoxicity in primary cultured cortical neurons through OPRM1 and mTOR activation. Primary cultured rat cerebral cortical neurons were incubated with 1 μM morphine 2 h before 1 μM Aβ‐(1‐40) oligomers was added for another 24 h. 10 μM naloxone, 5 μM CTAP, or 1 μM rapamycin was added 10 min before the administration of morphine, respectively. *P < 0.05; **P < 0.01; ***P < 0.001.
The Attenuation of Aβ Oligomers‐Induced Neurotoxicity by OPRM1 Activation was through mTOR Activation
To investigate whether mTOR activation is involved in protective effect of morphine, mTOR inhibitor rapamycin was applied. As shown in Figure 2A, 1 μM rapamycin reduced the cell viability to 56.4 ± 3.6% of control. To further evaluate the role of mTOR signaling pathway in these processes, the activation of mTOR and p70 S6k was examined. Aβ‐(1‐40) oligomers induced a progressive reduction of phosphorylated mTOR (0.44 ± 0.09‐fold of control) and p70 S6k (0.50 ± 0.12‐fold of control), meanwhile, the total forms of mTOR and p70 S6k slightly decreased but without statistically significance (Figure 3A and B). This inhibition of mTOR and p70 S6k was rescued by 1 μM morphine pretreatment (Figure 3A and B, 1.02 ± 0.17‐fold of control for phosphorylated mTOR; 0.90 ± 0.11‐fold of control for phosphorylated p70 S6k). Moreover, the effects of morphine were prohibited by OPRM1 selective antagonist CTAP (Figure 3A and B), indicating the neuroprotective effect of morphine through modulating mTOR signaling pathway was mediated by receptor activation. Taken together, these results revealed that pharmacological activation of mTOR by OPRM1 activation provided definite protective effects against Aβ oligomers‐induced cytotoxicity in primary cultured neurons.
Figure 3.
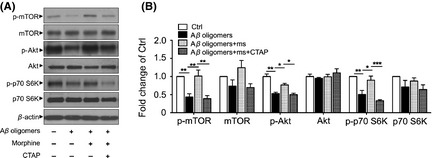
Morphine activated mTOR signaling through OPRM1 activation. Primary cultured rat cerebral cortical neurons were incubated with 1 μM morphine 2 h before 1 μM Aβ‐(1‐40) oligomers was added for another 24 h. 5 μM CTAP was added 10 min before the administration of morphine. Then cell extracts were analyzed by immunoblotting with antibodies against mTOR phosphorylated at Ser2448 (p‐mTOR), total mTOR, Akt phosphorylated at Thr308, total Akt, p70 S6K phosphorylated at Thr389 (p‐p70 S6K), total p70 S6k (A), and the relative densities were quantified (B). *P < 0.05; **P < 0.01; ***P < 0.001. ms, morphine.
PI3K/Akt Mediated the mTOR Activation by OPRM1 Activation
Next, the signaling pathway leading to mTOR activation was investigated by employing specific inhibitors. As shown in Figure 4, blockade of PI3K or Akt signaling pathway abolished morphine‐mediated cell toxicity improvement against Aβ oligomers treatment (43.4 ± 2.0% and 37.2 ± 2.9% of control, respectively, P < 0.001 vs. Aβ oligomers treatment), while blockade of MEK1/2 and PKC did not affect the action of morphine in primary cortical neurons.
Figure 4.
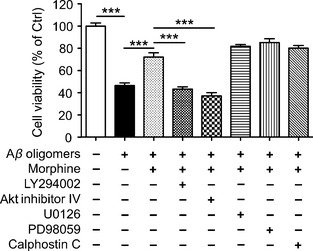
PI3K‐Akt signaling pathway was involved in the morphine‐induced attenuation of Aβ oligomers‐induced neurotoxicity in rat cerebral cortical neurons. Primary cultured rat cerebral cortical neurons were pretreated with morphine (1 μM) alone or with PI3K inhibitor LY294002 (10 μM), Akt inhibitor IV (0.2 μM), MEK1/2 inhibitor U0126 (1 μM), MEK1 inhibitor PD98059 (10 μM), or PKC inhibitor Calphostin C (1 μM) as indicated for 2 h. Cultures were then exposed to 1 μM Aβ‐(1‐40) oligomers for 24 h, then cell viability was measured by CCK‐8 assay. ***P < 0.001.
Moreover, the activation of Akt was assessed. As shown in Figure 3A and B, Aβ‐(1‐40) oligomers induced a progressive reduction of phosphorylated Akt (0.53 ± 0.03‐fold of control), and 1 μM morphine pretreatment could block such effect (0.77 ± 0.04‐fold of control). Further, OPRM1 selective antagonist CTAP could reverse the effect of morphine. All these results demonstrated that PI3K/Akt signaling but not MEK1/2 and PKC played a role in morphine's attenuation of Aβ oligomers‐induced neurotoxicity.
Endogenous Opioid Peptides Attenuated Aβ Oligomers‐Induced Neurotoxicity
Similar to morphine, endogenous opioids including endorphins, enkephalins (ENKs), and endomorphins can activate OPRM1. We further investigated whether two forms of ENKs—Met‐ENK and Leu‐ENK could attenuate Aβ oligomers‐induced toxicity. As expected and shown in Figure 5, co‐incubation of Aβ‐(1‐40) oligomers with 10 μM Met‐ENK and 0.1 μM Leu‐ENK increased the cell viability to 73.7 ± 1.8% and 72.7 ± 4.6% of control, respectively (P < 0.001 vs. Aβ oligomers treatment), and the protection could be eliminated by blockade of opioid receptor antagonist naloxone, 50.6 ± 5.5% of control (P < 0.001 vs. Met‐ENK treatment) and 53.4 ± 3.5% of control (P < 0.05 vs. Leu‐ENK treatment), respectively. The protection could also be eliminated using mTOR inhibitor rapamycin which decreased cell viability to 58.0 ± 3.0% of control (P < 0.05 vs. Met‐ENK treatment) and 50.2 ± 2.5% of control (P < 0.001 vs. Leu‐ENK treatment), respectively. These data suggested that both Met‐ENK and Leu‐ENK possessed beneficial effect against toxic Aβ and the protective function was dependent on opioid receptor activation and cascade mTOR activation.
Figure 5.
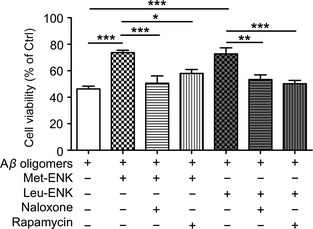
Neuroprotective effects of endogenous opioid peptides enkephalins in Aβ oligomers‐induced neurotoxicity involved opioid receptor and mTOR activation. Primary cultured rat cerebral cortical neurons were pretreated with 10 μM Met‐ENK or 0.1 μM Leu‐ENK, opioid receptor antagonist naloxone (10 μM), mTOR inhibitor rapamycin (1 μM) as indicated for 2 h, cultures were then exposed to Aβ‐(1‐40) oligomers for 24 h. Then, cell viability was measured by CCK‐8 assay. *P < 0.05, **P < 0.01, ***P < 0.001.
Discussion
In the present study, we demonstrated that morphine dose‐dependently attenuated Aβ oligomers‐induced neurotoxicity. Morphine's effect was shown to be dependent on activation of OPRM1 and was mediated by reversal of Aβ oligomers’ downregulation of mTOR signaling. Moreover, such effect is also crucial in the attenuation of Aβ oligomers‐induced neurotoxicity by endogenous opioids ENKs.
As one of the most indispensable translational conductors in the eukaryotic system, mTOR and its regulation have been intensely investigated in the pathogenesis of AD. It is widely acknowledged that decreased synaptic plasticity caused by soluble Aβ oligomers emerges well before the senile plague formation in the brain and correlates much better with the presence and degree of subtle, intermittent impairment of hippocampal neuronal function as well as episodic cognitive deficits 40. mTOR and its downstream signaling pathways are significantly involved in modulating long‐lasting synaptic plasticity 18, 41, 42 and consolidation of long‐term learning and memory 18, 43, 44. Both of these processes are dramatically impaired in the progress of AD. A line of evidence has demonstrated that mTOR signaling is impaired in AD mouse models or in cells exposed to Aβ 21, 22, 23, 24, 25, 26. Moreover, decreased mTOR signaling is observed in the lymphocytes of Alzheimer's patients, and it is statistically correlated with Mini Mental Status Examination (MMSE) scores 22. However, mTOR signaling is reported to be upregulated in postmortem human AD brains, especially in tangle‐bearing neurons 45 as well as in a mouse model of AD containing a tau mutation 46. Significant increase of the mTOR signaling positively correlates with total tau and phospho‐tau in the medial temporal cortex in AD patients 47, 48. Specifically, there is a report that Aβ‐(1‐42) oligomers activates Akt and mTOR in primary cortical neurons and inhibition of mTOR and PI3K/Akt prevents its toxic effects 49. In current study, we observed apparent downregulation of mTOR signaling following 24 h of Aβ exposure in primary cultured neurons. We also confirmed our findings in vivo by assessing mTOR expression in hippocampus of SAMP8 mice (data not shown), a senescence accelerated mouse model of AD which featured with early deposition of Aβ 50. The disparity between our study and the increased Akt, mTOR levels in Aβ‐(1‐42) oligomers‐treated neurons 49 could be attributed to the different Aβ peptides (Aβ‐(1‐40) in our study). Besides, the different preparation methods for oligomers could also contribute to the disparity. Aβ‐(1‐40) was dissolved in water and aged for 48 h in our study, whereas Aβ‐(1‐42) was dissolved in sodium phosphate buffer, and the formation of oligomers was monitored in their study. Nevertheless, our data strongly support the view that Aβ impairs mTOR signaling and are consistent with a previous report that rapamycin exacerbates neurotoxicity of Aβ 21.
mTOR is mainly linked to the PI3K/Akt signaling pathway, but its activity can be modulated by other signaling molecules such as ERK1/2, and it can regulate the activities of other signaling molecules such as PKC. Our results demonstrated that PI3K/Akt was exclusively involved in the activation of mTOR by morphine. One of the main translation effectors of mTOR is the p70 S6k. Activation of p70 S6k mediated by mTOR results in the translation of mRNAs that encodes both ribosomal proteins and translational elongation factors 51, which may directly contributed to the neuroprotective effects of morphine.
Besides binding to and activating OPRM1, morphine is also a κ‐opioid and δ‐opioid receptor agonist. Specific antagonist of OPRM1, CTAP, blocked morphine's effect to attenuate Aβ oligomers‐induced neurotoxicity and activation of mTOR and p70 S6k. Further, mTOR activation could not be elicited in cells lacking endogenous OPRM1 (data not shown). These data indicate that morphine activated mTOR signaling mainly through OPRM1.
Met‐ENK, an endogenous opioid peptide, has been demonstrated to be elevated in dentate gyrus of human AD brains and contributes to the cognitive impairments 52. Our study demonstrated that Met‐ENK reduced Aβ oligomers‐induced neurocytoxicity, suggesting that the elevated Met‐ENK in AD brain could be a compensatory process and the peptide's effect on cognitive impairments could reflect other opioid receptor function such as inhibiting adult neurogenesis 53.
In summary, our current study demonstrated that OPRM1 activation attenuated Aβ oligomers‐induced neurotoxicity in primary cultured cortical neurons through mTOR signaling. Our results may provide better understanding of the pathological process of AD and could provide a possible target for modulating neuroprotection.
Conflict of Interest
The authors declare no conflict of interest.
Acknowledgments
This research was supported by National Natural Science Foundation of China (81173044), International Science & Technology Cooperation Program of China (2011DFA33180), National Great Basic Science Project of China (2010CB529806), and Shanghai Pujiang Program (11PJ1406200), Shanghai Natural Science foundation (10ZR1417000) and in parts by National Institutes of Health grants (DA007339, DA011806).
References
- 1. Woolf CJ. Mu and delta opioid receptors diverge. Cell 2009;137:987–988. [DOI] [PubMed] [Google Scholar]
- 2. Cui Y, Zhang XQ, Xin WJ, Jing J, Liu XG. Activation of phosphatidylinositol 3‐kinase/Akt‐mammalian target of Rapamycin signaling pathway in the hippocampus is essential for the acquisition of morphine‐induced place preference in rats. Neuroscience 2010;171:134–143. [DOI] [PubMed] [Google Scholar]
- 3. Liu H, Li H, Guo L, et al. Mechanisms involved in phosphatidylinositol 3‐kinase pathway mediated up‐regulation of the mu opioid receptor in lymphocytes. Biochem Pharmacol 2010;79:516–523. [DOI] [PubMed] [Google Scholar]
- 4. McQuiston AR, Saggau P. Mu‐opioid receptors facilitate the propagation of excitatory activity in rat hippocampal area CA1 by disinhibition of all anatomical layers. J Neurophysiol 2003;90:1936–1948. [DOI] [PubMed] [Google Scholar]
- 5. Berrios I, Castro C, Kuffler DP. Morphine: axon regeneration, neuroprotection, neurotoxicity, tolerance, and neuropathic pain. P R Health Sci J 2008;27:119–128. [PubMed] [Google Scholar]
- 6. Sargeant TJ, Miller JH, Day DJ. Opioidergic regulation of astroglial/neuronal proliferation: where are we now? J Neurochem 2008;107:883–897. [DOI] [PubMed] [Google Scholar]
- 7. Sanchez‐Simon FM, Arenzana FJ, Rodriguez RE. In vivo effects of morphine on neuronal fate and opioid receptor expression in zebrafish embryos. Eur J Neurosci 2010;32:550–559. [DOI] [PubMed] [Google Scholar]
- 8. Qian L, Tan KS, Wei SJ, et al. Microglia‐mediated neurotoxicity is inhibited by morphine through an opioid receptor‐independent reduction of NADPH oxidase activity. J Immunol 2007;179:1198–1209. [DOI] [PubMed] [Google Scholar]
- 9. Rambhia S, Mantione KJ, Stefano GB, Cadet P. Morphine modulation of the ubiquitin‐proteasome complex is neuroprotective. Med Sci Monit 2005;11:BR386–BR396. [PubMed] [Google Scholar]
- 10. Ammon‐Treiber S, Stolze D, Schroder H, Loh H, Hollt V. Effects of opioid antagonists and morphine in a hippocampal hypoxia/hypoglycemia model. Neuropharmacology 2005;49:1160–1169. [DOI] [PubMed] [Google Scholar]
- 11. Zhao P, Huang Y, Zuo Z. Opioid preconditioning induces opioid receptor‐dependent delayed neuroprotection against ischemia in rats. J Neuropathol Exp Neurol 2006;65:945–952. [DOI] [PubMed] [Google Scholar]
- 12. Wullschleger S, Loewith R, Hall MN. TOR signaling in growth and metabolism. Cell 2006;124:471–484. [DOI] [PubMed] [Google Scholar]
- 13. Jacinto E, Hall MN. Tor signalling in bugs, brain and brawn. Nat Rev Mol Cell Biol 2003;4:117–126. [DOI] [PubMed] [Google Scholar]
- 14. Costa‐Mattioli M, Monteggia LM. mTOR complexes in neurodevelopmental and neuropsychiatric disorders. Nat Neurosci 2013;16:1537–1543. [DOI] [PubMed] [Google Scholar]
- 15. Smith ED, Prieto GA, Tong L, et al. Rapamycin and interleukin‐1beta impair brain‐derived neurotrophic factor ‐ dependent neuron survival by modulating autophagy. J Biol Chem 2014;289:20615–20629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Park KK, Liu K, Hu Y, et al. Promoting axon regeneration in the adult CNS by modulation of the PTEN/mTOR pathway. Science 2008;322:963–966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Urbanska M, Gozdz A, Swiech LJ, Jaworski J. Mammalian target of rapamycin complex 1 (mTORC1) and 2 (mTORC2) control the dendritic arbor morphology of hippocampal neurons. J Biol Chem 2012;287:30240–30256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Stoica L, Zhu PJ, Huang W, Zhou H, Kozma SC, Costa‐Mattioli M. Selective pharmacogenetic inhibition of mammalian target of Rapamycin complex I (mTORC1) blocks long‐term synaptic plasticity and memory storage. Proc Natl Acad Sci USA 2011;108:3791–3796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Palop JJ, Mucke L. Amyloid‐beta‐induced neuronal dysfunction in Alzheimer's disease: from synapses toward neural networks. Nat Neurosci 2010;13:812–818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Weiner MW. Dementia in 2012: further insights into Alzheimer disease pathogenesis. Nat Rev Neurol 2013;9:65–66. [DOI] [PubMed] [Google Scholar]
- 21. Lafay‐Chebassier C, Perault‐Pochat MC, Page G, et al. The immunosuppressant rapamycin exacerbates neurotoxicity of Abeta peptide. J Neurosci Res 2006;84:1323–1334. [DOI] [PubMed] [Google Scholar]
- 22. Lafay‐Chebassier C, Paccalin M, Page G, et al. mTOR/p70S6k signalling alteration by Abeta exposure as well as in APP‐PS1 transgenic models and in patients with Alzheimer's disease. J Neurochem 2005;94:215–225. [DOI] [PubMed] [Google Scholar]
- 23. Marwarha G, Prasanthi JR, Schommer J, Dasari B, Ghribi O. Molecular interplay between leptin, insulin‐like growth factor‐1, and beta‐amyloid in organotypic slices from rabbit hippocampus. Mol Neurodegener 2011;6:41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Paccalin M, Pain‐Barc S, Pluchon C, et al. Activated mTOR and PKR kinases in lymphocytes correlate with memory and cognitive decline in Alzheimer's disease. Dement Geriatr Cogn Disord 2006;22:320–326. [DOI] [PubMed] [Google Scholar]
- 25. Damjanac M, Rioux BA, Paccalin M, et al. Dissociation of Akt/PKB and ribosomal S6 kinase signaling markers in a transgenic mouse model of Alzheimer's disease. Neurobiol Dis 2008;29:354–367. [DOI] [PubMed] [Google Scholar]
- 26. Avrahami L, Farfara D, Shaham‐Kol M, Vassar R, Frenkel D, Eldar‐Finkelman H. Inhibition of glycogen synthase kinase‐3 ameliorates beta‐amyloid pathology and restores lysosomal acidification and mammalian target of rapamycin activity in the Alzheimer disease mouse model: in vivo and in vitro studies. J Biol Chem 2013;288:1295–1306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Shang YC, Chong ZZ, Wang S, Maiese K. Prevention of beta‐amyloid degeneration of microglia by erythropoietin depends on Wnt1, the PI 3‐K/mTOR pathway, Bad, and Bcl‐xL. Aging (Albany NY) 2012;4:187–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Polakiewicz RD, Schieferl SM, Gingras AC, Sonenberg N, Comb MJ. mu‐Opioid receptor activates signaling pathways implicated in cell survival and translational control. J Biol Chem 1998;273:23534–23541. [DOI] [PubMed] [Google Scholar]
- 29. Favata MF, Horiuchi KY, Manos EJ, et al. Identification of a novel inhibitor of mitogen‐activated protein kinase kinase. J Biol Chem 1998;273:18623–18632. [DOI] [PubMed] [Google Scholar]
- 30. Durkin JP, Tremblay R, Chakravarthy B, et al. Evidence that the early loss of membrane protein kinase C is a necessary step in the excitatory amino acid‐induced death of primary cortical neurons. J Neurochem 1997;68:1400–1412. [DOI] [PubMed] [Google Scholar]
- 31. Lange‐Asschenfeldt C, Raval AP, Dave KR, Mochly‐Rosen D, Sick TJ, Perez‐Pinzon MA. Epsilon protein kinase C mediated ischemic tolerance requires activation of the extracellular regulated kinase pathway in the organotypic hippocampal slice. J Cereb Blood Flow Metab 2004;24:636–645. [DOI] [PubMed] [Google Scholar]
- 32. Vianna MR, Barros DM, Silva T, et al. Pharmacological demonstration of the differential involvement of protein kinase C isoforms in short‐ and long‐term memory formation and retrieval of one‐trial avoidance in rats. Psychopharmacology 2000;150:77–84. [DOI] [PubMed] [Google Scholar]
- 33. Mandel SA, Avramovich‐Tirosh Y, Reznichenko L, et al. Multifunctional activities of green tea catechins in neuroprotection. Modulation of cell survival genes, iron‐dependent oxidative stress and PKC signaling pathway. Neurosignals 2005;14:46–60. [DOI] [PubMed] [Google Scholar]
- 34. Iglesias M, Segura MF, Comella JX, Olmos G. Mu‐opioid receptor activation prevents apoptosis following serum withdrawal in differentiated SH‐SY5Y cells and cortical neurons via phosphatidylinositol 3‐kinase. Neuropharmacology 2003;44:482–492. [DOI] [PubMed] [Google Scholar]
- 35. Zheng H, Loh HH, Law PY. Beta‐arrestin‐dependent mu‐opioid receptor‐activated extracellular signal‐regulated kinases (ERKs) Translocate to Nucleus in Contrast to G protein‐dependent ERK activation. Mol Pharmacol 2008;73:178–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Wang Y, Xia Z, Xu JR, et al. Alpha‐mangostin, a polyphenolic xanthone derivative from mangosteen, attenuates beta‐amyloid oligomers‐induced neurotoxicity by inhibiting amyloid aggregation. Neuropharmacology 2012;62:871–881. [DOI] [PubMed] [Google Scholar]
- 37. Majno G, Joris I. Apoptosis, oncosis, and necrosis. An overview of cell death. Am J Pathol 1995;146:3–15. [PMC free article] [PubMed] [Google Scholar]
- 38. Earnshaw WC. Nuclear changes in apoptosis. Curr Opin Cell Biol 1995;7:337–343. [DOI] [PubMed] [Google Scholar]
- 39. Liepins A. Morphological, physiological and biochemical parameters associated with cell injury: a review. Immunopharmacol Immunotoxicol 1989;11:539–558. [DOI] [PubMed] [Google Scholar]
- 40. Haass C, Selkoe DJ. Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer's amyloid beta‐peptide. Nat Rev Mol Cell Biol 2007;8:101–112. [DOI] [PubMed] [Google Scholar]
- 41. Tang SJ, Reis G, Kang H, Gingras AC, Sonenberg N, Schuman EM. A rapamycin‐sensitive signaling pathway contributes to long‐term synaptic plasticity in the hippocampus. Proc Natl Acad Sci USA 2002;99:467–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Jaworski J, Sheng M. The growing role of mTOR in neuronal development and plasticity. Mol Neurobiol 2006;34:205–219. [DOI] [PubMed] [Google Scholar]
- 43. Tischmeyer W, Schicknick H, Kraus M, et al. Rapamycin‐sensitive signalling in long‐term consolidation of auditory cortex‐dependent memory. Eur J Neurosci 2003;18:942–950. [DOI] [PubMed] [Google Scholar]
- 44. Parsons RG, Gafford GM, Helmstetter FJ. Translational control via the mammalian target of rapamycin pathway is critical for the formation and stability of long‐term fear memory in amygdala neurons. J Neurosci 2006;26:12977–12983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. An WL, Cowburn RF, Li L, et al. Up‐regulation of phosphorylated/activated p70 S6 kinase and its relationship to neurofibrillary pathology in Alzheimer's disease. Am J Pathol 2003;163:591–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Caccamo A, Majumder S, Richardson A, Strong R, Oddo S. Molecular interplay between mammalian target of rapamycin (mTOR), amyloid‐beta, and Tau: effects on cognitive impairments. J Biol Chem 2010;285:13107–13120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Tang Z, Bereczki E, Zhang H, et al. Mammalian target of rapamycin (mTor) mediates tau protein dyshomeostasis: implication for Alzheimer disease. J Biol Chem 2013;288:15556–15570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Li X, Alafuzoff I, Soininen H, Winblad B, Pei JJ. Levels of mTOR and its downstream targets 4E‐BP1, eEF2, and eEF2 kinase in relationships with tau in Alzheimer's disease brain. FEBS J 2005;272:4211–4220. [DOI] [PubMed] [Google Scholar]
- 49. Bhaskar K, Miller M, Chludzinski A, Herrup K, Zagorski M, Lamb BT. The PI3K‐Akt‐mTOR pathway regulates Abeta oligomer induced neuronal cell cycle events. Mol Neurodegener 2009;4:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Del Valle J, Duran‐Vilaregut J, Manich G, et al. Early amyloid accumulation in the hippocampus of SAMP8 mice. J Alzheimers Dis 2010;19:1303–1315. [DOI] [PubMed] [Google Scholar]
- 51. Proud CG. Role of mTOR signalling in the control of translation initiation and elongation by nutrients. Curr Top Microbiol Immunol 2004;279:215–244. [DOI] [PubMed] [Google Scholar]
- 52. Meilandt WJ, Yu GQ, Chin J, et al. Enkephalin elevations contribute to neuronal and behavioral impairments in a transgenic mouse model of Alzheimer's disease. J Neurosci 2008;28:5007–5017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Eisch AJ, Harburg GC. Opiates, psychostimulants, and adult hippocampal neurogenesis: insights for addiction and stem cell biology. Hippocampus 2006;16:271–286. [DOI] [PubMed] [Google Scholar]