

Abstract
Objectives: The Wnt signalling pathway has been shown to play an important role in tooth development, however its effects with stem cells from the apical papilla (SCAP) have remained unclear. The purpose of this study was to determine effects of Wnt/β‐catenin on proliferation and differentiation of SCAP in vitro.
Materials and methods: SCAP were obtained, identified and cultured. Cell proliferation, alkaline phosphatase (ALP) activity, mRNA expression of mineralization‐related genes and mineralized nodule formation were measured in presence or absence of various concentrations of lithium chloride.
Results: MTT assay and flow cytometry demonstrated that Wnt/β‐catenin activity could promote proliferation of SCAP. Real‐time PCR analysis found that Wnt/β‐catenin strongly upregulated expression of dentine sialophosphoprotein, osteocalcin and ALP in SCAP after incubation with mineralization induction medium, while ALP and alizarin red staining indicated that Wnt/β‐catenin enhanced ALP activity and formation of mineralized nodules.
Conclusion: Our results suggest that canonical Wnt/β‐catenin signalling promotes proliferation and odonto/osteogenic differentiation of SCAP.
Introduction
External signals and extracellular factors constitute the external micro‐environment of cells and are responsible for maintaining balance between cell proliferation and differentiation, according to requirements of the host tissue (1). Wnt signalling is involved in distinct stages of cell‐fate decision, maturation and differentiation. Wnt pathways have been divided into four main categories (2, 3), with the best characterized being Wnt/β‐catenin signalling, also known as the canonical pathway. In cells’ resting state, very little β‐catenin is present in either cytoplasm or nuclei as it is rapidly degraded by the proteasome. This degradation is promoted by the cytoplasmic complex containing adenomatous polyposis coli (APC) protein, glycogen synthase kinase‐3β (GSK‐3β), axin and casein kinase I. As part of this complex, GSK‐3β constitutively phosphorylates β‐catenin, resulting in its ubiquitination and degradation by the 26S proteasome. Binding of Wnt ligands to the Frizzled (FZD) receptor and low‐density lipoprotein‐related receptor protein (LRP) 5/6 causes inactivation of the Axin/APC/GSK‐3β/CKI complex and stabilization of cytoplasmic β‐catenin; β‐catenin then activates the canonical Wnt/β‐catenin pathway (4). Canonical Wnt/β‐catenin signalling dynamically results in accumulation of cytoplasmic β‐catenin that can translocate to the nucleus, where it forms a complex with members of T‐cell factor (Tcf)/lymphoid enhancer factor (LEF) family of transcription factors. This complex then activates expression of Wnt target genes, which regulate cell proliferation, cell‐fate determination, apoptosis and axis polarity induction (5).
Previous studies that promoted Wnt/β‐catenin signalling, either by overexpressing Wnt or by inducing deficiency in Wnt antagonists have been associated with increased bone formation in mice and humans (6, 7, 8). In contrast, several further studies showed that activation of the canonical Wnt pathway promoted proliferation, but inhibited osteogenic differentiation of human adipose‐derived mesenchymal stem cells (MSCs), BMSCs and dental stem cells, such as PDLSCs and DPSCs (9, 10, 11, 12). Interestingly, one study has revealed that canonical Wnt signalling stimulated hMSC proliferation at low doses, but inhibited it at high doses (13). The dual effects of Wnt signalling suggest that intensity of Wnt signalling can result in differing or even opposing biological functions. However, these controversial findings concerning complex regulation of canonical Wnt signalling may also result from differences in culture conditions and cell stage.
Recent advances in oral‐tissue research have revealed essential roles for the Wnt/β‐catenin signalling pathway, which is dynamically active in tooth‐forming regions at every stage of tooth development, and plays multiple roles in these events (14, 15, 16). Stem cells from the apical papilla (SCAP), which are positioned at the apex of immature teeth and at the site of root development processes, have previously been isolated and characterized as a further type of adult MSC. These cells exhibit strong propensity for dentine‐like tissue formation and proliferation activity when compared to DPSCs (17, 18). Thus, we have hypothesized that Wnt/β‐catenin signalling plays a role in root dentine formation. The present study was designed to culture SCAP in vitro, profile expression of Wnt components in SCAP and explore whether Wnt/β‐catenin signalling would induce proliferation and mineralization of SCAP.
Materials and methods
Isolation and characterization of stem cells from the apical papilla
Normal impacted third molars (n = 8) were collected from adults (18–20 years of age) at the Shanghai Ninth People’s Hospital according to approved guidelines set by the Shanghai Jiao Tong University School of Medicine. Root apical papillae were gently separated from the apex of the root, minced and digested in a solution of 3 mg/ml type I collagenase (Sigma‐Aldrich, St Louis, MO, USA) and 4 mg/ml dispase (Roche Diagnostic/Boehringer Mannheim Corp, Indianapolis, IN, USA) for 30 min at 37 °C. Single‐cell suspensions of SCAP were obtained by passing cells through a 70 μm strainer (BD Labware, Franklin Lakes, NJ, USA). SCAP were cultured in Dulbecco’s modified Eagle’s medium (DMEM; Gibco, Grand Island, NY, USA) supplemented with 20% foetal bovine serum (FBS; Gibco), 100 U/ml penicillin and 100 mg/ml streptomycin at 37 °C in a humidified atmosphere with 5% CO2. Single‐cell‐derived colony cultures were obtained using the limiting dilution technique (17), to obtain homogeneous populations of SCAP. Confluent cells were detached with 0.20% trypsin and 0.02% EDTA and seeded into new culture dishes with DMEM and 10% FBS. Cell cultures of third to fifth passages were used for all experiments.
Odonto/osteogenic, adipogenic and chondrogenic differentiation of the stem cells were performed as previously described (17, 18). Briefly, for the odonto/osteogenic differentiation study, 2 × 104 cells/well were seeded in six‐well dishes with culture medium for 1 day. Cells were then incubated in odonto/osteogenic‐inducing medium containing 10 mmβ‐glycerophosphate, 50 mg/ml ascorbic acid and 10 nm dexamethasone (Sigma‐Aldrich), for an additional period of 21 days. Adipogenic/chondrogenic differentiation media were purchased from Cyagen Bioscience (Sunnyvale, CA, USA) and differentiation was performed according to the manufacture’s manual; cells were induced for 14 days. At the end of the culture period, cells were fixed in 75% ethanol and stained with alizarin red, oil red O solution or 2% alcian blue (Sigma‐Aldrich).
For identification of the SCAP phenotype, approximately 5 × 105 cells were incubated with anti‐Stro‐1 (Santa Cruz Biotechnology, Santa Cruz, CA, USA), anti‐CD24 (Labvision, Lappeenranta Finland) and fluorescein isothiocyanate (FITC)‐conjugated monoclonal antibodies for human CD146 (Santa Cruz Biotechnology), or isotype‐matched control IgGs. For Stro‐1 and CD24 staining, cells were incubated in primary antibody, on ice, for 1 h. After washing in 5% heat‐inactivated FBS, cells were incubated in FITC‐conjugated goat anti‐mouse IgM (R&D Systems, Minneapolis, MN, USA) on ice, for an additional 30 min. Cells were subjected to flow cytometric analysis using a Beckman Coulter Epics XL flow cytometer (Beckman Coulter, Brea, CA, USA).
Cell proliferation and cell‐cycle assay
Cell viability was determined using the MTT [3‐(4, 5‐dimethylthiazol‐2‐yl)‐2, 5‐diphenyl‐2H‐tetrazolium bromide] assay. Plating was at 1000 cells per well in 96‐well plates and cells were allowed to recover for 24 h before exposing them to culture medium containing a variety of concentrations of lithium chloride (LiCl; Sigma‐Aldrich) for 7 days. On days 1, 3, 5 and 7, 20 μl MTT at 5 mg/ml concentration was added to each well and incubation continued for 4 h. Formazan crystals resulting from mitochondrial enzymatic activity on the MTT substrate were solubilized with 150 μl of dimethyl sulphoxide, and absorbance was measured at 490 nm using a SpectraMAX microplate reader (Molecular Devices, Sunnyvale, CA, USA). Each combination of cells and LiCl concentration was set up in four parallel wells, and the experiment was repeated three times; results are presented as mean ± SD.
Cell‐cycle analysis was performed using flow cytometry. SCAP were plated in six‐well plates at 2 × 105 density, overnight. After expansion in serum‐free medium and starvation for 24 h, culture medium was replaced with 10% FBS DMEM containing the various concentrations of LiCl, and plates were incubated for 7 days. On days 1, 3 and 5, cells were harvested and fixed in 70% ethanol overnight. After washing three times in phosphate‐buffered saline (PBS), cells were stained in 300 μl of PI/RNase staining buffer (BD Biosciences, Franklin Lakes, NJ, USA) at room temperature for 15 min. Cells (104 cells/analysis) were examined by flow cytometry (Becton Dickinson and Company, Franklin Lakes, NJ, USA).
Mineralization assay
Cells (1 × 104 cells/cm2) were cultured in DMEM containing 10% FBS, 10 mmβ‐glycerophosphate, 50 mg/ml ascorbic acid and 10 nm dexamethasone (Sigma‐Aldrich) and incubated with or without LiCl, Wnt1 or DKK1 for up to 21 days; during this period, medium was changed every 3 days. Alkaline phosphatase (ALP) staining was performed according to product manufacturer’s instructions (Beytine, Hanmen, China) on day 14, and level of ALP activity was determined on days 4, 7 and 14 after incubation. Briefly, cells were rinsed in PBS and suspended in lysis buffer with 0.2% NP‐40. ALP activity was determined by measuring absorbance at 405 nm using p‐nitrophenyl phosphate (pNPP; Sigma‐Aldrich) as substrate. Each sample was mixed with pNPP (1 mg/ml) in 1 m diethanolamine buffer and incubated at 37 °C for 15 min. The reaction was stopped by addition of 3 N NaOH to the reaction mixture. Enzyme activity was quantified by measuring absorbance at 405 nm (Bio‐Tek, Winooski, VT, USA). Total protein content was determined using the Bradford method on aliquots of the same samples using Bio‐Rad protein assay kit (Bio‐Rad, Hercules, CA, USA). Absorbance was read at 630 nm and protein concentration was calculated based on a series of BSA (Sigma‐Aldrich) standards. ALP activity was expressed as absorbance at 405 nm (OD value) per milligram of total cellular protein. All experiments were performed in triplicate.
Alizarin red staining was performed on day 21. Cells were fixed in ice‐cold 95% ethanol for 30 min at room temperature. Then they were stained in alizarin red (40 mm, pH 4.2) to detect calcification, at 37 °C for 30 min. Unbound alizarin red was removed using water (five washes). PBS was added for an additional 10 min to further reduce non‐specific staining.
Quantitative real‐time reverse‐transcription polymerase chain reaction
Total RNA was extracted from SCAP using RNAiso plus (Takara, Tokyo, Japan) according to the manufacturer’s protocols. RNA quantity and purity were analysed using a spectrophotometer (ND‐1000, NanoDrop; NanoDrop Technologies, Wilmington, DE, USA). For each sample, 500 ng of total RNA was reverse transcribed using PrimeScript RT reagent kit (Perfect Real Time) (Takara) and RT reaction was carried out in a water bath at 37 °C for 15 min and 85 °C for 15 s. Real‐time PCR assays were performed using SYBR Premix Ex TaqTM II (Perfect Real Time) (Takara) in the MyiQ Real‐time PCR detection system (Bio‐Rad) according to the manufacturer’s protocol. Preliminary experiments were carried out to determine annealing temperature that yielded greatest amounts of specific products; oligonucleotide primer sequences are shown in Table 1. PCR solution (10 μl SYBR green real‐time PCR Premix, 0.4 μl forward primer, 0.4 μl reverse primer, 1 μl cDNA and 8.2 μl ddH2O) was amplified under MyiQ conditions for PCR, which consisted of incubation at 95 °C for 3 min, followed by 40 cycles of 95 °C for 10 s, 60 °C for 30 s, then 95 °C for 1 min, 58 °C for 1 min and 81 cycles of extension at 55 °C for 10 s. Negative control without cDNA template was run with each assay to assess overall specificity. β‐actin was used as control for amount of input RNA to normalize genes tested from the same cDNA sample. A standard melting curve was used to check quality of amplification and to ensure specificity. A selection of amplified samples were visualized on 2.0% agarose gels stained with ethidium bromide and photographed under UV light.
Table 1.
Primer sequence used for polymerase chain reaction amplifications
| Gene | Primer sequences forward/reverse | Products (bp) |
|---|---|---|
| OCN | CCCAGGCGCTACCTGTATCAA | 212 |
| GGTCAGCCAACTCGTCACAGTC | ||
| DSPP | AGTGACAGCCAGAGCAAG | 156 |
| CCTATCCCATTACCA AACT | ||
| ALP | CCGTGGAACATTCTGGATCTGAC | 173 |
| CCCTGGTGGTCTTGGAGTGAGT | ||
| β‐catenin | GCCAAGTGGGTGGTATAGAGG | 173 |
| GTGGGATGGTGGGTGTAAGAG | ||
| GSK‐3β | AGACGCTCCCTGTGATTTATGT | 196 |
| TACGAAACATTGGGTTCTCCTC | ||
| Tcf1 | GATGAGCTACCAACCAAGAAGG | 141 |
| CCTATTGCACTCCTCCACTAGC | ||
| Tcf3 | AGTTCGGAGGTTCAGGTCTTG | 152 |
| CAGGAATGTGGATGAAGAGAGG | ||
| Tcf4 | TTTCAGCCAACAGACATTCACT | 196 |
| GAGACACTCTGCCCCTGTAGTC | ||
| LRP5 | GACTCCTTCCCCGACTGTATC | 127 |
| AGAGAGAGAGGATGATGCCAAT | ||
| LRP6 | AGCCTGTGGGACTTACTGTGTT | 186 |
| CACAAGGGTGCTGTCTGTATTC | ||
| β‐actin | CTTAGTTGCGTTACACCCTTTCTTG | 146 |
| ACTGCTGTCACCTTCACCGTTC |
Western blot analysis
Cells were cultured in 60‐mm dishes supplemented with DMEM (10% FBS). When they were 80% confluent, medium was replaced with serum‐free DMEM for 12 h, and cells were then grown in DMEM (5% FBS) containing various concentrations of LiCl for 24 or 48 h. Cytoplasmic and nuclear proteins were extracted with the appropriate extraction kit, according to manufacturer’s protocol (Sangon Biotech Co. Ltd., Shanghai, China). Protein concentrations of the extracted lysates were determined by measuring their absorbance at 590 nm using BCA assay (Beyotine, Hanmen, China). An equal amount of protein from each sample (50 μg) was electrophoresed on 10% SDS–PAGE gel then transferred to PVDF membrane. After blocking at room temperature with 5% (w/v) skimmed milk in TBST [Tris–HCl 50 mm, NaCl 150 mm, 0.05% (w/v) Tween20, pH 7.4] for at least 1 h, membranes were incubated in primary antibody, anti‐β‐catenin mAb (1:1000; Sigma‐Aldrich, St Louis, MO, USA), anti‐β‐catenin mAb (1:1000; Abmart, Shanghai, China), anti‐Ref‐1 (1:1000; Earth Ox, San Francisco, CA,), overnight at 4 °C. After removal of primary antibody, membranes were washed in TBST four times for 10 min each. Membranes were then incubated in secondary antibody: anti‐mouse fluorescence labelled Ab (1:1000; Sigma‐Aldrich) for 1 h, followed by fluorescent band detection using Odyssey Infrared Imaging System (LI‐COR Biotechnology, Lincoln, NE, USA).
Statistical analysis
Results are reported as mean ± SD data. Statistical analyses were carried out using Student’s t‐test with spss (Version 13, IBM, NY, USA) and P‐value <0.05 was considered statistically significant. All experiments were performed at least in triplicate.
Results
Isolated SCAP had stem‐cell properties
SCAP were harvested from apical papillae of immature impacted third molars (Fig. 1a); the cells were of typical fibroblastic spindle shape (Fig. 1b). To obtain monoclonal cultures, cells were seeded at low density (2000 cells per plate) in 10‐cm dishes then CFU‐F were generated from single cells as shown (Fig. 1c). Dental tissue‐derived MSCs are characterized by their multipotentiality and ability to differentiate into several cell‐restricted lineages when grown under defined culture conditions. To investigate multilineage differentiation potential of SCAP, cells were cultured in odonto/osteogenic‐inducing medium for 21 days, or in adipogenic‐inducing or in chondrogenic‐inducing media for 14 days. Thus, alizarin red staining labelled mineralized nodules formed (Fig. 1d), and oil red O staining showed that cells had formed lipid droplets (Fig. 1e). In addition, alcian blue staining of cultured pellets indicated that SCAP underwent chondrogenic differentiation (Fig. 1f). Furthermore, flow cytometry indicated that SCAP expressed stem‐cell markers Stro‐1 (25.32%), CD146 (95.35%) and CD24 (21.97%) (Fig. 1g–i).
Figure 1.
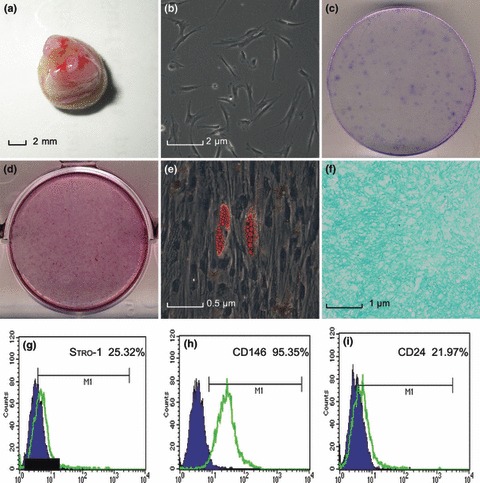
Isolation and characterization of stem cells from the apical papilla (SCAP). (a) Root apical papilla from an extracted human third molar of developmental stage; (b) Primary outgrowth culture of SCAP (100×); (c) Representative images of colonies formed by SCAP at low seeding density (2000 cells/plate); (d) Alizarin red S staining of mineralized nodules after SCAP culture in odontogenic/osteogenic‐inducing medium for 3 weeks; (e) SCAP formed oil red O‐positive lipid clusters after 2 weeks culture in adipogenic‐inducing medium (400×); (f) Alcian blue staining of pellet of SCAP after chondrogenic differentiation for 2 weeks (200×); Flow cytometric analysis of SCAP, (g) Stro‐1: 25.32%; (h) CD146: 95.35%; (i) CD24: 21.97%.
Wnt/β‐catenin signalling components were expressed by SCAP
To discover effects of Wnt/β‐catenin on SCAP, initially activation of Wnt/β‐catenin was investigated after cells had been treated with LiCl for 24 h. Levels of β‐catenin increased in both cytosolic and nuclear fractions in a dose‐dependent manner (Fig. 2a). RT‐PCR detected expression of Wnt signalling components (Fig. 2b) and genes encoding cell membrane receptors (LRP5/6), cytosolic signalling molecules (GSK‐3β and β‐catenin), and nuclear transcription factors (Tcf1, Tcf3 and Tcf4) were identified, suggesting that Wnt/β‐catenin signalling could be activated by LiCl, and that the cells possibly responded to Wnt signalling.
Figure 2.
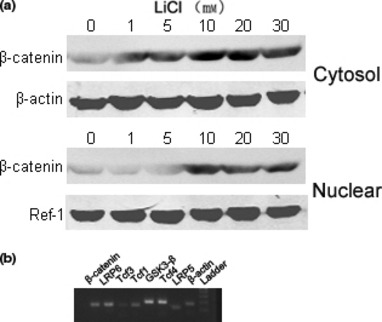
Canonical Wnt signalling components were functionally expressed by SCAP. (a) Cells were incubated with and without LiCl for 24 h. LiCl‐mediated nuclear translocation of β‐catenin in SCAP was detected by immunoblotting. A representative result from three independent experiments is shown. β‐actin and Ref‐1 were used as cytosolic and nuclear control proteins, respectively; (b) RT‐PCR was used to detect expression of Wnt signalling components. Total cellular RNA was extracted from confluent SCAP, and transcripts were analysed by RT‐PCR. Data representative of three independent experiments.
Effects of mimicking Wnt/β‐catenin signalling on SCAP proliferation
Cells were cultured over a 7‐day period in presence or absence of LiCl. Figure 3A shows that cell proliferation significantly increased in groups treated with 5 or 10 mm LiCl compared to negative controls (P < 0.05), but there was only slight (not significant), increase in the group treated with 1 mm LiCl (P > 0.05). Cell proliferation was significantly suppressed when the dose was increased to 20 or 30 mm (P < 0.05). To explore the influence of Wnt/β‐catenin on the cell cycle, freshly isolated SCAP were cultured in basic medium containing various concentrations of LiCl, and cell cycle was analysed by flow cytometry. As shown in Table 2, proliferation index (PI) increased after LiCl treatment, in a dose‐dependent manner. Numbers of cells in G2/M phase of the cell cycle increased, accompanied by reduction in numbers of cells in G1, in a time‐dependent manner, with greatest effect seen at 30 mm (Fig. 3B). After treatment with LiCl for 24 h, cells seemed to be enlarged, with more spread‐out and flattened appearance – indicating G2/M cell‐cycle arrest (Fig. 3C).
Figure 3.
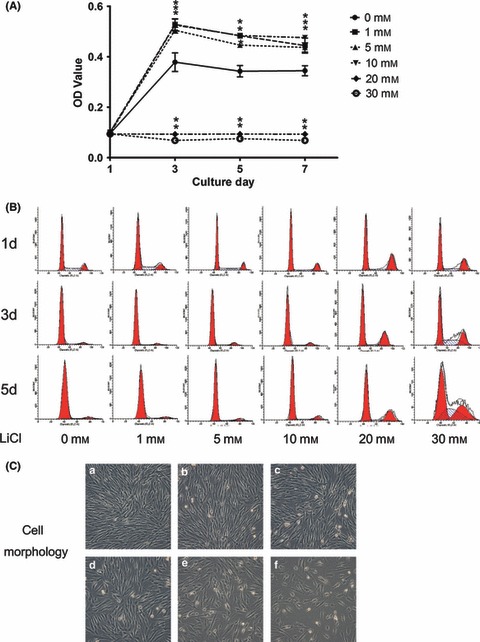
Effects of mimicking Wnt/β‐catenin effects on SCAP proliferation. (A) MTT assays showed effects of different concentrations of LiCl on SCAP proliferation, over 7 days. Values shown are mean ± SD of triplicate samples from a representative experiment. Student’s t‐tests were performed to compare treatments. *P < 0.05; (B) Cell‐cycle profiles of SCAP treated with various concentrations of LiCl for indicated times were analysed by flow cytometry; (C) Cell morphology of SCAP treated with various concentrations of LiCl for 24 h (a, Control; b, 1 mm; c, 5 mm; d,10 mm; e, 20 mm; f, 30 mm) (reverse phase contrast microscope, 100×).
Table 2.
Flow cytometric analysis of proliferation index (PI = S+G2/M, %) of SCAP treated with various concentrations of LiCl
| LiCl (mm) | ||||||
|---|---|---|---|---|---|---|
| 0 | 1 | 5 | 10 | 20 | 30 | |
| 1 day | 36.36 ± 0.97 | 35.29 ± 1.37 | 37.34 ± 1.10 | 38.51 ± 0.58 | 41.38 ± 1.15 | 43.56 ± 0.68 |
| 3 days | 6.72 ± 0.87 | 7.51 ± 0.16 | 12.40 ± 1.13 | 13.09 ± 0.10 | 37.03 ± 0.80 | 54.37 ± 1.00 |
| 5 days | 7.38 ± 0.54 | 9.26 ± 0.47 | 10.84 ± 0.68 | 14.85 ± 0.99 | 35.60 ± 1.05 | 53.67 ± 1.55 |
Values are expressed as Mean ± SD.
Wnt/β‐catenin had a stimulating effect on SCAP differentiation
To evaluate whether Wnt/β‐catenin had any positive effect on SCAP, cells were incubated in odonto/osteogenic medium with LiCl (1, 5, or 10 mm) or NaCl (10 mm, control group). ALP activity, an early marker of both odontoblastic and osteoblastic differentiation, was detected after 4 days culture in differentiation medium. On days 7 and 14, ALP activity was higher in groups treated with 5 or 10 mm LiCl or NaCl, specially LiCl‐treated cells (P < 0.05) (Fig. 4a); Figure 4b shows dose‐dependent increase in ALP staining. After prolonged induction of differentiation, cells were stained in alizarin red to determine level of calcium mineral deposition; dose‐dependent increase in calcium nodule formation and matrix mineralization was seen after LiCl treatment in a manner similar to that demonstrated above with ALP (Fig. 4c). Mineralized matrix produced in SCAP‐induced cultures was positive for markers of both osteogenic and odontogenic differentiation, including ALP, osteocalcin (OCN) and dentine sialophosphoprotein (DSPP). mRNA expression of mineralization‐related markers was measured using real‐time PCR, on day 14. There was no significant difference in ALP, OCN or DSPP expression between groups treated with 1 mm LiCl and the control group (P > 0.05); however, 5 and 10 mm LiCl promoted mRNA expression of their genes in a dose‐dependent manner (P < 0.05) (Fig. 4d). In all experiments, NaCl was used as negative control for LiCl, and had no effect on β‐catenin levels, ALP activity, or mineralization, irrespective of differentiation conditions.
Figure 4.
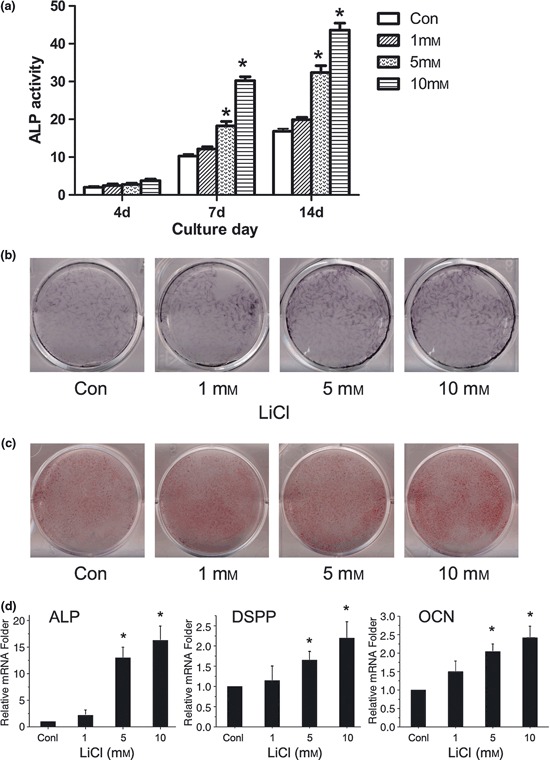
Wnt/β‐catenin had a stimulating effect on SCAP differentiation. (a) Cultures were treated with various concentrations of LiCl for 14 days. ALP activity was determined on days 4, 7 and 14; (b) Cells were cultured in odontogenic/osteogenic‐inducing medium in presence or absence of LiCl for 14 days. Resulting mineralization was assessed by ALP staining; (c) After incubation for up to 3 weeks, calcium mineral deposition was detected by alizarin red. Each image is representative of three separate experiments; (d) Real‐time PCR was used to measure effects of Wnt/β‐catenin on mRNA expression of genes of ALP, DSPP and OCN after SCAP were treated with LiCl or NaCl for 14 days. Gene expression was normalized to β‐actin and is presented as percentage of relative expression levels of untreated cells at the same time points. ALP, alkaline phosphatase; DSPP, dentine sialophosphoprotein; OCN, osteocalcin. Values expressed as mean ± SD. *P < 0.05 versus control values.
DKK1 had an inhibitory effect on Wnt1‐induced proliferation and differentiation of SCAP
Previously, Wnt‐1 has been shown to signal through β‐catenin. Thus, the cells were incubated with Wnt‐1 (500 ng/ml) to determine whether β‐catenin was sufficient to facilitate their proliferation and differentiation. As seen with LiCl‐treated cells, those treated with Wnt‐1 had increased proliferation and differentiation compared to induced control cells. To confirm these effects, cells were co‐treated with Wnt‐1 (500 ng/ml) and DKK1 (100 ng/ml), an inhibitor of canonical Wnt signalling, that binds to LRP5/6 co‐receptors. As shown in Fig. 5a, MTT assays demonstrated that DKK1 significantly decreased cell viability and replication compared to negative controls, whereas Wnt‐1 promoted proliferation (P < 0.05). Figure 5b shows ALP staining on day 14, and Fig. 5c shows alizarin red staining on day 21, when obvious reduction in matrix mineralization and calcium nodule formation could be seen after DKK1 treatment. Each image is representative of three independent experiments. Real‐time PCR showed that DKK1 markedly suppressed mRNA expression of ALP, OCN and DSPP compared to Wnt1‐treatment, on day 14 (P < 0.05) (Fig. 5d).
Figure 5.
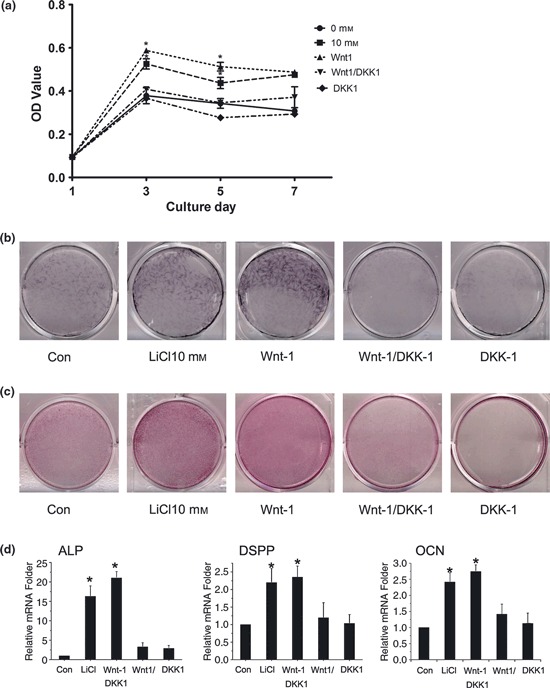
DKK1 had inhibitory effect on Wnt1‐induced proliferation and differentiation of SCAP. (a) SCAP were cultured with LiCl (10 mm), Wnt1 (500 ng/ml) alone or with 100 ng/ml DKK1, and NaCl (10 mm); MTT assays were performed to examine the inhibitory effect of DKK1 on Wnt1‐induced proliferation of SCAP after 7 days; (b) Cultures were induced with different components for 2 weeks and resulting mineralization was assessed by ALP staining; (c) Following incubation for up to 3 weeks, calcium mineral deposition was detected using alizarin red staining. Each image is representative of three separate experiments; (d) After 14‐days incubation, mRNA levels of ALP, DSPP and OCN were measured by real‐time PCR. Results are mean ± SD of three independent experiments. *P < 0.05 versus control values.
Discussion
After the bell stage of tooth development, location of the dental papilla surrounded by dentine becomes the apex of pulp tissue, termed the apical papilla (18, 19). Many studies have made clear distinction between ‘primary odontoblasts’ from the apical papilla, which make primary and secondary dentine, and ‘replacement odontoblasts’ derived from pulp, which replace primary odontoblasts and make tertiary dentine, or more specifically, reparative dentine (19). In 2006, MSCs isolated from apical papillae of immature human permanent teeth were described as SCAP. These cells have potential for osteogenesis, chondrogenesis, adipogenesis and self‐renewal, thus representing a potential source of dental regeneration (17, 18). In the study described here, cells isolated from tooth apical papillae expressed stem‐cell markers STRO‐1 and CD146 and specific marker CD24, and possessed the capacity for both self‐renewal and differentiation into odonto/osteoblast‐like cells, adipocytes and chondrocyte lineages; these results were consistent with the findings of Sonoyama (17).
Previously, immunohistochemistry has been used to show that β‐catenin expression in human tooth germ development plays a pivotal role in transcriptional activation of genes likely to be involved in mesenchyme–epithelial interactions upon which human tooth development is based; high percentage of dental papilla had intense nuclear expression of β‐catenin (15). We found that elements of canonical Wnt/β‐catenin signalling were present in cell cytoplasm and nuclei. LiCl, a drug used for decades to treat bipolar disorder (20, 21), elevated β‐catenin levels in both cytoplasm and the nucleus (Fig. 2a), demonstrating that Wnt/β‐catenin‐dependent signals could be activated, as seen previously (21).
Cell proliferation is a key property during development, and consists of DNA synthesis and cytodieresis. Canonical Wnt signalling has been reported to stimulate cell proliferation by activation of cell‐cycle genes (such as c‐Myc and cyclin D1) in hMSCs (9, 11), however, effective doses of LiCl used varied from 1 to 30 mm (21, 22, 23). According to our preliminary results, SCAP were exposed to LiCl concentrations of 1, 5, 10, 20 and 30 mm in this study. Compared to controls, cell viability increased in a time‐ and dose‐dependent manner after treatment with 5 and 10 mm LiCl (P < 0.05); however, viability was inhibited by treatment with 20, 30 mm LiCl (P < 0.05). Cell‐cycle analysis and cell morphology changes indicated that high‐dose lithium induced G2/M cell‐cycle arrest. Similarly, Wang et al. found that high‐dose lithium arrested hepatocellular carcinoma cell line SMMC‐7721 cells at the G2/M checkpoint by inducing cdc2 phosphorylation (24). This effect was not associated with inhibition of GSK‐3β and inositol monophosphatase, two well‐documented targets of lithium, but increase of Chk1 activity was seen (24). Therefore, LiCl should be used at concentrations no higher than 10 mm. Wnt‐1 was also capable of boosting Wnt signalling to sufficiently high levels to enhance proliferation in SCAP, while DKK‐1 abrogated enhancement of proliferation by Wnt‐1, consistent with other reports (25, 26).
Odontoblast‐like differentiation of dental stem cells shares a similar process to osteogenesis in bone marrow MSCs: ALP expression increases, followed by deposition of collagenous matrix (27, 28). Thus, differentiation and mineralization of SCAP in this study were considered with reference to the osteogenic mode. ALP has pronounced expression 1 week after induction of differentiation and can be considered to be an early marker of osteogenic differentiation and hard tissue formation. OCN is regarded as a marker of late stages of osteoblast differentiation, and its production denotes onset of matrix deposition, seen only in odontoblasts of normal pulp and non‐mineralized predentine during formation of intertubular dentine (29). DSPP has been considered to be a representative marker of dental tissues, and its expression has been detected in bone in the ratio of 1/400 (30). It has been found that DSPP plays a crucial role during dentinogenesis and in the carious process (1). In our study, expression of ALP, OCN and DSPP increased from day 4 up to day 14 following induction of mineralization differentiation (4, 5). These data suggest that mineralized matrix product had characteristics of dentine matrix (probably osteodentin, without typical tubular organization).
Recent experiments in vitro have confirmed that β‐catenin suppresses chondrogenic potential of osteo‐chondroprogenitors, a bipotential precursor of osteoblast lineage, but promotes its differentiation into osteoblasts. It has been proposed that β‐catenin concentrations elevates capability of differentiation into osteoblasts (31). Likewise, Wnt signalling had a positive effect on osteoblasts when Wnt was overexpressed or there was a deficiency of Wnt antagonists, in human MSCs (6, 7). In our study, ALP activity and calcified nodule formation were augmented by mimicking Wnt/β‐catenin signalling in vitro. Moreover, assessing mRNA expression of ALP, DSPP and OCN further supported the hypothesis that Wnt/β‐catenin signalling promoted odonto/osteogenic differentiation. Upregulation of their genes indicated that SCAP underwent differentiation, deposition of collagenous matrix and formation of predentine. DKK1, a well‐defined Wnt antagonist, was included in our analysis to confirm effects of Wnt/β‐catenin (7, 32). Our results showed that DKK1 negatively regulated canonical Wnt signalling and blocked odonto/osteogenic differentiation, which completely prevented Wnt1‐mediated odontogenic events from occurring. These data suggest a positive role for Wnt/β‐catenin in differentiation and mineralization of SCAP.
Wnt‐1 and Wnt‐3a however, have been found to strongly inhibit osteogenic differentiation of MSCs (9, 10, 11). Scheller, who transduced DPSCs with canonical Wnt‐1 and the active form of β‐catenin, found that overexpression of β‐catenin suppressed odontoblast‐like differentiation and mineralization of DPSCs (12). These discrepancies may be related to cell type, culture conditions, differentiation state of the cells, or make‐up of Wnts and Wnt antagonists in the pericellular microenvironment. Previous studies that explored the function of Wnt signalling at different stages of osteoblast development showed that it stimulated early osteoblasts to differentiate, whereas mature osteoblasts were strongly inhibited in their capacity to induce mineralization (16, 33). Consequently, as biological characteristics of SCAP indicate that this cell population has early stem‐cell/progenitor‐cell properties, it is possible that Wnt/β‐catenin signalling may help enhance dentinogenic potential of SCAP, thereby inhibiting odontoblast‐like differentiation of DPSCs.
Our data support the idea that Wnt/β‐catenin significantly promotes SCAP proliferation and odonto/osteogenic differentiation. Unfortunately, the multitude of processes involved and complexity of their regulation posed a challenge to our analysis. Understanding how downstream biological events are individually regulated would allow higher degree of target specificity to be achieved. To determine differentiation ability of SCAP, we used the relatively simple and widely used differentiation method for hard tissue formation. This method provides results that correlate with in vivo dentine‐forming ability, but it is not absolutely specific for odontoblast differentiation. In addition, there are disadvantages with using an in vitro study to mimic canonical Wnt/β‐catenin signalling. During dental development in vivo, the tissue microenvironment (niche) also plays an important role in maintenance of stemness, and regulates interactions with surrounding molecules, and new isolation and detection techniques have been used to optimize stem cells (34). Moreover, recent investigations into optimal odontogenic factors, their spatial and temporal activities, and integration with scaffolds have led to enhanced odonto/osteogenesis as well as angiogenesis in human DPSCs (34, 35, 36); this will enable obtainable dentine‐pulp complex tissues. Further studies in vivo and use of scaffolds may allow us to promote proliferation or mineralization of SCAP, which will help us understand comprehensive networks of interacting signalling pathways controlling this process, and thus improve dental tissue engineering.
References
- 1. Chaussain C, Eapen AS, Huet E, Floris C, Ravindran S, Hao J et al. (2009) MMP2‐cleavage of DMP1 generates a bioactive peptide promoting differentiation of dental pulp stem/progenitor cell. Eur. Cell Mater. 18, 84–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Gordon MD, Nusse R (2006) Wnt signaling: multiple pathways, multiple receptors, and multiple transcription factors. J. Biol. Chem. 281, 22429–22433. [DOI] [PubMed] [Google Scholar]
- 3. Logan CY, Nusse R (2004) The Wnt signaling pathway in development and disease. Annu. Rev. Cell Dev. Biol. 20, 781–810. [DOI] [PubMed] [Google Scholar]
- 4. Aberle H, Bauer A, Stappert J, Kispert A, Kemler R (1997) Beta‐catenin is a target for the ubiquitin‐proteasome pathway. EMBO J. 16, 3797–3804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Reya T, Clevers H (2005) Wnt signalling in stem cells and cancer. Nature 434, 843–850. [DOI] [PubMed] [Google Scholar]
- 6. Bennett CN, Ouyang H, Ma YL, Zeng Q, Gerin I, Sousa KM et al. (2007) Wnt10b increases postnatal bone formation by enhancing osteoblast differentiation. J. Bone Miner. Res. 22, 1924–1932. [DOI] [PubMed] [Google Scholar]
- 7. Morvan F, Boulukos K, Clement‐Lacroix P, Roman S, Suc‐Royer I, Vayssiere B et al. (2006) Deletion of a single allele of the Dkk1 gene leads to an increase in bone formation and bone mass. J. Bone Miner. Res. 21, 934–945. [DOI] [PubMed] [Google Scholar]
- 8. An JH, Yang JY, Ahn BY, Cho SW, Jung JY, Cho HY et al. (2010) Enhanced mitochondrial biogenesis contributes to Wnt induced osteoblastic differentiation of C3H10T1/2 cells. Bone 47, 140–150. [DOI] [PubMed] [Google Scholar]
- 9. Cho HH, Kim YJ, Kim SJ, Kim JH, Bae YC, Ba B et al. (2006) Endogenous Wnt signaling promotes proliferation and suppresses osteogenic differentiation in human adipose derived stromal cells. Tissue Eng. 12, 111–121. [DOI] [PubMed] [Google Scholar]
- 10. De Boer J, Siddappa R, Gaspar C, van Apeldoorn A, Fodde R, van Blitterswijk C (2004) Wnt signaling inhibits osteogenic differentiation of human mesenchymal stem cells. Bone 34, 818–826. [DOI] [PubMed] [Google Scholar]
- 11. Boland GM, Perkins G, Hall DJ, Tuan RS (2004) Wnt 3a promotes proliferation and suppresses osteogenic differentiation of adult human mesenchymal stem cells. J. Cell. Biochem. 93, 1210–1230. [DOI] [PubMed] [Google Scholar]
- 12. Scheller EL, Chang J, Wang CY (2008) Wnt/beta‐catenin inhibits dental pulp stem cell differentiation. J. Dent. Res. 87, 126–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. De Boer J, Wang HJ, Van Blitterswijk C (2004) Effects of Wnt signaling on proliferation and differentiation of human mesenchymal stem cells. Tissue Eng. 10, 393–401. [DOI] [PubMed] [Google Scholar]
- 14. Liu F, Millar SE (2010) Wnt/beta‐catenin signaling in oral tissue development and disease. J. Dent. Res. 89, 318–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lo Muzio L, Lo Russo L, Pannone G, Santoro A, Leonardi R, Serpico R et al. (2009) Expression of beta‐catenin in human tooth germ. Anal. Quant. Cytol. Histol. 31, 324–331. [PubMed] [Google Scholar]
- 16. Galli C, Passeri G, Macaluso GM (2010) Osteocytes and WNT: the mechanical control of bone formation. J. Dent. Res. 89, 331–343. [DOI] [PubMed] [Google Scholar]
- 17. Sonoyama W, Liu Y, Fang D, Yamaza T, Seo B‐M, Zhang C et al. (2006) Mesenchymal stem cell‐mediated functional tooth regeneration in swine. PLoS ONE 1, e79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Huang GT, Sonoyama W, Liu Y, Liu H, Wang S, Shi S (2008) The hidden treasure in apical papilla: the potential role in pulp/dentin regeneration and bioroot engineering. J. Endod. 34, 645–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. D’Souza R (2002) Development of the pulpodentin complex In: Hargreaves KM, Goodis HE, eds. Seltzer and Bender’s Dental Pulp, pp. 13–40. Chicago: Quintessence Pub. Co. Inc. [Google Scholar]
- 20. Hedgepeth CM, Conrad LJ, Zhang J, Huang HC, Lee VM, Klein PS (1997) Activation of the Wnt signaling pathway: a molecular mechanism for lithium action. Dev. Biol. 185, 82–91. [DOI] [PubMed] [Google Scholar]
- 21. Hiyama A, Sakai D, Arai F, Nakajima D, Yokoyama K, Mochida J (2011) Effects of a glycogen synthase kinase 3beta inhibitor (LiCl) on c‐myc protein in intervertebral disc cells. J. Cell. Biochem. 112, 2974–2986. [DOI] [PubMed] [Google Scholar]
- 22. Tang GH, Xu J, Chen RJ, Qian YF, Shen G (2011) Lithium delivery enhances bone growth during midpalatal expansion. J. Dent. Res. 90, 336–340. [DOI] [PubMed] [Google Scholar]
- 23. Nemoto E, Koshikawa Y, Kanaya S, Tsuchiya M, Tamura M, Somerman MJ et al. (2009) Wnt signaling inhibits cementoblast differentiation and promotes proliferation. Bone 44, 805–812. [DOI] [PubMed] [Google Scholar]
- 24. Wang X‐M, Li J, Feng X‐C, Wang Q, Guan D‐Y, Shen Z‐H (2008) Involvement of the role of Chk1 in lithium‐induced G2/M phase cell cycle arrest in hepatocellular carcinoma cells. J. Cell. Biochem. 104, 1181–1191. [DOI] [PubMed] [Google Scholar]
- 25. Holmen SL, Zylstra CR, Mukherjee A, Sigler RE, Faugere M‐C, Bouxsein ML et al. (2005) Essential role of β‐catenin in postnatal bone acquisition. J. Biol. Chem. 280, 21162–21168. [DOI] [PubMed] [Google Scholar]
- 26. Heo JS, Lee SY, Lee JC (2010) Wnt/beta‐catenin signaling enhances osteoblastogenic differentiation from human periodontal ligament fibroblasts. Mol. Cells 30, 449–454. [DOI] [PubMed] [Google Scholar]
- 27. Jaiswal N, Haynesworth SE, Caplan AI, Bruder SP (1997) Osteogenic differentiation of purified, culture‐expanded human mesenchymal stem cells in vitro. J. Cell. Biochem. 64, 295–312. [PubMed] [Google Scholar]
- 28. Wang J, Ma H, Jin X, Hu J, Liu X, Ni L et al. (2011) The effect of scaffold architecture on odontogenic differentiation of human dental pulp stem cells. Biomaterials 32, 7822–7830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Reichert T, Storkel S, Becker K, Fisher LW (1992) The role of osteonectin in human tooth development: an immunohistological study. Calcif. Tissue Int. 50, 468–472. [DOI] [PubMed] [Google Scholar]
- 30. Qin C, Brunn JC, Cadena E, Ridall A, Tsujigiwa H, Nagatsuka H et al. (2002) The expression of dentin sialophosphoprotein gene in bone. J. Dent. Res. 81, 392–394. [DOI] [PubMed] [Google Scholar]
- 31. Day TF, Guo X, Garrett‐Beal L, Yang Y (2005) Wnt/beta‐catenin signaling in mesenchymal progenitors controls osteoblast and chondrocyte differentiation during vertebrate skeletogenesis. Dev. Cell 8, 739–750. [DOI] [PubMed] [Google Scholar]
- 32. Qiang YW, Barlogie B, Rudikoff S, Shaughnessy JD Jr (2008) Dkk1‐induced inhibition of Wnt signaling in osteoblast differentiation is an underlying mechanism of bone loss in multiple myeloma. Bone 42, 669–680. [DOI] [PubMed] [Google Scholar]
- 33. Eijken M, Meijer IM, Westbroek I, Koedam M, Chiba H, Uitterlinden AG et al. (2008) Wnt signaling acts and is regulated in a human osteoblast differentiation dependent manner. J. Cell. Biochem. 104, 568–579. [DOI] [PubMed] [Google Scholar]
- 34. Tirino V, Paino F, d’Aquino R, Desiderio V, De Rosa A, Papaccio G (2011) Methods for the identification, characterization and banking of human DPSCs: current strategies and perspectives. Stem Cell Rev. 7, 608–615. [DOI] [PubMed] [Google Scholar]
- 35. d’Aquino R, Graziano A, Sampaolesi M, Laino G, Pirozzi G, De Rosa A et al. (2007) Human postnatal dental pulp cells co‐differentiate into osteoblasts and endotheliocytes: a pivotal synergy leading to adult bone tissue formation. Cell Death Differ. 14, 1162–1171. [DOI] [PubMed] [Google Scholar]
- 36. d’Aquino R, De Rosa A, Lanza V, Tirino V, Laino L, Graziano A et al. (2009) Human mandible bone defect repair by the grafting of dental pulp stem/progenitor cells and collagen sponge biocomplexes. Eur. Cell Mater. 18, 75–83. [DOI] [PubMed] [Google Scholar]